
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeep insight into reasons for the mechanoluminescence phenomenon of triphenylamine-substituted imidazoles and their distinct mechano-fluorochromic behaviours†
Xiao-Jing Liu,
Guan-Lei Gao,
Hao Jiang,
Yan-Rong Jia and
Min Xia *
*
Department of Chemistry, Zhejiang Sci-Tech University, Hangzhou 310018, P. R. China. E-mail: xiamin@zstu.edu.cn
First published on 17th June 2020
Abstract
Three imidazoles with different numbers of fused aromatic rings have been prepared by the respective introduction of triphenylamine and 4-cyanophenyl at the N1 and C2 positions in the imidazole ring. Each imidazole effectively exhibits positive solvatochromism, and that of benzo[d]imidazole is the most significant. Although these imidazole crystals have centrosymmetric space groups, they are all ML-active. It was verified by DFT calculations based on X-ray crystallography that some molecular couples with strong intermolecular interactions possess large net dipole moments that should be dominantly responsible for the ML behaviours of these crystals. Moreover, the considerably high molecular dipole moments of the three imidazoles also make a great contribution to good ML effects. Based on this triphenylamine-substituted imidazole system, the relationships among space groups, molecular dipole moments, polar molecular couples and the ML phenomenon are made clear for the first time. Unlike the remarkable MFC activities on imidazole and benzo[d]midazole crystals, phenanthro[9,10-d]imidazole is MFC-inert, and this may well be attributed to strong intramolecular C–H⋯π interactions, which make the rotation of triphenylamine nearly impossible under force stimuli.
Introduction
Mechanoluminescence (ML), an interesting optical phenomenon in which a flash of light is produced by mechanical action performed on materials,1 was first recorded by Francis Bacon in 1605. Since then, various types of ML-active molecules including inorganic, organic, metal chelates, and polymers have been discovered or designed. Due to their potential applications, which include non-destructive stress or crack sensors,2 internal light sources for bioimaging and phototherapy3 or photocatalysis,4 intelligent solid lubricants,5 fraud protection,6 solar energy converters,7 smart skin8 and so on, the study of ML-active compounds has once again come to the centre stage of material chemistry. Compared to ML-active materials involved with inorganic, polymeric, or metal chelated molecules, those composed of purely organic molecules that have advantages like single components, ready preparation, wide-range emission wavelength, low toxicity and simple processing have been the focus in recent years. For the vast majority of purely organic ML-active crystals, the ML phenomenon is induced by fracture under force stimuli and is also closely dependent on molecular packing. Hence, such reported ML-active systems have, so far, been isolated or serendipitous events since packing modes in crystals cannot be well predicted. Any systematic strategy that can guide people to produce purely organic ML-active crystals is still urgently needed.Although the ML phenomenon more frequently occurs on crystals with non-centrosymmetric space groups and the piezoelectric effect is, therefore, widely believed to be the feasible reason, more and more centrosymmetric crystals have been reported to display outstanding ML performance.9 Conversely, ML inactivity also emerges on some crystals with non-centrosymmetric space groups;9a hence, there is no full correlation between ML activity and the piezoelectric effect. Substantial experiments and detailed investigations have revealed10 that the ML effect, in most cases, can be induced by electron discharge released from freshly formed surfaces when crystals are fractured. The recombination of these electrons with such surfaces, motivated by the neutralization of charges, will lead to electron bombardment, whose energy is strong enough to excite molecules on newly cracked surfaces and produce the subsequent intense emission of those excited molecules. Hence, whether crystals have non-centrosymmentric or centrosymmetric space groups is not yet critical, as long as freshly cracked surfaces are polar and electron discharge can take place on them. However, in spite of the above mechanism, which clearly elucidates the elementary physical process of the ML phenomenon, it does not present any clue regarding how to obtain polar cracked surfaces in view of the packing mode and molecular features.
Nishida's group11 proposed that molecules with large dipole moments are very helpful in building ML-active crystals, but they still claimed that non-centrosymmetric space groups should be the indispensable condition. Although it was Li's group12 who firstly connected ML phenomena with the net dipole moments of molecular couples in one of their papers, they also attributed the ML activity of their crystal to its piezoelectric effect, even if the crystal had a centrosymmetric P21/n space group that must not possess such an effect. Obviously, the above considerations of the relationship among ML activity, space groups, molecular dipole moments and polar molecular couples are still quite equivocal. In fact, to our best knowledge, there has not yet been any clear understanding of such relationships so far in reported works. Herein, we present our deep insight into the mechanism for the ML effect on purely organic crystals packed by polar molecules, and hope that this pioneering knowledge could effectively guide people to improve the probability in the acquisition of ML-active crystals. According to our hypothesis, we assume that both the arrangement pattern of molecules in molecular couples with strong interactions and the dipole moment of the molecule itself are actually the significant elements for ML phenomena rather than the space group or piezoelectric effect. If some molecular couples involving two molecules in a non-anti-parallel arrangement really exist in a crystal, whether the crystal has a non-centrosymmetric or centrosymmetric space group, the polar cracked surfaces can surely be formed to produce electron discharge on them, only if the crystal fracture can trigger the separation of the two molecules from each other. From a statistical point of view, such force-induced disassembly of molecular couples must take place when crystals are fractured. Of course, the high molecular dipole moment is favourable for couples to hold large net dipole moments, which are preferable for generating many highly polar cracked surfaces.
In order to prove that the above hypothesis is reliable, we developed three imidazoles by respectively introducing 4-cyanophenyl and triphenylamine (TPA) at the C2 and N1 positions on the imidazole core (Fig. 1), and the molecular dipole moments of IMTPA, BIMTPA and PHIMTPA are 8.08, 6.40 and 7.11 debye, which are quite larger than other common organic small molecules. Although each crystal is centrosymmetric, it was found that the involved double molecules in some molecular couples with strong interactions were still arranged in a non-anti-parallel way. Thanks to the very high molecular dipole moments and the formation of non-centrosymmetric molecular couples, the three crystals expectedly displayed readily observable ML effects. Therefore, the occurrence of ML-active crystals seemed to change from the unpredictable to the predictable event.
 | ||
| Fig. 1 Structures of IMTPA, BIMTPA, PHIMTPA and their molecular dipole moments in the crystalline phase. | ||
Another solid-state photophysical property that is closely related to packing modes and intermolecular interactions is mechanofluorochromism (MFC),13a–e and materials with the MFC effect have been widely applied as pressure or chemical sensors,14 memory chips,15 security inks16 or papers,17 and optical data storage devices.18 Similar to many other reported imidazoles,19 IMTPA and BIMTPA also display good MFC activities. However, PHIMTPA is MFC-inactive, as force stimuli can only trigger the disappearance of fine vibration structures rather than the wavelength shift of emission spectra. X-ray crystallography revealed that strong intramolecular interactions occur between the phenanthrenyl and the phenyl group at the triphenylamine directly linked to the imidazole ring, which hinder the rotation of the phenyl group from offering a more planarized conformation and red-shifted emission wavelength when PHIMTPA lattices collapse under force stimuli. In this work, we investigate the photophysical behaviours of the three imidazoles in both the solution and solid states, presenting deep insight into the nature of these behaviours at the single molecular and supramolecular levels.
Results and discussion
Photophysical properties in solution
The absorption and emission behaviours of the three imidazoles in different solvents were surveyed (Fig. 2). Both the curve profiles and the peak positions of the absorption spectra are almost independent of solvents. The molar extinction coefficients of IMTPA, BIMTPA and PHIMTPA in CH2Cl2 are respectively 1.85, 2.68 and 1.96 × 104 M−1 cm−1. However, as the solvent polarity increased, the emission wavelengths gradually red-shifted and the positive solvatochromic effect occurred on each imidazole, which means that considerable intramolecular charge transfer (ICT) should take place on these molecules when they are excited. The Lippert–Mataga plots of the three imidazoles (Fig. S18–S20, ESI†) possess good linear correlation and the dipole moments of the excited molecules were respectively enhanced by 5.21 D for IMTPA, 4.27 D for BIMTPA and 5.06 D for PHIMTPA. Moreover, the different ICT extents can be distinguished by the direct observation of emission intensity in highly polar solvents via the naked eyes. In acetone and acetonitrile, the fluorescence of IMTPA was still very strong, whereas it was dramatically diminished for PHIMTPA or even fully quenched for BIMTPA. The measured quantum yields in acetonitrile were 0.245 for IMTPA, 0.093 for PHIMTPA and 0.012 for BIMTPA. Accordingly, the order of the extent of ICT should be BIMTPA > PHIMTPA > IMTPA. The DFT calculation results (Fig. S1, ESI†) also indicated this order. It was illustrated that a complete charge transfer from the TPA moiety on the HOMO to the 4-cyanophenyl-benzimidazole moiety on the LUMO appeared in BIMTPA, while the electrons were partly shifted between the HOMO and LUMO in IMTPA, since the electrons on the HOMO are dislocated across the entire molecule. Very differently in PHIMTPA, neither the HOMO nor LUMO was filled by the electrons from the TPA moiety, and the electron shift only occurred between the HOMO and LUMO at the 2-(4-cyanophenyl)phenanthrol[9,10-d]imidazole moiety. Also, we prepared the analogue PHIMCN without TPA at the N1 position, and its emission spectrum in n-hexane is almost identical to that of PHIMTPA (Fig. S2, ESI†). Such consistence indicates that some stable vibration levels are the same for both PHIMTPA and PHIMCN, while the TPA indeed does not act as the electron-donating group for PHIMTPA in solution. These stable vibrations could have originated from the phenanthrol [9,10-d]imidazole core (PHIM), as the emission curve profile of PHIM in n-hexane (Fig. S2, ESI†) is exactly like those of both PHIMTPA and PHIMCN, and the red-shifted wavelengths of PHIMTPA and PHIMCN could be ascribed to the π-conjugation of 4-cyanophenyl with the PHIM core. As the solvent polarity increases, the energy gaps between vibration levels are reduced due to strong solvation, dispersing the fine structures into a wide and continuous emission band. Apparently, as the fused polycyclic system was enlarged, the stability of the vibration levels correspondingly improved, turning the emission band from structureless in IMTPA to having several small shoulder peaks in BIMTPA or even some separated sharp peaks in PHIMTPA.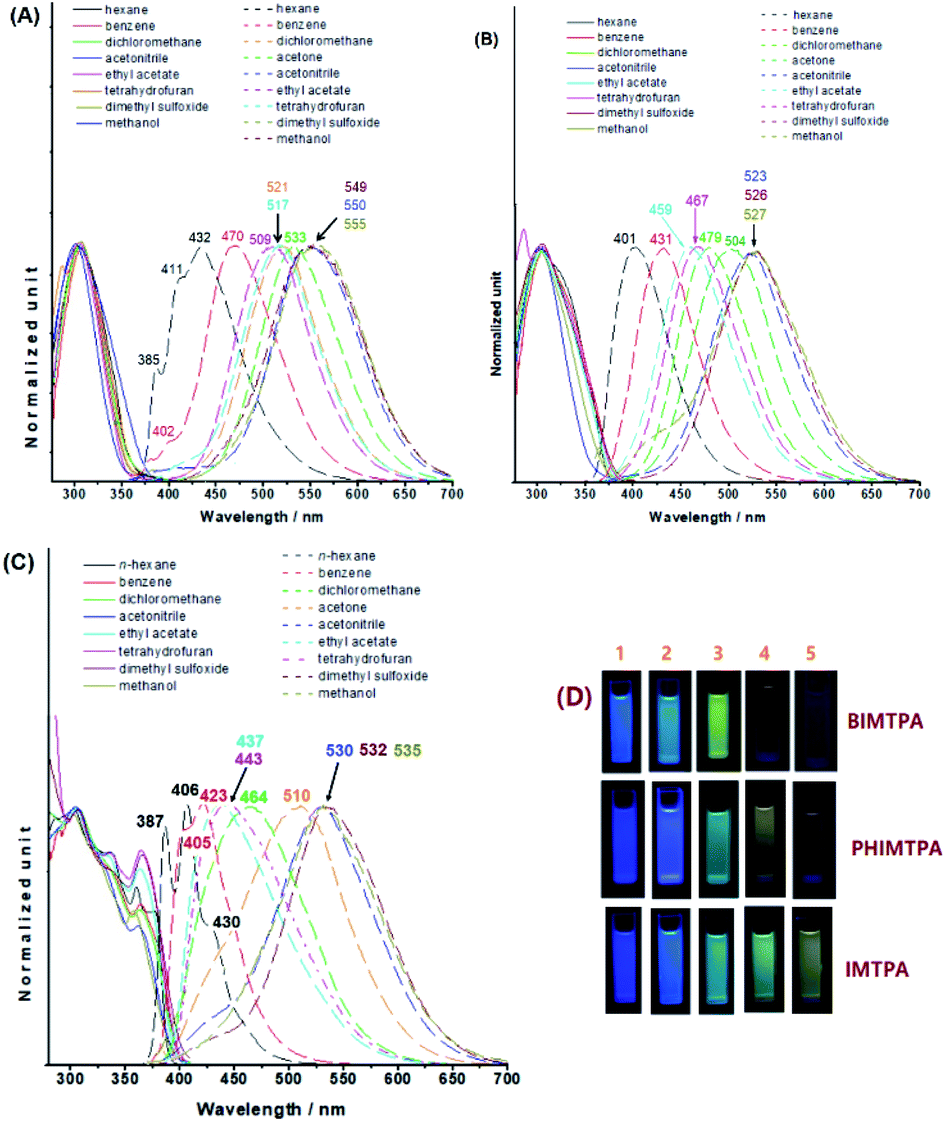 | ||
| Fig. 2 Absorption (solid lines) and emission spectra (dashed lines) of BIMTPA (A), IMTPA (B) and PHIMTPA (C) in different solvents; photos of imidazoles in different solvents (D) ((1) n-hexane; (2) benzene; (3) CH2Cl2; (4) acetone; (5) acetonitrile). | ||
X-ray crystallographic analysis
Since the solid-state physical properties of a fluorophore are closely related to the conformation, packing mode and intermolecular interactions, it is necessary to reveal the crystalline structures before the investigation of optical features. At the single molecular level, it was demonstrated that the conformation of each imidazole molecule is highly twisted (Fig. S3, ESI†), which makes the packing quite loose. Hence, each imidazole crystal is strongly emissive due to the lack of dense intermolecular interactions. Compared with the large torsion angles θ2 in IMTPA (82.51°) and PHIMTPA (76.08°), it is much smaller in BIMTPA (48.95°). The differences in the torsion angles θ1 and the dihedral angles ϕ between each phenyl on TPA are relatively small for the molecules in the three crystals.At the supramolecular level, molecules are packed into non-polar P21/n, P21/c and P21/n space groups, respectively, for crystals of IMTPA, BIMTPA and PHIMTPA. Due to the highly twisted conformations of imidazole molecules, there is no π⋯π overlap in each crystal. It was found in the IMTPA crystal that C–H⋯π interactions (3.403 Å) emerged between 4- and 5-phenyls while C–H⋯N interactions (3.088 Å) occur between the 5-phenyl and N3 atom on the imidazole ring (Fig. S4, ESI†). The other H-bonds (3.867 Å) appeared between the P2 phenyl on TPA and the N1 atom on the imidazole ring. In the BIMTPA crystal (Fig. S5, ESI†), three types of H-bonds were found without any strong C–H⋯π interactions, which were 2.468 Å between the cyanophenyl and N3 atom on the benzimidazole ring, 2.774 Å between the cyano and P2 phenyl groups on TPA, and 3.299 Å between cyanophenyl and the N atom on TPA. In the PHIMTPA crystal (Fig. S6, ESI†), three types of intermolecular C–H⋯N interactions were built, which are 3.191 Å between the double phenanthrol[9,10-d]imidazole rings, 3.821 Å between the P3 phenyl on TPA and the N1 atom on the phenanthrol[9,10-d]imidazole ring and 3.042 Å between the cyano and the P3 phenyl group on TPA. More significantly, the strong intramolecular C–H⋯π interaction (3.009 Å) takes place between the phenanthrol[9,10-d] imidazole ring and the P1 phenyl on TPA. Such intermolecular interactions will have a profound impact on some of the photophysical properties of the PHIMTPA crystal (vide infra).
Although each of the three crystals has a centrosymmetric space group, the molecules are not centrosymmetrically arrayed in all of the molecular couples built by the above strong intermolecular interactions. There are three types of molecular couples in each crystal (Fig. 3) and two of them are arranged in non-anti-parallel modes. We carried out DFT calculations for each couple and obtained the net dipole moment along with the HOMO–LUMO levels and energy gap. For each crystal, the net dipole moments of the two non-centrosymmetrically arranged couples were much larger than the corresponding molecular dipole moment. Such net dipole moments are large enough for heavy electron discharge to happen when the molecules in these two types of couples on freshly cracked surfaces are separated under force stimuli. Moreover, when molecules in couples are excited, the charge transfers occur between the HOMOs and LUMOs of both involved molecules in centrosymmetric couples, while the electrons shifted from the HOMO of one molecule to the LUMO of the other in non-centrosymmetric couples. Hence, the intermolecular charge transfers occur on the latter couples. It is believed that the intermolecular charge transfer effect can remarkably reduce the HOMO–LUMO energy gap and induce long-wavelength emission.9c,20,24 Hence, the emission wavelengths of molecules in crystals were obviously red-shifted as compared to those in n-hexane (negligible solvatochromic effect).
 | ||
| Fig. 3 Electron distributions, HOMO–LUMO levels and net dipole moments of molecular couples in IMTPA (A and B), BIMTPA (C and D) and PHIMTPA (E, F and G) crystals based on DFT/B3LYP/6-311+G** calculations. | ||
Mechanofluorochromic properties
In the solid-state, the three imidazole crystals are intensely emissive and the emission spectra demonstrate that the maximum wavelengths of the pristine samples are 410 nm for IMTPA, 435 nm for BIMTPA and 450 nm for PHIMTPA (Fig. 4). The quantum yields of the pristine crystals were 0.23 for IMTPA, 0.35 for BIMTPA and 0.33 for PHIMTPA. Unlike the broad structureless emission bands of IMTPA and BIMTPA, several fine vibrations appeared on the spectrum of PHIMTPA. However, the curve profile is dramatically different from that of the emission spectrum in n-hexane. Importantly, a broad structureless emission band is also presented by the as-prepared PHIMCN sample (Fig. S2, ESI†). Additionally, the DFT calculations (Fig. 3G) plainly indicate that the TPA group is deeply involved in the heavy charge transfers inside couple II and III of PHIMTPA crystals when molecules in these couples are excited. All these results imply that the TPA group on PHIMTPA must participate in the formation of the excited state under the crystalline phase, which is totally different from the case in solution. The decay lifetimes of the shoulder peaks at 428, 450, 478 and 514 nm were determined to be 3.82, 2.33, 3.06 and 4.79 ns, respectively (Fig. S7–S10, ESI†), which could be assigned to fluorescence rather than phosphorescence, or a fluorescence and phosphorescence mixture. It is assumed that the immobilization of the TPA group in the crystal by the strong intramolecular interactions may well account for the generation of several stable vibration levels that are dramatically different from those in solution.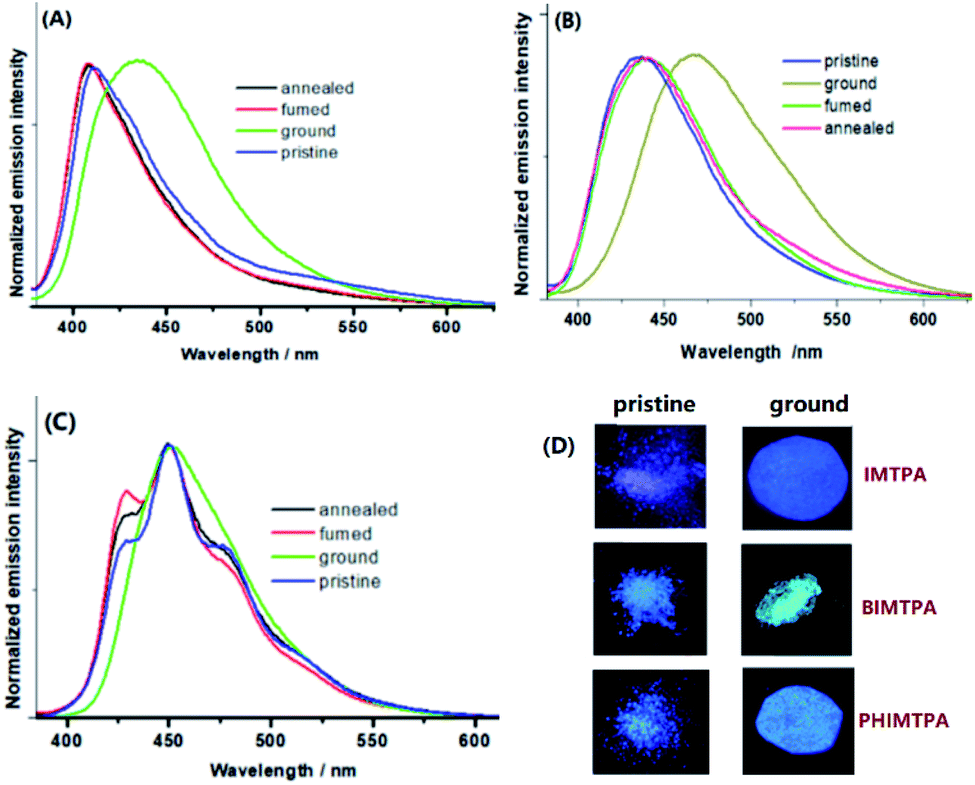 | ||
| Fig. 4 Solid-state emission spectra of IMTPA (A), BIMTPA (B) and PHIMTPA (C) under different conditions; photos of samples under 356 nm UV light (D). | ||
When the as-prepared samples were vigorously ground (30 min), the emission wavelengths of IMTPA and BIMTPA were remarkably red-shifted from 410 to 436 nm and from 435 to 467 nm, respectively (Fig. 3A and B), while the quantum yields were slightly reduced to 0.17 for IMTPA and 0.28 for BIMTPA. However, the colour change of the PHIMTPA sample after grinding for the same time could hardly be discerned by the naked eyes, since only the disappearance of the fine structures rather than the shift of the emission wavelength is present on the emission spectrum of the ground sample (Fig. 3C). The measured quantum yield for the ground PHIMTPA sample was 0.25. Before and after grinding, the emission lifetimes of the samples were prolonged from 2.56 ns to 4.07 ns for IMTPA, from 4.54 ns to 6.31 ns for BIMTPA and from 2.33 ns to 4.89 ns for PHIMTPA. The extension of the emission lifetime implies that the intermolecular interactions were strengthened due to the relatively more planarized conformations of molecules in the amorphous phase (Fig. S21–S25, ESI†).
The powder X-ray diffraction (PXRD) patterns (Fig. S11–S13, ESI†) together with the differential scanning calorimetry (DSC) curves (Fig. S15–S17, ESI†) illustrate that the morphological alternation from the crystalline to the amorphous phase really takes place on each sample in response to force stimuli, and such alternation is surely reversible. There were two observable exothermal peaks on the DSC curve of each ground sample before it melted, which means that cold recrystallization could be responsible for the reorganization of molecules in the amorphous phase in the original crystalline structures under heat treatment. The gradually increased melted temperature and cold recrystallization among the three ground samples correspondingly imply that the intermolecular interactions and force-induced rotation of the TPA group respectively became stronger and more difficult as the number of fused rings increased. Therefore, the emission shifts of both IMTPA and BIMTPA after force applications could be attributed to the morphologic changes in the samples. When the intermolecular interactions are broken under external force, the planarized conformations induced by the reduction of θ1 and θ2 may well be provided, which would considerably extend the π-conjugation areas on molecules in the amorphous phase to longer emission wavelength. Since the emission wavelengths of the PHIMCN sample after vigorous grinding (Fig. S14, ESI†) were just bathochromically shifted by 2 nm (Fig. S2, ESI†), the considerably red-shifted emission of the ground IMTPA and BIMTPA samples should be most dominantly contributed by the reduction of θ2 under force stimuli. However, such force-induced reduction of θ2 can hardly happen on the ground PHIMTPA sample due to the strong intramolecular C–H⋯π interactions (3.009 Å) between the P1 phenyl on TPA and the phenanthrol[9,10-d] imidazole ring, as intramolecular interactions are much less susceptible to damage than intermolecular ones. Consequently, molecular conformations locked by intermolecular interactions can be readily adjusted when lattices are broken by force stimuli. However, such adjustments cannot happen on those immobilized by intramolecular interactions. Hence, it becomes understandable that the PHIMTPA crystal is MFC-inactive. In addition to P1 phenyl, the DFT calculations demonstrated that P2 and P3 phenyls on TPA are also heavily involved in the production of stable vibration levels under their crystalline conformations. Accordingly, due to the locking of P1 phenyl in the PHIMTPA crystal, an external force had to be partly applied to trigger the rotations of P2 and P3 phenyls, which resulted in the destruction of the stable vibration levels and the disappearance of the fine structure in the emission spectrum.
Mechanoluminescence properties
In the dark and with no UV light irradiation, IMTPA, BIMTPA and PHIMTPA crystals give off a bright flash of light when they are rubbed against the inner walls of a container. Such phenomena indicate that the crystals are ML-active. It has been shown that the ML emission wavelengths of these crystals are very similar to the fluorescence emission wavelengths of the corresponding amorphous samples (Fig. 5). Such similarities also appear in most reported ML-active organic crystals. In order to release enough electrons that can effectively excite molecules on freshly cracked surfaces, sufficient discharge surfaces should be produced by breaking crystals into tiny particles. Under such circumstances, conformations of molecules separated from molecular couples on newly cracked surfaces on exposure to force stimuli will be more planarized due to the lack of lattice constraint, making the emission of these molecules by mechano-excitation very close to those in the amorphous phase by photo-excitation. Hence, it may be safe to claim that the similarity between the ML spectra and PL spectra of amorphous samples is a characteristic of ML activity caused by polar molecular couples. Of course, when the size of crystalline particles is too small, it will be quite difficult for them to display ML phenomenon, since particles can hardly be further fractured under force stimuli.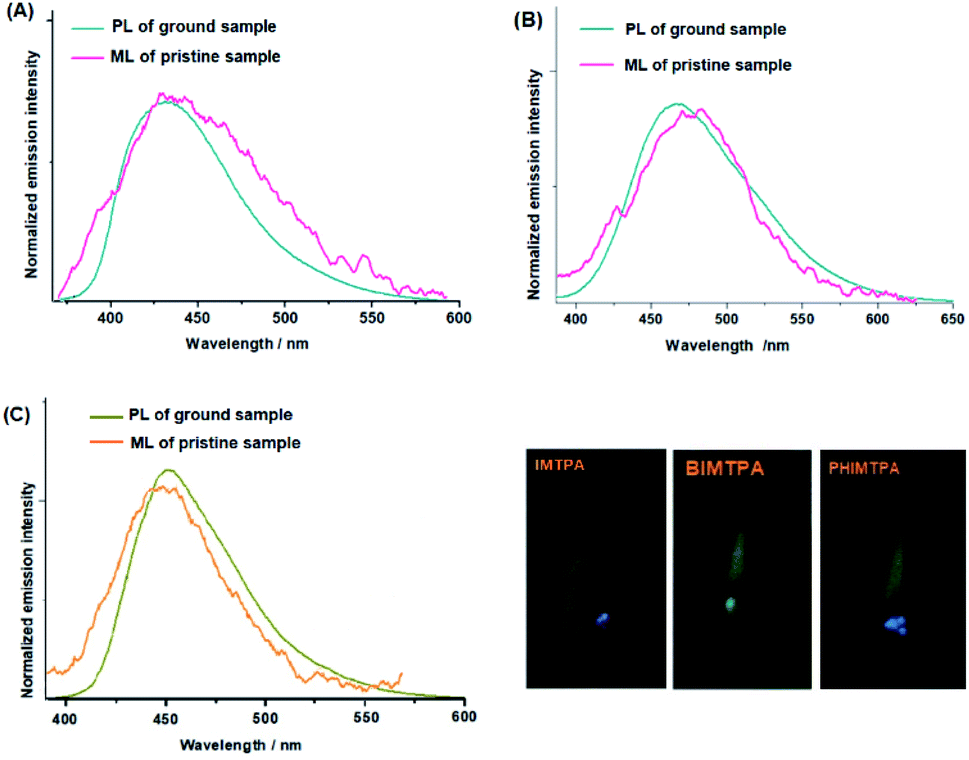 | ||
| Fig. 5 PL and ML spectra of IMTPA (A), BIMTPA (B) and PHIMTPA (C) (inserted: photos of ML phenomena on three crystals). | ||
It is well known that the occurrence of electron discharge depends on not only the voltages of the charged species but also the distances among them. It is believed that the discharges occur simultaneously when molecules in polar couples on freshly cracked surfaces are separated by force. For couples with weak intermolecular interactions, even if they are still endowed with large net dipole moments, the electron discharges have to be far less strong as the distances between molecules in these couples are much longer than those with strong intermolecular interactions. Therefore, for the couple analysis in this work, only couples with strong intermolecular interactions were involved.
Although IMTPA, BIMTPA and PHIMTPA crystals have centrosymmetric space groups, they remain highly ML-active; some molecules in them are still arranged into couples with strong intermolecular interactions in non-anti-parallel ways, resulting in considerably large net dipole moments and intermolecular charge transfers on these couples (Fig. 3). Therefore, the existence of molecular couples with enough high net dipole moments seems to be the most significant element for crystals to possess the ML effect, regardless of non-centrosymmetric or centrosymmetric space groups. Crystallography text books indicate that all molecules in crystals with non-centrosymmetric space groups must be packed in non-anti-parallel ways, making all couples in such crystals certainly non-centrosymmetric. Piezoelectric crystals, which have to possess non-centrosymmetric space groups, are much more prone to having ML activity due to largely charge-accumulated cracked surfaces caused by the non-zero net dipole moments of all couples. This is the basic reason why ML phenomena are more frequently observed on crystals with non-centrosymmetric space groups. Overall, in order to obtain highly polar cracked surfaces, two factors seem to be most essential: one is non-centrosymmetrically arrayed couples with strong intermolecular interactions and the other is molecules with large dipole moments. Molecules with large dipole moments are quite helpful in forming couples having high net dipole moments. As the formation of non-centrosymmetric couples is unpredictable at the supramolecular level, the purposeful design of molecules with large dipole moments becomes the only controllable way at the single molecular level for people to possibly produce ML-active crystals.
Our polar molecular couple hypothesis can also effectively elucidate the reason for the ML activity of some other reported crystals. Although crystal system dependence was proposed by the authors to explain the different ML behaviours on crystals of benzaldehye-substituted triphenylamines (TPA-1BA, TPA-2BA and TPA-3BA),21 it seems to be quite a strained interpretation since any crystal will finally be fractured if the external force is strong enough. By analysis of the molecular couples provided in this paper, it was clearly illustrated that molecules in all couples of TPA-1BA crystals were arrayed in either a parallel or cross-parallel pattern, whereas those in all couples of TPA-2BA and TPA-3BA crystals were arranged in anti-parallel. Such different molecular orientations inside couples cause large net dipole moments and close-to-zero ones to respectively appear on couples in the former ML-active crystal and the latter two ML-inactive crystals. Additionally, the inhibition of non-radiative decay resulting from molecular slippage resistance due to strong intermolecular interactions was suggested by the authors to explain the distinct ML performance in the tPBI-B1/tPBI-B2/tPBI system,22 pp-TPE(PI)2/mm-TPE(PI)2 system12 and p-TPE-NH2/m-TPE-NH2 system.23 Indeed, both non-radiative and radiative decay of excited molecules must occur long after molecules on cracked surfaces are excited by electron discharge due to the force-induced crystal fracture. Hence, molecular slippage resistance may only influence the difficulty of crystal fracture rather than the non-radiative decay of excited molecules. Once again, by checking the molecular packing in these papers, non-centrosymmetric and centrosymmetric molecular couples were found in corresponding ML-active and ML-inactive crystals in the above systems. Interestingly, it was reported that the ML activity of N-hydroxyethylcarbazole was switched off under a period of UV irradiation,24 and such deactivation could be attributed to the improved interaction strength inside molecular couples induced by UV light. Accordingly, the molecular slippage resistance caused by strong intermolecular interactions seems to be quite unreliable for the ML mechanism, and exceedingly strong interactions are sometimes detrimental to ML performance. Overall, polar molecular couples with large enough net dipole moments may well be much safer to reveal the nature of ML phenomena on many purely organic crystals in view of molecular packing. We believe that there should be more reported crystals in literature whose ML effect can be ascribed to high net dipole moments on molecular couples, if characteristics of those couples are re-examined, regardless of whether the space groups of crystals are non-centrosymmetric or centrosymmetric.
Of course, the above couple analysis does not work for understanding the ML mechanism of all crystals. Very recently, we reported two polymorphs TBIMB and TBIMG of 4-(2-(4-(diphenylamino)phenyl)-1H-benzo[d]imidazol-1yl)benzonitrile, which respectively exhibited brightly blue and green ML flashes of light.25 To our best knowledge, the examples that the ML effect with different emission wavelengths originated from molecules with the same chemical structure are truly very rare.9c,23,26 Crystallographic analysis indicated that molecules inside all the molecular couples in both crystals were arrayed in anti-parallel and the two crystals were quite fragile to be readily cleaved under force stimuli. Furthermore, the almost identical emission wavelengths and curve profiles appeared on the ML and the fluorescence spectra (FL) of the crystalline samples. This is dramatically different from most other reported ML-active crystals whose ML spectra are more similar to the FL spectra of amorphous samples. This implies that molecules in both ML-active polymorphs were very possibly excited by internal triboelectrification from the relative movements among the cleavage surfaces on broken crystals rather than electron discharge from cracked surfaces.
Hence, in order to learn the ML mechanism of a crystal packed by purely organic molecules, we recommend that the net dipole moments of molecular couples be checked, especially when molecules with large dipole moments are involved. For many organic crystals having centrosymmetric or non-centrosymmetric space groups, such a couple analysis method can be valid. If this analysis cannot be effective for understanding the ML behaviours of some crystals, other mechanisms should be considered.
Conclusion
Herein, three TPA- and 4-cyanophenyl-substituted imidazoles with different numbers of fused rings were prepared and their photophysical properties were investigated. In solution, as the number of fused rings increased, some vibration levels became more and more stable, causing the emission bands to gradually split into several sharp peaks. Unlike the complete exclusion of TPA on PHIMTPA from the π-conjugation with the PHIM core in solution, TPA is deeply involved in the formation of some stable vibration levels together with the PHIM core in the PHIMTPA crystal. Due to the strong intramolecular C–H⋯π interactions that make the rotation of P1 phenyl on TPA difficult, the PHIMTPA crystal is MFC-inactive. This is the opposite of the case with IMTPA and BIMTPA crystals since their good MFC performances are most dominantly induced by such rotation under force stimuli. Although centrosymmetric space groups are possessed by the three crystals, they are still all ML-active. X-ray crystallographic analysis and DFT calculations revealed that some molecular couples with strong intermolecular interactions can present quite large net dipole moments and heavy intermolecular ICT due to the non-anti-parallel arrangements of molecules in these couples. Therefore, strong electron discharge can emerge on freshly formed surfaces when crystals are fractured under force stimuli. Herein, the unusually large molecular dipole moments of IMTPA, BIMTPA and PHIMTPA are also significant for the couples to have high net dipole moments. In view of molecular packing, the existence of polar molecular couples seems to be a plausible reason for the ML activity of crystals. Meanwhile, the purposeful design of molecules with large dipole moments is the only human-controllable approach at the single molecular level to predict the possible formation of ML-active crystals built by purely organic molecules. Through such a strategy, some other ML-active crystals constructed by molecules with large dipole moments have been obtained in our lab, and their photophysical properties are being investigated and will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the Natural Science Fund of Zhejiang Province (LY19B020015). We thank Dr Jiyong Liu very much for his kindly help on X-ray crystallographic measurements.Notes and references
- (a) Y. Xie and Z. Li, Chem, 2018, 4, 943–971 CrossRef CAS; (b) Z. Yang, Z. Chi, Z. Mao, Y. Zhang, S. Liu, J. Zhao, M. P. Aldred and Z. Chi, Mater. Chem. Front., 2018, 2, 861–890 RSC; (c) Z. Monette, A. K. Kasar and P. L. Menezes, J. Mater. Sci., 2019, 30, 19675–19690 CAS; (d) S. Mukherjee and P. Thilagar, Angew. Chem., Int. Ed., 2019, 58, 7922–7932 CrossRef CAS PubMed; (e) E. Ubba, Y. Tao, Z. Yang, J. Zhao, L. Wang and Z. Chi, Chem.–Asian J., 2018, 13, 3106–3121 CrossRef CAS PubMed; (f) M. Liu, Q. Wu, H. Shi, Z. An and W. Huang, Acta Chim. Sin., 2018, 76, 246–258 CrossRef CAS; (g) H. Zhang, Y. Wei, X. Huang and W. Huang, J. Lumin., 2019, 207, 137–148 CrossRef CAS.
- (a) A. Ciniero, J. L. Rouzic, I. Baikie and T. Reddyhoff, Wear, 2017, 374, 113–119 CrossRef; (b) C.-N. Xu, T. Watanabe, M. Akiyama and X.-G. Zheng, Appl. Phys. Lett., 1999, 74, 2414–2416 CrossRef CAS; (c) I. Sage, R. Badcock, L. Humberstone, N. Geddes, M. Kemp and G. Bourhill, Smart Mater. Struct., 1999, 8, 504–510 CrossRef.
- N. Terasaki, H. Zhang, H. Yamada and C.-N. Xu, Chem. Commun., 2011, 47, 8034–8036 RSC.
- N. Terasaki, H. Yamada and C.-N. Xu, Catal. Today, 2013, 201, 203–208 CrossRef CAS.
- H. Xu, F. Wang, Z. Wang, H. Zhou, G. Zhang, J. Zhang, J. Wang and S. Yang, Tribol. Lett., 2019, 67, 1–10 CrossRef CAS.
- X. Wang, H. Zhang, R. Yu, L. Dong, D. Peng, A. Zhang, Y. Zhang, H. Liu, C. Pan and Z. L. Wang, Adv. Mater., 2015, 27, 2324–2331 CrossRef CAS PubMed.
- (a) S. M. Jeong, S. Song, K. I. Joo, J. Kim, S. H. Hwang, J. Jeong and H. Kim, Energy Environ. Sci., 2014, 7, 3338–3346 RSC; (b) D. K. Patel, B.-E. Cohen, L. Etgar and S. Magdassi, Mater. Horiz., 2018, 5, 708–714 RSC.
- (a) C.-N. Xu, T. Watanabe, M. Akiyama and X. G. Zheng, Appl. Phys. Lett., 1999, 74, 1236–1238 CrossRef CAS; (b) X. Qian, Z. Cai, M. Su, F. Li, W. Fang, Y. Li, X. Zhou, Q. Li, X. Feng, W. Li, X. Hu, X. Wang, C. Pan and Y. Song, Adv. Mater., 2018, 30, 1800291 CrossRef PubMed.
- (a) C. Wang, B. Xu, M. Li, Z. Chi, Y. Xie, Q. Lia and Z. Li, Mater. Horiz., 2016, 3, 220–225 RSC; (b) J. Tu, Y. Fan, J. Wang, X. Li, F. Liu, M. Han, C. Wang, Q. Li and Z. Li, J. Mater. Chem. C, 2019, 7, 12256–12262 RSC; (c) Z. Xie, T. Yu, J. Chen, E. Ubba, L. Wang, Z. Mao, T. Su, Y. Zhang, M. P. Aldred and Z. Chi, Chem. Sci., 2018, 9, 5787–5794 RSC.
- (a) J. I. Zink, Acc. Chem. Res., 1978, 11, 289–295 CrossRef CAS; (b) J. I. Zink, G. E. Hardy and J. E. Sutton, J. Phys. Chem., 1976, 80, 248–249 CrossRef CAS; (c) B. P. Chandra, V. K. Chandra, P. Jha, R. Patel, S. K. Shende, S. Thaker and R. N. Baghel, J. Lumin., 2012, 132, 2012–2022 CrossRef CAS; (d) J. T. Dickinson, L. B. Brix and L. C. Jensen, J. Phys. Chem., 1984, 88, 1698–1701 CrossRef CAS; (e) M. Zhenyi, S. C. Langford, J. T. Dickinson, M. H. Engelhard and D. R. Baer, J. Mater. Res., 1991, 6, 183–195 CrossRef CAS.
- (a) H. Nakayama, J. Nishida, N. Takada, H. Sato and Y. Yamashita, Chem. Mater., 2012, 24, 671–676 CrossRef CAS; (b) J. Nishida, H. Ohura, Y. Kita, H. Hasegawa, T. Kawase, N. Takada, H. Sato, Y. Sei and Y. Yamashita, J. Org. Chem., 2016, 81, 433–441 CrossRef CAS PubMed.
- F. Liu, J. Tu, X. Wang, J. Wang, Y. Gong, M. Han, X. Dang, Q. Liao, Q. Peng, Q. Li and Z. Li, Chem. Commun., 2018, 54, 5598–5601 RSC.
- (a) D. Xu, D. Cheng, Y. Wang, H. Zhou, X. Liu, A. Han and C. Zhang, Dyes Pigm., 2020 DOI:10.1016/j.dyepig.2019.107786; (b) Y. Wang, D. Cheng, H. Zhou, X. Liu, Y. Wang, A. Han and C. Zhang, Dyes Pigm., 2019 DOI:10.1016/j.dyepig. 2019.107689; (c) Y. Wang, D. Cheng, H. Zhou, J. Liu, X. Liu, J. Cao, A. Han and C. Zhang, Dyes Pigm., 2019 DOI:10.1016/j.dyepig.2019.107739; (d) D. Cheng, D. Xu, Y. Wang, H. Zhou, Y. Zhang, X. Liu, A. Han and C. Zhang, Dyes Pigm., 2019 DOI:10.1016/j. dye. pig. 2019.107934; (e) Y. Wang, D. Xu, H. Gao, Y. Wang, X. Liu, A. Han, C. Zhang and L. Zang, J. Phys. Chem. C, 2018, 122, 2297–2306 CrossRef CAS.
- (a) P. Shi, Y. Duan, W. Wei, Z. Xu, Z. Li and T. Han, J. Mater. Chem. C, 2018, 6, 2476–2482 RSC; (b) C. Feng, K. Wang, Y. Xu, L. Liu, B. Zou and P. Lu, Chem. Commun., 2016, 52, 3836–3839 RSC; (c) L. Wang, K. Wang, B. Zou, K. Ye, H. Zhang and Y. Wang, Adv. Mater., 2015, 27, 2918–2922 CrossRef CAS PubMed; (d) W.-Z. Xie, H.-C. Zheng and Y.-S. Zheng, J. Mater. Chem. C, 2017, 5, 10462–10468 RSC.
- (a) S. Hirata and T. Watanabe, Adv. Mater., 2006, 18, 2725–2729 CrossRef CAS; (b) S. J. Lim, B. K. An, S. D. Jung, M. A. Chung and S. Y. Park, Angew. Chem., Int. Ed., 2004, 43, 6346–6350 CrossRef CAS PubMed; (c) C. E. Olson, M. J. R. Previte and J. T. Fourkas, Nat. Mater., 2002, 1, 225–228 CrossRef CAS PubMed; (d) M. Irie, T. Fukaminato, T. Sasaki, N. Tamai and T. Kawai, Nature, 2002, 420, 759–760 CrossRef CAS PubMed.
- (a) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS PubMed; (b) X. Zhu, R. Liu, Y. Li, H. Huang, Q. Wang, D. Wang, X. Zhu, S. Liu and H. Zhu, Chem. Commun., 2014, 50, 12951–12954 RSC; (c) Q. Qi, Y. Liu, X. Fang, Y. Zhang, P. Chen, Y. Wang, B. Yang, B. Xu, W. Tian and S. X.-A. Zhang, RSC Adv., 2013, 3, 7996–8002 RSC; (d) P. Kumar, J. Dwivedi and B. K. Gupta, J. Mater. Chem. C, 2014, 2, 10468–10475 RSC; (e) X.-L. Lu and M. Xia, J. Mater. Chem. C, 2016, 4, 9350–9358 RSC.
- (a) W. Z. Yuan, Y. Tan, Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. Feng, H. Y. Sung, Y. Lu, I. D. Williams, J. Z. Sun, Y. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed; (b) C. Li, X. Tang, L. Zhang, C. Li, Z. Liu, Z. Bo, Y. Q. Dong, Y.-H. Tian, Y. Dong and B. Z. Tang, Adv. Opt. Mater., 2015, 3, 1184–1190 CrossRef CAS; (c) K. C. Naeem, A. Subhakumari, S. Varughese and V. C. Nair, J. Mater. Chem. C, 2015, 3, 10225–10231 RSC; (d) J. Sun, J. Han, Y. Liu, Y. Duan, T. Han and J. Yuan, J. Mater. Chem. C, 2016, 4, 8276–8283 RSC; (e) P. Xue, Z. Yang and P. Chen, J. Mater. Chem. C, 2018, 6, 4994–5000 RSC.
- (a) Y. Sagara and T. Kato, Angew. Chem., Int. Ed., 2011, 50, 9128–9132 CrossRef CAS PubMed; (b) S.-J. Yoon, J. W. Chung, J. Gierschner, K. S. Kim, M.-G. Choi, D. Kim and S. Y. Park, J. Am. Chem. Soc., 2010, 132, 13675–13683 CrossRef CAS PubMed; (c) H. Sun, S. Liu, W. Lin, K. Y. Zhang, W. Lv, X. Huang, F. Huo, H. Yang, G. Jenkins, Q. Zhao and W. Huang, Nat. Commun., 2014, 5, 3601–3609 CrossRef PubMed; (d) K. Y. Zhang, S. Liu, Q. Zhao and W. Huang, Coord. Chem. Rev., 2016, 319, 180–195 CrossRef CAS; (e) X. Chen, G. Sun, T. Zhang, S. Liu, Q. Zhao and W. Huang, Adv. Mater., 2016, 28, 7137–7142 CrossRef PubMed; (f) Q. Zhao, W. Xu, H. Sun, J. Yang, K. Y. Zhang, S. Liu, Y. Ma and W. Huang, Adv. Opt. Mater., 2016, 4, 1167–1173 CrossRef CAS; (g) W. Lin, Q. Zhao, H. Sun, K. Y. Zhang, H. Yang, Q. Yu, X. Zhou, S. Guo, S. Liu and W. Huang, Adv. Opt. Mater., 2015, 3, 368–375 CrossRef CAS; (h) J. Han, J. Sun, Y. Li, Y. Duan and T. Han, J. Mater. Chem. C, 2016, 4, 9287–9293 RSC.
- (a) Z.-Y. Wang, J.-W. Zhao, P. Li, T. Feng, W.-J. Wang, S.-L. Tao and Q.-X. Tong, New J. Chem., 2018, 42, 8924–8932 RSC; (b) T. Jadhav, J. M. Choi, B. Dhokale, S. M. Mobin, J. Y. Lee and R. Misra, J. Phys. Chem. C, 2016, 120, 18487–18495 CrossRef CAS; (c) T. Jadhav, J. M. Choi, J. Shinde, J. Y. Lee and R. Misra, J. Mater. Chem. C, 2017, 5, 6014–6020 RSC; (d) T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2015, 3, 9981–9988 RSC; (e) A. Ekbote, S. H. Han, T. Jadhav, S. M. Mobin, J. Y. Lee and R. Misra, J. Mater. Chem. C, 2018, 6, 2077–2087 RSC; (f) R. Misra, T. Jadhav, B. Dhokale and S. M. Mobin, Chem. Commun., 2014, 50, 9076–9078 RSC; (g) Y. Zhan, Y. Xu, Z. Jin, W. Ye and P. Yang, Dyes Pigm., 2017, 140, 452–459 CrossRef CAS; (h) Z. Gao, K. Wang, F. Liu, C. Feng, X. He, J. Li, B. Yang, B. Zou and P. Lu, Chem.Eur. J., 2017, 23, 773–777 CrossRef CAS PubMed.
- Z. Xie, Q. Huang, T. Yu, L. Wang, Z. Mao, W. Li, Z. Yang, Y. Zhang, S. Liu, J. Xu, Z. Chi and M. P. Aldred, Adv. Funct. Mater., 2017, 27, 1703918 CrossRef.
- M. Fang, J. Yang, Q. Liao, Y. Gong, Z. Xie, Z. Chi, Q. Peng, Q. Lia and Z. Li, J. Mater. Chem. C, 2017, 5, 9879–9885 RSC.
- Y. Yu, Y. Fan, C. Wang, Y. Wei, Q. Liao, Q. Li and Z. Li, J. Mater. Chem. C, 2019, 7, 13759–13763 RSC.
- Q. Huang, X. Mei, Z. Xie, D. Wu, S. Yang, W. Gong, Z. Chi, Z. Lin and Q. Ling, J. Mater. Chem. C, 2019, 7, 2530–2534 RSC.
- Y. Jiang, J. Wang, G. Huang, Z. Li, B. S. Li and B. Z. Tang, J. Mater. Chem. C, 2019, 7, 11790–11796 RSC.
- X.-J. Liu, Y.-R. Jia, H. Jiang, G.-L. Gao and M. Xia, Acta Chim. Sin., 2019, 77, 1194–1202 CrossRef.
- J. Yang, J. Qin, P. Geng, J. Wang, M. Fang and Z. Li, Angew. Chem., Int. Ed., 2018, 57, 14174–14178 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, MS spectra, absorption and emission spectra, PXRD patterns, DSC curves, X-ray crystallography and others. CCDC 1959200, 1959202 and 1959204. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra02737e |
| This journal is © The Royal Society of Chemistry 2020 |