
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dimers of pyrrolo-annelated indenofluorene-extended tetrathiafulvalenes – large multiredox systems†
Line Broløs and
Mogens Brøndsted Nielsen *
*
Department of Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark. E-mail: mbn@chem.ku.dk
First published on 16th April 2020
Abstract
Novel scaffolds of indenofluorene (IF)-extended tetrathiafulvalenes (TTF) were synthesized starting from a new pyrrolo-annelated IF-TTF monomer. Rigid para- and meta-phenylene linked dimers were obtained via N-arylation reactions of the monomer, and their optical and redox properties were elucidated by UV-Vis absorption spectroscopy and cyclic and differential pulse voltammetries.
Tetrathiafulvalene (TTF) is a redox-active unit that reversibly undergoes two sequential one-electron oxidations, forming first a radical cation (TTF+) and subsequently a dication (TTF2+) containing two aromatic 1,3-dithiolium rings, and it is due to these redox properties that it is an attractive unit for materials and supramolecular chemistries.1
Extension of the conjugated system, leading to so-called extended TTFs, has successfully been used as a tool to finely tune the redox properties and geometries of the various redox states.2 For example, introduction of an indeno[1,2-b]fluorene (IF) core3 has provided indenofluorene-extended TTFs of the general structure IF-TTF shown in Fig. 1. X-Ray crystallographic and computational studies reveal that all three redox states (0, +1, +2), generated in sequential and reversible steps, take a fully planar structure, and spectroelectrochemical studies have shown that the individual redox states exhibit significantly redshifted absorptions relative to those of TTF, TTF+, and TTF2+, respectively.2
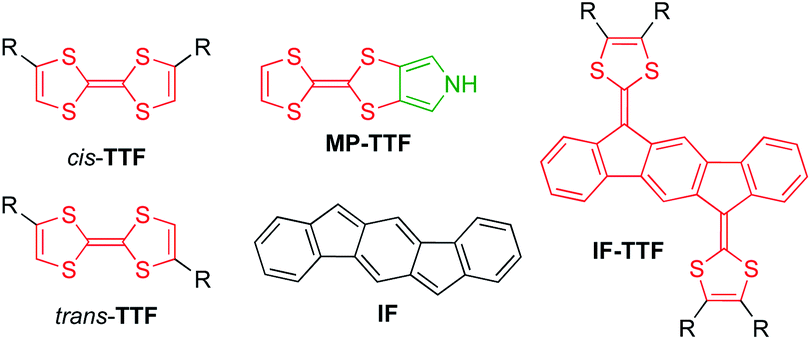 | ||
| Fig. 1 Structures of cis/trans-isomeric TTFs, mono-pyrrolo-TTF (MP-TTF), indenofluorene (IF) and an indenofluorene-extended TTF (IF-TTF). | ||
Recently, we developed synthetic protocols for linking together two IF-TTF units via anchoring at a peripheral position of each dithiafulvene unit.4 Such dimers unfortunately exist as unseparable mixtures of cis and trans isomers (cf., the disubstituted TTFs shown in Fig. 1), and to avoid this problem of isomerism we decided to develop a synthetic protocol for fusing a pyrrole unit to one of the dithiole rings of IF-TTF as in target molecule 1 shown in Fig. 2. Indeed, the related mono-pyrrolo-annelated TTF (MP-TTF, Fig. 1) and bis-pyrrolo-annelated TTF have proven important as versatile π-donor building blocks in macromolecular and supramolecular chemistry.5
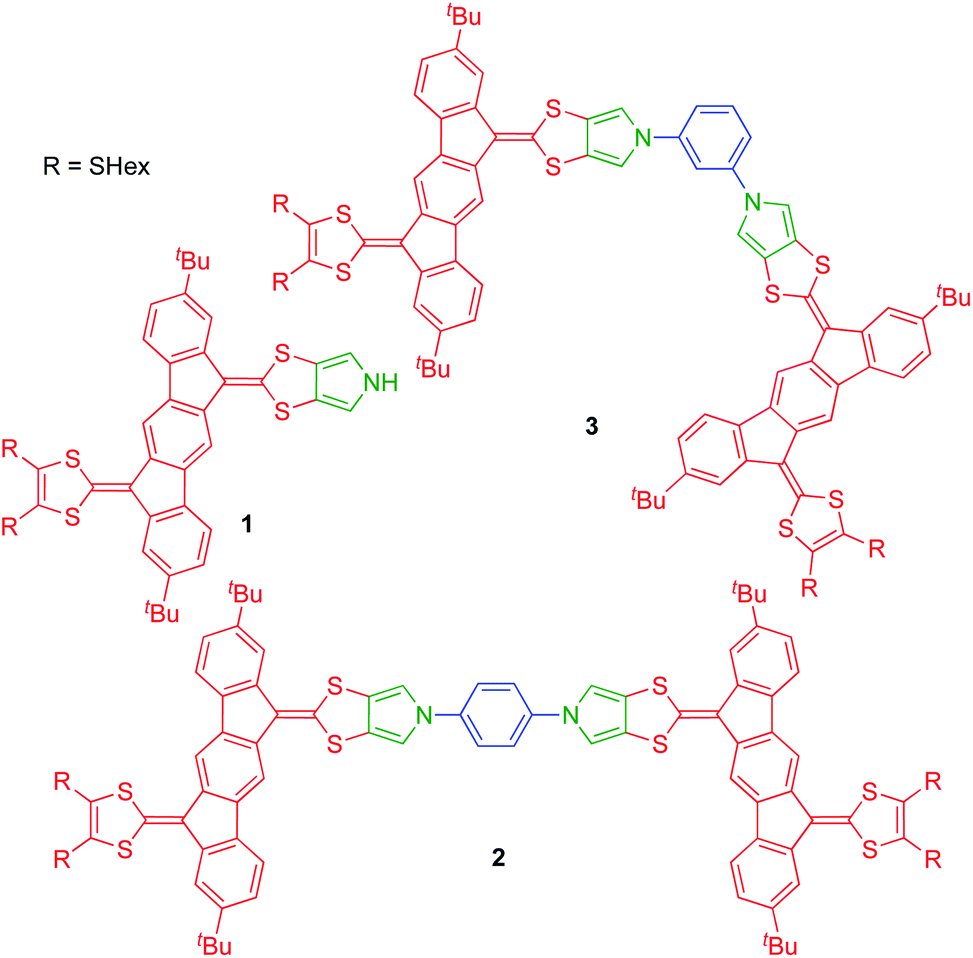 | ||
| Fig. 2 New pyrrolo-annelated IF-TTF mono- and dimers. | ||
Dimerization of two units 1 via its nitrogen atom and a suitable linker would prevent formation of isomers. As linkers we decided to explore rigid phenylene units as in target molecules 2 (para-phenylene bridge) and 3 (meta-phenylene bridge) shown in Fig. 2. Previously prepared cis/trans isomeric IF-TTF dimers had flexible linkers and showed intramolecular associations upon oxidation,4 which would be prevented by these rigid linkers. Moreover, intermolecular interactions are to a large extent prevented by the peripheral tert-butyl substituents that were chosen as substituent groups to enhance solubility of the dimers.
Synthesis of 1 proceeds according to Scheme 1, employing the known diketone 4,4 the N-tosyl-protected pyrrolo-annelated 1,3-dithiole-2-thione I5a and the phosphonate ester II3b as precursors. A phosphite-mediated coupling between 4 and I was carried out to give mono-olefinated product 5 in a yield of 61%. This compound was next subjected to a Horner–Wadsworth–Emmons olefination with compound II, deprotonated by sodium hexamethyldisilazide (NaHMDS), providing the tosyl-protected mono-pyrrolo IF-TTF 6 in good yield (76%). This compound was subsequently deprotected using NaOMe to give in almost quantitative yield the monopyrrolo IF-TTF 1 with a pyrrole N–H unit available for further reactions.
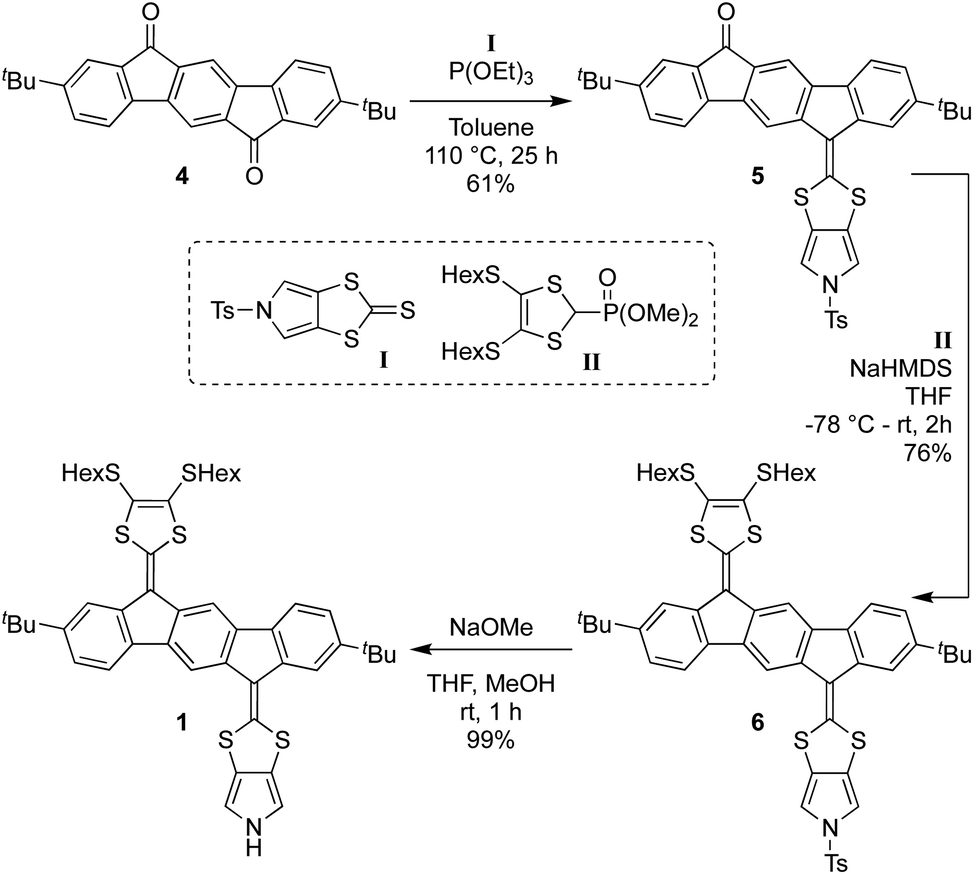 | ||
| Scheme 1 Synthesis of monomer 1 by stepwise olefination reactions. NaHMDS = sodium hexamethyldisilazide. | ||
With monomer 1 in hand, N-arylation reactions with 1,4- and 1,3-diiodobenzene, catalyzed by an excess of CuI and (±)-1,2-trans-diaminocyclohexane, were conducted in THF at reflux (Scheme 2). This procedure, previously applied for N-arylation of pyrrolo-annelated TTFs,6 yielded dimers 2 and 3, respectively. The procedure worked best for 1,3-diiodobenzene. For the arylation reaction with 1,4-diiodobenzene it was observed that the first arylation progressed rather willingly, as the mono-arylated product 7 could be isolated in 91% yield after only 3 hours. Only when compound 7 was subjected to significantly longer reaction time, however, formation of dimer 2 was observed, and the compound was isolated in 9% yield after 18 hours of reaction time. This result indicates that the substitution of an iodide on the benzene ring with one IF-TTF unit, in the para position, decreases the reactivity of the second iodide significantly, possibly due to the strongly electron-donating character of the pyrrolo-TTF. Albeit inconvenient in the current work, it could be a potential advantage for stepwise construction of unsymmetrical scaffolds. For the corresponding reaction with 1,3-diiodobenzene smooth formation of the dimer 3 was observed, and this product was isolated in 23% after 16 hours, while no mono-arylated intermediate could be isolated.
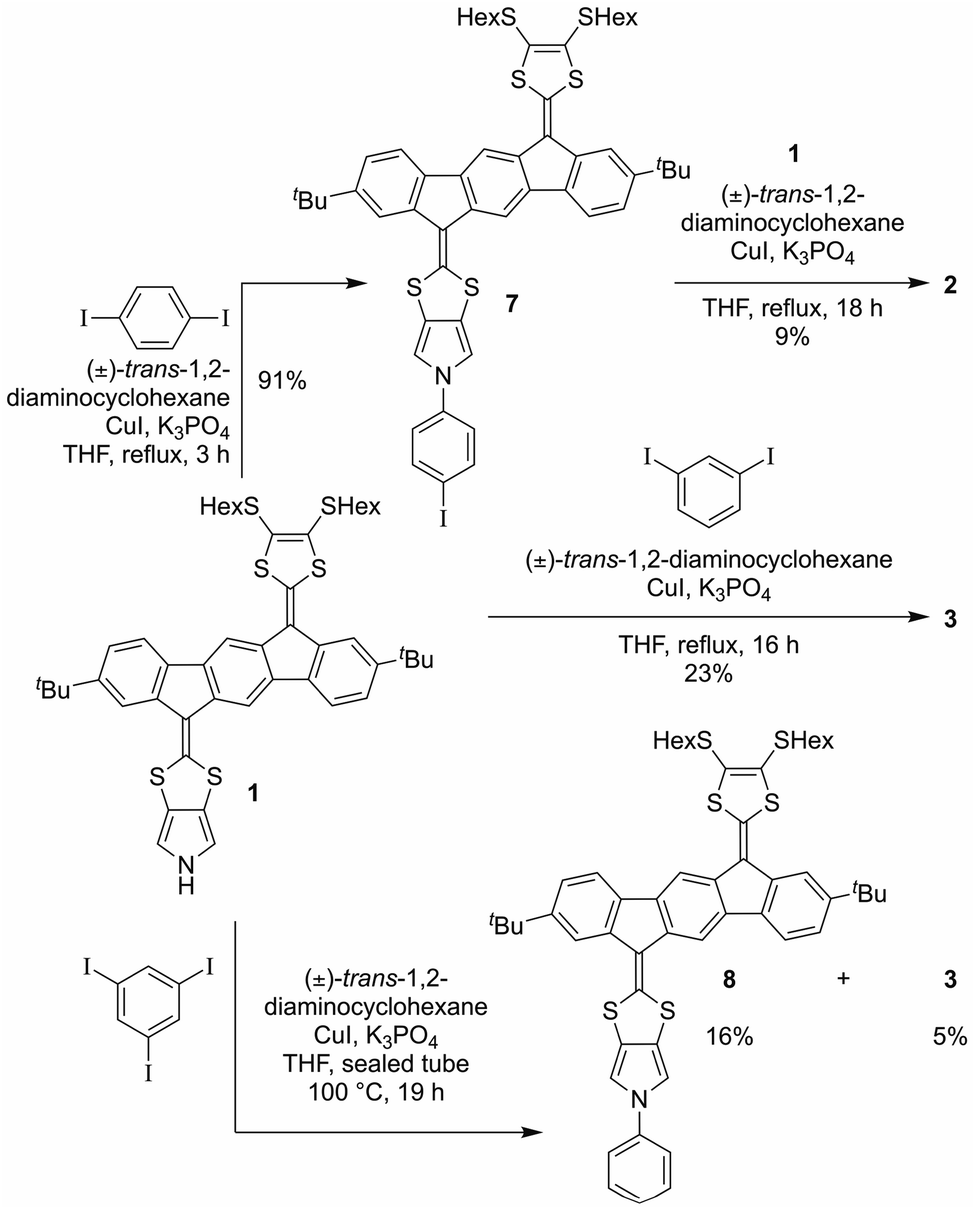 | ||
| Scheme 2 Synthesis of dimers 2 and 3, and of mono-arylated species 7 and 8. | ||
Synthesis of a trimer consisting of three IF-TTF units around one central benzene ring was also attempted by coupling of monomer 1 and 1,3,5-triiodobenzene. However, when employing the conditions proven successful for synthesis of the dimers, no reaction was observed; instead, 86% of the starting material was re-isolated. Nevertheless, when conducting the reaction in a sealed vial and heating to 100 °C, full conversion of monomer 1 was observed. However, the isolated products were mono-arylated monomer 8 and previously isolated dimer 3, and not the desired trimer. This result signals again that the reactivity of the iodides in the arylation reaction decreases upon introduction of IF-TTF units. Upon elevated pressure, as applied in the attempt to achieve the desired trimer, a competing reaction by which the iodides are substituted for hydrogen atoms is observed to exceed the arylation reaction. The mono-arylated compound 8 was used as a reference compound in subsequent studies of the synthesized dimers.
The photophysical properties of monomers 1 and 8 as well as dimers 2 and 3 were investigated by UV-Vis absorption spectroscopy in CH2Cl2 at 25 °C (Fig. 3). Absorption maxima and extinction coefficients are listed in Table 1. The longest-wavelength absorption maximum of 1 (468 nm) is close to that of the related IF-TTF with four peripheral SEt substituents (473 nm; R = SEt in Scheme 1).3a Expansion of the π-system with a benzene ring (compound 8) had little effect on the longest-wavelength absorption maximum (471 nm). Linking the monomeric units by phenylene linkers, dimers 2 and 3, did not change the longest-wavelength absorption maxima significantly either, but the intensity of the absorption was expectedly doubled (or slightly more than doubled).
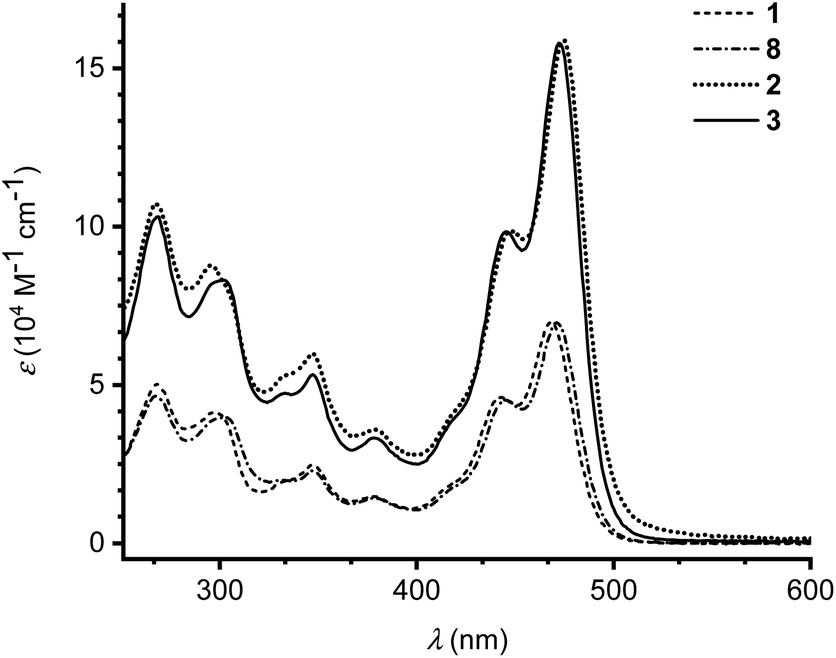 | ||
| Fig. 3 UV-Vis absorption spectra of monomer 1 (dashed line), monomer 8 (dot dash dot) and dimers 2 (dotted line) and 3 (full line) in CH2Cl2 at 25 °C. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CH2Cl2/C6H5Cl, for compounds 1, 2, 3 and 8
:1 CH2Cl2/C6H5Cl, for compounds 1, 2, 3 and 8
| Compound | λmax/nm (ε/104 M−1 cm−1) | Eox (V vs. Fc/Fc+) |
|---|---|---|
| 1 | 468 (6.99), 443 (4.61), 378 (1.47), 348 (2.46), 298 (4.13), 267 (5.00) | +0.18 (1e), +0.36 (1e) |
| 2 | 475 (15.9), 448 (9.88), 379 (3.59), 347 (5.98), 295 (8.78), 267 (10.7) | +0.11 (1e), +0.44 (2e) |
| 3 | 472 (15.8), 445 (9.84), 379 (3.32), 347 (5.32), 302 (8.31), 269 (10.3) | +0.14 (1e), +0.20 (1e), +0.41 (2e) |
| 8 | 471 (6.97), 444 (4.52), 381 (1.41), 347 (2.29), 302 (3.98), 268 (4.65) | +0.22 (1e), +0.33 (1e) |
Electrochemical studies of the synthesized compounds were conducted in 1:1 mixture of CH2Cl2/C6H5Cl containing 0.1 M NBu4PF6 as supporting electrolyte, and the cyclic voltammograms (CVs) and differential pulse voltammograms (DPVs) are shown in Fig. 4. Chlorobenzene was needed as co-solvent due to limited solubility of the dimers in neat CH2Cl2. Oxidation potentials are listed in Table 1 (taken from the DPVs), referenced against the ferrocene/ferrocenium (Fc/Fc+) redox couple (recorded in a separate experiment). For monomer 1 two reversible, one-electron oxidations were observed, at +0.18 and +0.36 V vs. Fc/Fc+, forming the radical cation and the dication, respectively. Similarly, monomer 8 was found to undergo two reversible one-electron oxidations at +0.22 and +0.33 V vs. Fc/Fc+. For dimer 3 two reversible, one-electron oxidations were observed, at +0.14 and +0.20 V vs. Fc/Fc+, forming the radical cation and the dication, respectively, followed by a reversible two-electron oxidation, at 0.41 V vs. Fc/Fc+, forming the tetracation. The electrochemistry of dimer 2 is, however, more complicated. It exhibits a reversible one-electron oxidation at +0.11 V vs. Fc/Fc+, hence at lower potential than for dimer 3 in accordance to the large linearly conjugated system provided by a para-phenylene bridge. The second oxidation seemed, however, to occur over a very broad potential range. As known from literature,7 the isolated 1,4-di(N-pyrrolyl)benzene unit itself undergoes an irreversible oxidation, and in the case of dimer 2 it seems that the redox properties are affected significantly by this structural unit of the molecule; dimer 2 thereby acts less like a ‘classical’ extended TTF. The CV may as well be complicated by intermolecular interactions despite the bulky tert-butyl groups present on the IF cores. A reversible two-electron oxidation, possibly due to formation of the tetracation or higher oxidation states, is observed at +0.44 V vs. Fc/Fc+, indicating that while the second oxidation wave is significantly broadened the reversibility is intact.
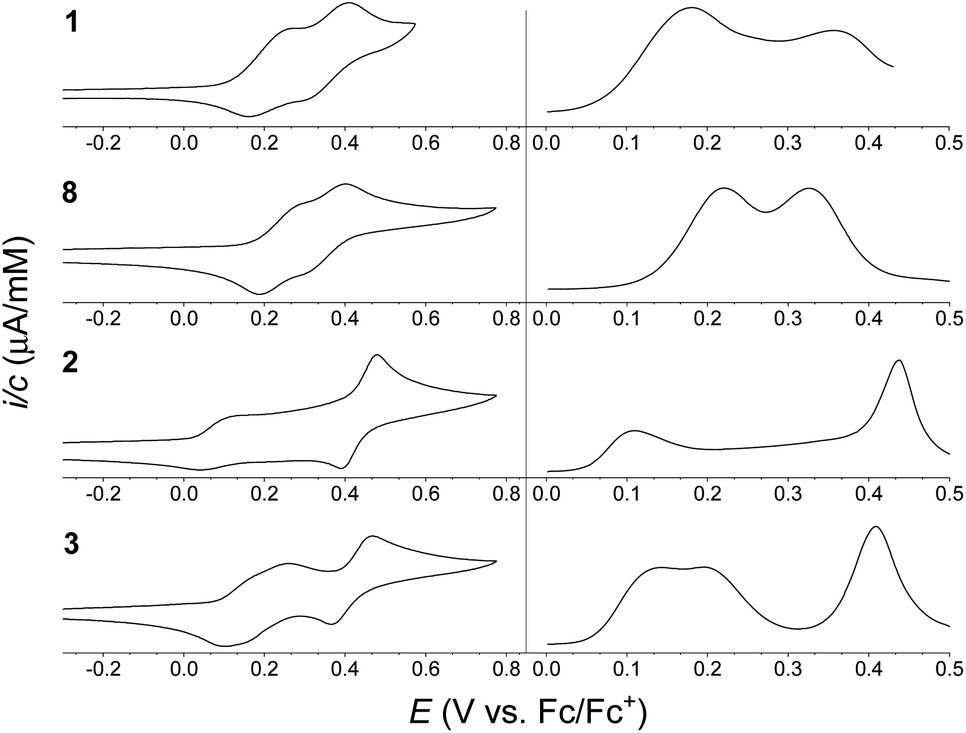 | ||
| Fig. 4 Cyclic voltammograms (CVs) (left) and differential pulse voltammograms (DPVs) (right) of (from the top) 1 (0.38 mM), 8 (0.38 mM), 2 (0.37 mM) and 3 (0.43 mM); potentials vs. Fc/Fc+ (solvent: 1:1 CH2Cl2/C6H5Cl; supporting electrolyte: 0.1 M NBu4PF6; scan rate: 0.1 V s−1). Oxidation potentials listed in Table 1 are based on DPVs. | ||
In conclusion, we have developed a convenient synthetic procedure to obtain the first pyrrolo-annelated IF-extended TTF that was successfully dimerized by N-arylation reactions. The resulting rigid dimers present new interesting multiredox systems to be explored further in future work. The possibility to perform functionalization at the α-carbon atoms of the pyrrole unit of these new scaffolds will also be interesting to pursue, taking advantage of the elaborate chemistry that has been developed for the parent mono- and bis-pyrrolo-annelated TTFs.5
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Emeritus Ole Hammerich for fruitful discussions regarding the electrochemical studies.Notes and references
- (a) M. B. Nielsen, C. Lomholt and J. Becher, Chem. Soc. Rev., 2000, 29, 153 RSC; (b) P. Batail, Special issue on Molecular Conductors, Chem. Rev., 2004, 104, 4887 CrossRef CAS; (c) M. Mas-Torrent and C. Rovira, J. Mater. Chem., 2006, 16, 433 RSC; (d) D. Canevet, M. Sallé, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245 RSC; (e) M. Hasegawa and M. Iyoda, Chem. Soc. Rev., 2010, 39, 2420 RSC; (f) H. Kobayashi, A. Kobayashi and H. Tajima, Chem.–Asian J., 2011, 6, 1688 CrossRef CAS PubMed; (g) N. Martín, Chem. Commun., 2013, 49, 7025 RSC; (h) P. J. Skabara and M. Sallé, Tetrathiafulvalene chemistry, Beilstein J. Org. Chem., 2015, 11, 1528 CrossRef CAS PubMed; (i) J. J. Bergkamp, S. Decurtins and S.-X. Liu, Chem. Soc. Rev., 2015, 44, 863 RSC; (j) V. A. Azov, Tetrahedron Lett., 2016, 57, 5416 CrossRef CAS; (k) A. Jana, M. Ishida, J. S. Park, S. Bähring, J. O. Jeppesen and J. L. Sessler, Chem. Rev., 2017, 117, 2641 CrossRef CAS PubMed; (l) H. Yamada, M. Yamashita, H. Hayashi, M. Suzuki and N. Aratani, Chem.–Eur. J., 2018, 24, 18601 CrossRef CAS PubMed; (m) H. V. Schröder and C. A. Schalley, Beilstein J. Org. Chem., 2018, 14, 2163 CrossRef PubMed.
- (a) J. Roncali, J. Mater. Chem., 1997, 7, 2307 RSC; (b) A. Gorgues, P. Hudhomme and M. Sallé, Chem. Rev., 2004, 104, 5151 CrossRef CAS PubMed; (c) P. Frère and P. J. Skabara, Chem. Soc. Rev., 2005, 34, 69 RSC; (d) M. B. Nielsen and O. Hammerich, J. Mater. Chem. C, 2019, 7, 2809 RSC.
- (a) M. A. Christensen, C. R. Parker, T. J. Sørensen, S. de Graaf, T. J. Morsing, T. Brock-Nannestad, J. Bendix, M. M. Haley, P. Rapta, A. Danilov, S. Kubatkin, O. Hammerich and M. B. Nielsen, J. Mater. Chem. C, 2014, 2, 10428 RSC; (b) M. Mansø, M. Koole, M. Mulder, I. J. Olavarria-Contreras, C. L. Andersen, M. Jevric, S. L. Broman, A. Kadziola, O. Hammerich, H. S. J. van der Zant and M. B. Nielsen, J. Org. Chem., 2016, 81, 8406 CrossRef PubMed; (c) J. F. Petersen, C. K. Frederickson, J. L. Marshall, G. E. Rudebusch, L. N. Zakharov, O. Hammerich, M. M. Haley and M. B. Nielsen, Chem.–Eur. J., 2017, 23, 13120 CrossRef CAS PubMed.
- L. Broløs, M. D. Kilde, O. Hammerich and M. B. Nielsen, J. Org. Chem., 2020, 85, 3277–3286 CrossRef PubMed.
- (a) J. O. Jeppesen, K. Takimiya, F. Jensen and J. Becher, Org. Lett., 1999, 1, 1291 CrossRef CAS; (b) C. P. Collier, J. O. Jeppesen, Y. Luo, J. Perkins, E. W. Wong, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2001, 123, 12632 CrossRef CAS PubMed; (c) J. O. Jeppesen and J. Becher, Eur. J. Org. Chem., 2003, 3245 CrossRef CAS; (d) L. J. O'Driscoll, S. S. Andersen, M. V. Solano, D. Bendixen, M. Jensen, T. Duedal, J. Lycoops, C. van der Pol, R. E. Sørensen, K. R. Larsen, K. Myntman, C. Henriksen, S. W. Hansen and J. O. Jeppesen, Beilstein J. Org. Chem., 2015, 11, 1112 CrossRef PubMed; (e) A. Jana, S. Bähring, M. Ishida, S. Goeb, D. Canevet, M. Sallé, J. O. Jeppesen and J. L. Sessler, Chem. Soc. Rev., 2018, 47, 5614 RSC.
- (a) Y. Salinas, M. V. Solano, R. E. Sørensen, K. R. Larsen, J. Lycoops, J. O. Jeppesen, R. Martínez-Máñez, F. Sancenón, M. D. Marcos, P. Amorós and C. Guillem, Chem.–Eur. J., 2014, 20, 855 CrossRef CAS PubMed; (b) M. V. Solano, E. A. Della Pia, M. Jevric, C. Schubert, X. Wang, C. van der Pol, A. Kadziola, K. Nørgaard, D. M. Guldi, M. B. Nielsen and J. O. Jeppesen, Chem.–Eur. J., 2014, 20, 9918 CrossRef CAS PubMed.
- P. E. Just, K. I. Chane-Ching, J. C. Lacroix and P. C. Lacaze, J. Electroanal. Chem., 1999, 479, 3 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, NMR and UV-Vis absorption spectra. See DOI: 10.1039/d0ra02787a |
| This journal is © The Royal Society of Chemistry 2020 |