
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbout the selectivity and reactivity of active nickel electrodes in C–C coupling reactions†
Sebastian B. Beil ab,
Manuel Breinera,
Lara Schulza,
Aaron Schülla,
Timo Müllera,
Dieter Schollmeyera,
Alexander Bommc,
Michael Holtkampd,
Uwe Karstc,
Wolfgang Schaded and
Siegfried R. Waldvogel*ab
ab,
Manuel Breinera,
Lara Schulza,
Aaron Schülla,
Timo Müllera,
Dieter Schollmeyera,
Alexander Bommc,
Michael Holtkampd,
Uwe Karstc,
Wolfgang Schaded and
Siegfried R. Waldvogel*ab
aDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10–14, 55128 Mainz, Germany
bGraduate School of Excellence Material Science in Mainz (MAINZ), Johannes Gutenberg University Mainz, Duesbergweg 10–14, 55128 Mainz, Germany. E-mail: waldvogel@uni-mainz.de
cFiber Optical Sensor Systems, Fraunhofer Heinrich-Hertz Institute, Am Stollen 19H, 38640 Goslar, Germany
dInstitute of Inorganic and Analytical Chemistry, University of Münster, Corrensstr. 30, 48149 Münster, Germany
First published on 8th April 2020
Abstract
Active anodes which are operating in highly stable protic media such as 1,1,1,3,3,3-hexafluoroisopropanol are rare. Nickel forms, within this unique solvent, a non-sacrificial active anode at constant current conditions, which is superior to the reported powerful molybdenum system. The reactivity for dehydrogenative coupling reactions of this novel active anode increases when the electrolyte is not stirred during electrolysis. Besides the aryl–aryl coupling, a dehydrogenative arylation reaction of benzylic nitriles was found while stirring the mixture providing quick access to synthetically useful building blocks.
Organic electro-synthesis evolved most recently as one of the major research areas in the toolbox of organic chemists.1–5 The development of new chemistry and reactivity requires innovative approaches, such as material science aspects, to advance this purely synthetic habit. Usually, platinum and carbon allotropes perform best in anodic conversions. In particular, boron-doped diamond (BDD) facilitates anodic coupling reactions due to the inert behaviour and clean electron transfer,6–8 exploiting inherent reactivity of substrates, such as phenols, arenes, anilines and various heterocycles and leading to high yields of the desired coupling products.9–13 In particular, solvent control enables distinct selectivity, wherein protic media, such as 1,1,1,3,3,3-hexafluoroisopropanol (HFIP), outperform others.14,15 To improve the morphology and allowing other design options instead of the limited geometries of BDD, together with a less costly anode material, we investigated different transition metals and their alloys with regards to their applicability in electrochemical conversions. Leaded bronze enabled the selective and high yielding dehalogenation reaction of cyclopropanes.16–20 These electrocatalytically active metal electrodes represent elegant alternatives to commonly applied materials which turned out to be unstable. Since the electro-catalytic species (mediator) remains immobilized at the electrode surface, the electrolyte is not contaminated which significantly facilitates work-up and down-stream processing (Fig. 1).
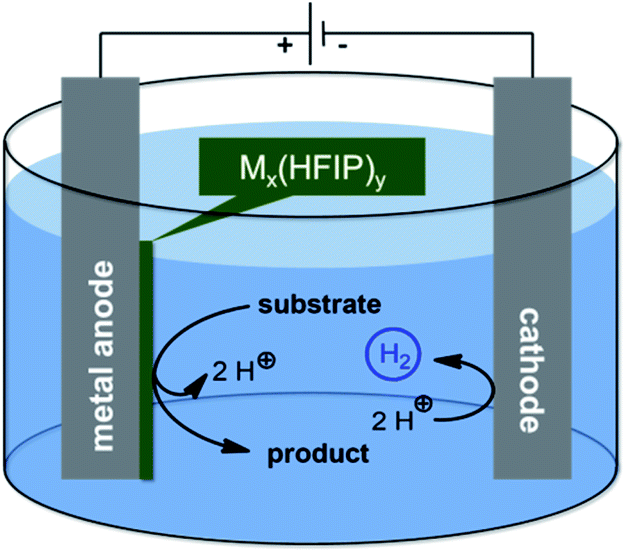 | ||
| Fig. 1 Schematic representation of an active anode within dehydrogenative electrolysis. | ||
Combining this elegant approach with the high performance of HFIP based electrolytes resulted in an active and non-sacrificial molybdenum anode, which accomplished a variety of dehydrodimerization and oxidative cyclization reactions.21–23 Obviously, these results represent only a starting point and much more common and abundant metals are favoured. Nickel is advantageous due to its inexpensive nature as well as many geometries being commercially available due to battery applications. Noteworthy, nickel seems to play an outstanding role as active anode in oxidations within alkaline media and anhydrous HF.24–29 In addition, nickel phosphides and other nickel salt coatings have been described as electrocatalytically active systems.30–35
During this work we observed a distinct influence of mechanical stirring on the formation of either homo- or cross-coupling products, which we attribute to the interaction of the vortex motion and the electrode double layer as described by Huang et al.36 In the absence of stirring, local substrate concentration is high and facilitates dimerization, whereas cross-coupling is enhanced due to charge distribution in stirred electrolytes.
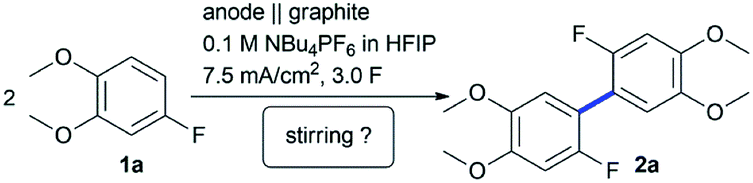
In our previous study,23 4-fluoroveratrole (1a) turned out to be a challenging substrate at the active molybdenum anode, since the corresponding biaryl 2a was only obtained in 12% isolated and 21% GC yield, respectively (Table 1, Entry 1). Other metal electrodes (see ref. 23 for a full collection), such as nickel, were barely superior and the yield of 2a was only slightly increased (Table 1, Entry 2). Surprisingly, the conversion and yield of substrate could be drastically improved when stirring was avoided. An isolated yield of 42 to 47% was obtained in high purity without dehalogenation processes observed in the undivided cell set-up for both metals (Table 1, Entries 3 and 4). Since both results were in the same range, we evaluated the mechanical stability of the electrodes. By subjecting the crude electrolyte mixtures to ICP-OES (inductively coupled plasma optical emission spectroscopy, see ESI, Section 4 for details†) we determined the amount of trace metals. The amount of 62 ± 5 ppm of nickel in solution (equals corrosion of 0.19 nmol C−1) was at least one order of magnitude lower compared to our previously reported protocol employing molybdenum as active anode material (740 ± 10 ppm),23 based on the higher mechanical stability of nickel or the lower solubility of nickel species on the surface. We therefore continued to use nickel and tried to optimize the condition. Various electrolysis parameters were investigated (see ESI, Section 9†), but interestingly the previous conditions23 matched the best. The applied charge was kept at 3.0 F per mole 2a. The substrate concentration was optimal at 0.2 M, and at room temperature the highest yield was detected by GC (for calibration details see ESI, Sections 7 and 8†). Similar to the molybdenum system, the use of additives like water or methanol resulted in diminished yield or complete failure.37 Other common alloys, such as Monel or Hastelloy, as well as several geometries25,27,38 were not able to outperform the simple nickel electrode making this protocol even more practical (see ESI, Sections 1 and 3†).
To obtain a better understanding of the electrode surface we applied the same nickel electrode in many consecutive runs (Fig. S3†). During the first three runs, the GC yield was constant at around 55%. Afterwards, the yield eroded constantly down to 10% after nine cycles. Within subsequent transformations the electro-active layer grows to a certain extend leading to a deactivated and therefore diminished active surface. This kind of deactivation is known for NiOOH electrodes, whereby nickelates are formed.39 Nevertheless, this inactivating layer can easily be removed by simple polishing (see ESI, Section 9†). The spontaneous formation of the active electrode was observed during CV studies where it acts as a redox-filter. The same redox behaviour was observed for either the blank electrolyte or together with the substrate, representing only the electrochemical window of the electro-active layer (see ESI, Section 10†).
We conclude that by using nickel in HFIP a more stable anode material was found, which works without stirring and still gives higher yields for challenging substrates such as 1a. With this novel active electrode system and handling, the scope was evaluated with regards to anisole and veratrole substrates (Scheme 1). Most substrates gave a higher yield on the non-sacrificial nickel anode compared to the molybdenum one. Various 2,5-dihalo-substituted anisoles were coupled and the dehydrodimers 2c–g were obtained in good yields up to 76%. For the first time, we could convert 2,6-substituted anisoles, which gave almost no conversion at the active molybdenum anode.23 A small library of 4-substituted veratroles was coupled to achieve good yields up to 76% for 2a–b and 2h–i, respectively. Even labile O-alkoxycarbonylmethyl protective groups40 could be applied in the dehydrodimerization in an acceptable yield of 56% (2j), in agreement with previous stoichiometric MoCl5-mediated transformations.41 The 1,3-dimethoxy arene 1k could be transformed in a synthetically useful yield of 11% and was obtained together with a by-product in 31%, which could be assigned to 4,6-dibromo-1,3-dimethoxybenzene (2kb). For the first time a halogen scrambling reaction was observed and could be due to the high reactivity of the active nickel electrode in a HFIP electrolyte. Phenylacetonitrile 1l was also subjected to the anodic coupling at the active nickel electrodes, but resulted in the benzyl-aryl cross-coupled product 2l in 51% isolated yield.
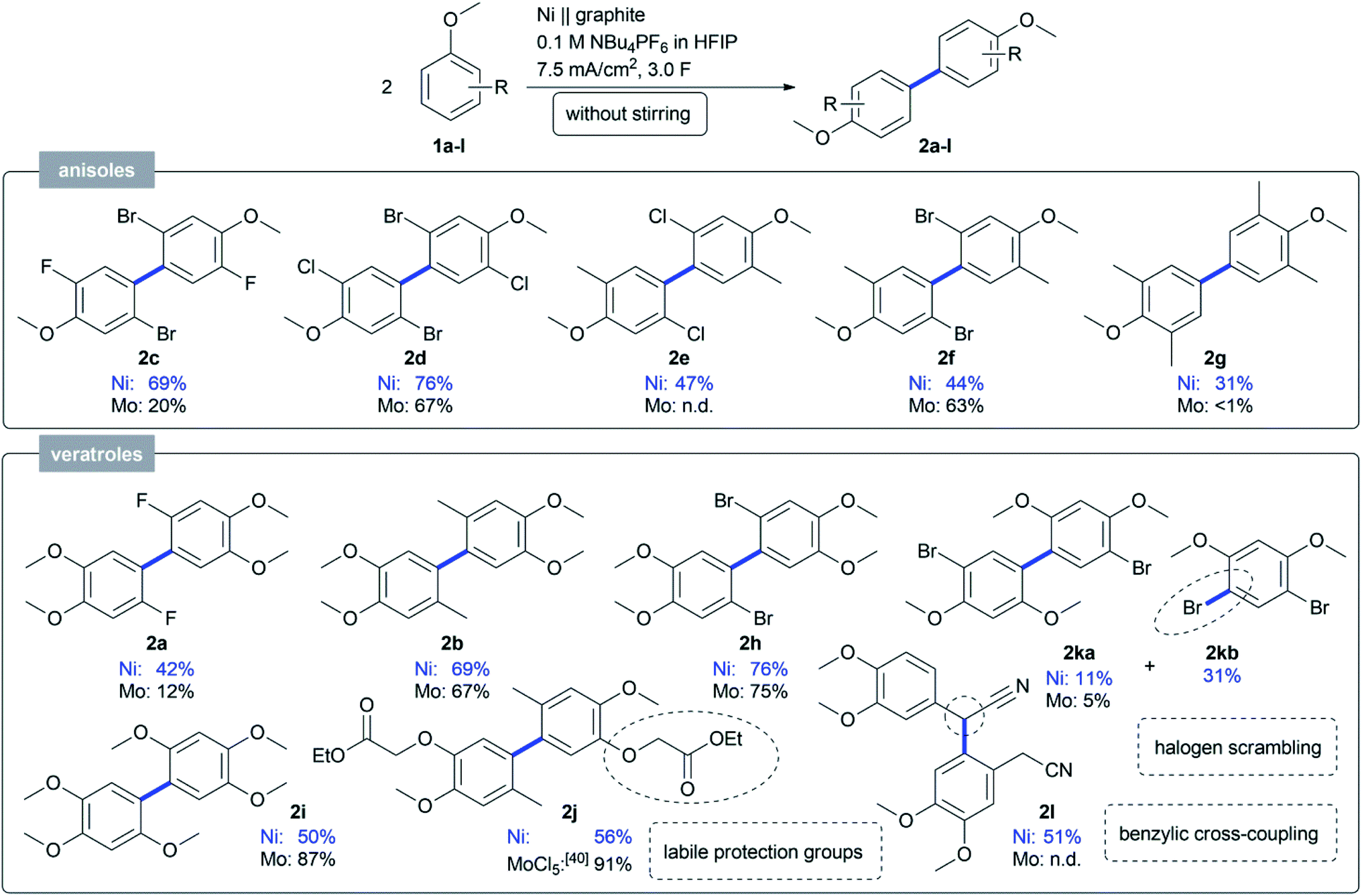 | ||
| Scheme 1 Scope obtained by anodic coupling of anisoles and veratroles 1 on an active nickel electrode. Isolated yields are displayed for all compounds. | ||
This observation inspired us to investigate benzylic cross-coupling reactions on active nickel anodes of such α-aryl acetonitrile derivatives, which lead to highly substituted nitriles common in natural products like cherylline dimethyl ether.42 Most reported approaches towards such targets apply transition-metal catalysis and require leaving groups.43–49 Only few examples described a direct synthesis of α-aryl benzylcyanides.50–52 Electrochemical dehydrodimerization of benzylcyanides was also described.53 None of them was observed in this electrochemical transformation. The reasonable stability of benzylic radical(-cationic) intermediates was recently described in polar solvents,54 and led here to successful benzylic cross-coupling on active nickel electrodes. We found, that biphenyl 3a can be coupled selectively to the benzylic position of 1l in 33% yield (4a, Scheme 2). Anisoles are also suitable cross-coupling partners and selective ortho- as well as para-coupling is accessible in yields up to 32% (4b–d). In all these cases, stirring was crucial for successful benzylic cross-coupling reactions, enabling enhanced convection. Sometimes, the formation of additional HFIP ethers was observed,55–57 which could be isolated in lower yields of 12% (4a′). Single crystals were successfully obtained for derivative 4d proving the formation of 2,2-biaryl acetonitrile compounds (CCDC: 1976461).
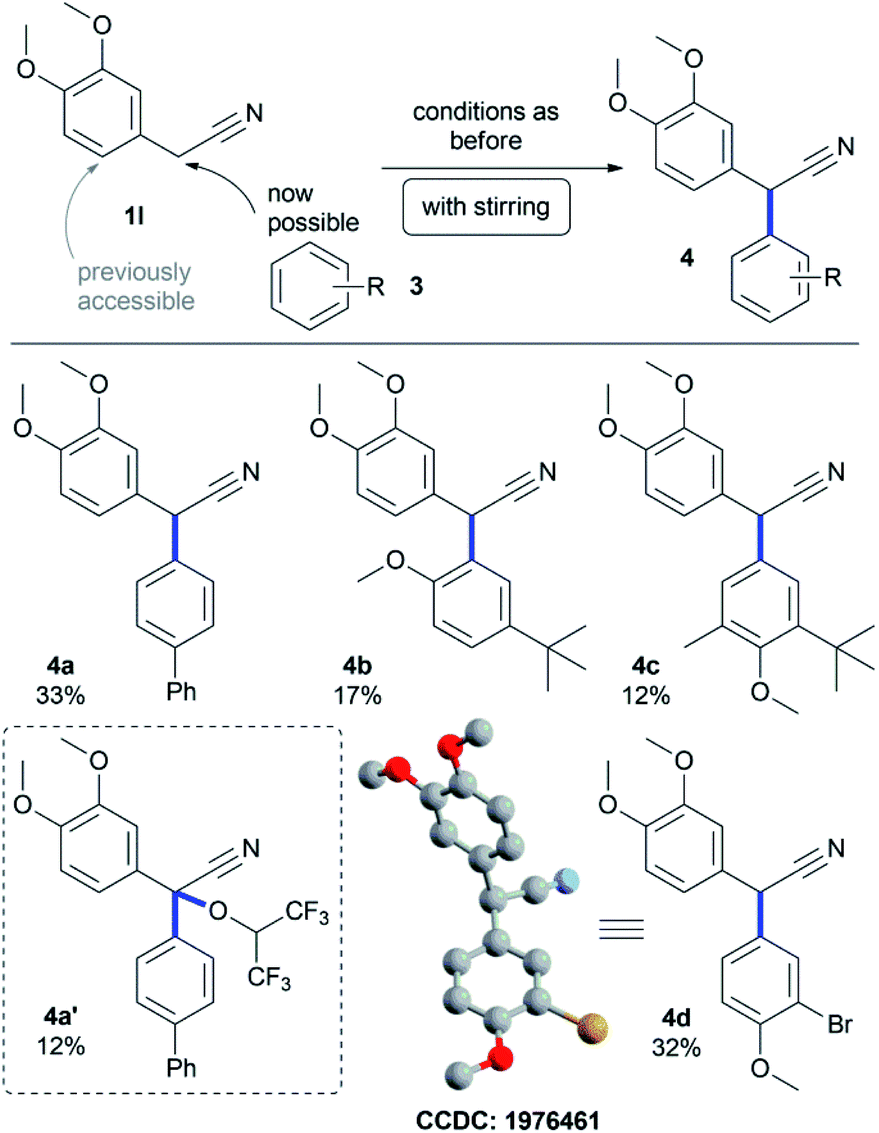 | ||
| Scheme 2 Benzylic dehydrogenative cross-coupling of 2-aryl acetonitrile (1l) and arenes 3 by anodic treatment. Hydrogen atoms of the X-ray single crystal structure analysis of 4d were removed for clarity. Conditions: Ni‖graphite 0.1 M NBu4PF6 in HFIP 7.5 mA cm−2, 3.0 F. | ||
In conclusion, an active and non-sacrificial nickel electrode system, which forms spontaneously in situ in the HFIP electrolyte was established. The successful dehydrodimerization and benzylic cross-coupling of arenes was performed. The expedient nature of this abundant material became obvious when different geometries and alloys were tested, and the crucial effect of stirring was observed. On the basis of these findings further investigations on the utility of active electrodes on valuable substrates are on-going.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support of the Advanced Lab of Electrochemistry and Electrosynthesis ELYSION (Carl-Zeiss-Stiftung) is gratefully acknowledged. A DFG fellowship through the Excellence Initiative by the Graduate School Materials Science in Mainz (GSC 266) is highly appreciated.Notes and references
- M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed.
- S. Tang, Y. Liu and A. Lei, Chem, 2018, 4, 27–45 CAS.
- A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594–5619 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- T. A. Ivandini and Y. Einaga, Chem. Commun., 2017, 53, 1338–1347 RSC.
- S. Lips and S. R. Waldvogel, ChemElectroChem, 2019, 6, 1649–1660 CrossRef CAS.
- S. R. Waldvogel, S. Mentizi and A. Kirste, in Radicals in Synthesis III, ed. M. Heinrich and A. Gansäuer, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 1–31 Search PubMed.
- S. Lips, B. A. Frontana-Uribe, M. Dörr, D. Schollmeyer, R. Franke and S. R. Waldvogel, Chem.–Eur. J., 2018, 24, 6057–6061 CrossRef CAS PubMed.
- S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 13325–13329 CrossRef CAS PubMed.
- L. Schulz, M. Enders, B. Elsler, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 4877–4881 CrossRef CAS PubMed.
- A. Wiebe, S. Lips, D. Schollmeyer, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2017, 56, 14727–14731 CrossRef CAS PubMed.
- A. Wiebe, D. Schollmeyer, K. M. Dyballa, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2016, 55, 11801–11805 CrossRef CAS PubMed.
- I. Colomer, A. E. R. Chamberlain, M. B. Haughey and T. J. Donohoe, Nat. Rev. Chem., 2017, 1, 0088 CrossRef CAS.
- R.-J. Tang, T. Milcent and B. Crousse, J. Org. Chem., 2018, 83, 930–938 CrossRef CAS PubMed.
- M. d. J. Gálvez-Vázquez, P. Moreno-García, H. Guo, Y. Hou, A. Dutta, S. R. Waldvogel and P. Broekmann, ChemElectroChem, 2019, 6, 2324–2330 CrossRef.
- V. Grimaudo, P. Moreno-García, A. Riedo, S. Meyer, M. Tulej, M. B. Neuland, M. Mohos, C. Gütz, S. R. Waldvogel, P. Wurz and P. Broekmann, Anal. Chem., 2017, 89, 1632–1641 CrossRef CAS PubMed.
- C. Gütz, M. Bänziger, C. Bucher, T. R. Galvão and S. R. Waldvogel, Org. Process Res. Dev., 2015, 19, 1428–1433 CrossRef.
- C. Gütz, V. Grimaudo, M. Holtkamp, M. Hartmer, J. Werra, L. Frensemeier, A. Kehl, U. Karst, P. Broekmann and S. R. Waldvogel, ChemElectroChem, 2018, 5, 247–252 CrossRef.
- C. Gütz, M. Selt, M. Bänziger, C. Bucher, C. Römelt, N. Hecken, F. Gallou, T. R. Galvão and S. R. Waldvogel, Chem.–Eur. J., 2015, 21, 13878–13882 CrossRef PubMed.
- S. B. Beil, P. Franzmann, T. Müller, M. M. Hielscher, T. Prenzel, D. Pollok, N. Beiser, D. Schollmeyer and S. R. Waldvogel, Electrochim. Acta, 2019, 302, 310–315 CrossRef CAS.
- S. B. Beil, S. Möhle, P. Enders and S. R. Waldvogel, Chem. Commun., 2018, 54, 6128–6131 RSC.
- S. B. Beil, T. Müller, S. B. Sillart, P. Franzmann, A. Bomm, M. Holtkamp, U. Karst, W. Schade and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 2450–2454 CrossRef CAS PubMed.
- L. Conte and G. Gambaretto, J. Fluorine Chem., 2004, 125, 139–144 CrossRef CAS.
- M. A. García-Contreras, S. M. Fernández-Valverde and J. R. Vargas-García, J. Alloys Compd., 2007, 434, 522–524 CrossRef.
- M. Okimoto, K. Ohashi, H. Yamamori, S. Nishikawa, M. Hoshi and T. Yoshida, Synthesis, 2012, 44, 1315–1322 CrossRef CAS.
- F. D. Popp and H. P. Schultz, Chem. Rev., 1962, 62, 19–40 CrossRef CAS.
- A. B. Sheremetev, B. V. Lyalin, A. M. Kozeev, N. V. Palysaeva, M. I. Struchkova and K. Y. Suponitsky, RSC Adv., 2015, 5, 37617–37625 RSC.
- J. H. Simons and W. J. Harland, J. Electrochem. Soc., 1949, 95, 55–59 CrossRef.
- M. M. Haring and E. G. V. Bosche, J. Phys. Chem., 1928, 33, 161–178 CrossRef.
- X. X. Liang, W. Weng, D. Gu and W. Xiao, J. Mater. Chem. A, 2019, 7, 10514–10522 RSC.
- F. S. Omar, A. Numan, S. Bashir, N. Duraisamy, R. Vikneswaran, Y.-L. Loo, K. Ramesh and S. Ramesh, Electrochim. Acta, 2018, 273, 216–228 CrossRef CAS.
- Y.-L. Shih, C.-L. Wu, T.-Y. Wu and D.-H. Chen, Nanotechnology, 2019, 30, 115601 CrossRef CAS PubMed.
- S. Surendran, S. Shanmugapriya, S. Shanmugam, L. Vasylechko and R. Kalai Selvan, ACS Appl. Energy Mater., 2018, 1, 78–92 CrossRef CAS.
- Z. Zhang, S. Liu, J. Xiao and S. Wang, J. Mater. Chem. A, 2016, 4, 9691–9699 RSC.
- C.-I. Huang, H. J. Huang and K. L. Cheng, in Advances in the Applications of Membrane-Mimetic Chemistry, ed. T. F. Yen, R. D. Gilbert and J. H. Fendler, Springer US, Boston, MA, 1994, pp. 227–240 Search PubMed.
- A. Kirste, B. Elsler, G. Schnakenburg and S. R. Waldvogel, J. Am. Chem. Soc., 2012, 134, 3571–3576 CrossRef CAS PubMed.
- L.-K. Wu, W.-Y. Wu, J. Xia, H.-Z. Cao, G.-Y. Hou, Y.-P. Tang and G.-Q. Zheng, J. Mater. Chem. A, 2017, 5, 10669–10677 RSC.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- A. Spurg and S. R. Waldvogel, Eur. J. Org. Chem., 2008, 337–342 CrossRef CAS.
- B. Kramer, R. Fröhlich, K. Bergander and S. R. Waldvogel, Synthesis, 2003, 1, 0091–0096 Search PubMed.
- A. S. Kumar, S. Ghosh, K. Bhima and G. N. Mehta, J. Chem. Res., 2009, 2009, 482–484 CrossRef.
- X. Cheng, H. Lu and Z. Lu, Nat. Commun., 2019, 10, 3549 CrossRef PubMed.
- Z. Jiao, K. W. Chee and J. S. Zhou, J. Am. Chem. Soc., 2016, 138, 16240–16243 CrossRef CAS PubMed.
- N. Rad and M. Mąkosza, Eur. J. Org. Chem., 2018, 376–380 CrossRef CAS.
- H. M. Refat, A. A. Faddo and E. Biehl, J. Fluorine Chem., 1996, 76, 99–103 CrossRef CAS.
- R. Shang, D.-S. Ji, L. Chu, Y. Fu and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 4470–4474 CrossRef CAS PubMed.
- G.-H. Yang, M. Liu, N. Li, R. Wu, X. Chen, L.-L. Pan, S. Gao, X. Huang, C. Wang and C.-M. Yu, Eur. J. Org. Chem., 2015, 616–624 CrossRef CAS.
- P. Y. Yeung, K. H. Chung and F. Y. Kwong, Org. Lett., 2011, 13, 2912–2915 CrossRef CAS PubMed.
- A. V. Aksenov, N. A. Aksenov, Z. V. Dzhandigova, D. A. Aksenov and M. Rubin, RSC Adv., 2015, 5, 106492–106497 RSC.
- M. H. Al-Huniti, Z. B. Sullivan, J. L. Stanley, J. A. Carson, I. F. D. Hyatt, A. C. Hairston and M. P. Croatt, J. Org. Chem., 2017, 82, 11772–11780 CrossRef CAS PubMed.
- D. K. Singh, S. S. Prasad, J. Kim and I. Kim, Org. Chem. Front., 2019, 6, 669–673 RSC.
- M. N. Elinson, A. I. Ilovaisky, V. M. Merkulova, T. A. Zaimovskaya, P. A. Belyakov and G. I. Nikishin, Mendeleev Commun., 2010, 20, 207–208 CrossRef CAS.
- J. P. Peterson and A. H. Winter, J. Am. Chem. Soc., 2019, 141, 12901–12906 CrossRef CAS PubMed.
- Y. Imada, J. L. Röckl, A. Wiebe, T. Gieshoff, D. Schollmeyer, K. Chiba, R. Franke and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 12136–12140 CrossRef CAS PubMed.
- J. L. Röckl, A. V. Hauck, D. Schollmeyer and S. R. Waldvogel, ChemistryOpen, 2019, 8, 1167–1171 CrossRef PubMed.
- J. L. Röckl, Y. Imada, K. Chiba, R. Franke and S. R. Waldvogel, ChemElectroChem, 2019, 6, 4184–4187 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal structure of 4d. CCDC 1976461. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra02673e |
| This journal is © The Royal Society of Chemistry 2020 |