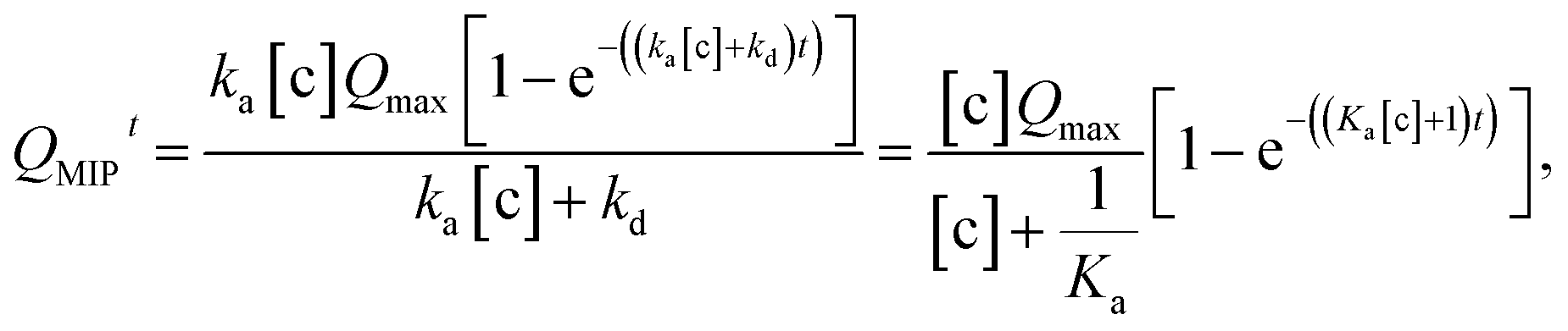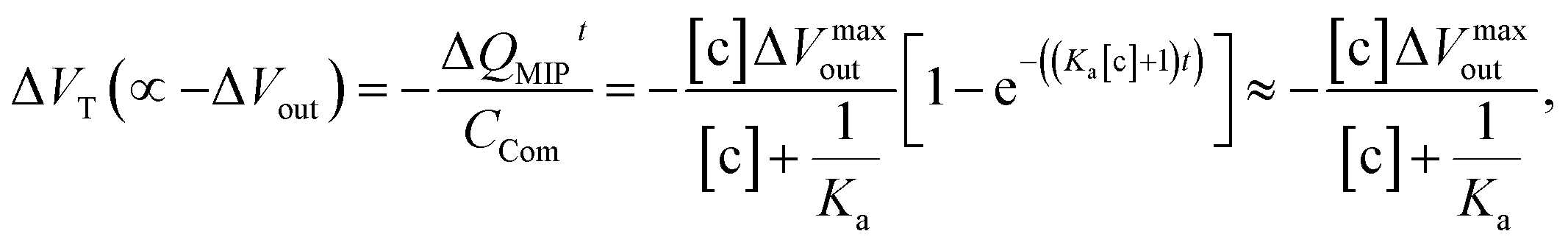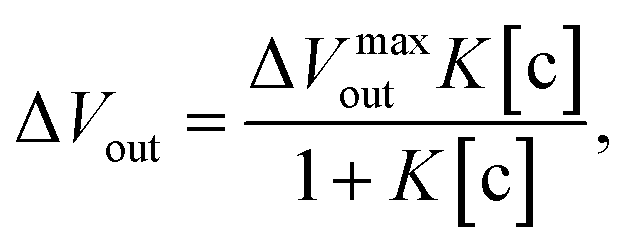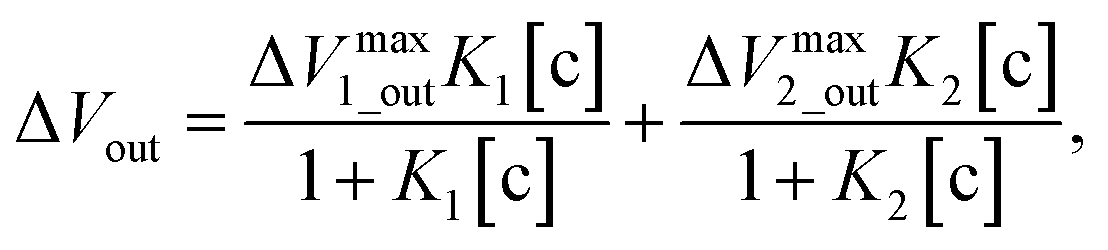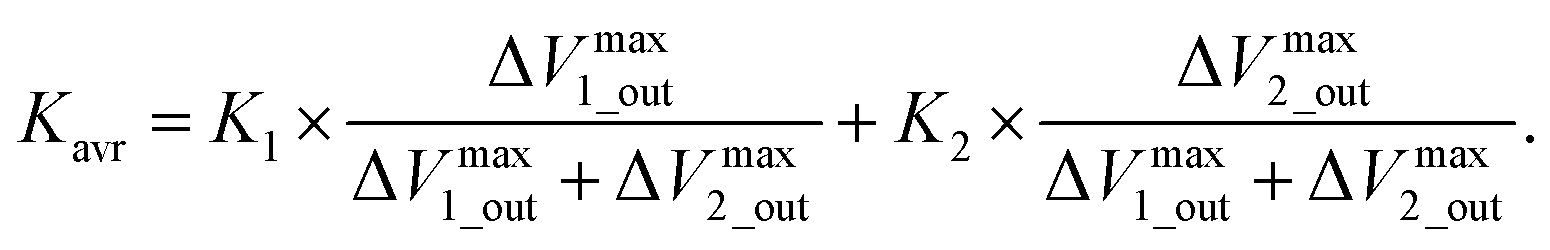DOI:
10.1039/D0RA02793F
(Review Article)
RSC Adv., 2020,
10, 16999-17013
Molecularly imprinted polymer-based bioelectrical interfaces with intrinsic molecular charges
Received
26th March 2020
, Accepted 23rd April 2020
First published on 30th April 2020
Abstract
For enzyme-/antibody-free and label-free biosensing, a molecularly imprinted polymer (MIP)-based membrane with phenylboronic acid (PBA) molecules, which induces the change in the density of molecular charges based on the small biomolecule–PBA diol binding, has been demonstrated to be suitable for the bioelectrical interface of biologically coupled gate field-effect transistor (bio-FET) sensors. MIP-coated gate FET sensors selectively detect various small biomolecules such as glucose, dopamine, sialic acid, and oligosaccharides without using labeled materials. In particular, the well-controlled MIP film by surface-initiated atom transfer radical polymerization (SI-ATRP) contributes to the quantitative analysis of small biomolecule sensing, resulting in potentiometric Langmuir isotherm adsorption analysis by which the parameters such as the binding affinity between small biomolecules and MIP cavities are evaluated. Also, the output electrical signal of even a random MIP-coated gate FET sensor is quantitatively analyzed using the bi-Langmuir adsorption isotherm equation, showing the adsorption mechanism of small biomolecules onto the template-specific MIP membrane. Thus, a platform based on the MIP bioelectrical interface for the bio-FET sensor is suitable for an enzyme-/antibody-free and label-free biosensing system in the fields of clinical diagnostics, drug discovery, the food industry, and environmental research.
1. Introduction
The first concept of an enzyme electrode was proposed by Updike et al. in 1967.1 Glucose oxidase (GOx) was immobilized in a gel, which has often been utilized for glucose sensors such as for self-monitoring blood glucose (SMBG) and continuous glucose monitoring (CGM) systems for diabetic patients.2,3 The enzymatic reaction between GOx and glucose generates hydroxy peroxide (H2O2) resulting in a redox reaction with a platinum electrode, which is electrochemically detected by amperometric measurement. Thereafter, various enzymes such as lactate dehydrogenase and penicillinase were utilized for the electrochemical detection of biomolecules.4–6 These enzymes seem to be biomimetic, which originated from biofunctions. However, the use of enzymes is problematic owing to their lack of stability, high-cost and time-consuming production, and the difficulty of quality control of their production.
Lateral flow immunochromatographic assays, which are carried out using a cellulose-based device, are suitable as part of a simple system to detect target analytes.7,8 Biological sample solutions easily flow through a paper sheet, and then biomolecular recognition events such as an antigen–antibody reaction are colorimetrically detected on the sheet without specialized equipment, where antibody-conjugated tags such as gold nanoparticles are included and reacted with the antigen at the test line. However, this measurement method is not quantitative and cannot continuously monitor vital signs. In particular, homogeneously controlled monoclonal antibodies are not available for various antigens owing to high-cost and time-consuming production.
A bioelectrical interface indicates a sample solution/electrode interface, where not only enzyme membranes but also polymeric functional films and probe biomolecules such as antibodies and single-stranded oligonucleotides are immobilized on the electrode. A molecularly imprinted polymer (MIP) film is artificially modified to selectively capture target biomolecules on electrodes, including the main monomer, a cross linker, and a functional monomer.9–13 A target biomolecule specifically binds to the functional groups around a template cavity in the MIP membrane, which should contribute to a change in molecular charge as an electrical signal. MIP-coated gate field-effect transistor (FET) sensors directly and selectively detect small biomolecules such as glucose, dopamine, lactic acid, histamine, and oligosaccharides in an enzyme-/antibody-free and label-free manner.14–18 In particular, the MIP films on the gate electrode were functionalized by phenylboronic acid (PBA) molecules, which caused the boronic acid–diol binding to glucose accompanied by the ionization of boronic acids.19–21 Moreover, the MIP-based bioelectrical interface was adopted for electrochemical impedance spectroscopy.22,23 Thus, the MIP films can be effectively adopted as an artificial bioelectrical interface for enzyme-/antibody-free and label-free biosensing.
Recently, biosensing devices with an MIP interface have been proposed for the selective detection of biomolecules; however, the novel functionalities of the biointerface of such devices should also be evaluated. A surface plasmon resonance (SPR) sensor or a quartz crystal microbalance (QCM) sensor has an attractive detection principle for biomolecular recognition such as antigen–antibody reactions, but generally has limitations for use in small-molecule analysis because the signals obtained depend on the molecular weight of the targets to be determined.24–27 On the other hand, fluorescent or chemiluminescent dyes are used as labels to detect biomolecules by fluorescence microscopy and enzyme-linked immunosorbent assay (ELISA), but are generally difficult to use as labels for small molecules.28,29 Therefore, new analytical methods are required to directly and quantitatively evaluate biointerfacial characteristics, such as the use of MIPs for small-biomolecule recognition. However, MIP compositions often have to be optimized in a bulk state before MIPs are applied to sensors. Such processes are not only time-consuming, but may also lead to differences in adhesion properties between the bulk and the sensor; therefore, a direct analytical methodology for MIPs is highly desirable.
Biological phenomena are interestingly related to the behaviors of ions and biomolecules. This is why biosensing devices that enable the detection of ionic and biomolecular charges contribute to the direct analysis of biological phenomena in a label- and enzyme-free manner. Potentiometric biosensors such as biologically coupled gate FET (bio-FET) sensors, which allow the direct detection of these charges based on the field effect, have this capability.30–32 Bio-FET sensors conceptually have three components: biological targets, bioelectrical interfaces, and semiconducting materials.33 In particular, bioelectrical interfaces contribute to the selective and specific detection of targets based on intrinsic molecular charges. Thus, the change in the density of molecular charges should be induced by a target–MIP interaction on the gate electrode.14–18,34–37
In this review paper, we provide an outline of the MIP-based bioelectrical interface for the selective and specific detection of small biomolecules using the bio-FET sensors. Such a bioelectrical interface is applied to different types of bio-FET sensors composed of various semiconducting materials as the channel (e.g., nanotubes, nanowires, 2D sheets, and organic and inorganic materials).
2. Enzyme-/antibody-free bioelectrical interface for small biomolecule sensing
2.1. Concept of MIP-coated gate FETs
An MIP is a biomimetic polymer designed for selective molecular recognition.9–13 The selective recognition is realized by a rigid, highly crosslinked polymer matrix holding target-specific binding sites generated by a strong target–functional monomer interaction in the prepolymer complex. Owing to its simple procedure and versatility, MIPs have been incorporated into various biosensors to enhance target selectivity on such sensors. In particular, they have been applied to bio-FET sensors for the selective detection of small biomolecules. Here, small biomolecules with diol groups such as glucose, dopamine, sialic acid, and oligosaccharides were focused as the targets of detection using MIP-coated FET biosensors.14,15,17,18
To design a diol-compound-selective MIP interface on the gate surface, PBA was utilized as the target-interacting functional monomer.14–18 PBA has attracted considerable attention in the field of molecular recognition as it can form stable esters with various biomolecules containing 1,2 or 1,3 cis-diol/polyol groups, such as oligosaccharides, in aqueous systems (Scheme 1A).14 Moreover, the esterification of PBA/diol is a reversible reaction that can be controlled by adjusting the pH of the solution. Thus, the template can be easily extracted from the polymer in the preparation of MIP by simply adjusting the pH of the solution (Scheme 1B). Moreover, in the esterification, PBA switches from a non-ionic form to an anionic form (Scheme 1A). Hence, the change in the density of molecular charges induced by the diol compound/PBA binding can be detected by bio-FET sensors. In addition, methacrylic acid (MAA) is the most commonly used functional monomer in the preparation of MIPs because of its versatility in the interaction. MAA was utilized to interact with amino groups in target small biomolecules such as catecholamines and oligosaccharides, to further improve the selectivity of the biosensor. Also, N-[3-(dimethylamino)propyl]methacrylamide (DMAPM) was utilized in MIP films with PBA to control the pH owing to its basicity, and 2-hydroxyethylmethacrylate (HEMA) was occasionally used as the main chain monomer to improve the hydrophilicity of MIP films, where small biomolecules and electrolytes were included. The polymer was basically crosslinked by N,N-methylenebisacrylamide (MBA) or ethylene glycol dimethacrylate (EGDMA). On the other hand, a nonimprinted polymer (NIP) was prepared on the gate electrode as a control polymer, which was prepared by the same method as that for MIP except for adding a small biomolecule as the template.
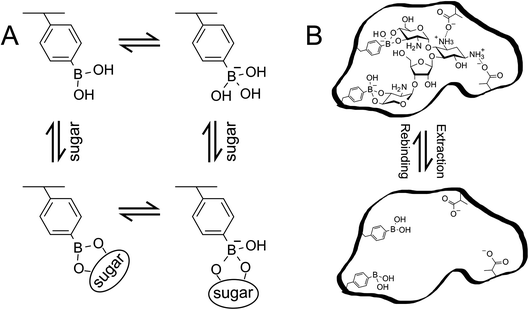 |
| | Scheme 1 (A) Phenylboronic acid equilibrium with sugar in aqueous environment. (B) Schematic illustration of MIP. The illustration is drawn with paromomycin template as an example. These schemes have been reproduced from ref. 15 with permission from American Chemical Society, copyright 2018. | |
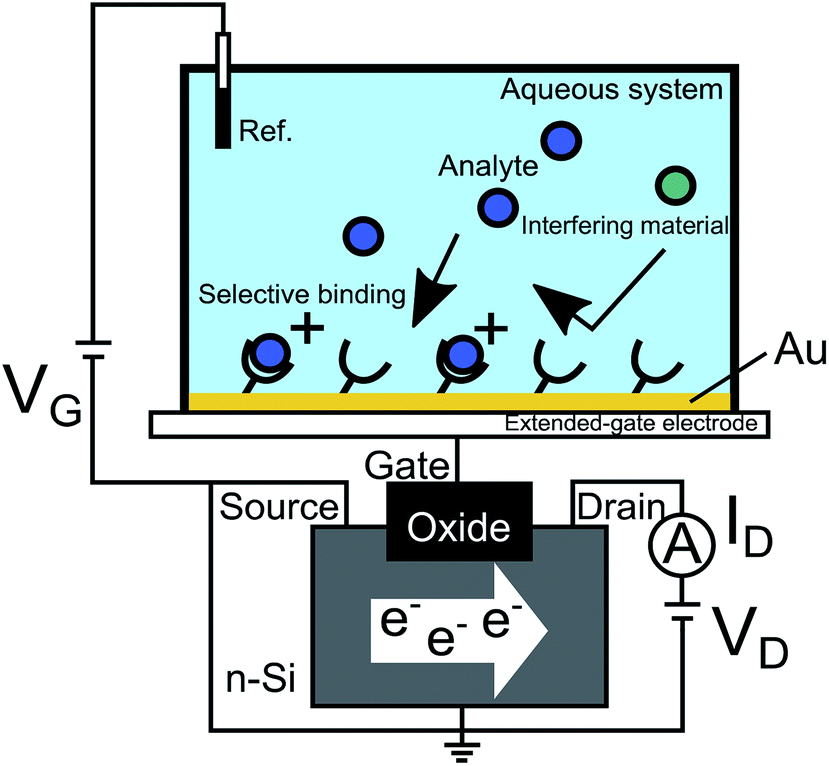
The MIP films were randomly copolymerized as poly(HEMA-ran-DMAPM-ran-VPBA-ran-AA) for glucose sensing for example,18 and the thickness and adhesiveness of the MIP layer at the gate electrode surface were precisely controlled by surface-initiated atom transfer radical polymerization (SI-ATRP), which directly forms a hydrogel nanolayer on the substrate, to quantify the affinity and binding constant of the target–MIP interaction.14,17 As shown in Fig. 1, the gate electrode such as Au-sputtered films was connected to the extended gate of a silicon-based n-channel FET, and gate voltage (VG) was applied through the Ag/AgCl reference electrode. Functional probe membranes such as MIP, which selectively bind to analytes but not impurities, are tethered on the gate electrode of the FET device. Molecular charges of analytes at the solution/gate electrode interface induce a change in VG at a constant source–drain current (ID) and drain voltage (VD), that is, a change in threshold voltage (ΔVT). Moreover, a change in electrolyte/gate electrode interfacial potential (ΔVout) was output with a source follower circuit.38 As a result, ΔVout corresponded to −ΔVT.
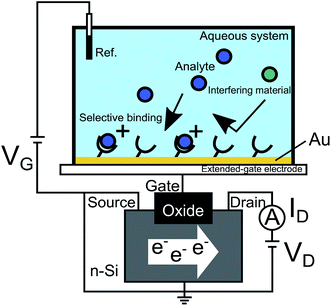 |
| | Fig. 1 Conceptual design of FET biosensor. The Au gate electrode is connected to the extended gate of a silicon-based n-channel FET, and VG is applied through the Ag/AgCl reference electrode. Probe molecules such as MIP, which selectively bind to analytes but not to impurities, are tethered on the gate electrode of the FET device. Molecular charges of analytes at the solution/gate electrode interface induce a change in VG at a constant ID and VD. This figure has been reproduced from ref. 15 with permission from American Chemical Society, copyright 2018. | |
2.2. Electrical response of randomly copolymerized MIP bioelectrical interface
A monomer solution containing HEMA as the main chain monomer and vinyl-PBA (VPBA) as the sugar recognition monomer was directly placed on the Au surface and copolymerized in an inert gas atmosphere. The conceptual structure of the glucose-template–MIP–FET (glu–MIP–FET) is shown in Fig. 2A; the hydrogel was randomly copolymerized as poly(HEMA-ran-DMAPM-ran-VPBA-ran-AA) (random MIP).18 Before copolymerization, VPBA was expected to have negative charges upon binding to glucose. Glucose molecules were washed out with an acid solution after copolymerization, resulting in cavities formed in the MIP. The glucose responsivity of the glu–MIP–FET was analyzed from the VG–ID electrical characteristics. Fig. 2B shows ΔVT analyzed from the VG–ID electrical characteristics of glu–MIP–FETs at various glucose concentrations. ΔVT shifted in the positive direction at ID of 1 mA with increasing glucose concentration from 10 μM to 10 mM. The positive shift of ΔVT means the increase of negative charges at the gate of FET devices; therefore, the increase in the density of negative charges caused by the formation of the glucose/PBA complex was detected using the glu–MIP–FET on the basis of the increase in glucose concentration. In a previous report, the VG–ID electrical characteristics were evaluated using the stimulus-responsive polymer-gel-modified glucose FET sensor (Fig. 2C).39 In this case, ΔVT shifted in the negative direction after the addition of 5 g L−1 glucose, which was opposite to our results. This is because a previous work demonstrated the change in the capacitance of the gel, which was induced by the deswelling–swelling of the polymer gel.39 That is, the change in capacitance as the output was largely determined using FET devices (Fig. 2D), although the change in the density of the molecular charges of the gel was included in the output signal. In the case of the MIP interface, the capacitance of the glu–MIP hydrogel hardly changed after adding glucose (Fig. 2E); therefore, the increase in the density of negative charges induced by the formation of the glucose/PBA complex was detected as larger signals on the basis of the principle of the field effect than those resulting from the swelling of the glu–MIP hydrogel after glucose addition. Considering the above, the MIP membrane is regarded as one of the bioelectrical interfaces that can induce molecular charges on the basis of the glucose–PBA interaction, whereas such molecular charges of boronic acids cause the change in capacitance induced by the structural change caused by swelling–deswelling in the stimulus-responsive polymer gel, depending on the polymer components. The stimulus-responsive polymer membrane is also available for the bioelectrical interface.
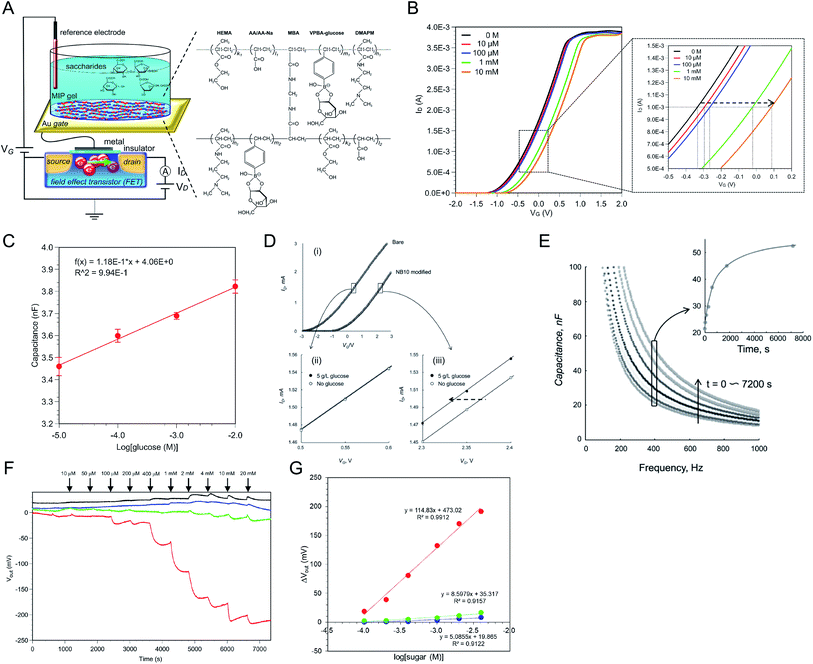 |
| | Fig. 2 (A) Schematic diagram of glu–MIP–FET and chemical structure of copolymerized glucose–imprinted MIP hydrogel. The Au electrode with the glu–MIP interface was extended from the gate of FET. (B) ΔVT upon adding glucose at 0 (black), 10 μM (red), 100 μM (blue), 1 mM (green), and 10 mM (orange) was analyzed from VG–ID electrical characteristics. (C) Change in capacitance of glu–MIP interface upon adding glucose in the range from 10 μM to 10 mM. (D) VG–ID electrical characteristics of FETs with and without phenylboronic acid/N-isopropylacrylamide-based glucose-responsive polymer gel (NB10) gate modification (i); separately shown in (ii) and (iii) are enlarged areas, as indicated. (E) Change in capacitance of NB10 gel layer during phase transition upon addition of 3 g L−1 glucose in a pH 9 N-cyclohexyl-2-aminoethanesulfonic acid (CHES) buffer solution at 20 °C. The inset shows the time-course change in capacitance measured at 400 Hz. (F) ΔVout of glu–MIP–FET upon adding sugars (red, glucose; blue, fructose; green, sucrose). Each sugar was added at the time indicated by arrows in the range from 10 μM to 20 mM. As a control sensor, NIP-coated FET was prepared (black). (G) ΔVout values were plotted against the changes in semilogarithmic sugar concentrations. The figures (A), (B), (C), (F), and (G) have been reproduced from ref. 18 with permission from American Chemical Society, copyright 2018. The figures (D) and (E) have been reproduced from ref. 39 with permission from Wiley, copyright 2009. | |
Moreover, glucose responsivity was monitored in real time as the change in interfacial potential (ΔVout) between the measurement solution and the MIP-coated gate electrode using the glu–MIP–FET. In Fig. 2F, ΔVout clearly decreased upon adding glucose from 10 μM to 20 mM, while other sensors showed no response upon adding each sugar (e.g., fructose, sucrose). In this case, the detection sensitivities of the glu–MIP–FET to the sugars in the range of 100 μM to 4 mM were 114.8 mV per decade for glucose, 8.6 mV per decade for fructose, and 5.1 mV per decade for sucrose, as shown in Fig. 2G. Thus, the detection sensitivity to glucose was about 15–20 times higher than those to other sugars owing to the well-designed artificial MIP interface that provided glucose specificity. On the other hand, ΔVout for the NIP–FET shifted in the positive direction upon adding glucose, although the signal shifts were very small. The positive shifts could be due to the increase in the capacitance of NIP, because the swelling of NIP was induced upon the addition of glucose solutions, resulting in the increase in the permittivity of NIP. This means that the response of the glu–MIP–FET to glucose may include the change in the capacitance of the polymer, although signal contamination is assumed to be small. Note that NIP did not have the glucose template supported by PBA but it included PBA. Thus, the change in the shape of the MIP membrane depending on its swelling–deswelling was not very large owing to the more dense crosslinking (MBA), which prevented more water molecules from invading into the MIP hydrogel. However, the glu–MIP–FET detected the increase in the density of negative charges through the glucose/PBA complexes with high sensitivity.
Such random MIPs were also prepared for the selective detection of oligosaccharides on the gate electrode.15 The thicknesses of the random MIP interfaces were about 200 nm for the glucose-template–MIP and 10 μm at most for the oligosaccharide-template–MIP. This means that random copolymers yield weak quantitative signals owing to the randomness of composition, thickness, and so forth. Therefore, the thickness and adhesiveness of bioelectrical interfaces such as MIPs at substrates should be precisely controlled by some grafting methods such as SI-ATRP. However, we believe that the resulting electrical signals in sensors based on random MIP–FETs are reliable, since they are prepared using the same procedure, resulting in a similar thickness and adhesiveness on the gate electrodes. In particular, the hydrogel sufficiently includes electrolytes and water molecules even before target–MIP interactions, so that the change in the capacitance of the MIP interface is negligible while the change in the density of molecular charges can be detected with the FETs.
2.3. Controlled polymerization of MIP bioelectrical interface on gate surface by SI-ATRP and electrical characteristics
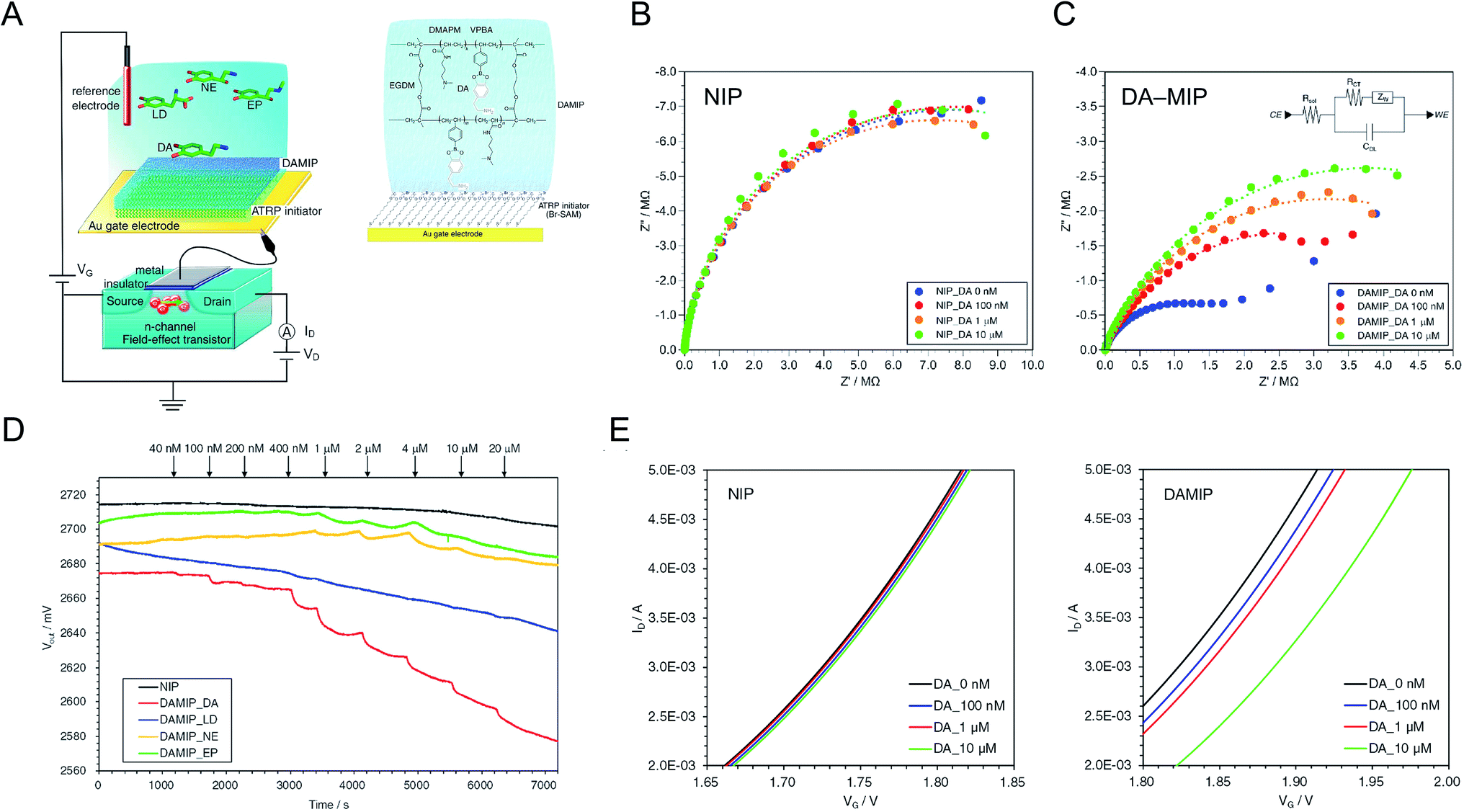
A nanolayer of dopamine (DA)-template–MIP was fabricated by SI-ATRP, which is a controlled living radical polymerization method.17 To fabricate the FET with the DA–MIP-coated gate electrode, a MIP hydrogel was synthesized on the surface of the Au electrode, following the modification of bromo-terminated alkanethiol SAMs as the ATRP initiator. Fig. 3A shows the FET with the DA–MIP-coated gate and the chemical structure of the DA–MIP obtained by SI-ATRP. To prepare a thin layer of DA–MIP, the DA template molecule was mixed into a solution containing a functional monomer (VPBA). Similarly to other catecholamines, DA rapidly self-polymerizes while forming the ring structure of 5,6-dihydroxyindole by oxidation.40,41 To prevent such self-oxidation of DA, the DA–MIP was polymerized in a nitrogen atmosphere with all air completely removed. Thus, the thickness and adhesiveness of the MIP interface were controlled by SI-ATRP, and the electrical and interfacial properties were quantitatively analyzed as follows.
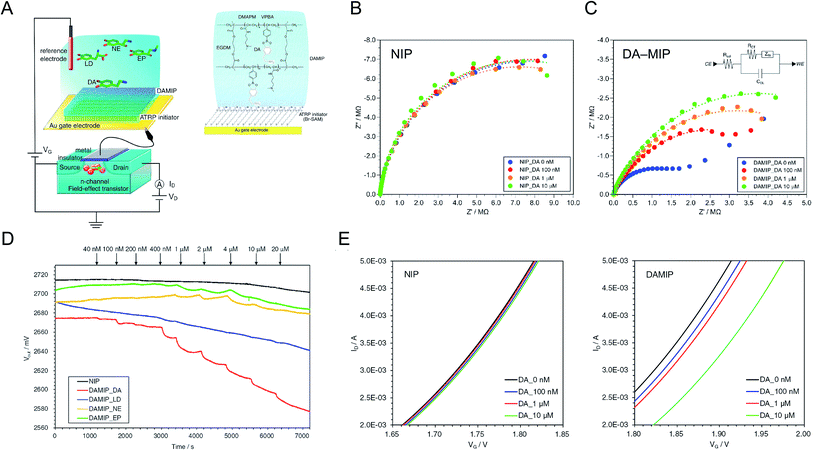 |
| | Fig. 3 (A) Conceptual structure of DA–MIP-coated gate FET. The DA–MIP-coated Au gate electrode was extended from MOSFET. Bromo-terminated alkanethiol SAM was tethered as the ATRP initiator on the Au electrode, and then DA–MIP was synthesized with the formation of VPBA–DA complexes. Nyquist plots of (B) NIP- and (C) DA–MIP-coated Au electrodes at different concentrations of DA from 0 nM to 10 μM. The inset in (C) shows the equivalent circuit of DA–MIP-coated Au surface (CE, counter electrode, Rsol, resistance of solution, RCT, resistance of charge transfer, ZW, Warburg impedance, CDL, capacitance of electric double layer, WE, working electrode). (D) Vout of DA–MIP-coated gate FET with different concentrations of catecholamines at a constant ID (100 μA). Four types of catecholamine, DA (red line), LD (blue line), NE (orange line), and EP (green line), were added at each time indicated by arrows. The NIP-coated gate FET was used as a control upon adding DA (black line). (E) VG–ID electrical characteristics of NIP-coated (left) and DA–MIP-coated (right) gate FET at different concentrations of DA added from 100 nM to 10 μM. An n-channel MOSFET device was used, and ΔVT at ID of 4 mA and VD of 2 V was measured using a semiconductor parameter analyzer. These figures have been reproduced from ref. 17 with permission from Elsevier, copyright 2018. | |
The surface of each Au electrode modified with the SI-ATRP initiator-SAM and DA–MIP was analyzed by ellipsometry and capacitance measurements. The experimental spectra obtained by ellipsometry fitted well to the spectra simulated using the Cauchy model.17 From these data, the thicknesses of the ATRP-initiator SAMs, NIP, and DA–MIP (d) were determined as 1.6 ± 0.0 nm, 57.3 ± 1.3 nm, and 61.3 ± 4.1 nm, respectively. Furthermore, the capacitance of each organic layer on the Au surface (C) decreased from 1.60 μF (electric double-layer on the bare Au surface) to 1.24 μF in the ATRP-initiator-SAM-modified Au electrode, 450 nF in the NIP-coated Au electrode, and 190 nF in the DA–MIP-coated Au electrode. The theoretical relative permittivities (ε) calculated from the thicknesses, surface area (S), and capacitances were approximately 3 in the Br–SAM layer, 58 in the NIP/Br–SAM layer, and 20 in the DA–MIP/Br–SAM layer on the Au electrode on the basis of 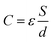 . The relative permittivity was lower in DA–MIP than in NIP despite the nearly identical thicknesses of the two layers. The different dielectric constants of DA–MIP and NIP might be explained by their morphological differences, whether with or without template cavities, in the hydrogels of a molecularly imprinted polymer and a nonimprinted random copolymer.
. The relative permittivity was lower in DA–MIP than in NIP despite the nearly identical thicknesses of the two layers. The different dielectric constants of DA–MIP and NIP might be explained by their morphological differences, whether with or without template cavities, in the hydrogels of a molecularly imprinted polymer and a nonimprinted random copolymer.
The DA–MIP-coated Au electrode was electrochemically characterized by electrochemical impedance spectroscopy (EIS). The impedance of each Au surface was measured at various DA concentrations. According to the modulus of impedance in the NIP- and DA–MIP-coated Au electrodes, which increases with increasing frequency in the low frequency range (below Hz order), the redox reaction of the ferricyanide/ferrocyanide mediator proceeded at the Au electrode. Fig. 3B and C show Nyquist plots of the NIP- and DA–MIP-coated Au electrodes, which resolve the impedance into real and imaginary parts, in the frequency range from 10–1 MHz. For the DA–MIP-coated Au electrode in the absence and presence of 100 nM DA, the diagonal line at low frequencies corresponds to the diffusion-limited Warburg impedance (Fig. 3C). Therefore, the equivalent circuit may be a series connection of the charge transfer resistance (RCT) and the Warburg impedance (ZW), in parallel with the electric double-layer capacitance of the Au electrode (Fig. 3C, inset). Increasing the DA concentration enlarged the capacitive semicircle for the DA–MIP-coated Au electrode, but not for the NIP-coated Au electrode. Table 1 summarizes the parameters of the resistance–capacitance (RC) circuits of the NIP- and DA–MIP-coated Au electrodes at different DA concentrations. As the DA concentration increased, RCT, which was estimated from the intersection of the semicircle plot with the X-axis, increased in the DA–MIP-coated Au electrode but not in the NIP-coated Au electrode, whereas the electric double-layer capacitance (CEDL), which was calculated from the frequency corresponding to the vertex of the semicircle [fmax = 1/(2πRCTCEDL)] and RCT, remained unchanged in both electrodes. This indicates the gradual inhibition of redox reactions by the binding of DA and PBA molecules in the MIP, which blocks the DA–MIP cavities and prevents the redox mediator from passing through the DA–MIP membrane. On the other hand, RCT was several times larger in the NIP-coated Au electrode than in the DA–MIP-coated Au electrode, and remained unchanged after DA addition. This behavior might occur because the redox mediator cannot easily reach the Au surface through the NIP membrane. EIS analysis clarified and electrochemically characterized the polymer thin film on the Au electrode of the NIP- and DA–MIP-coated Au electrodes, and the DA binding behavior in DA–MIP was observed as an impedance change.
Table 1 Impedimetric parameters in RC circuit of NIP- and DA–MIP-coated Au electrodes at different concentrations of DA. This table has been reproduced from ref. 17 with permission from Elsevier, copyright 2018
| Dopamine concentration (μM) |
Parameters |
| fmax (mHz) |
CEDL (μF) |
RCT (MΩ) |
| NIP |
0 |
14.7 |
0.70 |
15.5 |
| 0.1 |
14.7 |
0.70 |
15.5 |
| 1 |
14.7 |
0.75 |
14.5 |
| 10 |
14.7 |
0.73 |
14.9 |
| DA–MIP |
0 |
56.2 |
1.37 |
2.07 |
| 0.1 |
21.5 |
1.43 |
5.17 |
| 1 |
14.7 |
1.70 |
6.36 |
| 10 |
14.7 |
1.44 |
7.52 |
Fig. 3D plots the ΔVout values of the FET with the DA–MIP-coated gate at different concentrations of catecholamines added and the FET with the NIP-coated gate in the presence of DA. In the DA–MIP-coated gate FET, the ΔVout decreased gradually as the DA concentration increased from 40 nM to 20 μM. The time for the DA template removal affected the ΔVout performance. It was proved that immersion in a 0.1 M HCl/MeOH solution for 3 days affected the sensitivity at the nanomolar level, although almost the entire template was removed after immersion for 18 h at the micromolar level. Owing to the field effect, the negatively charged PBA–DA complexes formed by incorporating DA in the DA–MIP membrane affected the electron density at the source–drain channel in the MOSFET. As a result, this device detected DA concentrations as low as 40 nM, the gate surface potential of which decreased with increasing DA concentrations at a constant ISD. This result was supported by the semiconductor parameter analysis of the NIP- and DA–MIP-coated gate FETs in the presence of different DA concentrations (Fig. 3E). On the other hand, the ΔVout values of the DA–MIP-coated gate FET were markedly lower after LD, NE, and EP addition than after DA addition. ΔVout was also lower in the NIP-coated gate FET after DA addition. From the results of the EIS analysis and FET measurements of the DA–MIP and NIP FETs, we infer that the ATRP-based DA–MIP successfully imprinted the shape of DA at the interface of the Au electrode, enabling the electrochemical detection of the charge density changes as DA binds to PBA.
The MIP interface well controlled by SI-ATRP was also modified on the gate electrode for the detection of sialic acids.14 The chemical structures of 3′-sialyllactose (3SLac) and 6′-sialyllactose (6SLac) used in a previous work were very similar. Note that sialyl Lewis and sialyllactose share a common structure, the sialic acid unit, which is the most important target for recognizing cancer cells.42,43 For MIP preparation, multiple diol groups were targeted for PBA–SLac binding, including the diol group on the sialic acid unit, whose binding affinity has already been investigated.44–48 ΔVout shifted in the negative direction when using the 3SLac–MIP-coated gate FET, depending on the concentration of 3SLac (Fig. 4A). ΔVout was induced by the negative charges of PBA–3SLac complexes, that is, 3SLac should have entered the template cavity in the polymer matrix. On the other hand, ΔVout of the NIP-coated gate FET remained constant, although a slight drift of the signal was observed because of only a few background noises such as the changes in temperature and ion strength upon the addition of sample solutions (Fig. 4B). The polymer was highly crosslinked by EGDMA; therefore, the target molecules were blocked from entering the polymer matrix. Real-time electrical measurements were also performed during the addition of 6SLac onto the 3SLac– and 6SLac–MIP-coated gate FETs or 3SLac onto the 3SLac– and 6SLac–MIP-coated gate FETs. As part of the results, the calibration curves for 3SLac– and 6SLac–MIP-coated gate FETs were evaluated in terms of the change in the concentration of added 3SLac, as shown in Fig. 4C. Thus, the molecular imprinting contributed to the increase in selectivity for the molecule detected using the FET sensors, and the weak signals observed at 1 × 10−5 M for both sensors were regarded as background noises, such as the changes in temperature and ion strength by the addition of sample solutions.
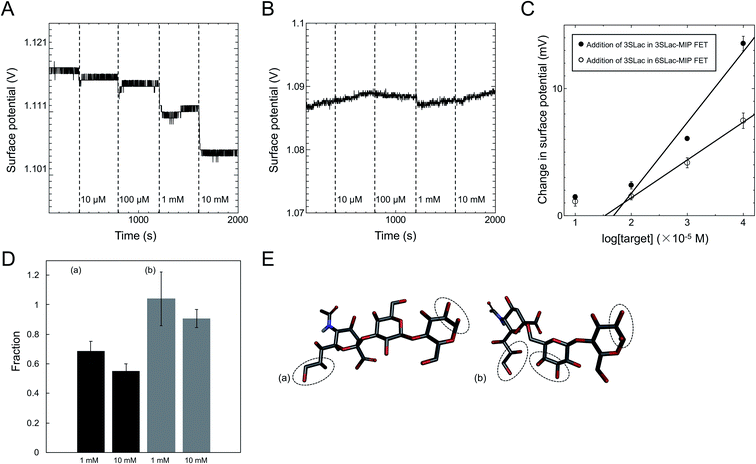 |
| | Fig. 4 ΔVout when 3SLac was added to (A) 3SLac–MIP-gate FET system and (B) NIP-gate FET system. (C) Calibration curve for 3SLac detection using 3SLac–MIP-gate FET and 6SLac–MIP-gate FET. (D) Ratio of surface potential change for (a) 3SLac in 6SLac–MIP/3SLac in 3SLac–MIP and (b) 6SLac in 3SLac–MIP/6SLac in 6SLac–MIP. (E) Structural model of 3SLac and 6SLac, showing the different numbers of VPBA binding sites per molecule. These figures have been reproduced from ref. 14 with permission from The Japan Society of Applied Physics, copyright 2017. | |
According to the above real-time monitoring, the selectivity of the sensors was evaluated using the fractions shown in Fig. 4D, which were calculated using the following equations:
| |
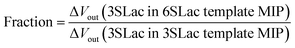 | (1) |
and
| |
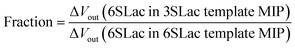 | (2) |
The first fraction was calculated using eqn (1) to be approximately 0.6. When considering the intensity of the signal of 3SLac entering the 3SLac-template–MIP cavity and binding to PBA as maximum, the signal intensity decreased by about 40% when 3SLac entered the 6SLac-template–MIP system. This indicates that the 6SLac-template–MIP has selectivity to the template target to some extent. However, the second fraction was calculated using eqn (2) to be approximately 1, which indicates that 6SLac could easily enter even into the cavities of the 3SLac-template–MIP; thus, the 3SLac-template–MIP has low selectivity. This result could be analyzed on the basis of the molecular structure. The target-selective cavities of sugar-template–MIP are formed by the covalent bonding of the PBA–sugar complex in the prepolymer solution. Since PBA has a stronger affinity to cis-diol than to trans-diol,49 it can be inferred that 3SLac has two binding sites per molecule, whereas 6SLac has three binding sites per molecule, as shown by the three-dimensional molecular structure in Fig. 4E. Thus, 6SLac–MIP likely has more template-specific cavities than 3SLac–MIP, which led to the results shown in Fig. 4D. Although 3SLac and 6SLac are similar in structure, the difference in the structure of template molecules may have significantly affected the selectivity of MIPs.
3. Potentiometric adsorption isotherm analysis of MIP bioelectrical interface
3.1. Concept of potentiometric adsorption isotherm analysis
To understand the chemical basis of interactions between the MIP and the target small biomolecules underlying the electrical responses of the MIP–FET sensor to such biomolecules, further quantitative analysis was required. In general, the characteristics of the binding of a target molecule to MIP are quantified using adsorption isotherm equations, because the binding process involves the reversible adhesion of the target molecule to the target-selective membrane.50 Moreover, the homogeneity and heterogeneity of binding sites distributed in MIPs are critical to the effective increase in selectivity. In most cases, the binding sites in MIPs are heterogeneously distributed because of the randomness of copolymerization and the template–functional monomer interaction;51,52 therefore, MIPs include both nonselective and highly selective binding sites at a certain ratio. In this study, Langmuir and bi-Langmuir adsorption isotherm equations, which are often used for MIP characterization,53,54 were utilized as the homogeneous and heterogeneous binding models, respectively, and to derive the corresponding equations for the analysis of the electrical properties of the MIP–FET system. In this way, the potentiometric analyses based on the FET sensor can directly characterize the MIP interface without the batch rebinding process, which is often required for MIP characterization. According to a previous study,50 the Langmuir adsorption isotherm and bi-Langmuir adsorption isotherm equations for a bulk rebinding system are respectively expressed by| |
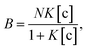 | (3) |
| |
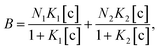 | (4) |
where B refers to a signal observed at equilibrium for the MIP-bound template, [c] to the free concentration of the template at equilibrium, N to the number of available active centers in the MIP per unit volume, and K to the binding constant. Eqn (3) assumes homogeneously distributed binding sites with a constant binding constant K. On the other hand, eqn (4) assumes two main types of binding site with different affinities distributed at a ratio of N1/N2 in the polymer, that is, a heterogeneous system.
The operation of a silicon-based FET in the unsaturated region can generally be described by
| |
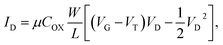 | (5) |
where
ID is the drain current,
μ is the electron mobility in the channel,
COX is the gate oxide capacitance,
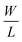
is the channel width-to-length ratio,
VD and
VG are the applied drain–source and gate–source voltages, respectively, and
VT is the threshold voltage, which can be expressed by
55| |
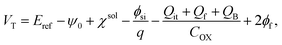 | (6) |
where
Eref is the reference electrode potential relative to a vacuum, (−
ψ0 +
χsol) describes the interfacial potential at the electrolyte/Au gate electrode interface (the factor
χsol is the surface dipole moment of the solution, which can be considered to be constant),
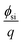
is the silicon electron work function,
Qit,
Qf, and
QB are the charge of the interface traps, the fixed oxide charge, and the bulk depletion charge per unit area, respectively, and
ϕf is the Fermi potential difference between the doped bulk silicon and the intrinsic silicon.
Considering the MIP membrane on the Au gate electrode of the extended-gate FET, the capacitance and charge in the MIP membrane should be added to eqn (6), and can be expressed by
| |
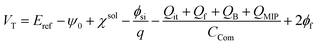 | (7) |
with
| |
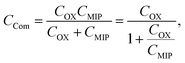 | (8) |
where
QMIP is the charge in the MIP membrane and
CCom is the combined capacitance of the FET gate oxide (
COX) and the MIP membrane (
CMIP) on the Au gate electrode. In this study, it is assumed that
CMIP hardly changed even after the addition of targeted molecules, on the basis of our previous results for a similar hydrogel;
56 therefore,
CCom was nearly constant regardless of the adsorption of small molecules because
COX was also constant. Moreover, the interfacial potential (Δ
ψ0) at the electrolyte/Au gate electrode interface should not change because the ionic concentration (
i.e., pH) was basically maintained by using the buffer solution. Also,
Eref,
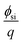
,
Qit,
Qf,
QB, and
ϕf should be the same before and after the molecular recognition events at the MIP interface. Thus, the signal response obtained using the FET sensor is based on the change in
VT (Δ
VT); therefore, Δ
QMIP should be evaluated in this study, in accordance with
eqn (7) and the above considerations.
The binding affinity of PBA to a diol is pH-dependent, but it is generally understood that the B(OH)3− complex is much more stable than the B(OH)2 complex, as shown in a previous work.57 For the reversible interaction between a diol compound (diol) and PBA in the MIP membrane (Scheme 1A),
| | |
diol + PBA ↔ diol·PBA−,
| (9) |
the rate of formation of the diol·PBA
− complex at time
t is written as
| |
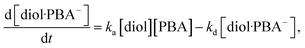 | (10) |
where
ka is the association rate constant and
kd is the dissociation rate constant. At time
t, [PBA] = [PBA]
0 − [diol·PBA
−], where [PBA]
0 is the concentration of PBA at
t = 0. This is substituted into
eqn (10) to give
| |
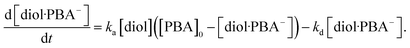 | (11) |
In this study, the charge QMIP is derived from reaction (9); therefore, it is proportional to the rate of formation of the diol·PBA− complex in the MIP membrane. Additionally, Qmax is proportional to the concentration of PBA in the MIP membrane ([PBA]0 at t = 0), which indicates the capacity of the immobilized ligand. Therefore, eqn (11) is modified to
| |
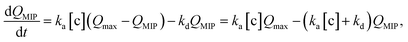 | (12) |
where
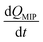
is the rate of formation of the associated complex (diol·PBA
−) in the MIP membrane (on the Au gate) and [c] is the concentration of the analyte (diol) in the solutions. Moreover, integrating
eqn (12) gives
| |
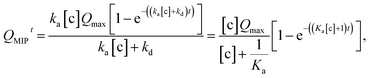 | (13) |
where
Ka is the stability constant (binding affinity) of diol and PBA (
ka/
kd) in the MIP membrane. From
eqn (13),
Qt=0MIP = 0. Considering
eqn (7),
| |
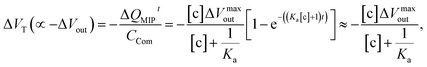 | (14) |
which is estimated after a certain reaction time
t. Here, Δ
Vmaxout is the maximum change in surface potential induced by Δ
Qmax, which is proportional to the number of binding sites. In this study, Δ
Vout at the gate was measured at a constant
ID using the source follower circuit. The detected Δ
Vout was regarded as the change in
VGS, which was proportional to −Δ
VT at a constant
ID.
According to the above considerations, the electrical signal in the entire FET circuit should obey the Langmuir adsorption model. By modifying eqn (3) and (4) in accordance with eqn (14), we can obtain the adsorption isotherm equations for the MIP–FET system as
| |
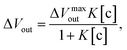 | (15) |
| |
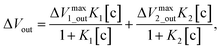 | (16) |
where [c] is determined as the concentration of the target biomolecule at equilibrium, which is obtained from the saturated electrical signal in real-time measurement.
3.2. Quantification of MIP effect using adsorption isotherm equations
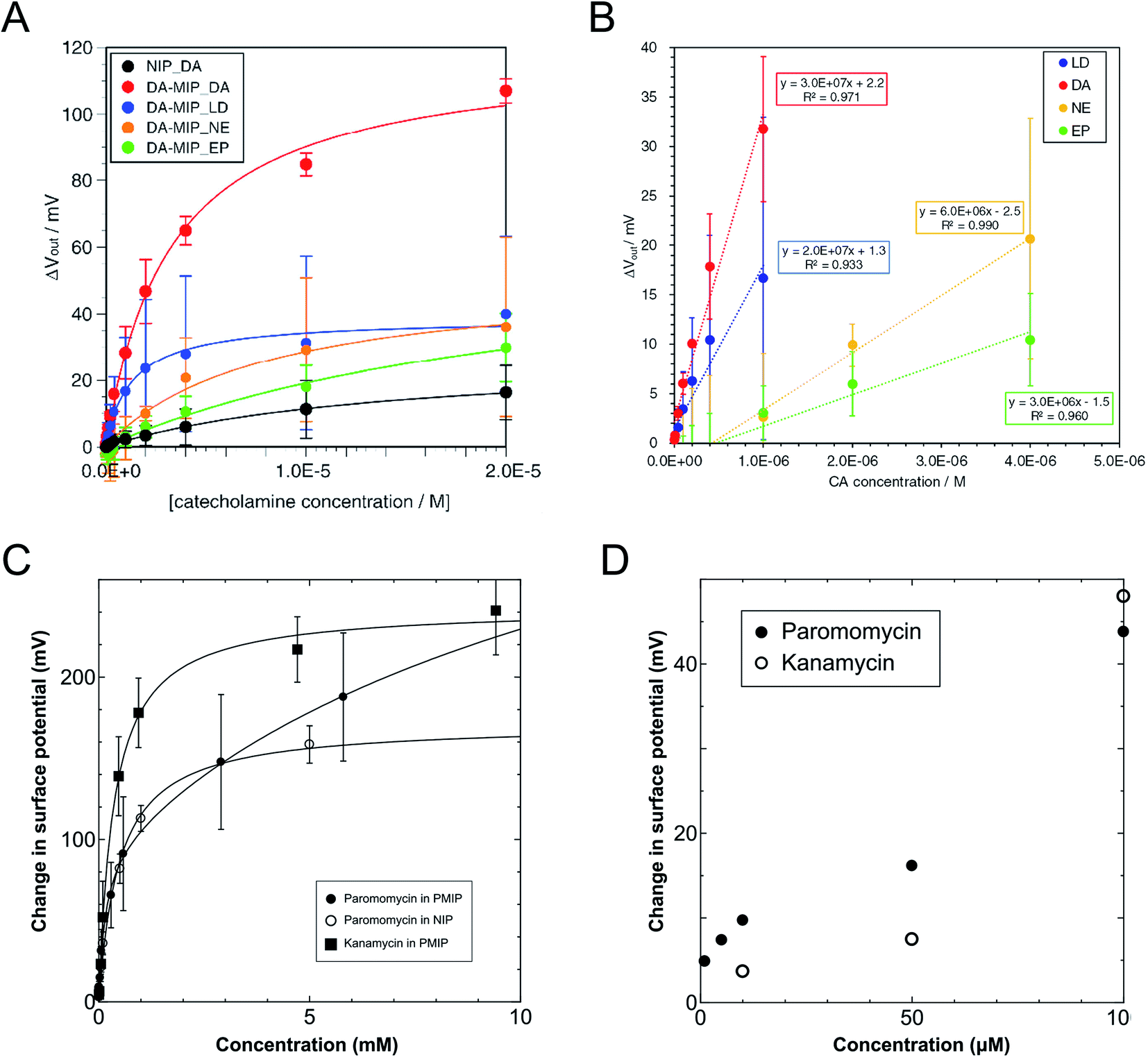
On the basis of the above considerations, the electrical signal in the whole FET circuit should obey the Langmuir adsorption model. In the case of the DA–MIP and NIP FETs, which are shown in Fig. 3, ΔVLout was determined from ΔVout after 300 s each time DA was added. That is, the substrate specificity of DA–MIP to each catecholamine was quantitatively evaluated using the equation of adsorption equilibrium. Fig. 5A shows the ΔVLout versus concentration plots for each catecholamine from the data shown in Fig. 3D, which were sufficiently fitted using eqn (15). The parameters of the Langmuir fitting curve are given in Table 2. The dependence of output voltage on DA concentration clearly differs between DA–MIP and NIP with DA addition. The saturated amount of DA adsorbed (recorded as ΔVLmax) and Ka were 4.3 and 4.4 times higher, respectively, in the DA–MIP-coated gate FET than in the NIP-coated gate FET. In the DA–MIP-coated gate FET, the ΔVLmax was clearly higher after DA addition than after LD addition, although the binding constant of LD was twice that of DA. In LD, the carboxyl group in the vicinity of a catechol provides many diester–PBA binding sites, so the ΔVLmax appears to be higher for LD than for NE and EP.58,59 The Ka of PBA was determined in the presence of different catecholamines using a fluorescence chemosensor in a previous work, where the Ka values were 1.5 and 2 times higher for NE and EP than for DA, respectively, suggesting that the affinity of PBA to DA was lower than that to other catecholamines.60 However, from the results shown in Fig. 5A, the PBA in the DA–MIP-coated gate FET showed higher affinity to DA than to NE and EP. From the quantitative evaluation of the DA–MIP-coated gate FET, we concluded that a DA–MIP-coated gate FET can potentially select DA from a mixture of catecholamines.
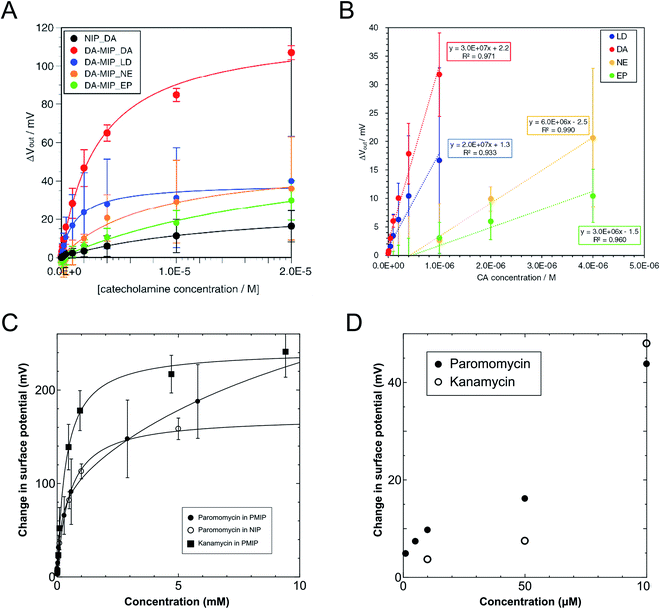 |
| | Fig. 5 (A) Plots of ΔVout at different concentrations of catecholamines added based on the data shown in Fig. 3D. Approximate curves were fitted by Langmuir adsorption isotherm. ΔVout was analyzed for the DA–MIP-coated gate FETs, where four types of catecholamine, DA (red), LD (blue), NE (orange), and EP (green), were added with different concentrations. The NIP-coated gate FET was used as a control upon adding DA (black). (B) Plots of output voltage shift and their approximate straight lines of DA–MIP–FET in response to each catecholamine in the low concentration region in Fig. 3D. Approximated formula and the square values of correlation factor of the plots in each catecholamine addition are also indicated. (C) Change in surface potential as a function of target concentrations up to 10 mM. Calibration curves were determined by solving eqn (15) and (16) (filled circle, open circle, and filled square show the additions of paromomycin into the PMIP–FET, paromomycin into the NIP–FET, and kanamycin into the PMIP–FET, respectively). (D) Comparison of the addition of paromomycin into PMIP–FET with that of kanamycin at concentrations of lower than 100 μM. The figures (A) and (B) have been reproduced from ref. 17 with permission from Elsevier, copyright 2018. The figures (C) and (D) have been reproduced from ref. 15 with permission from American Chemical Society, copyright 2018. | |
Table 2 Kinetic parameters calculated from the approximate curves shown in Fig. 3D, which were based on the Langmuir model. This table has been reproduced from ref. 17 with permission from Elsevier, copyright 2018
| Polymer-additive catecholamine |
Kinetic parameters |
| ΔVLmax (mV) |
Ka (105 M−1) |
R2 |
| NIP_DA |
27.5 |
0.71 |
0.996 |
| DA–MIP_DA |
119 |
3.11 |
0.995 |
| DA–MIP_LD |
63.6 |
7.75 |
0.986 |
| DA–MIP_NE |
51.5 |
1.26 |
0.974 |
| DA–MIP_EP |
38.8 |
4.33 |
0.981 |
To assess the sensitivity of the DA–MIP-coated gate FET to different catecholamines, we calculated the limit of detection (LOD) for each catecholamine in terms of the Kaiser limit.61 The LOD was calculated by regression analysis of the linear region, which maximizes the square of the correlation factor (R2) of the ΔVout versus catecholamine concentration (Fig. 5B). The estimated LODs of DA–MIP were 96 nM for DA, 150 nM for LD, 179 nM for NE, and 355 nM for EP. We compared these results with the reported values based on the electrochemical detection systems using MIP (Table 3).62–69 The DA–MIP-coated gate FET could measure the direct charge changes induced by DA binding to PBA without affecting the MIP gel by applying voltage and without using any mediators, which provided a comparable or even higher sensitivity than the sensitivities reported for the electrochemical devices. In previous HPLC and fluorescence spectroscopy analyses, the dopamine concentrations were determined to be from sub-nM order to 100 nM in plasma, saliva, and tears, and μM order in urine.70,71 The DA–MIP-coated gate FET in this study has sufficient DA sensitivity to monitor analytes in urine, and nearly sufficient DA sensitivity to monitor other analytes. Therefore, the DA–MIP-coated gate FET can potentially measure DA in various body fluids with high sensitivity and specificity in the future.
Table 3 Comparison with other electrochemical sensors for the determination of DA using the MIP technique. This table has been reproduced from ref. 17 with permission from Elsevier, copyright 2018
| Interface materials |
Detection method |
Dynamic range/M |
Limit of detection/M |
Reference |
| Multi-walled carbon nanotube–molecularly imprinted polymer–glassy carbon electrode. Graphene oxide. Polypyrrole MIP. Nanoporous Au–Ag alloy microrod. |
| MWCNT–MIP–GCEa |
Cyclic voltammetry |
5.0 × 10−7 to 2.0 × 10−4 |
5.0 × 10−7 |
Kan et al. (2008)62 |
| MIP–Au |
Cyclic voltammetry |
2.3 × 10−7 to 1.4 × 10−4 |
2.3 × 10−7 |
Lakshmi et al. (2009)63 |
| MIP–Au |
Cyclic voltammetry |
2.0 × 10−8 to 2.5 × 10−7 |
2.0 × 10−9 |
Li et al. (2009)64 |
| GOb–MIP–GCE |
Cyclic voltammetry |
1.0 × 10−7 to 8.3 × 10−4 |
1.0 × 10−7 |
Mao et al. (2011)65 |
| Au@SiO2 MIPs–GCE |
Cyclic voltammetry |
4.8 × 10−8 to 5.0 × 10−5 |
2.0 × 10−8 |
Yu et al. (2012)66 |
| MWCNT–pPyMIPc–GCE |
Cyclic voltammetry |
6.3 × 10−7 to 1.0 × 10−4 |
6.0 × 10−8 |
Kan et al. (2012)67 |
| MIP–GO–GCE |
Cyclic voltammetry |
5.0 × 10−8 to 1.6 × 10−4 |
3.0 × 10−8 |
Zeng et al. (2013)68 |
| MIP/NPAMRd |
Cyclic voltammetry |
2.0 × 10−13 to 2.0 × 10−8 |
7.6 × 10−14 |
Li et al. (2016)69 |
| MIP–Au |
FET |
4.0 × 10−8 to 2.0 × 10−5 |
9.6 × 10−8 |
Kajisa et al. (2018)17 |
Moreover, potentiometric adsorption isotherm analysis was performed for the oligosaccharide-template–MIP using the FET sensor.15 Here, the paromomycin-template–MIP (PMIP) was coated on the Au gate electrode for the selective detection of paromomycin, whereas kanamycin, which has a similar chemical structure to paromomycin, was utilized as a non-target oligosaccharide. Similarly to the analysis of dopamine detection using the DA–MIP-coated gate FET, ΔVout was analyzed using eqn (15) and (16). First, ΔVLout was calculated for each concentration of paromomycin by subtracting Vout at t = 0 from Vout at each concentration. Then, the results (ΔVLout) were plotted vs. the paromomycin concentration, as shown in Fig. 5C. ΔVLout was the average of 10 data plots taken 5 min after the addition of the target. The best-fit adsorption isotherm equations were determined by optimizing K and ΔVmaxout using the application software to minimize R2 (in Microsoft Excel). Then, ΔVmaxout and the average binding affinity Kavr were respectively expressed as
| | |
ΔVmaxout = ΔVmax1_out + ΔVmax2_out,
| (17) |
| |
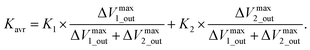 | (18) |
Table 4 shows the optimized values for the paromomycin/PMIP, paromomycin/NIP, and kanamycin/PMIP interactions in the MIP/NIP–FET measurement systems. The fitting curves are also shown in Fig. 5C. From Table 4, R2 shows that the adsorption isotherm equations were successfully applied to the PMIP/NIP–FET measurement systems; thus, the assumptions made in the derivation were valid. K2 and ΔVmax2_out in the case of adding paromomycin to NIP were zero, which indicated that the result fitted the Langmuir adsorption isotherm, indicating in turn that the binding sites were homogeneously distributed in NIP. However, there were no paromomycin-selective binding sites in NIP. Thus, the signal probably originated from nonspecific adsorption on the surface of NIP. On the other hand, the paromomycin–PMIP interaction fitted the bi-Langmuir adsorption isotherm equation, indicating the heterogeneous distribution of binding sites. That is, from Table 4, two different types of paromomycin binding sites were found, one with high binding affinity (K1 = 6970 M−1) and the other with low binding affinity (K2 = 73 M−1). Moreover, the number of binding sites (proportional to ΔVmax1_out) corresponding to K1 was much smaller even for the paromomycin–PMIP interaction, as similarly observed previously in the MIP produced by noncovalent interactions.50 In designing the PMIP for its interaction with paromomycin, PBA was assumed to be a covalently interacting functional monomer and MAA was assumed to be a noncovalently interacting one. Considering the equilibrium reaction shown in Scheme 1B, moreover, some PBAs might be used for the interaction with template–paromomycin in the PMIP membrane, but not for rebinding to target–paromomycin even upon adding it, resulting in noncovalent interactions in the PMIP membrane. These noncovalently interacting monomers should be heterogeneously distributed and randomly functionalized in the PMIP membrane, then noncovalent, hydrogen bonding may be screened by water molecules in an aqueous solution; thus, the number of well-bound complexes was assumed to be small. In the FET measurement, the largest difference between the PMIP and NIP interfaces that interacted with paromomycin was found in ΔVmaxout, which was proportional to the total number of binding sites. As shown in Fig. 5C, the concentration of paromomycin added to NIP–FET reached a maximum of approximately 5 mM (170 mV), but the PMIP–FET showed a much higher ΔVmaxout [415 mV (according to eqn (17)) at 850 mM in calculation] upon adding paromomycin. Thus, the difference between MIP and NIP was clearly demonstrated using the potentiometric adsorption isotherm equations derived from the MIP–FET measurement system.
Table 4 Summary of optimized values of K and ΔVout determined on the basis of the bi-Langmuir adsorption isotherm equation. This table has been reproduced from ref. 15 with permission from American Chemical Society, copyright 2018
| |
K1 (M−1) |
ΔVmax1_out (mV) |
K2 (M−1) |
ΔVmax2_out (mV) |
Kavr (M−1) |
ΔVmaxout (mV) |
R2 |
| P/PMIP |
6970 |
95 |
73 |
320 |
1070 |
415 |
0.998 |
| P/NIP |
2060 |
170 |
0 |
0 |
2060 |
170 |
0.994 |
| K/PMIP |
2800 |
240 |
0 |
0 |
2800 |
240 |
0.998 |
A similar trend was observed in the comparison between paromomycin and kanamycin added to the PMIP–FET devices. Although the binding affinity of kanamycin was higher on average (Kavr = 2800 M−1 according to eqn (18)), the selectivity of PMIP for paromomycin was better than expected at concentrations of less than 100 μM, as shown in Fig. 5D. Initially, the addition of kanamycin to the PMIP–FET system fitted the Langmuir adsorption isotherm equation, similarly to the addition of paromomycin to the NIP–FET system. This indicated that the detection of kanamycin using the PMIP–FET system was also based on the nonspecific adsorption of kanamycin to the binding sites with low affinity (K2 and ΔVmax2_out in the case of adding kanamycin to PMIP were zero). On the other hand, the binding sites with higher affinity were crucial at lower concentrations of target molecules. Fig. 5D shows the change in surface potential at paromomycin and kanamycin concentrations of lower than 100 μM when using the PMIP–FET sensors. From this result, paromomycin was detected more sensitively than kanamycin at the lower concentrations. That is, a target molecule at a lower concentration will first bind to high-affinity binding sites. Thus, the effect of K1 on the selectivity of PMIP for paromomycin at low concentrations was very important until the signal reached ΔVmax1_out. Additionally, the LOD for paromomycin using the PMIP–FET sensor in this study was predicted to be 2.3 μM from the semilogarithmic plots in the range of 100 μM to 5.8 mM shown in Fig. 5C, which was obtained on the basis of the Kaiser limit theory.61 This means that the higher selectivity of PMIP for paromomycin than for kanamycin at concentrations of less than 100 μM should be ensured down to 2.3 μM. Therefore, the potentiometric adsorption isotherm analysis using the MIP–FET device can elucidate the formation of selective binding sites at the MIP interface. The electrochemical analysis of the functional bioelectrical interface used in this study is expected to support the design and construction of sensors for small biomarkers.
4. Conclusions and outlook
In this review paper, the MIP-based membrane with PBA, which induced the change in the density of molecular charges based on the small biomolecule–PBA diol binding, was demonstrated to be available for the bioelectrical interface of bio-FET sensors. The MIP-coated gate FET sensors selectively detected various small biomolecules such as glucose, dopamine, sialic acid, and oligosaccharides without using labeled materials. In particular, the well-controlled MIP film by SI-ATRP contributed to the quantitative analysis of small biomolecule sensing, resulting in potentiometric Langmuir isotherm adsorption analysis by which the parameters such as the binding affinity between small biomolecules and MIP cavities were evaluated. Also, the output electrical signal of even the random MIP-coated gate FET sensor was quantitatively analyzed using the bi-Langmuir adsorption isotherm equation, showing the adsorption mechanism of oligosaccharides onto the template-specific MIP membrane. Moreover, the LODs for the small biomolecules using the MIP-coated gate FET sensor in this study were comparable to those shown in previous works. Thus, a platform based on the MIP-coated gate FET sensor is suitable for a label-free and enzyme-/antibody-free biosensing system, enabling the miniaturization of healthcare devices.
However, note that the binding affinities of MIPs for small biomolecules (around 105 M−1 order), as shown in this review paper, remain inferior to those of monoclonal antibodies (105 to 1012 M−1). This may be the fate of artificially produced devices. However, the MIP concept as a bioelectrical interface contributed to not only the increase in the binding affinity between the target small biomolecule (the diol compound) and the PBA molecule but also the sufficient detection sensitivity and LOD for the target small biomolecule. Surely, the artificially produced MIP interface may solve the problems inherent in enzymes and antibodies, such as their lack of stability, high-cost and time-consuming production, and the difficulty of quality control of their production. Therefore, we consider other functional monomers as well as PBA in the MIP membrane for the development of the platform of the MIP-based bioelectrical interface for potentiometric biosensors such as bio-FETs in the future.
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
We would like to thank Professor T. Takeuchi of Kobe University and members of Sakata Laboratory for their help and useful discussion.
References
- S. J. Updike and G. P. Hicks, The Enzyme Electrode, Nature, 1967, 214, 986–988 CrossRef CAS PubMed
 .
. - R. M. Bergenstal, Continuous Glucose Monitoring: Transforming Diabetes Management Step by Step, Lancet, 2018, 391, 1334–1336 CrossRef .
- E. L. Klatman, A. J. Jenkins, M. Y. Ahmedani and G. D. Ogle, Blood Glucose Meters and Test Strips: Global Market and Challenges to Access in Low-Resource Settings, Lancet Diabetes Endocrinol., 2019, 7, 150–160 CrossRef PubMed .
- B. Danielsson, I. Lundström, K. Mosbach and L. Stiblert, On a New Enzyme Transducer Combination: The Enzyme Transistor, Anal. Lett., 1979, 12, 1189–1199 CrossRef CAS .
- S. Caras and J. Janata, Field Effect Transistor Sensitive to Penicillin, Anal. Chem., 1980, 52, 1935–1937 CrossRef CAS .
- Y. Miyahara, T. Moriizumi and K. Ichimura, Integrated Enzyme FETs for Simultaneous Detections of Urea and Glucose, Sens. Actuators, 1985, 7, 1–10 CrossRef CAS .
- A. H. Free, E. C. Adams, M. L. Kercher, H. M. Free and M. H. Cook, Simple Specific Test for Urine Glucose, Clin. Chem., 1957, 3, 163–168 CrossRef CAS .
- D. E. Charlton, Test Device and Method for Colored Particle Immunoassay, US Pat. 6485982B1, 2002 .
- G. Wulff and A. Sarhan, Über die Anwendung von enzymanalog gebauten Polymeren zur Racemattrennung, Angew. Chem., 1972, 84, 364 CrossRef .
- L. Andersson, B. Ekberg and K. Mosbach, Imprinting of Amino Acid Derivatives in Macroporous Polymers, Tetrahedron Lett., 1985, 26, 3623–3624 CrossRef CAS .
- B. Sellergren, B. Ekberg and K. Mosbach, Molecular Imprinting of Amino Acid Derivatives in Macroporous Polymers: Demonstration of Substrate- and Enantio-Selectivity by Chromatographic Resolution of Racemic Mixtures of Amino Acid Derivatives, J. Chromatogr. A, 1985, 347, 1–10 CrossRef CAS .
- G. Wulff and S. Schauhoff, Racemic Resolution of Free Sugars with Macroporous Polymers Prepared by Molecular Imprinting. Selectivity Dependence on the Arrangement of Functional Groups Versus Spatial Requirements, J. Org. Chem., 1991, 56, 395–400 CrossRef CAS .
- T. Takeuchi, T. Hayashi, S. Ichikawa, A. Kaji, M. Masui, H. Matsumoto and R. Sasao, Molecularly Imprinted Tailor-Made Functional Polymer Receptors for Highly Sensitive and Selective Separation and Detection of Target Molecules, Chromatography, 2016, 37, 43–64 CrossRef CAS .
- S. Nishitani, Y. Kajisa and T. Sakata, Development of Molecularly Imprinted Polymer-Based Field Effect Transistor for Sugar Chain Sensing, Jpn. J. Appl. Phys., 2017, 56, 04CM02 CrossRef .
- S. Nishitani and T. Sakata, Potentiometric Adsorption Isotherm Analysis of a Molecularly Imprinted Polymer Interface for Small-Biomolecule Recognition, ACS Omega, 2018, 3, 5382–5389 CrossRef CAS PubMed .
- H. Yang, S. Nishitani and T. Sakata, Potentiometric Langmuir Isotherm Analysis of Histamine-Selective Molecularly Imprinted Polymer-Based Field-Effect Transistor, ECS J. Solid State Sci. Technol., 2018, 7, Q3079–Q3082 CrossRef CAS .
- T. Kajisa, W. Li, T. Michinobu and T. Sakata, Well-Designed Dopamine-Imprinted Polymer Interface for Selective and Quantitative Dopamine Detection among Catecholamines Using a Potentiometric Biosensor, Biosens. Bioelectron., 2018, 117, 810–817 CrossRef CAS PubMed .
- T. Kajisa and T. Sakata, Molecularly Imprinted Artificial Biointerface for an Enzyme-Free Glucose Transistor, ACS Appl. Mater. Interfaces, 2018, 10, 34983–34990 CrossRef CAS PubMed .
- R. J. Ferrier, A. J. Hannaford, W. G. Overend and B. C. Smith, Boric Acid Derivatives as Reagents in Carbohydrate Chemistry: Part IV. The Interaction of Phenylboronic Acid with Hexopyranoid Compounds, Carbohydr. Res., 1965, 1, 38–43 CrossRef CAS .
- G. Springsteen and B. Wang, A Detailed Examination of Boronic Acid–Diol Complexation, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS .
- W. Yang, X. Gao and B. Wang, Boronic Acid Compounds as Potential Pharmaceutical Agents, Med. Res. Rev., 2003, 23, 346–368 CrossRef CAS PubMed .
- V. Ratautaite, S. D. Janssens, K. Haenen, M. Nesládek, A. Ramanaviciene, I. Baleviciute and A. Ramanavicius, Molecularly Imprinted Polypyrrole Based Impedimentric Sensor for Theophylline Determination, Electrochim. Acta, 2014, 130, 361–367 CrossRef CAS .
- V. Ratautaite, D. Plausinaitis, I. Baleviciute, L. Mikoliunaite, A. Ramanaviciene and A. Ramanavicius, Characterization of Caffeine-Imprinted Polypyrrole by A Quartz Crystal Microbalance and Electrochemical Impedance Spectroscopy, Sens. Actuators, B, 2015, 212, 63–71 CrossRef CAS .
- E. H. Gillis, J. P. Gosling, J. M. Sreenan and M. Kane, Development and Validation of a Biosensor-Based Immunoassay for Progesterone in Bovine Milk, J. Immunol. Methods, 2002, 267, 131–138 CrossRef CAS PubMed .
- J. S. Mitchell, Y. Wu, C. J. Cook and L. Main, Sensitivity Enhancement of Surface Plasmon Resonance Biosensing of Small Molecules, Anal. Biochem., 2005, 343, 125–135 CrossRef CAS PubMed .
- J. Mitchell, Small Molecule Immunosensing Using Surface Plasmon Resonance, Sensors, 2010, 10, 7323–7346 CrossRef CAS PubMed .
- T. Sakata and R. Fukuda, Simultaneous Biosensing with Quartz Crystal Microbalance with Dissipation Coupled-Gate Semiconductor Device, Anal. Chem., 2013, 85, 5796–5800 CrossRef CAS PubMed .
- P. Pradelles, J. Grassi, C. Creminon, B. Boutten and S. Mamas, Immunometric Assay of Low Molecular Weight Haptens Containing Primary Amino Groups, Anal. Chem., 1994, 66, 16–22 CrossRef CAS PubMed .
- G. Giraudi, L. Anfossi, I. Rosso, C. Baggiani, C. Giovannoli and C. Tozzi, A General Method to Perform a Noncompetitive Immunoassay for Small Molecules, Anal. Chem., 1999, 71, 4697–4700 CrossRef CAS PubMed .
- P. Bergveld, Development of an Ion-Sensitive Solid-State Device for Neurophysiological Measurements, IEEE Trans. Biomed. Eng., 1970, 17, 70–71 CAS .
- T. Matsuo and K. D. Wise, An Integrated Field-Effect Electrode for Biopotential Recording, IEEE Trans. Biomed. Eng., 1974, 21, 485–487 Search PubMed .
- M. Esashi and T. Matsuo, Integrated Micro-Multi-Ion Sensor Using Field Effect of Semiconductor, IEEE Trans. Biomed. Eng., 1978, 25, 184–192 CAS .
- T. Sakata, Biologically coupled gate field-effect transistors meet in vitro diagnostics, ACS Omega, 2019, 4, 11852–11862 CrossRef CAS PubMed .
- K. Haupt and K. Mosbach, Molecularly Imprinted Polymers and Their Use in Biomimetic Sensors, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed .
- Z. Iskierko, M. Sosnowska, P. S. Sharma, T. Benincori, F. D'Souza, I. Kaminska, K. Fronc and K. Noworyta, Extended-Gate Field-Effect Transistor (EG-FET) with Molecularly Imprinted Polymer (MIP) Film for Selective Inosine Determination, Biosens. Bioelectron., 2015, 74, 526–533 CrossRef CAS PubMed .
- M. Dabrowski, P. S. Sharma, Z. Iskierko, K. Noworyta, M. Cieplak, W. Lisowski, S. Oborska, A. Kuhn and W. Kutner, Early Diagnosis of Fungal Infections Using Piezomicrogravimetric and Electric Chemosensors Based on Polymers Molecularly Imprinted with D-Arabitol, Biosens. Bioelectron., 2016, 79, 627–635 CrossRef CAS PubMed .
- Z. Iskierko, A. Checinska, P. S. Sharma, K. Golebiewska, K. Noworyta, P. Borowicz, K. Fronc, V. Bandi, F. D'Souza and W. Kutner, Molecularly Imprinted Polymer Based Extended-Gate Field-Effect Transistor Chemosensors for Phenylalanine Enantioselective Sensing, J. Mater. Chem. C, 2017, 5, 969–977 RSC .
- T. Sakata, M. Kamahori and Y. Miyahara, DNA Analysis Chip Based on Field Effect Transistors, Jpn. J. Appl. Phys., 2005, 44(4B), 2854–2859 CrossRef CAS .
- A. Matsumoto, N. Sato, T. Sakata, R. Yoshida, K. Kataoka and Y. Miyahara, Chemical-to-Electrical-Signal Transduction
Synchronized with Smart Gel Volume Phase Transition, Adv. Mater., 2009, 21, 4372–4378 CrossRef CAS PubMed .
- J. Liebscher, R. Mrowczynski, H. A. Scheidt, C. Filip, N. D. Hadade, R. Turcu, A. Bende and S. Beck, Structure of Polydopamine: A Never-Ending Story?, Langmuir, 2013, 29, 10539–10548 CrossRef CAS PubMed .
- L. Shi, S. Santhanakrishnan, Y. S. Cheah, M. Li, C. L. Chai and K. G. Neoh, One-Pot UV-Triggered o-Nitrobenzyl Dopamine Polymerization and Coating for Surface Antibacterial Application, ACS Appl. Mater. Interfaces, 2016, 8, 33131–33138 CrossRef CAS PubMed .
- B. Adamczyk, T. Tharmalingam and P. M. Rudd, Glycans as Cancer Biomarkers, Biochim. Biophys. Acta, 2012, 1820, 1347–1353 CrossRef CAS PubMed .
- M. M. Fuster and J. D. Esko, The Sweet and Sour of Cancer: Glycans as Novel Therapeutic Targets, Nat. Rev. Cancer, 2005, 5, 526–542 CrossRef CAS PubMed .
- A. Kugimiya, T. Takeuchi, J. Matsui, K. Ikebukuro, K. Yano and I. Kurabe, Recognition in Novel Molecularly Imprinted Polymer Sialic Acid Receptors in Aqueous Media, Anal. Lett., 1996, 29, 1099–1107 CrossRef CAS .
- H. Otsuka, E. Uchiyama, H. Koshino, T. Okano and K. Kataoka, Anomalous binding profile of phenylboronic acid with N-acetylneuraminic acid (Ner5Ac) in aqueous solution with varying pH, J. Am. Chem. Soc., 2003, 125, 3493–3502 CrossRef CAS PubMed .
- K. Djanashvili, L. Frullano and J. A. Peters, Molecular Recognition of Sialic Acid End Groups by Phenylboronates, Chem.–Eur. J., 2005, 11, 4010–4018 CrossRef CAS PubMed .
- S. Shinde, Z. El-Schich, A. Malakpour, W. Wan, N. Dizeyi, R. Mohammadi, K. Rurack, A. G. Wingren and B. Sellergren, Sialic Acid-Imprinted Fluorescent Core–Shell Particles for Selective Labeling of Cell Surface Glycans, J. Am. Chem. Soc., 2015, 137, 13908–13912 CrossRef CAS PubMed .
- S. Nishitani, Y. Maekawa and T. Sakata, Understanding Molecular Structure of Sialic Acid/Phenylboronic Acid Complex by a Combined Study of NMR and DFT: Point to Note Toward Sialic Acid Detection at Cell Membrane, ChemistryOpen, 2018, 7, 513–519 CrossRef CAS PubMed .
- T. Kajisa and T. Sakata, Fundamental Properties of Phenylboronic-Acid-Coated Gate Field-Effect Transistor for Saccharide Sensing, ChemElectroChem, 2014, 1, 1647–1655 CrossRef CAS .
- J. A. García-Calzón and M. E. Díaz-García, Characterization of Binding Sites in Molecularly Imprinted Polymers, Sens. Actuators, B, 2007, 123, 1180–1194 CrossRef .
- B. J. Stanley, P. Szabelski, Y.-B. Chen, B. Sellergren and G. Guiochon, Affinity Distributions of a Molecularly Imprinted Polymer Calculated Numerically by the Expectation-Maximization Method, Langmuir, 2003, 19, 772–778 CrossRef CAS .
- F. Lanza, M. Ruther, A. J. Hall, C. Dauwe and B. Sellergren, Studies on the Process of Formation, Nature and Stability of Binding Sites in Molecularly Imprinted Polymer, Mater. Res. Soc. Symp. Proc., 2002, 723, 93–103 CrossRef CAS .
- R. J. Umpleby, S. C. Baxter, Y. Chen, R. N. Shah and K. D. Shimizu, Characterization of Molecularly Imprinted Polymers with the Langmuir–Freundlich Isotherm, Anal. Chem., 2001, 73, 4584–4591 CrossRef CAS PubMed .
- P. Sajonz, M. Kele, G. Zhong, B. Sellergren and G. Guiochon, Study of the Thermodynamics and Mass Transfer Kinetics of Two Enantiomers on a Polymeric Imprinted Stationary Phase, J. Chromatogr. A, 1998, 810, 1–17 CrossRef CAS .
- P. Bergveld, The Impact of MOSFET-Based Sensors, Sens. Actuators, 1985, 8, 109–127 CrossRef CAS .
- T. Kajisa and T. Sakata, Glucose Responsive Hydrogel Electrode for Biocompatible Glucose Transistor, Sci. Technol. Adv. Mater., 2017, 18, 26–33 CrossRef CAS PubMed .
- Y. Furikado, T. Nagahata, T. Okamoto, T. Sugaya, S. Iwatsuki, M. Inamo, H. D. Takagi, A. Odani and K. Ishihara, Universal Reaction Mechanism of Boronic Acids with Diols in Aqueous Solution: Kinetics and the Basic Concept of a Conditional Formation Constant, Chem.–Eur. J., 2014, 20, 13194–13202 CrossRef CAS PubMed .
- S. Friedman, B. Pace and R. Pizer, Complexation of Phenylboronic Acid with Lactic acid. Stability Constant and Reaction Kinetics, J. Am. Chem. Soc., 1974, 96, 5381–5384 CrossRef CAS .
- S. Takahashi, S. Kurosawa and J.-i. Anzai, Electrochemical Determination of L-Lactate Using Phenylboronic Acid Monolayer-Modified Electrodes, Electroanalysis, 2008, 20, 816–818 CrossRef CAS .
- K. E. Secor and T. E. Glass, Selective Amine Recognition: Development of a Chemosensor for Dopamine and Norepinephrine, Org. Lett., 2004, 6, 3727–3730 CrossRef CAS PubMed .
- H. Kaiser, Die Berechnung der Nachweisempfindlichkeit, Spectrochim. Acta, 1947, 3, 40–67 CrossRef CAS .
- X. Kan, Y. Zhao, Z. Geng, Z. Wang and J.-J. Zhu, Composites of Multiwalled Carbon Nanotubes and Molecularly Imprinted Polymers for Dopamine Recognition, J. Phys. Chem. C, 2008, 112, 4849–4854 CrossRef CAS .
- D. Lakshmi, A. Bossi, M. J. Whitcombe, I. Chianella, S. A. Fowler, S. Subrahmanyam, E. V. Piletska and S. A. Piletsky, Electrochemical Sensor for Catechol and Dopamine Based on a Catalytic Molecularly Imprinted Polymer-Conducting Polymer Hybrid Recognition Element, Anal. Chem., 2009, 81, 3576–3584 CAS .
- J. Li, J. Zhao and X. Wei, A Sensitive and Selective Sensor for Dopamine Determination Based on a Molecularly Imprinted Electropolymer of o-Aminophenol, Sens. Actuators, B, 2009, 140, 663–669 CrossRef CAS .
- Y. Mao, Y. Bao, S. Gan, F. Li and L. Niu, Electrochemical Sensor for Dopamine Based on a Novel Graphene-Molecular Imprinted Polymers Composite Recognition Element, Biosens. Bioelectron., 2011, 28, 291–297 CrossRef CAS PubMed .
- D. Yu, Y. Zeng, Y. Qi, T. Zhou and G. Shi, A Novel Electrochemical Sensor for Determination of Dopamine Based on AuNPs@SiO2 Core–Shell Imprinted Composite, Biosens. Bioelectron., 2012, 38, 270–277 CrossRef CAS PubMed .
- X. Kan, H. Zhou, C. Li, Z. Xing and Z. Zhao, Imprinted Electrochemical Sensor for Dopamine Recognition and Determination Based on a Carbon Nanotube/Polypyrrole Film, Electrochim. Acta, 2012, 63, 69–75 CrossRef CAS .
- Y. Zeng, Y. Zhou, L. Kong, T. Zhou and G. Shi, A Novel Composite of SiO2-Coated Graphene Oxide and Molecularly Imprinted Polymers for Electrochemical Sensing Dopamine, Biosens. Bioelectron., 2013, 45, 25–33 CrossRef CAS PubMed .
- Y. Li, H. Song, L. Zhang, P. Zuo, B. Ye, J. Yao and W. Chen, Supportless Electrochemical Sensor Based on Molecularly Imprinted Polymer Modified Nanoporous Microrod for Determination of Dopamine at Trace Level, Biosens. Bioelectron., 2016, 78, 308–314 CrossRef CAS PubMed .
- J. L. Cuche, J. Prinseau, F. Selz, G. Ruget, J. Tual, L. Reingeissen, M. Devoisin, A. Baglin, J. Guédon and D. Fritel, Oral Load of Tyrosine or L-Dopa and Plasma Levels of Free and Sulfoconjugated Catecholamines in Healthy Men, Hypertension, 1985, 7, 81–89 CrossRef CAS PubMed .
- J. L. Cuche, J. Prinseau, F. Selz, G. Ruget and A. Baglin, Plasma Free, Sulfo- and Glucuro-Conjugated Catecholamines in Uremic Patients, Kidney Int., 1986, 30, 566–572 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Shoichi Nishitania and
Taira Kajisa
*a,
Shoichi Nishitania and
Taira Kajisa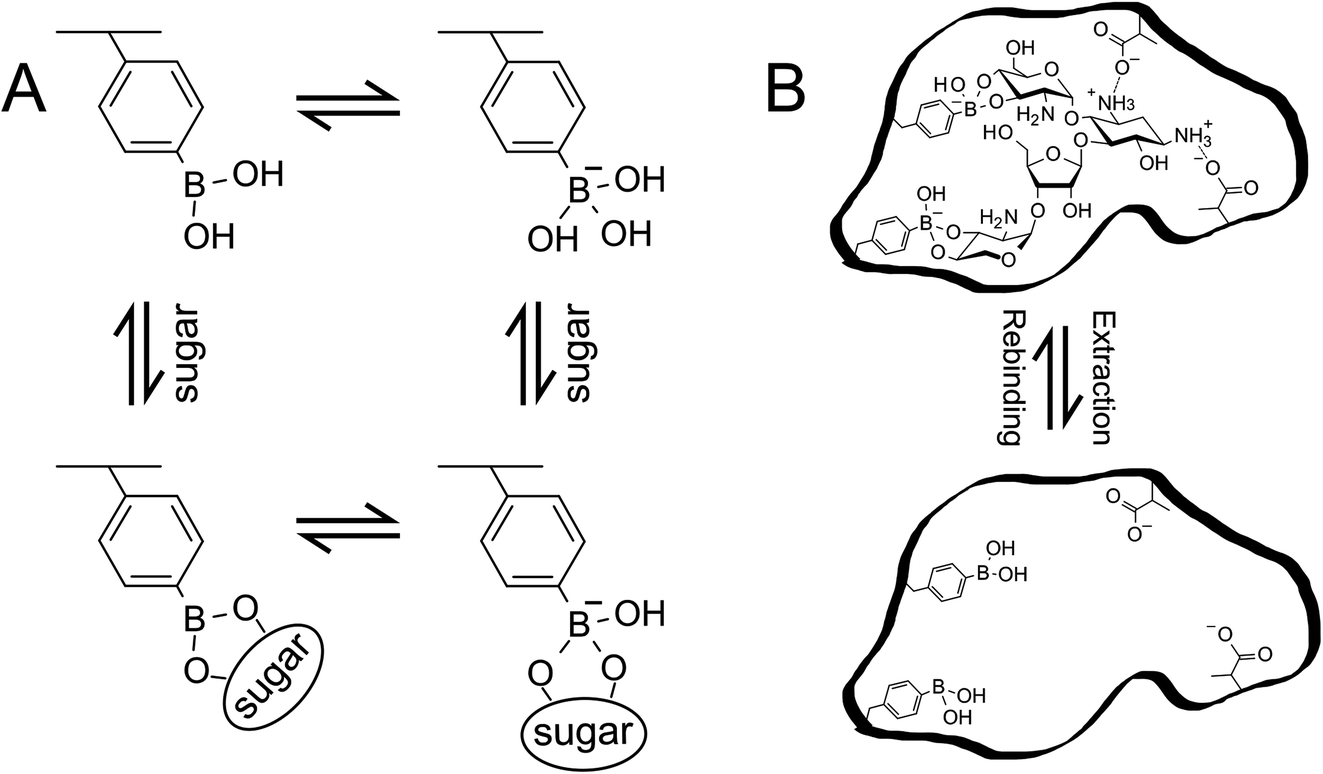

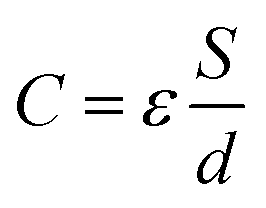 . The relative permittivity was lower in DA–MIP than in NIP despite the nearly identical thicknesses of the two layers. The different dielectric constants of DA–MIP and NIP might be explained by their morphological differences, whether with or without template cavities, in the hydrogels of a molecularly imprinted polymer and a nonimprinted random copolymer.
. The relative permittivity was lower in DA–MIP than in NIP despite the nearly identical thicknesses of the two layers. The different dielectric constants of DA–MIP and NIP might be explained by their morphological differences, whether with or without template cavities, in the hydrogels of a molecularly imprinted polymer and a nonimprinted random copolymer.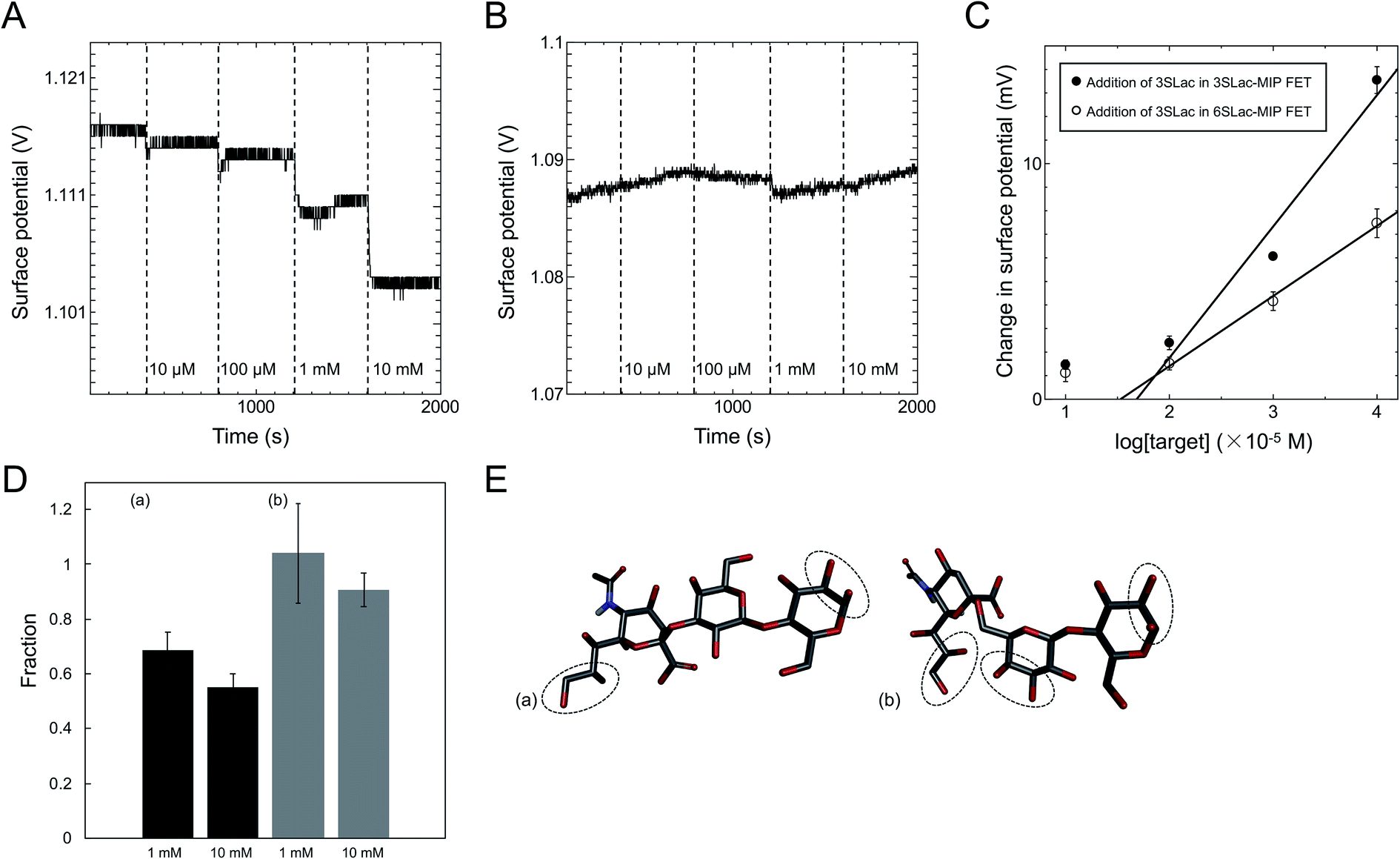
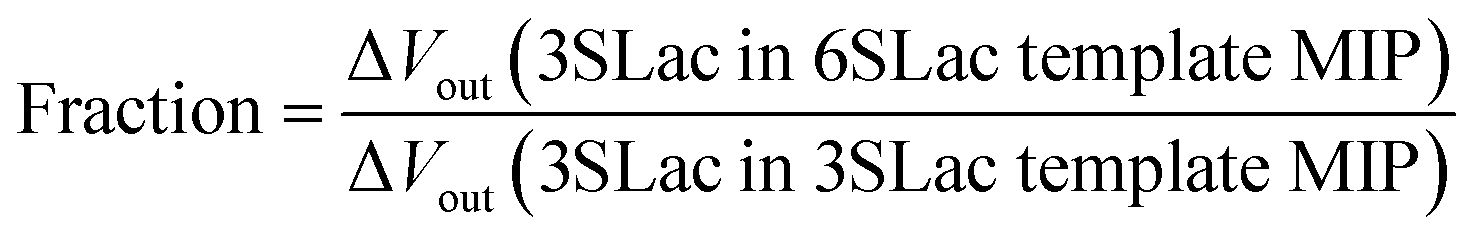
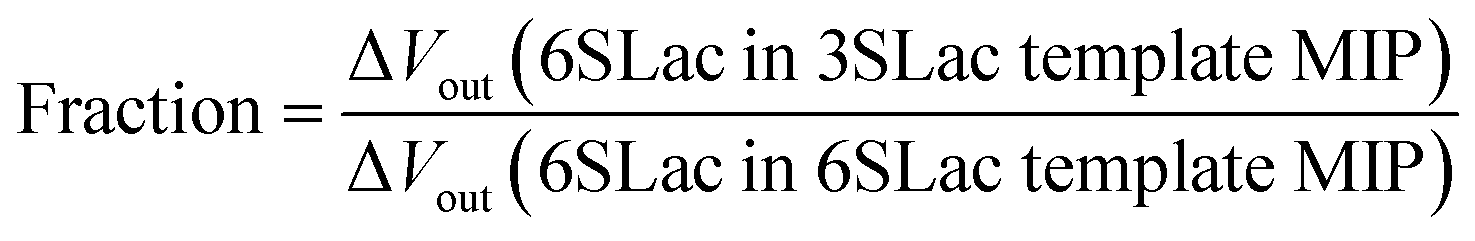
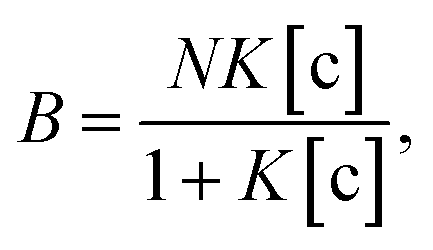
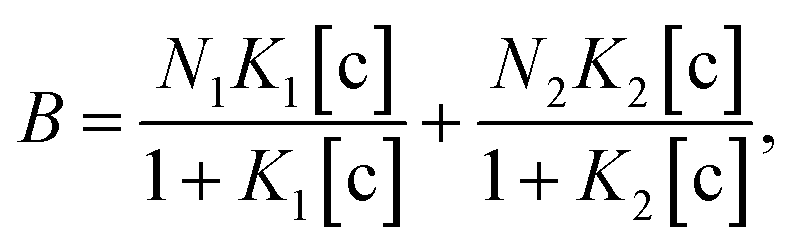
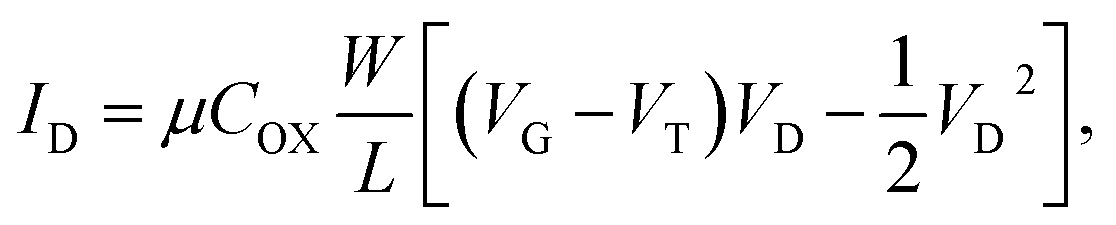
 is the channel width-to-length ratio, VD and VG are the applied drain–source and gate–source voltages, respectively, and VT is the threshold voltage, which can be expressed by55
is the channel width-to-length ratio, VD and VG are the applied drain–source and gate–source voltages, respectively, and VT is the threshold voltage, which can be expressed by55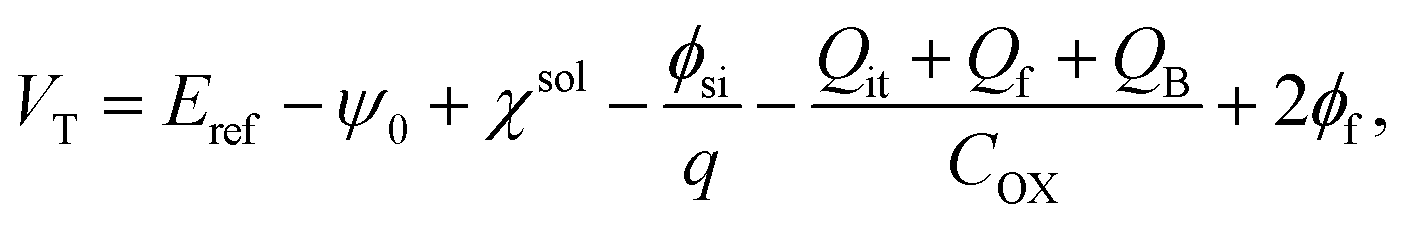
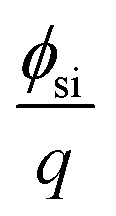 is the silicon electron work function, Qit, Qf, and QB are the charge of the interface traps, the fixed oxide charge, and the bulk depletion charge per unit area, respectively, and ϕf is the Fermi potential difference between the doped bulk silicon and the intrinsic silicon.
is the silicon electron work function, Qit, Qf, and QB are the charge of the interface traps, the fixed oxide charge, and the bulk depletion charge per unit area, respectively, and ϕf is the Fermi potential difference between the doped bulk silicon and the intrinsic silicon.
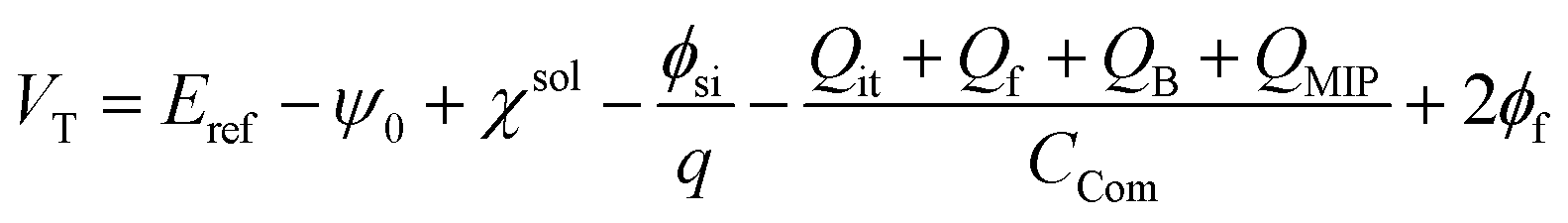
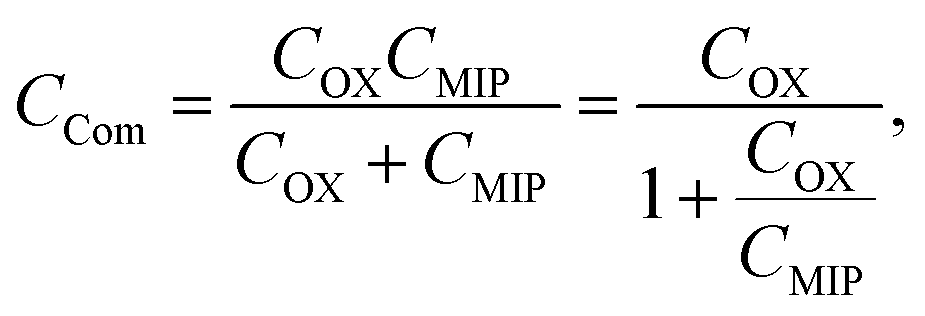
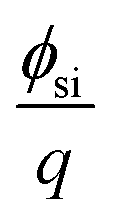 , Qit, Qf, QB, and ϕf should be the same before and after the molecular recognition events at the MIP interface. Thus, the signal response obtained using the FET sensor is based on the change in VT (ΔVT); therefore, ΔQMIP should be evaluated in this study, in accordance with eqn (7) and the above considerations.
, Qit, Qf, QB, and ϕf should be the same before and after the molecular recognition events at the MIP interface. Thus, the signal response obtained using the FET sensor is based on the change in VT (ΔVT); therefore, ΔQMIP should be evaluated in this study, in accordance with eqn (7) and the above considerations.
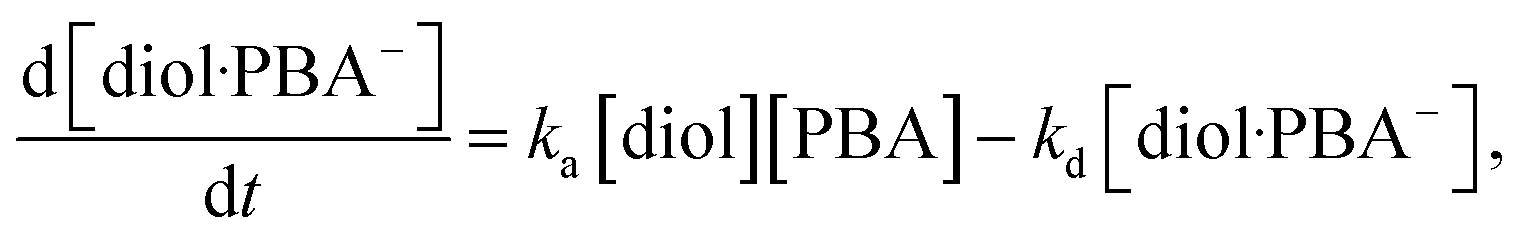
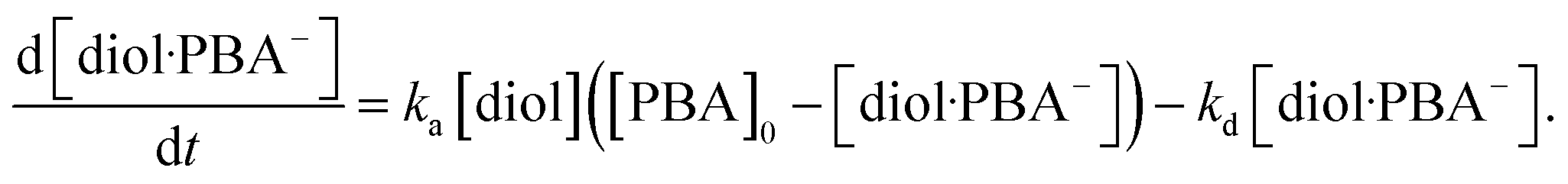
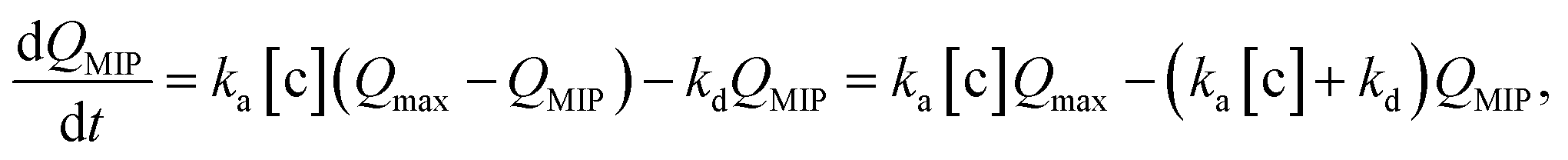
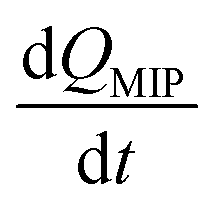 is the rate of formation of the associated complex (diol·PBA−) in the MIP membrane (on the Au gate) and [c] is the concentration of the analyte (diol) in the solutions. Moreover, integrating eqn (12) gives
is the rate of formation of the associated complex (diol·PBA−) in the MIP membrane (on the Au gate) and [c] is the concentration of the analyte (diol) in the solutions. Moreover, integrating eqn (12) gives