
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy of the electrochemical recovery of cobalt from spent cemented carbide
Hongguang Kang ,
Jidong Li*,
Chaogang Zhang,
Jinlin Lu,
Qian Wang and
Yiyong Wang
,
Jidong Li*,
Chaogang Zhang,
Jinlin Lu,
Qian Wang and
Yiyong Wang
School of Materials and Metallurgy, University of Science and Technology Liaoning, Anshan 114051, Liaoning Province, China. E-mail: kk20190710@163.com; lijidong2009@ustl.edu.cn
First published on 9th June 2020
Abstract
The massive accumulation of spent cemented carbide not only produces environmental pollution but also wastes resources such as tungsten and cobalt. To solve the problem, a low-temperature acid aqueous electrochemical method was used; cobalt was recycled on a stainless steel cathode, and at the same time, tungstic acid was enriched at a spent cemented carbide anode, achieving a high efficiency, low energy consumption, and low pollution separation and recovering spent cemented carbide. The transient electrochemical test results show the following: the reduction mechanism of cobalt is Co2+(aq) + 2e− → Co(s). The nucleation mechanism is close to instantaneous nucleation. The electrodeposition is irreversible and controlled by the diffusion step. The average diffusion coefficient of Co(II) is 2.16589 × 10−7 cm2 s−1. Electrodeposition experiments show that cobalt enters the electrolyte in the form of Co(II) and is reduced to elemental cobalt on the stainless steel electrode, and tungsten carbide (WC) is oxidized to tungstic acid (H2WO4) under the oxidizing atmosphere of the anode and enriched in the anode area. The investigation provides favorable electrochemical conditions for the recovery and separation of other valuable metals from spent alloys.
1 Introduction
The nonferrous metals tungsten and cobalt are important strategic resources that are often used in the military, aerospace, science and technology, and other industrial fields.1,2 Tungsten–cobalt alloy is widely used in cemented carbide because of its high melting point, high hardness, and good wear and corrosion resistance.3 With the increase in the use of cemented carbide in all walks of life, a large amount of spent cemented carbide, which is difficult to handle, has been generated, not only polluting the environment but also wasting metal resources such as tungsten and cobalt. Therefore, the efficient recovery of valuable metals in waste alloys is of great significance for promoting the development of a global circular economy.The methods for recycling spent cemented carbides mainly include the zinc melting method, electrochemical analysis, high-temperature treatment, and acid dissolution.4,5 Compared with other methods, the electrochemical recovery method can be used for many types of recovered metals, and the process is easy to control.6 Electrochemical recovery are divided into high-temperature molten salt electrochemistry and low-temperature aqueous solution electrochemistry. Xiaoli Xi7,8 and Liwen Zhang9 effectively separated tungsten and cobalt metals in cemented carbide by the molten salt electrochemical method. However, the purity of the metal recovered by the high-temperature molten salt is not very high, a large amount of energy is consumed, and the resultant pollution is significant. Xiaqi Liang10 and R. R. Srivastava11 described the ability to recover WC and cobalt in spent cemented carbide by aqueous solution electrochemistry. However, the purity of the WC obtained at the anode is not high, and the electrochemical conditions are difficult to control. To further develop the low-consumption and high-efficiency recovery technology of tungsten and cobalt in spent cemented carbide, this paper uses low-temperature aqueous solution electrochemical separation technology. The process mechanism of cobalt recovery by cathodic electrodeposition has successfully achieved a high-efficiency short-flow recovery process, same time H2WO4 enrichment at the anode, and similar studies have rarely been reported. At the same time, the relevant electrochemical behavior rules can provide theoretical guidance for the recovery and separation of other valuable metals from spent alloys.
2 Materials and methods
All the chemicals involved in the experiment were produced by Sinopharm Chemical Reagent Co., Ltd. with the expected purity (AR).In the electrochemical experiment, CoCl2·6H2O with a concentration of 0.05 mol L−1 was used as the electrolyte solution at a temperature of 298 K, and a three-electrode system was employed to study the mechanism of the electrochemical reaction. The working electrode was stainless-steel, and the electrode surface area was 0.785 cm2. The reference electrode was a saturated calomel electrode. The counter electrode was platinum, and the electrode surface area was 8 cm2. Before the experiment, the stainless-steel plates were sanded until smooth with different sizes of sandpaper. The alkaline solution was degreased by an electrochemical method. The plates were cleaned with alcohol (10 min) and deionized water (5 min) via an ultrasonic method and then dried (308 K, 4 h) for use.
In the electrodeposition experiment, the spent cemented carbide was the anode, the stainless-steel was the cathode, and the actual working areas of both electrodes were equal to 6 cm2. CoCl2·6H2O and H3BO3 at concentrations of 0.25 mol L−1 and 0.30 mol L−1, respectively, were used as electrolyte solutions. Electrodeposition occurred when two electrodes were placed opposite to each other. High-purity cobalt was obtained on the surface of the stainless-steel cathode, and high-purity tungstic acid was gathered in the region of the anode. The experimental process and mechanism are given in Fig. 1 and eqn (1)–(5).
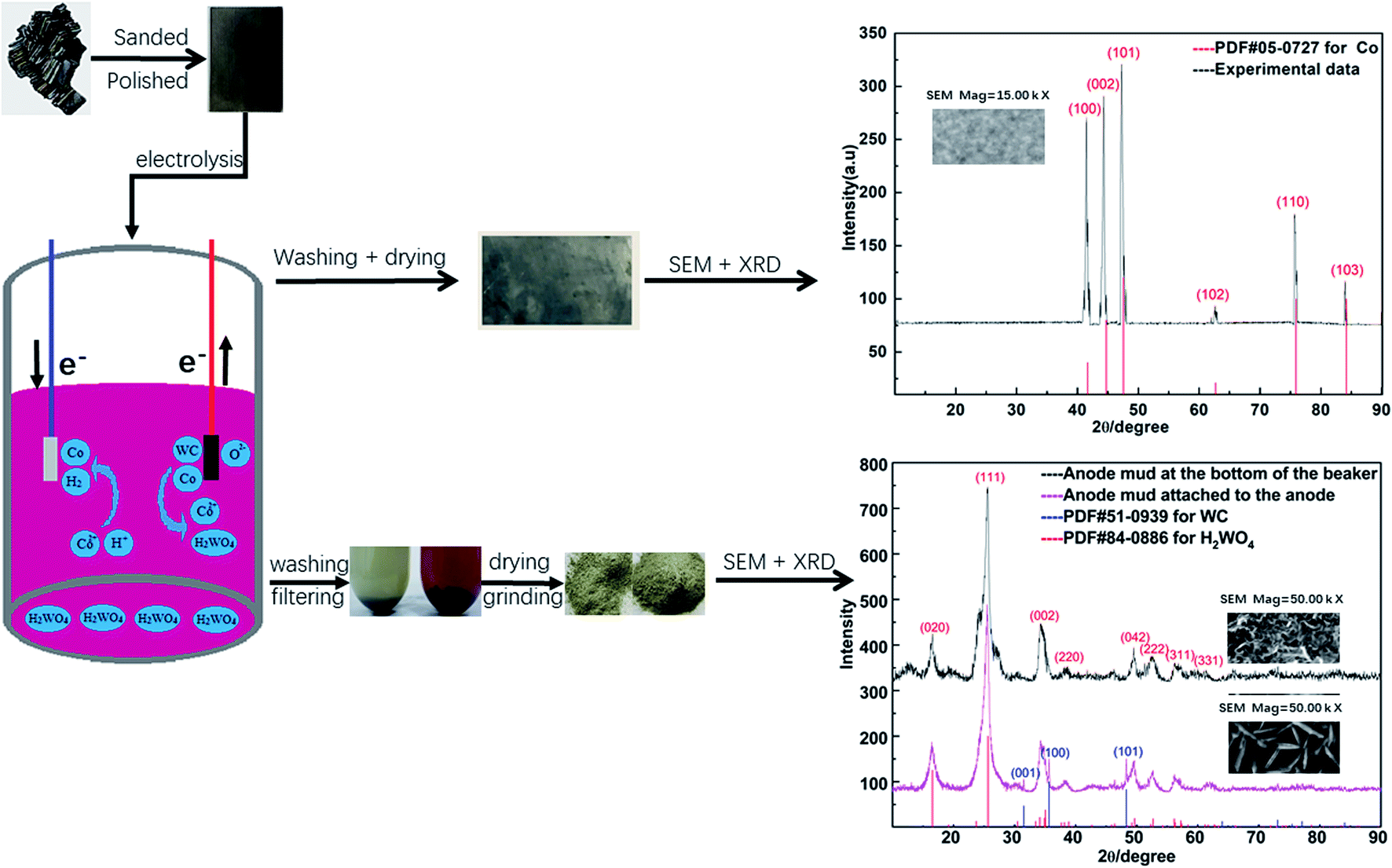 | ||
| Fig. 1 Experimental process. | ||
The experimental mechanism is as follows:
Anode:
| Co(s) − 2e− → Co2+(aq) | (1) |
| M(s) − ne− → Mn+(aq) | (2) |
| WC(s) + 6H2O − 6e− → CO2(g) + H2WO4(s) + 10H+(aq) | (3) |
Cathode:
| 2H+ + 2e− → H2(g) | (4) |
| Co2+ + 2e− → Co(s) | (5) |
3 Results and discussion
3.1 Voltammetry analysis
The cobalt in spent cemented carbides, the deposition potential and the deposition current in deionized water, and the reduction process on the cathode surface were studied with linear sweep voltammetry testing.12,13 The temperature was 298 K. The linear sweep voltage range was −0.15 to −1.70 V. The scan range was 100–115 mV s−1. The test results are shown in Fig. 2.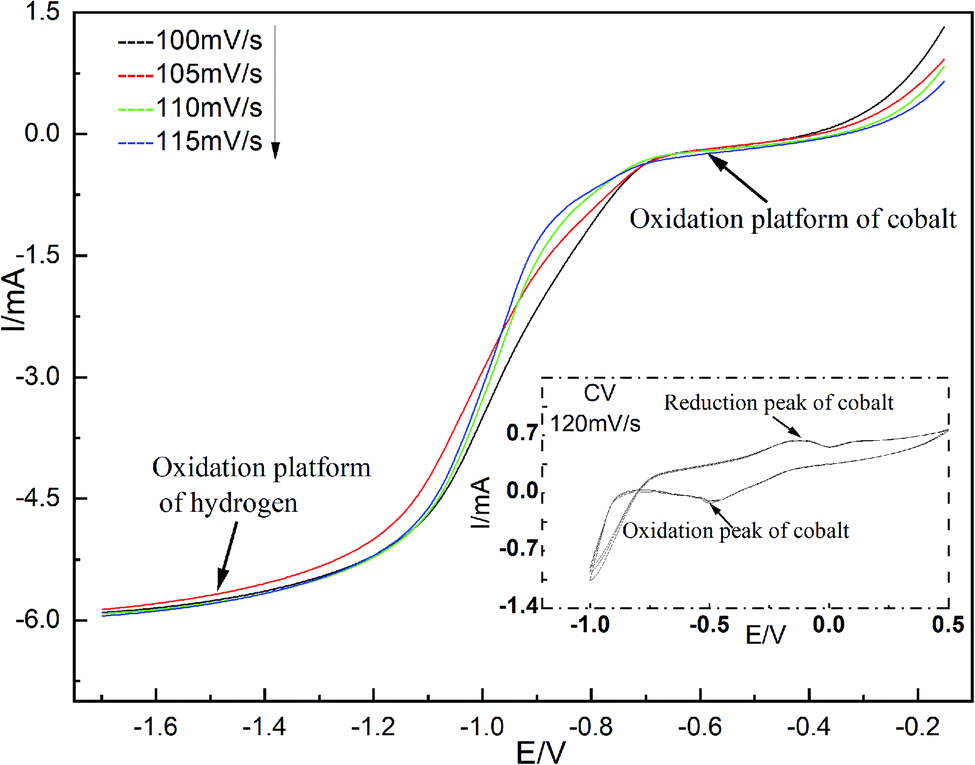 | ||
| Fig. 2 Voltammetry curves of Co(II) at different scanning rates (298 K). | ||
Fig. 2 shows that when the scan rate is 120 mV s−1, there are only two reduction peaks in the range of −1.0–0.6 V, corresponding to the reduction potential of cobalt and hydrogen, while there is only one oxidation peak corresponding to the oxidation of cobalt. On the basis of cyclic voltammetry, linear sweep voltammetry was carried out to study the reduction mechanism on the working electrode surface. The experiment began at −0.15 V and stopped at −1.70 V. Two reduction plateaus of cobalt and hydrogen appear in the curve. Although the deposition potential of cobalt is more negative than that of hydrogen, the values are very close. According to the Nernst equation,14 in a single-metal ion solution, the concentration increases 100 times every time the low ion potential increases by 0.06 V. An increase in the concentration of Co(II) can make the deposition potential of cobalt more positive than that of hydrogen. From −0.35 to −0.70 V, the plateau facilitates the reduction precipitation of Co(II) via a two-electron one-step reduction process. After −1.2 V, a hydrogen evolution plateau occurs. It can be concluded from Fig. 2 that the reduction of Co(II) to cobalt on the surface of the stainless-steel cathode is an irreversible process.15–17
3.2 Chronopotentiometric analysis
The diffusion coefficient DCo(II) was studied by chronopotentiometry in an acidic aqueous solution. The temperature was 298 K. The current intensity was 0.04–0.10 mA. The test results are shown in Fig. 3.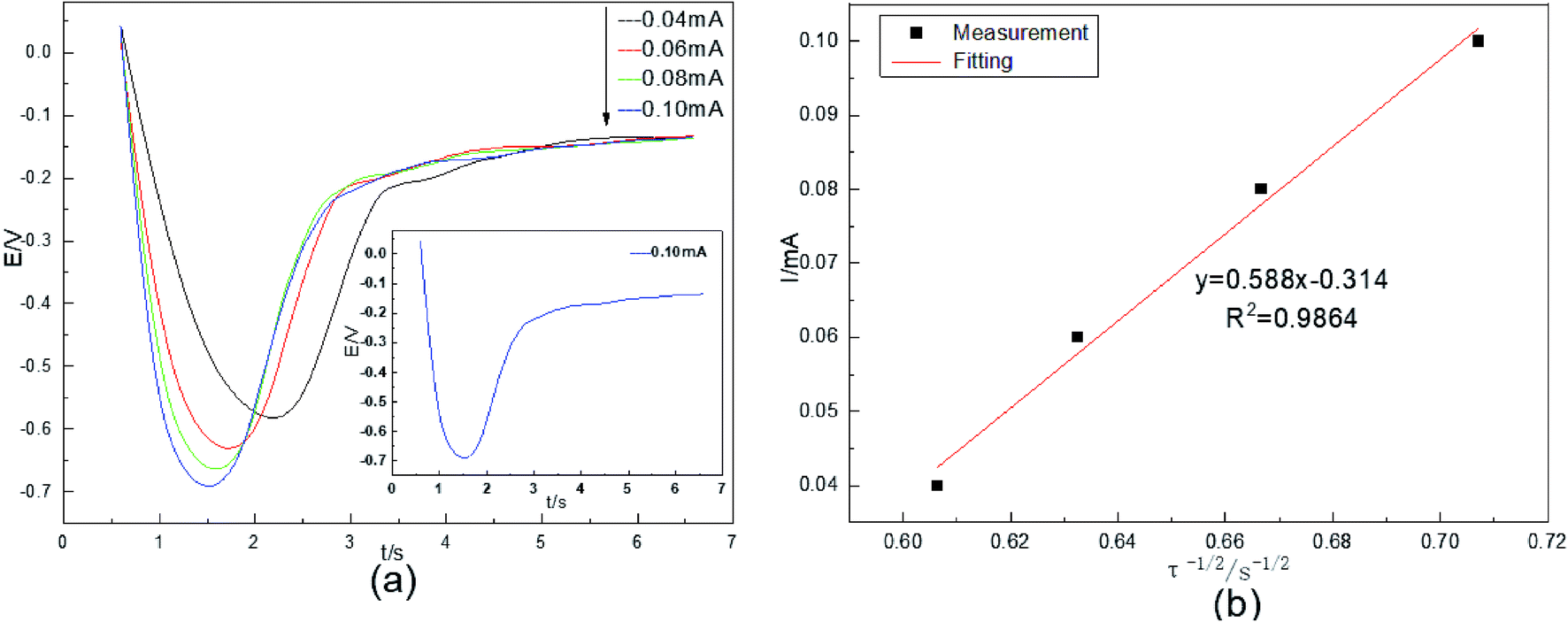 | ||
| Fig. 3 (a) Chronopotentiometric curves of Co(II) at different current intensities; (b) the relationship between the current intensity I(t) and the negative square root of the transition time (τ−1/2) obtained via chronopotentiometry (298 K). | ||
Fig. 3(a) shows that the electrode potential first decreases, then increases, and finally remains stable, and the 0.10 mA curve is given as an example. During the period of 0.5–1.5 s, the system suddenly applies a constant current, causing ohmic polarization of the solution on the cathode surface. This polarization greatly changes the electrode potential of the system, and the cathode potential decreases rapidly. From 1.5–1.7 s, electroactive Co(II) on the surface of the stainless-steel deposits at a constant rate because of the polarization of the current. From 1.7–3 s, the cobalt content of the working electrode surface increases as the electrolysis time increases. The reduction process on the electrode surface is controlled by concentration polarization. The electrode potential begins to change dramatically again and is accompanied by the occurrence of the HER. After 3 s, the electroactive Co(II) concentration drops to 0 on the surface of the stainless-steel working electrode, and the HER occurs completely, therefore, the electrode potential remains stable. The reaction potential of cobalt is gradually reduced from −0.58 V to −0.70 V as the current intensity increases. Co(II) undergoes a one-step two-electron irreversible reduction process on the surface of the stainless-steel. This reduction potential is very close to the Co(II) reduction potential in Fig. 1.
In chronopotentiometry, the electrochemical behavior of Co(II) can be further studied according to the relationship between I(t) and τ−1/2.18 This relationship is shown in Fig. 3(b).
Fig. 3(b) shows that I(t) and τ−1/2 have a good linear relationship. The results further indicate that the electrochemical reduction process is controlled by the diffusion step of Co(II) on the surface of the cathode in the spent cemented carbide. In an irreversible system, the diffusion coefficient DCo(II) of Co(II) can be calculated according to the Sand equation.19
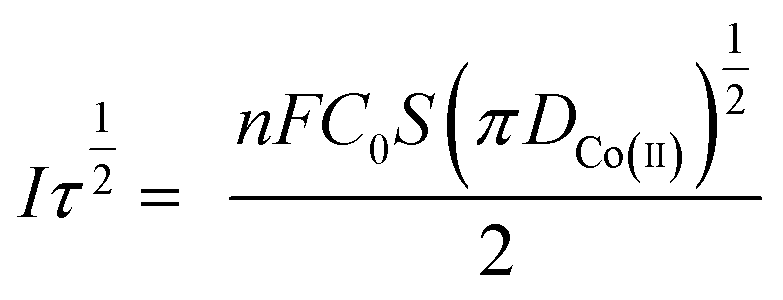 | (6) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485/C mol−1; π = 3.14; and n-electron transfer number.
485/C mol−1; π = 3.14; and n-electron transfer number.
According to eqn (6), when S = 0.785 cm2, Iτ1/2 = 0.588 mA s, F = 96485 C mol−1, C0 = 0.05 mol cm−3, n = 2, and π = 3.14, and it can be calculated that DCo(II) = 0.76776 × 10−8 cm2 s−1.
3.3 Chronoamperometric analysis
The nucleation process during electrodeposition in an aqueous acidic solution was studied by chronoamperometry. The temperature was 298 K. The electric potential was −0.85 to −1.05 V. The test results are shown in Fig. 4(a).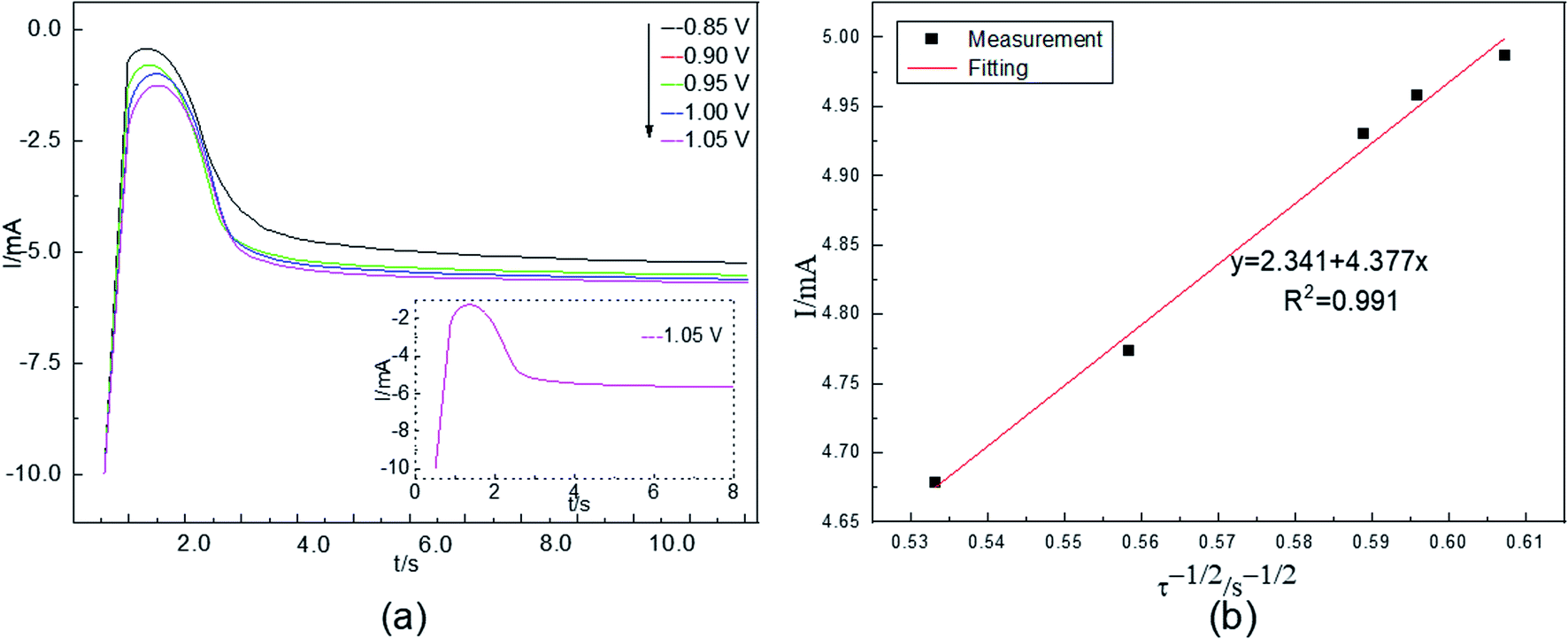 | ||
| Fig. 4 (a) Chronoamperometric curve of Co(II) at different electric potentials; (b) the relationship between the current intensity I(t) and the negative square root of the transition time (τ−1/2) obtained via chronoamperometry (298 K). | ||
Fig. 4(a) shows that the current intensities first increase, then decrease, and finally stabilize. For example, in the −1.05 V curve, in the beginning, the current rapidly increases to the maximum because of the charging of the electrical double layer on the electrode surface. Additionally, Co(II) is quickly reduced, and the cobalt content of the working electrode surface increases as the electrolysis time increases. The amount of Co(II) in the active nucleation sites cannot meet the formation rate of cobalt nuclei, and the reduction process of the electrode surface is controlled by concentration polarization. These factors cause the current to gradually decrease until it reaches a stable state. Afterwards, a new cobalt nucleus does not form; instead, the growth process of the nucleus on the surface of the cathode takes place. The stable current value is close to the current value caused by Co(II) at the corresponding voltage in linear scanning voltammetry. The electrochemical reduction process of Co(II) is controlled by the diffusion step on the surface of the cathode, as revealed by comparing the chronoamperometric curves at different potentials.
In chronoamperometry, the electrochemical behavior of Co(II) can also be further studied on the basis of the relationship between I(t) and τ−1/2. This relationship is shown in Fig. 4(b).
Fig. 4(b) shows that I(t) has a good linear relationship with τ−1/2. The results further indicate that the electrochemical reduction process of Co(II) is controlled by the diffusion step on the surface of the stainless-steel working electrode in the spent cemented carbide. This finding is consistent with the conclusions obtained above. In an irreversible system, the diffusion coefficient DCo(II) of Co(II) can be calculated according to the Sand equation. According to eqn (6), when S = 0.785 cm2, Iτ1/2 = 4.377 mA s, F = 96485 C mol−1, C0 = 0.05 mol cm−3, n = 2, and π = 3.14, the calculated result is DCo(II) = 4.25500 × 10−7 cm2 s−1. The diffusion coefficients obtained according to the two different curves are different. This difference occurred because as the electrodeposition time increases, cobalt is adsorbed on the surface of the working electrode, and the surface area of the working electrode continuously increases. However, when using the known working electrode surface area for the calculation, the value of the diffusion coefficient increases, leading to a deviation. To reduce the deviation in the experiments, the average value was calculated. The average diffusion coefficient of Co(II) was 2.16589 × 10−7 cm2 s−1.
The process by which cobalt is reduced to form a cobalt nucleus can be clearly obtained from the chronoamperometric curves at different potentials, and the nucleation mechanism of Co(II) can be obtained according to this process. By taking 4 different points randomly from the rising portion of the chronoamperometric curve at different electric potentials in Fig. 4, the Co(II) nucleation mechanism could be calculated from the equations of instantaneous nucleation and continuous nucleation.20
Instantaneous nucleation:
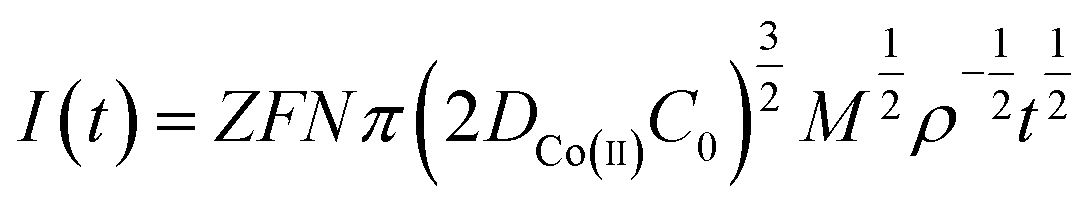 | (7) |
Continuous nucleation:
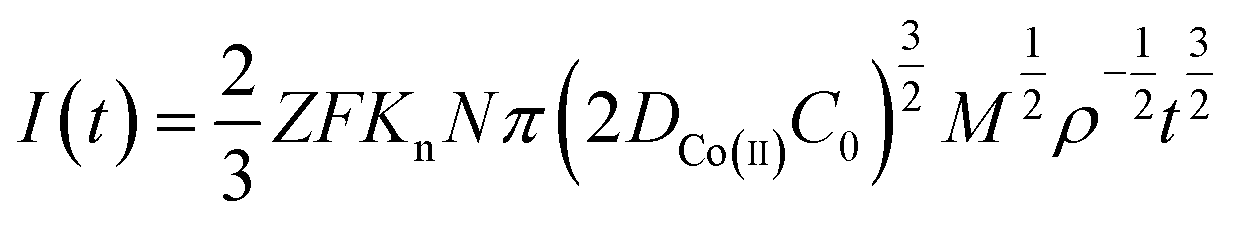 | (8) |
485 C mol−1; N-number density of crystal nucleus/cm−2; DCo(II)-diffusion coefficient/cm2 s−1; C0-concentration of Co(II)/mol cm−3; M-molar mass/g mol−1; ρ-density/g cm−3; Kn-nucleation rate constant/cm−2 s−1; t-time/s; and π = 3.14.
Eqn (7) and (8) can be expressed as follows:
Instantaneous nucleation:
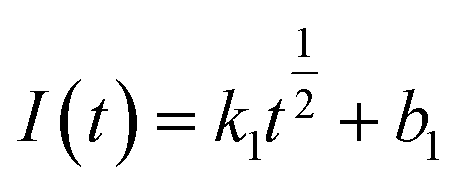 | (9) |
Continuous nucleation:
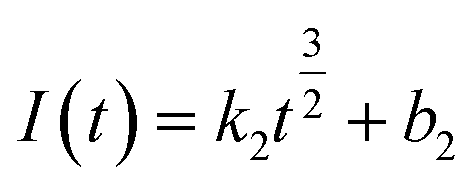 | (10) |
The relationship between 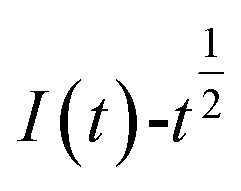 and
and 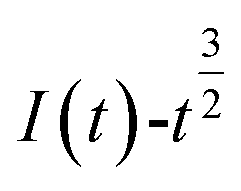 was plotted, according to which the nucleation mechanism of Co(II) was estimated. Different points (from the 3rd to the 6th points) from the rising portion of the chronoamperometric curve at 0.85 V were taken. The relationship of
was plotted, according to which the nucleation mechanism of Co(II) was estimated. Different points (from the 3rd to the 6th points) from the rising portion of the chronoamperometric curve at 0.85 V were taken. The relationship of 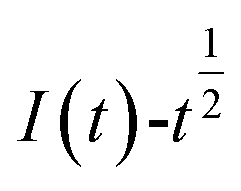 and
and 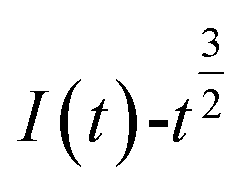 is shown in Fig. 5.
is shown in Fig. 5.
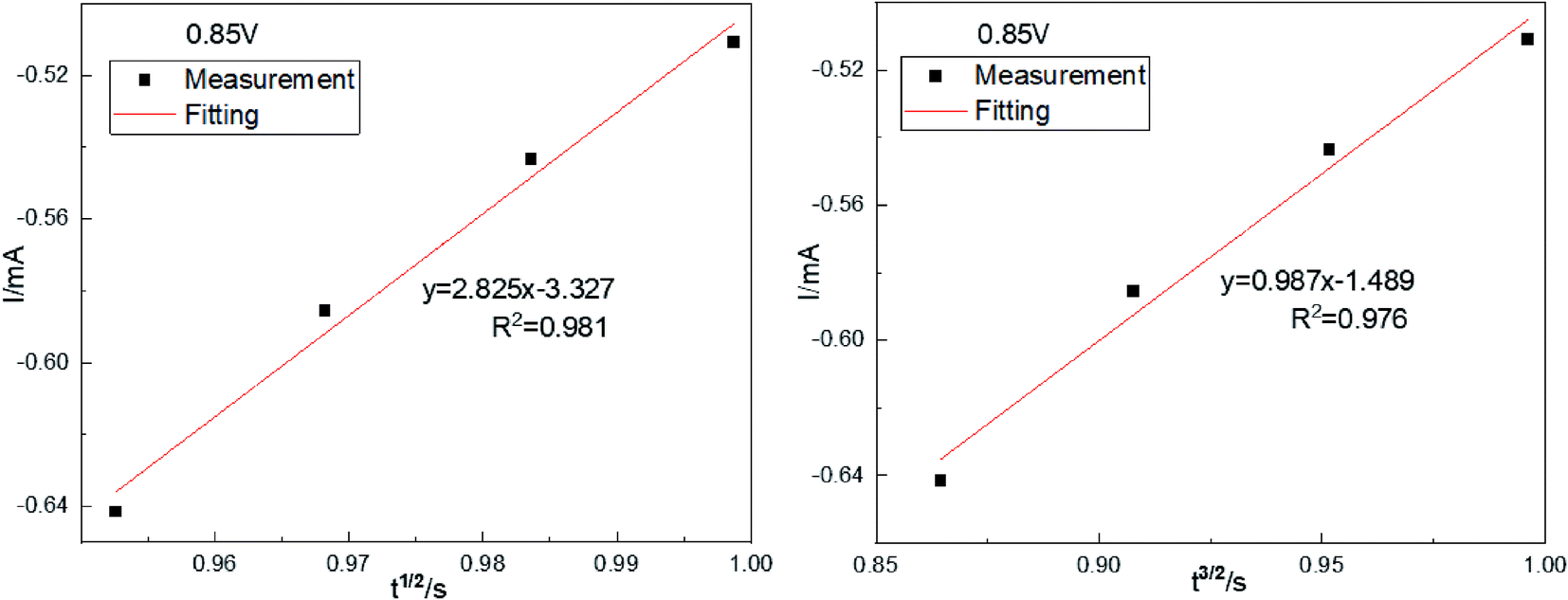 | ||
Fig. 5 The relationship of 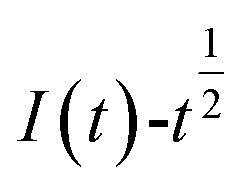 and and 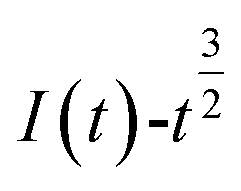 (0.85 V). (0.85 V). | ||
Fig. 5 shows that both fitted lines have a certain deviation. This deviation occurs because cobalt is reduced but adheres to the stainless-steel working electrode surface, which causes the actual working surface area of the electrode to increase rapidly, resulting in fluctuations in the current intensity on the working electrode surface. The goodness of fit suggests that the nucleation mechanism is closer to instantaneous nucleation on the working electrode surface. The fitting equations are shown in eqn (11) and (12).
Instantaneous nucleation:
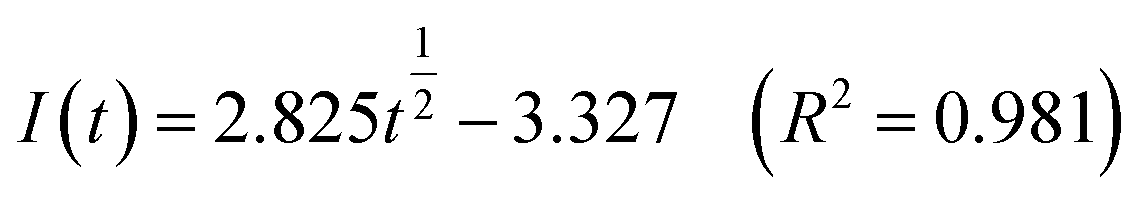 | (11) |
Continuous nucleation:
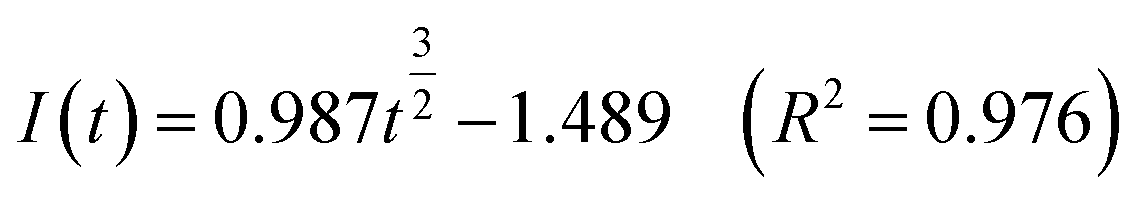 | (12) |
3.4 Test results analysis
To further verify that cobalt can be extracted and recovered from spent cemented carbide by electrodeposition in an acidic solution, deposition products were collected on the surface of the working electrode. The electrode was washed, dried, and then characterized by X-ray diffraction (XRD) and scanning electron microscopy (SEM). The characterization results are shown in Fig. 6.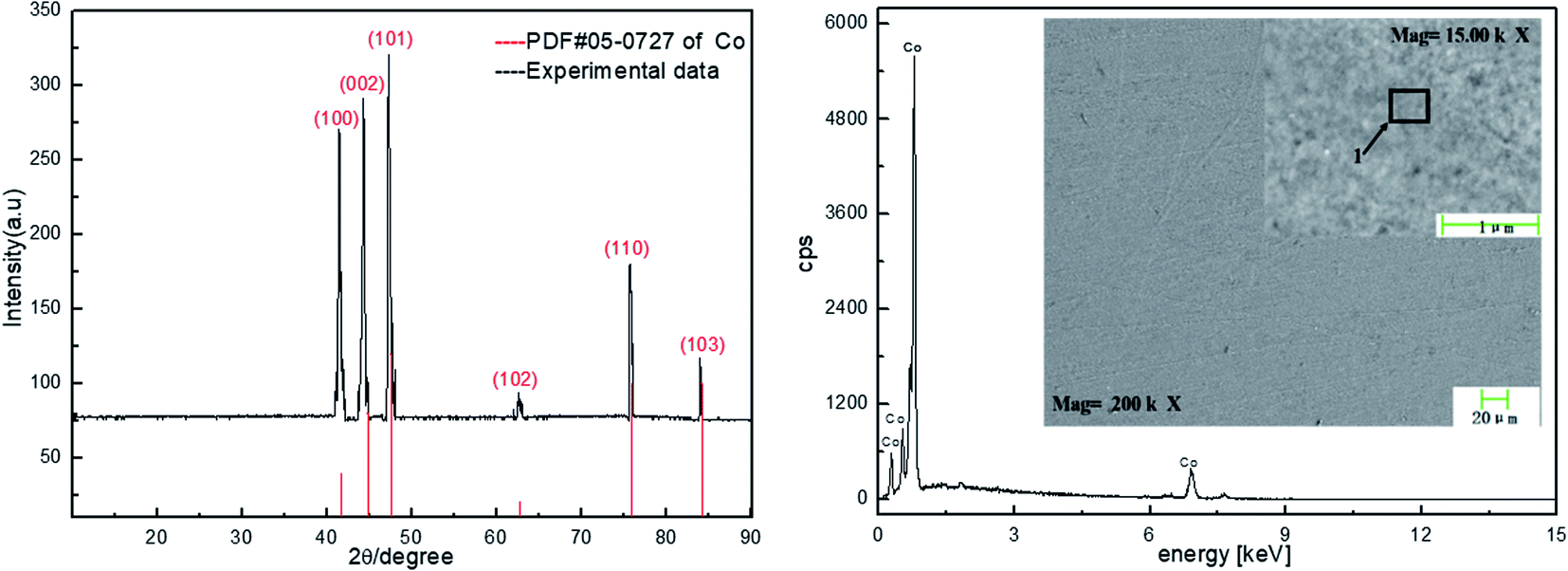 | ||
| Fig. 6 Characterization of Co by XRD and SEM. | ||
Fig. 6 shows that the cobalt obtained in this experiment is dominated by diffraction of the (101) surface. Among the surfaces represented by the diffraction peaks, this surface has the highest cobalt content and is the preferred orientation. The characterizations of Co by XRD and SEM show that the cathode product is cobalt.
To verify that this cobalt resulted from the skeleton metal of the spent cemented carbide, the anode mud was analyzed by XRD and SEM after being recovered, washed, filtered, dried, and ground. The anode mud was divided into two parts in the electrodeposition process: one part oxidized and fell into solution due to the anodizing atmosphere, and the other part adhered to the spent cemented carbide anode. The characterization results are shown in Fig. 7.
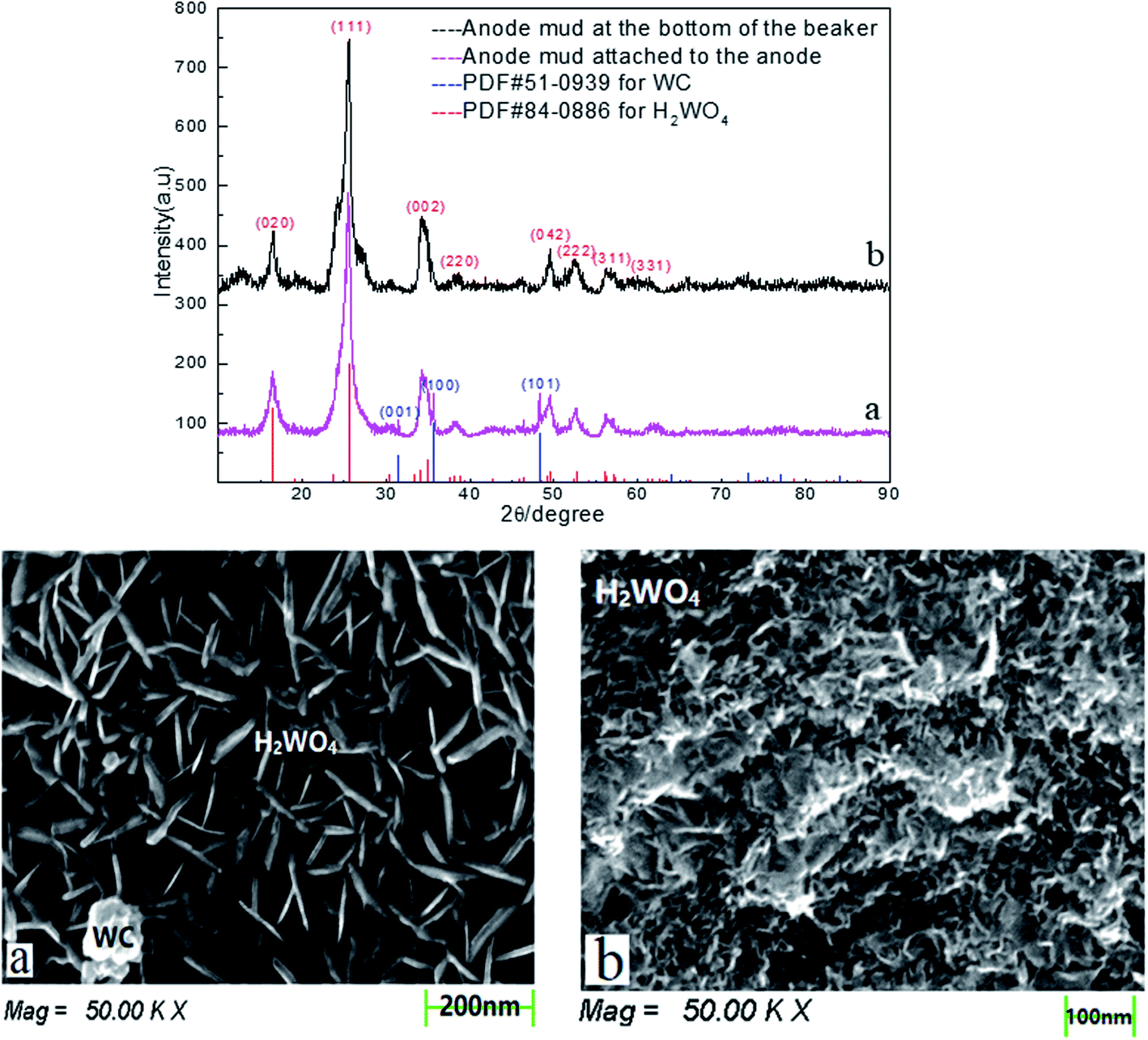 | ||
| Fig. 7 Characterization of anode mud by XRD and SEM. | ||
Fig. 7 shows that there is no cobalt phase in the anode mud characterized by XRD. The cobalt enters the solution under electrodeposition conditions and is then reduced on the surface of the stainless-steel cathode. Comparison of Fig. 7(a) and (b) shows only the tungstic acid (H2WO4) phase of the anode mud is present in solution, while a small amount of the WC phase of the anode mud adheres to the spent cemented carbide. This distribution occurs because a small amount of WC is carried by the anode slime attached to the spent carbide during the scraping process.
4 Conclusions
(1) On the stainless-steel electrode, the reduction mechanism of cobalt is Co2+ + 2e− → Co(s). The nucleation mechanism is close to instantaneous nucleation. The electrodeposition is controlled by the diffusion step and is irreversible. The average diffusion coefficient of Co(II) is 2.16589 × 10−7 cm2 s−1.(2) The cathode product obtained by electrodeposition in an acidic solution at 298 K was characterized by XRD. The results show that the cobalt in the cathode product comes from the skeleton metal of spent cemented carbide.
(3) Electrodeposition experiments under the guidance of transient electrochemical tests show that WC is oxidized to tungstic acid enrichment at the anode, and cobalt adheres to the cathode as a pure element.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
this work is partly supported by the Liaoning Province Natural Science Foundation of China (2019-ZD-0278); The Youth Backbone Talent Project of University of Science and Technology Liaoning (601011507-11); Liaoning BaiQianWan Talents Program (2017104); the Doctoral Scientific Research Startup Foundation of Liaoning Province of China (No. 20180540100); Liaoning University of Science and Technology Graduate Education Reform and Technology Innovation and Entrepreneurship Project (LKDYC201902).References
- P. S. Babu, Y. Madhavi and L. R. Krishna, et al. Thermally-Sprayed WC-Based Cermet Coatings for Corrosion Resistance Applications, Jom, 2018, 70(11), 2636–2649 CrossRef.
- A. B. G. Lansdown, The carcinogenicity of metals: human risk through occupational and environmental exposure, Royal Society of Chemistry, 2013 Search PubMed.
- P. K. Katiyar and N. S. Randhawa, A comprehensive review on recycling methods for cemented tungsten carbide scraps highlighting the electrochemical techniques, Int. J. Refract. Met. Hard Mater., 2020, 105251 CrossRef CAS.
- F. L. Sun, Z. W. Zhao and X. Y. Chen, Recovery of WC and Co from cemented carbide scraps by remelting and electrodissolution, Int. J. Refract. Met. Hard Mater., 2019, 80, 23–29 CrossRef CAS.
- C. S. Freemantle, N. Sacks, M. Topic and C. A. Pineda-Vargas, Impurity characterization of zinc-recycled WC-6 wt% Co cemented carbides, Int. J. Refract. Met. Hard Mater., 2014, 44, 94–102 CrossRef CAS.
- X. Long, R. Chen and J. Tan, et al. Electrochemical recovery of cobalt using nanoparticles film of copper hexacyanoferrates from aqueous solution, J. Hazard. Mater., 2020, 384, 121252 CrossRef CAS PubMed.
- X. Xiao, X. Xi and Z. Nie, et al. Direct electrochemical preparation of cobalt, tungsten, and tungsten carbide from cemented carbide scrap, Metall. Mater. Trans. B, 2017, 48(1), 692–700 CrossRef CAS.
- X. Xi, X. Xiao and Z. Nie, et al. Electrolytic separation of cobalt and tungsten from cemented carbide scrap and the electrochemical behavior of metal ions, J. Electroanal. Chem., 2017, 794, 254–263 CrossRef CAS.
- A. L. Zhang, B. Z. Nie and C. X. Xi, et al. Electrochemical separation and extraction of cobalt and tungsten from cemented scrap, Sep. Purif. Technol., 2018, 195, 244–252 CrossRef CAS.
- H. X. Liang and S. G. Chen, Electrochemical Treatment of spent Carbide for Recovery of Tungsten and Cobalt, J. Shanghai Univ., Nat. Sci. Ed., 1995, 12(2), 89–91 Search PubMed.
- R. R. Srivastava, J. Lee, M. Bae and V. Kumar, Reclamation of tungsten from carbide scraps and spent materials, J. Mater. Sci., 2019, 54(1), 83–107 CrossRef CAS.
- J. Wang, J. Liang, H. Li, et al. Reduction Mechanism of Metal Cobalt from Cathode Material of Waste Lithium Cobalt Oxide Battery, TMS 2020 149th Annual Meeting & Exhibition Supplemental Proceedings, Springer, Cham, 2020, pp. 1301–1307 Search PubMed.
- P. Patnaik, S. K. Padhy, B. C. Tripathy, I. N. Bhattacharya and R. K. Paramguru, Electrodeposition of cobalt from aqueous sulphate solutions in the presence of tetra ethyl ammonium bromide, Trans. Nonferrous Met. Soc. China, 2015, 25(6), 2047–2053 CrossRef CAS.
- V. M. López-Hirata and E. M. Arce-Estrada, Characterization of Co–Cu mechanical alloys by linear sweep voltammetry, Electrochim. Acta, 1997, 42(1), 61–65 CrossRef.
- M. P. Olson and W. R. LaCourse, Voltammetry, Ewing's Analytical Instrumentation Handbook, CRC Press, 2019, 4th edn, pp. 509–522 Search PubMed.
- I. L. A. Joshi, K. Khati, A. Bisht, R. Rikhari, S. Mehtab and M. G. H. Zaidi, Electrical and electrochemical properties of tungsten carbide, Int. J. Conserv. Sci., 2018, 6(3), 3600–3603 Search PubMed.
- N. Schubert, M. Schneider and A. Michaelis, Electrochemical Machining of cemented carbides, Int. J. Refract. Met. Hard Mater., 2014, 47, 54–60 CrossRef CAS.
- W. S. Choi, S. H. Cho and Y. J. Lee, et al. Separation behavior of nickel and cobalt in a LiCl–KCl–NiCl2 molten salt by electrorefining process, J. Electroanal. Chem., 2020, 114175 CrossRef CAS.
- C. Brubaker and Z. Liu, Diffusion couple study of the Mg-Al system, Magnesium Technology 2004, Minerals, Metals and Materials Society, 2004, pp. 229–234 Search PubMed.
- V. A. Isaev, O. V. Grishenkova and Y. P. Zaykov, Theoretical modeling of electrochemical nucleation and growth of a single metal nanocluster on a nanoelectrode, RSC Adv., 2020, 10(12), 6979–6984 RSC.
| This journal is © The Royal Society of Chemistry 2020 |