
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTantalum(V) 1,3-propanediolate β-diketonate solution as a precursor to sol–gel derived, metal oxide thin films†
Christopher Beale ab,
Stefanie Hamacherab,
Alexey Yakushenkoc,
Oumaima Bensaidab,
Sabine Willboldd,
Guillermo Beltramoe,
Sören Möllerf,
Heinrich Hartmannd,
Elmar Neumanng,
Gregor Musslerh,
Alexander Shkurmanovh,
Dirk Mayera,
Bernhard Wolfrumai and
Andreas Offenhäusser*a
ab,
Stefanie Hamacherab,
Alexey Yakushenkoc,
Oumaima Bensaidab,
Sabine Willboldd,
Guillermo Beltramoe,
Sören Möllerf,
Heinrich Hartmannd,
Elmar Neumanng,
Gregor Musslerh,
Alexander Shkurmanovh,
Dirk Mayera,
Bernhard Wolfrumai and
Andreas Offenhäusser*a
aIBI-3, Bioelectronics, Forschungszentrum Jülich GmbH, D-52425, Germany. E-mail: a.offenhaeusser@fz-juelich.de
bRWTH Aachen University, Templergraben 55, D-52062, Germany
cFraunhofer Research Institute for Microsystems and Solid State Techologies, D-80686 Munich, Germany
dZEA-3, Analytics, Forschungszentrum Jülich GmbH, D-52425, Germany
eIBI-2, Mechanobiology, Forschungszentrum Jülich GmbH, D-52425, Germany
fIEK-1, Materials Synthesis and Processing, Forschungszentrum Jülich GmbH, D-52425, Germany
gHelmholtz Nano Facility, Forschungszentrum Jülich GmbH, D-52425, Germany
hPGI-9, Semiconductor Nanoelectronics, Forschungszentrum Jülich GmbH, D-52425, Germany
iNeuroelectronics, Munich School of Bioengineering, Department of Electrical and Computer Engineering, Technical University of Munich (TUM), D-85748 Garching, Germany
First published on 3rd April 2020
Abstract
Tantalum oxide is ubiquitous in everyday life, from capacitors in electronics to ion conductors for electrochromic windows and electrochemical storage devices. Investigations into sol–gel deposition of tantalum oxide, and its sister niobium oxide, has accelerated since the 1980s and continues to this day. The aim of this study is to synthesize a near UV sensitive, air stable, and low toxicity tantalum sol–gel precursor solution for metal oxide thin films – these attributes promise to reduce manufacturing costs and allow for facile mass production. By utilizing 1D and 2D nuclear magnetic resonance, this study shows that by removing ethanol from the precursor solution at a relatively low temperature and pressure, decomposition of the photosensitive complex can be minimized while obtaining a precursor solution with sufficient stability for storage and processing in the atmosphere. The solution described herein is further modified for inkjet printing, where multiple material characterization techniques demonstrate that the solution can be utilized in low temperature, photochemical solution deposition of tantalum oxide, which is likely amorphous. Tested substrates include amorphous silica, crystalline silicon wafer, and gold/titanium/PET foil. The hope is that these results may spark future investigations into electronic, optical, and biomedical device fabrication with tantalum oxide, and potentially niobium oxide, based films using the proposed synthesis method.
Introduction
At the start of World War II, Lieutenant Commander O. Hugh Fulcher of the United States Navy performed the first successful cranioplasty with tantalum metal, claiming it as the most suitable material and as to “keep as many men at as many guns as many days as possible”.1 The success of tantalum in medical implants was known to be due to a biocompatible, corrosion resistant oxide film which forms on the surface of the metal.2 Modern medical research has additionally elucidated that implants having a porous tantalum coating upregulate osteogenic genes, resulting in greater osteointegration when compared to titanium implants without the coating.3 Aside from applications in orthopedic implants,4 tantalum and its sister niobium form oxides which have a variety of uses, including capacitors,5 batteries,6,7 electrochromic windows,8 pH and conductivity sensors,9–13 surface plasmon resonance sensors,14–16 ferroelectric memory devices,17–20 corrosion resistant coatings,21 and even gravity wave detectors.22During manufacturing of such devices, production inefficiencies and waste material lead to the addition of a recycling step,23 thereby complicating the fabrication of products containing tantalum. Efforts to deposit such oxides via additive processes at relatively low temperatures, which would save material and energy,24–26 still remain a challenge. Therefore, this study aims to further investigations into additive processing via inkjet printing and low temperature annealing of tantalum oxide, and potentially niobium oxide, with a focus on the sol–gel method – this method is a subset of chemical solution deposition (CSD) and photochemical solution deposition (PCSD). Advantages of the sol–gel method include lower costs, stoichiometry control, and feasible implementation into a roll-to-roll manufacturing line.27,28
Sol–gel synthesis with niobium and tantalum alkoxides
In order to produce metal oxide films free of halide impurities via the sol–gel route, a metal halide precursor can be converted to a metal alkoxide derivative before use in CSD or PCSD.29 Avoiding halides may also be necessary in some cases, while compounds such as hydrochloric acid can be formed during synthesis, thus creating an acidic environment which may be undesirable.30 However, niobium and tantalum alkoxides (dimers at room temperature) are relatively sensitive to atmospheric moisture via the following reaction31–34 (M = Nb, Ta; R = methyl, ethyl):| M2(OR)10 + 5H2O → M2O5 + 10ROH | (1) |
An option to minimize hydrolysis from water is by using 2-methoxyethanol as the solvent for the metal alkoxide precursor, due to 2-methoxyethanol's ability to prevent nucleophilic attack from water to the cation via a chelate complex.35 Although 2-methoxyethanol is often used as a solvent in CSD,36 2-methoxyethanol has been reported to have developmental and reproductive toxicity,35 and poses a danger via inhalation (with its relatively high vapor pressure) and skin exposure. Furthermore, in light of its inherent toxicity, use of 2-methoxyethanol has been restricted in some countries, thus making its use in manufacturing difficult.35 This calls for alternative strategies to prevent hydrolysis before solution deposition.
Sol–gel synthesis with glycolate solutions
One way to avoid 2-methoxyethanol is by using glycols as a solvent instead. Following the work of Mehrotra and Kapoor37,38 in 1965, where glycol substitution in niobium and tantalum ethoxides was described, Calzada and González17,18 reported in 2005 the substitution of ethoxy groups in tantalum(V) ethoxide with 1,3-propanediol via reflux and distillation. This resulted in a tantalum(V) 1,3-propanediolate solution, which is air stable for months:| Ta(OC2H5)5 + 5HOC3H6OH → Ta(OC3H6OH)5 + 5C2H5OH | (2) |
In this case, the substituting glycol also acts as the solvent, which simplifies the formulation of the solution. 1,3-Propanediol has also been used for niobium based solutions.19,20
The air stable, low toxic tantalum(V) 1,3-propanediolate solution produced by Calzada and González proves advantageous for high temperature CSD,17,18 and possibly even for far UV processing in PCSD where exposure of tantalum(V) ethoxide to these conditions has resulted in tantalum oxide films.39,40 However, the solution is likely not suitable for low temperature and near UV processing as its photoabsorption in this range would likely be too low.39,40
Sol–gel synthesis with β-diketonate complexes
One way to increase the photoabsorption for near UV processing is to chelate a β-diketone or β-ketoester to the metal cation.41–45 These ligands can absorb in the 200 nm to 380 nm range as a result of the π → π* transition (i.e., the π bonding to π antibonding transition), where photo-excitation can result in ligand dissociation and metal oxide formation,26,35,41 with the photochemical mechanism described in detail by Segawa et al.46 and Squibb et al.47After Whitley48,49 pioneered the synthesis of various tantalum(V) alkoxides for her PhD thesis in 1954 by following the experiments of Funk and Niederländer,50,51 she reported in her same work the chelation of benzoylacetone to an ethoxide of tantalum.48,52 This was later confirmed for various β-diketonate and β-ketoester compounds with niobium and tantalum53–56 (M = Nb, Ta; R = methyl, ethyl; AH = β-diketone or β-ketoester; A = chelated form; x = 1–3):
| M(OR)5 + xAH → (RO)5−xMAx + xROH | (3) |
Glycolate solutions with β-diketonate complexes
When mixing metal β-diketonate complexes with glycols, preliminary attempts at isolating metal glycolate β-diketonate complexes were reported in the 1990s. For example, Calzada and coworkers57 claim to have successfully produced titanium(IV) 1,3-propanediolate bis acetylacetonate complexes via reflux and discuss their potential use in PCSD.58However, Kemmitt and Daglish59 found that prolonged solution exposure to heat leads to the alcoholysis of acetylacetone and the acetylation of 1,3-propanediol, resulting in the decomposition of the titanium(IV) acetylacetonate complex and the formation of an insoluble Ti(IV)(OC3H6O)2 precipitate (both terminal oxygens being bound to the Ti(IV) cation). This indicates that attempts at creating metal glycolate β-diketonate solutions may result in unwanted side products, thus decreasing the amount of desired β-diketonate complexes, which are necessary for photosensitivity in the near UV range. Alcoholysis of other metal acetylacetonates, resulting in acetylation of alcohol groups for nanoparticle formation,60,61 has been known for some time.62
Thus, in order to combine the benefits of tantalum or niobium glycolate solutions, those being low toxicity and stability in ambient conditions, with the near UV photosensitivity of tantalum or niobium β-diketonate complexes, a procedure is needed which reduces the chance of complex alcoholysis when mixing glycols, such as 1,3-propanediol, with niobium or tantalum β-diketonate complexes. This would lead not only to safer, stable solutions that could be more easily used in manufacturing facilities, but would also result in a more efficient β-diketone chelation reaction while minimizing the formation of side products from alcoholysis.
Therefore, this study will characterize two solutions from a proposed synthesis method via nuclear magnetic resonance (NMR), where 1,3-propanediol is added to an ethanol solution containing tantalum(V) acetylacetonate or tantalum(V) benzoylacetonate complexes at a reduced temperature with subsequent ethanol removal at a reduced pressure, with the desired reaction (A = chelated β-diketone):
| TaA(OC2H5)4 + 4HOC3H6OH → TaA(OC3H6OH)4 + 4C2H5OH | (4) |
Solution synthesis characterization will be followed by examination of the tantalum oxide layers derived from these solutions.
Results and discussion
In preparing the tantalum(V) ethoxide β-diketonate precursor, anhydrous ethanol was chosen as the reflux solvent since lighter monoalcohols are less likely to undergo alcoholysis than heavier diols in the presence of a metal cation and a β-diketone.59 In support of this, exposure of the Calzada and González tantalum(V) 1,3-propanediolate precursor18 with acetylacetone to <80 °C over 72 hours demonstrates that alcoholysis with 1,3-propanediol is possible in the absence of ethanol (see ESI†). Therefore, higher temperatures with heavier diols should be avoided, as well as long reflux times with lighter monoalcohols where 8 hours should be sufficient.48,55,63 Reflux solvents such as benzene48,55 or tetrahydrofuran63 could be used, but the former is carcinogenic and the latter may present an explosion hazard.Proposed synthesis method
The proposed method for minimizing alcoholysis from 1,3-propanediol first involves acquiring or forming a tantalum(V) ethoxide β-diketonate complex, followed by mixing the complex with 1,3-propanediol at room temperature (rather than under reflux17,18,57,59). The mixing should lead to the substitution of the ethoxide groups with 1,3-propanediol, where the reaction is driven to completion by adjusting the system temperature and pressure so that ethanol distills without 1,3-propanediol also distilling or causing decomposition of the tantalum(V) β-diketonate complex.More particularly, the distillation temperature and pressure should be chosen such that alcoholysis of the tantalum(V) β-diketonate complex is minimized and that the vapor pressure of ethanol is greater than the pressure of the closed system, while the vapor pressure of 1,3-propanediol and the tantalum(V) 1,3-propanediolate β-diketonate complex is less than the pressure of the closed system. After ethanol removal, the tantalum(V) 1,3-propanediolate β-diketonate solution would be isolated in a liquid form at room temperature, ready for CSD or PCSD.
For example in Fig. S1,† at 23 °C ethanol has a vapor pressure of 70 mbar,64,65 while 1,3-propanediol has a vapor pressure of 0.1 mbar66,67 (1,3-propanediol data are extrapolated beyond the experimental data range via the literature Antoine parameters). By increasing the temperature mildly to 40 °C, ethanol's vapor pressure increases to 180 mbar64,65 and 1,3-propanediol's to 0.4 mbar66,67 (the latter still being a theoretical extrapolation). Thus, at 40 °C the system pressure should be set above 0.4 mbar and below 180 mbar in order to remove ethanol from the solution, which should then allow the tantalum(V) 1,3-propanediolate β-diketonate solution to remain.
Therefore, when starting with 1,3-propanediol and a tantalum(V) tetraethoxide β-diketonate complex, such as tantalum(V) tetraethoxide acetylacetonate or tantalum(V) tetraethoxide benzoylacetonate, under the optimal temperature and pressure the desired substitution may proceed as shown in Fig. 1. If the temperature is set too high and for too long, then undesired alcoholysis of the complex is more likely, with the proposed reaction mechanism62 also shown in Fig. 1.
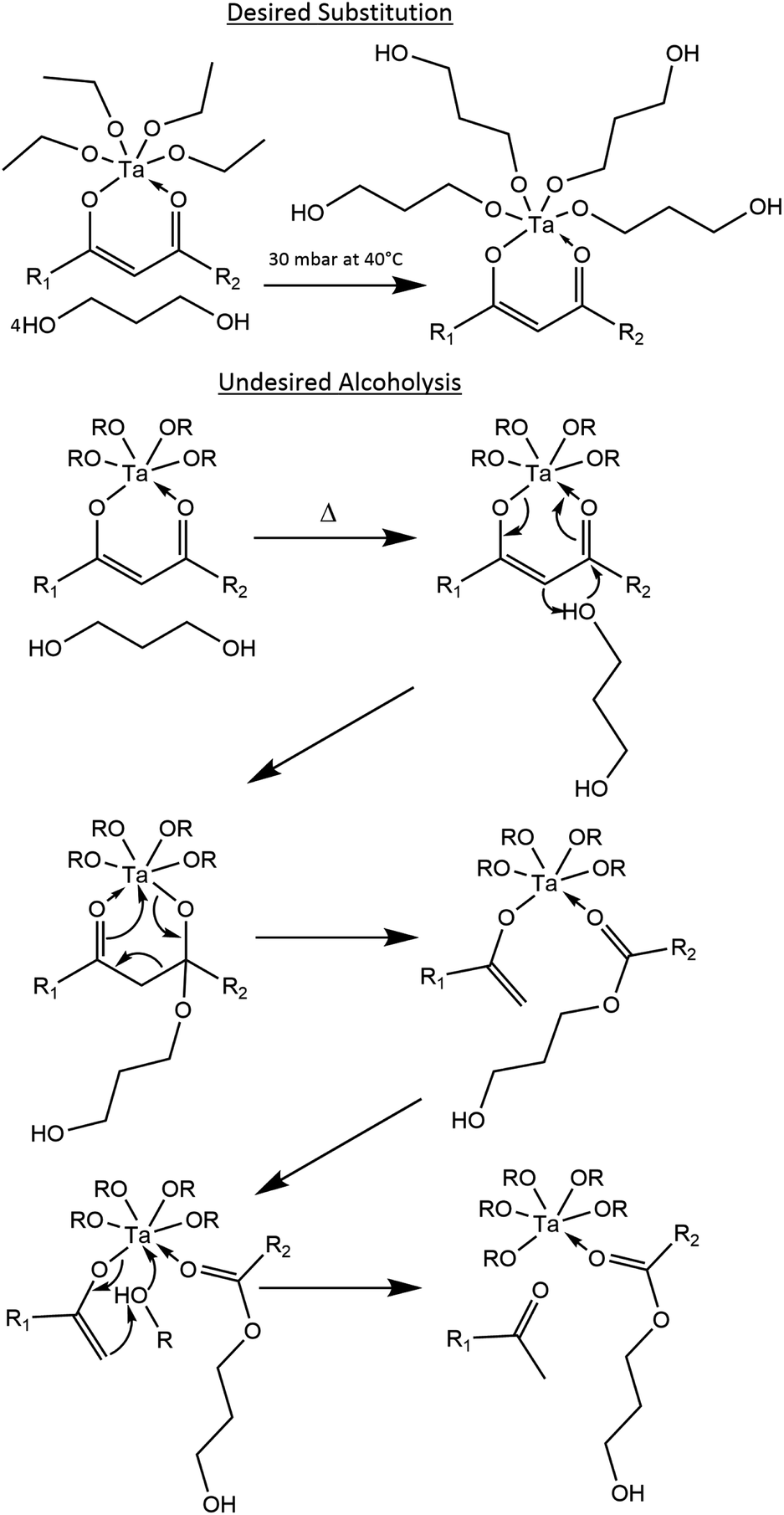 | ||
| Fig. 1 Desired substitution of ethanol with 1,3-propanediol, and the undesired mechanism of alcoholysis; acetylacetone: R1 = R2 = methyl; benzoylacetone: R1 ≠ R2, R1 = methyl or phenyl, R2 = methyl or phenyl. | ||
After removing the ethanol at 30 mbar and <40 °C in both the acetylacetone and the bezoylacetone solutions over 8 hours, the benzoylacetone solution from the proposed method in Fig. 2b appears to be redshifted from that of the acetylacetone solution in Fig. 2a. Furthermore, no visible, insoluble precipitates are present, as was reported for Ti(IV) and Zr(IV) acetylacetonate complexes in the presence of 1,3-propanediol (where M(IV)(OC3H6O)2 was formed).59 Additionally, when Ta(V) ethoxide is replaced with Ti(IV) isopropoxide, a turbid solution forms at room temperature after 1,3-propanediol addition (see ESI†). The lack of visible precipitates in the Ta(V) solutions may be due to the odd, +5 valency of the cation. UV/Vis spectrophotometry for the solutions was not performed, as this would require a significant dilution, thereby changing the product. Therefore, UV/Vis absorption of the printed layers was performed instead (described later), which is more relevant for applications in PCSD.
 | ||
| Fig. 2 Final solution products from the proposed method with acetylacetone (a), and the proposed method with benzoylacetone (b), where (b) appears redshifted relative to (a). | ||
NMR measurements
1H and 13C NMR spectra of the reagents are shown in Fig. S2–S9,† which aided in the peak assignments described in this section. Total correlation spectroscopy (TOCSY) and heteronuclear multiple bond correlation (HMBC) spectra were also used to assign peaks for some of the products. Fig. 3 serves as a visual aid for the discussion.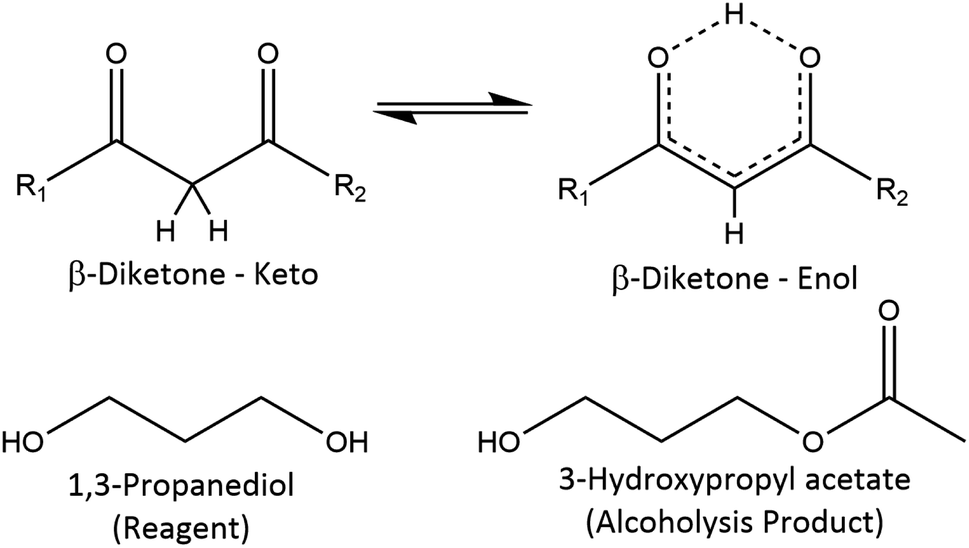 | ||
| Fig. 3 Reference structures with NMR data in the ESI† and literature. In the β-diketone enol tautomer, the upper proton can be replaced by a metal cation; acetylacetone: R1 = R2 = methyl; benzoylacetone: R1 ≠ R2, R1 = methyl or phenyl, R2 = methyl or phenyl. | ||
NMR: proposed synthesis – acetylacetone
The 1H NMR spectrum of the product (δ [ppm], Fig. S10 and S11†) shows the presence of the expected 1,3-propanediol peaks at δ 1.81 [HOCH2CH2CH2OH], δ 3.72 [HOCH2CH2CH2OH], and δ 5.21 [HOCH2CH2CH2OH]. Three separate methine acetylacetone-enol peaks appear at δ 5.96, δ 5.87, and δ 5.74 [H3C(CO)CH(CO)CH3]. The peaks from δ 2.30 to δ 2.14 overlap significantly (with δ 2.21 being the most prominent peak, see Fig. S11†), making the following methyl peak assignments tentative: acetylacetone-keto [H3C(CO)CH2(CO)CH3] or acetone [H3C(CO)CH3] methyl peaks (the latter from alcoholysis)59,68 occur at δ 2.30 and δ 2.26, and the acetylacetone-enol [H3C(CO)CH(CO)CH3] methyl peaks around δ 2.21 and δ 2.14. The multiple peaks for the same proton on the acetylacetone-enol tautomer may be due to the different ways in which acetylacetone can bind to the metal cation.62Further downfield, as the δ 4.21 peak resonates at a higher frequency than the alcoholic methylene 1,3-propanediol peak at δ 3.72, in addition to the presence of another overlapping methyl peak at δ 2.14, likely indicates the acetylation of 1,3-propanediol and the creation of 3-hydroxypropyl acetate,69 despite the extra steps taken to avoid alcoholysis of the complex. One of the overlapping peaks at δ 2.14 would then correspond to the methyl attached to the ester carbonyl [HOCH2CH2CH2O(CO)CH3], and the peak at δ 4.21 would be the protons on the methylene carbon bound to the ester oxygen [HOCH2CH2CH2O(CO)CH3]. Additionally, the TOCSY spectrum in Fig. S12† shows three distinct proton groups on three consecutive carbon atoms, and the 13C NMR and HMBC spectra corroborate these findings (discussed next), as well as the literature values for 3-hydroxypropyl acetate.70 It is not clear whether the δ 4.21 peak occurs as a triplet, and the peak at δ 4.65 is difficult to assign with the available data.
The 13C NMR spectrum of the product (δ [ppm], Fig. S13†) shows the expected 1,3-propanediol peaks at δ 35.2 [HOCH2CH2CH2OH] and δ 59.0 [HOCH2CH2CH2OH]. Acetylacetone-enol peaks around δ 192.0 [H3C(CO)CH(CO)CH3], around δ 102.0 [H3C(CO)CH(CO)CH3], and around δ 26.2 [H3C(CO)CH(CO)CH3] appear in the spectrum, along with some acetylacetone-keto [H3C(CO)CH2(CO)CH3] or acetone59,71 [H3C(CO)CH3] peaks at δ 209.2 and δ 204.3, and around δ 31.6 [H3C(CO)CH2(CO)CH3] or [H3C(CO)CH3]; there is possibly an acetylacetone-keto peak around δ 59.0 [H3C(CO)CH2(CO)CH3], which is difficult to assign due to the presence of 1,3-propanediol. The peak at δ 171.8 [HOCH2CH2CH2O(CO)CH3], not associated with any of the reagents, is located in the range typically found for ester carbonyl carbons and would support the idea that 1,3-propanediol has been acetylated and that 3-hydroxypropyl acetate has been formed.69 This is supported by literature70 and the HMBC spectrum in Fig. S14,† which shows cross-peaks at 1H δ 2.14–13C δ 171.8 and 1H δ 4.21–13C δ 171.8. HMBC also shows no visible cross-peak at 1H δ 4.65–13C δ 171.8, possibly indicating that the compound with the 1H δ 4.65 peak does not contain an acetylated alcohol.
NMR: proposed synthesis – benzoylacetone
The 1H NMR spectrum of the product (δ [ppm], Fig. S15 and S16†) shows the presence of the expected 1,3-propanediol peaks at δ 1.68 [HOCH2CH2CH2OH], δ 3.59 [HOCH2CH2CH2OH], and δ 5.11 [HOCH2CH2CH2OH]. Three separate methine benzoylacetone-enol peaks appear at δ 6.53, δ 6.44, and δ 6.24 (Ph = phenyl group) [Ph(CO)CH(CO)CH3]. The peaks from δ 2.50 to δ 2.00 overlap significantly (with δ 2.22 being the most prominent peak), with the following tentative assignments: δ 2.46 for the benzoylacetone-keto methyl [Ph(CO)CH2(CO)CH3] and around δ 2.22 for the enol methyl [PhC(CO)CH(CO)CH3]. Peaks in the δ 8.00 to δ 7.30 range belong to the phenyl group protons on benzoylacetone [PhC(CO)CH(CO)CH3]. Many unassigned peaks are present, possibly associated with different alcoholysis products. As for acetylacetone, benzoylacetone may be bound to the metal cation in different ways,62 resulting in multiple peaks for the same proton.The 13C NMR spectrum of the product (δ [ppm], Fig. S17†) shows the expected 1,3-propanediol peaks at δ 35.1 [HOCH2CH2CH2OH] and δ 58.9 [HOCH2CH2CH2OH]. Benzoylacetone-enol peaks around δ 193.0 [PhC(CO)CH(CO)CH3], around δ 182.0 [PhC(CO)CH(CO)CH3], around δ 100.0 [PhC(CO)CH(CO)CH3], peak(s) within the range δ 28.0 to δ 25.0 [Ph(CO)CH(CO)CH3] (difficult to assign exact peak), and the multiple phenyl ring carbon peaks (likely mixed in with those of the keto tautomer) in the range δ 140.0 to δ 125.0 [Ph(CO)CH(CO)CH3] appear in the spectrum, along with some benzoylacetone-keto peaks at δ 199.2 [PhC(CO)CH2(CO)CH3], a carbonyl peak likely overshadowed by enol peaks in the range δ 195.0 to 190.0 [PhC(CO)CH2(CO)CH3], around δ 59.0 [H3C(CO)CH2(CO)CH3] (difficult to assign exact peak), and peak(s) within the range δ 33.0 to δ 30.0 [Ph(CO)CH2(CO)CH3] (difficult to assign exact peak). The peak at δ 171.7, not associated with any of the reagents, is located in the range typically found for ester carbonyl carbons and would support the idea that acetylation of an alcohol group has occurred.69
Proposed synthesis product and ink rheology
For viscosity and surface tension measurements, separate syntheses of the proposed method (following the same procedures) were completed due to lack of material, and the reagent amounts are shown in Fig. S18.† Layers characterized by other methods described in this study were derived from the solutions which were produced for the NMR measurements.Dilutions were made with diethylene glycol monoethyl ether (DEGEE), which allows the target viscosity (∼10 mPa s) and surface tension (∼30 mN m−1) for inkjet printing to be reached. However, unlike how Matavž et al.72 achieved the target viscosity and surface tension by mixing 1,3-propanediol with highly toxic 2-methoxyethanol, DEGEE presents a reduced exposure hazard with its lower vapor pressure, and can be used in lieu of 2-methoxyethanol to achieve the target values. The results are shown in Table 1.
| Solutions at 23 °C | Viscosity (mPa s) | Surface tension (mN m−1) |
|---|---|---|
| 1,3-Propanediol | 48.1 | 44 |
| DEGEE | 4.60 | 31 |
| Acac product | 347 | 45 |
| 40 wt% Acac prod. | 15.4 | 34 |
| 30 wt% Acac prod. | 10.3 | 33 |
| Bzac product | 850 | 45 |
| 30 wt% Bzac prod. | 10.0 | 33 |
UV/Vis – proposed synthesis method on a-SiO2
For the acetylacetone based printed film, UV/Vis on a-SiO2 in Fig. 4a and b shows a local maximum at 319 nm before xenon flash lamp exposure (125 °C anneal – 0 shots), demonstrating chelation of acetylacetone to the tantalum cation within the layer.43,44 After firing 1000 shots on the layer, the peak at 319 nm disappears, and the global maximum redshifts to about 212 nm, which is similar to the maximum obtained in prior work via photoirradiation.39,40 This shows that despite the solution impurities and alcoholysis byproducts that appear in the NMR spectra, the solution is still viable for PCSD. | ||
| Fig. 4 UV/Vis spectra (a and b) of acetylacetone based layers on a-SiO2, with (b) zooming in on the metal ligand absorption peak for the 0 shots sample. Raman (c) and XRD (d) appear to indicate that xenon flash lamp treatment may result in amorphous tantalum oxide films; UV/Vis spectra are normalized to the maximum of the 0 shots sample, while Raman and XRD spectra have been arbitrarily shifted for clarity. | ||
For the benzoylacetone based printed film, UV/Vis on a-SiO2 in Fig. S19† shows an absorption peak at 348 nm before xenon flash lamp exposure, again demonstrating chelation of benzoylacetone to the tantalum cation.42–44 More than 1000 shots are required for the chelation peak to diminish, and for a global maximum around 212 nm to form. Again, despite possible alcoholysis byproducts, the solution can be implemented in PCSD applications.
In the case of both the acetylacetone and benzoylacetone based films, the π → π* transition metal ligand peaks are redshifted in comparison to the β-diketones' unbound form (319 nm in the film from ∼270 nm for acetylacetone, and 348 nm in the film from ∼310 nm for benzoylacetone),73 which is to be expected.41,43,44
The remaining characterization results were only performed for the acetylacetone based films from the proposed synthesis method. This was done in order to avoid carbon impurities in the photocured oxide layer, as acetylacetone is a smaller molecule than benzoylacetone.
Raman and XRD – acetylacetone based film on a-SiO2
The literature Raman spectra of Ta2O5 are very complex, containing multiple peaks which are dependent on whether the final compound is amorphous or crystalline.74 The bands between 400 cm−1 and 900 cm−1 are associated with the coupled modes involving mainly the stretching and bending of the various Ta–O bonds. In the case of amorphous Ta2O5, the band at 670 cm−1 corresponds to the bending and stretching of triple-coordinated oxygen (O–3Ta) and the band at 800 cm−1 is due to the stretching of double-coordinated oxygen (O–2Ta).22,74Fig. 4c shows the Raman spectra obtained from the thin tantalum oxide based layer on the a-SiO2 substrate. These spectra were achieved after a hyperspectral analysis of the measured interface and subsequent subtraction of the remnant a-SiO2 spectrum. Depth scans distinguishing the deposited and photocured tantalum oxide layer from the a-SiO2 substrate are shown in Fig. S20,† where the thickness of the layer observed in the Raman image is thicker than the actual thickness; this is due to the lower Z-resolution in optical microscopy.
The lower spectrum (0 shots) in Fig. 4c was measured after a 125 °C thermal anneal, and the upper spectrum is of the same sample after 1000 flash lamp shots. Both spectra show a broad peak around 670 cm−1 and a shoulder around 850 cm−1, where these peaks correspond to amorphous Ta2O5.22,74,75 X-ray diffraction (XRD) spectra in Fig. 4d supports inconclusively that photocuring at room temperature does not create crystalline Ta2O5, which is known to require temperatures above 600 °C.18,74,75
Before photocuring (0 shots), the Raman spectrum shows not only amorphous tantalum oxide peaks, but also organic peaks. These organic peaks are mainly observed around 2800 cm−1, which corresponds to the C–H stretching modes of organic compounds. Additionally, the spectra of the precursors in Fig. S21† show that the precursor peaks are largely absent after photocuring.
RBS/NRA – acetylacetone based films on c-Si
The Rutherford backscattering spectrometry (RBS) and nuclear reaction analysis (NRA) spectra of the 125 °C pre-annealed samples show sharp peak forms in Fig. 5a and b (0 shots and 1000 shots, respectively), and correspondingly all detected material lies in the first resolution bin of 5.5 × 1021 atoms per m2. The quantitative data is tabulated in Table 2, and the layer thicknesses are in the range of 1.0 × 1021 atoms per m2 to 1.5 × 1021 atoms per m2. Small amounts of carbon corresponding to a few atom% (at%) in the layer were found in both samples. The oxygen to tantalum ratios in the 1000 shots sample do not match the Ta2O5 stoichiometry within error, with an oxygen excess of O/Ta = 3.9 ± 0.5 (ideally O/Ta = 2.5) and exceeding that of anodic tantalum oxide NRA oxygen standards.76–78 The extra oxygen could be due to SiO2 formation, organics, or water. Due to the limited depth-resolution, it cannot be excluded that some of the oxygen is present near the surface of or in the c-Si substrate. Although the layer thicknesses vary, the O/Ta ratio stays constant within uncertainties. | ||
| Fig. 5 RBS/NRA spectra of samples on c-Si for 0 shots (a) and 1000 shots (b) with simulated element peaks superimposed on the experimental data – the 12C(d,p0)13C peak appears to decrease relative to the 16O(d,p0)17O peak after xenon flash lamp exposure; XPS O 1s spectra of samples on Au/Ti/PET foil for 0 shots (c) and 1000 shots (d) – the organic peak appears to decrease after xenon flash lamp exposure. | ||
| Sample | at% C | at% O | at% Ta | Thickness |
|---|---|---|---|---|
| (1) 0 shots | 10.9 | 70.7 | 18.4 | 149 |
| (2) 1000 shots | 3.3 | 76.7 | 19.9 | 143 |
The carbon content of the printed layers appears to decrease after flash lamp annealing, and the 12C(d,p0)13C peak decreases from the 0 shots to the 1000 shots samples relative to the 16O(d,p0)17O peak. The amount of detected carbon would correspond to a few monolayers of graphite; however, surface contamination may influence the presence of carbon.
XPS – acetylacetone based films on Au/Ti/PET
X-ray photoelectron spectroscopy (XPS) survey spectra of the samples are shown in Fig. S22 and S23† for the 0 shots and 1000 shots samples (both with 125 °C pre-anneal), respectively. The carbon content of the printed layers appears to decrease after flash lamp annealing, as shown in Table 3. However, the presence of carbon is also influenced by surface contamination, and therefore may not be an accurate indicator of layer carbon content. Nonetheless, xenon flash lamp treatment appears to reduce the amount of carbon present on the samples.| Sample | at% C 1s | at% O 1s | at% Ta 4f | at% Au 4f |
|---|---|---|---|---|
| (1) 0 shots | 40.9 | 47.9 | 10.7 | 0.4 |
| (2) 1000 shots | 16.5 | 64.2 | 17.4 | 1.9 |
On both the 0 shots and the 1000 shots samples, the binding energies at 26.8 eV for Ta 4f7/2 (57.1% Ta 4f area) and 28.7 eV for Ta 4f5/2 (42.9% Ta 4f area) are present, indicating the presence of one oxidation state which does not change after photocuring. This oxidation state would be +5 for Ta2O5.79 Ta 4f spectra for both samples can be seen in Fig. S24,† along with the C 1s spectra with fitted peaks.80 Although the detected ester groups could come from acetylated 1,3-propanediol, the presence of esters could also be due to organic contaminants.
For the O 1s binding energies in Fig. 5c for 0 shots and Fig. 5d for 1000 shots, an increase in oxide oxygen and a decrease in organic oxygen is visible after photocuring.81 The O/Ta oxide ratio in the 1000 shot sample is 3.0, showing an excess of oxygen like the RBS/NRA samples. However, there is a difficult to ascertain error from the overlapping integrals. Furthermore, as XPS is a surface characterization method, there could be different amounts of oxygen on the surface and in the bulk, which would also affect the O/Ta ratio.
LCR meter – acetylacetone based films on Au/Ti/PET foil
After deposition and 125 °C annealing on wet-etched, interdigitated structures, parallel capacitance increases and parallel resistance decreases with exposure to a xenon flash lamp, as shown in Fig. 6a. After about 500 shots, the parallel capacitance levels off at just over 80 pF, and the parallel resistance at roughly 200 kΩ. The leakage current could be due either to carbon impurities,42,82–84 or oxygen vacancies and other defects.40 | ||
Fig. 6 LCR meter measurements (a) of wet-etched, interdigitated gold structures on PET foil. The parallel capacitance appears to peak after 500 shots, and the parallel resistance levels off at roughly 200 kΩ. FIB sectioning and SEM (b) after a total of 5000 shots and a 2 hour 125 °C post-anneal shows a resulting tantalum oxide layer between the gold fingers being less than 10 nm thick (image: 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000×, 52° tilt). 000000×, 52° tilt). | ||
Elucidation of the exact leakage current mechanism would require an improved model, such as using a metal–insulator–metal (MIM) configuration and an impedance analyser.72 However, given that the oxide layer needs to be exposed for photocuring, such a configuration would prove challenging. One solution to this problem in future studies could be to produce many MIM samples while following the procedure of Matavž et al.,72 with each sample having a different amount of photoexposure. Furthermore, the layer geometry would need to be defect free and accurately ascertained for the model. Additionally, the layer thickness could affect the diffusion of carbon volatiles leaving the layer, as well as oxygen diffusion in filling oxygen vacancies in the layer.
FIB/SEM – acetylacetone based films on Au/Ti/PET foil
The printed films on Au/Ti/PET foils were further exposed to 4000 more flash lamp shots, in order to be sure the photosensitive compounds were destroyed. The foils were then exposed to another 125°C post-anneal (1 hour ramp, 2 hour hold) to condense the films, whereby FIB sectioning and SEM in Fig. 6b reveal a tantalum oxide layer between the gold fingers which is less than 10 nm thick. Such an oxide film deposited on interdigitated, gold structures could potentially be used in pH or conductivity sensors.10,13Conclusions
A method to reduce alcoholysis for photosensitive tantalum(V) β-diketonate complexes in an air stable, low-toxic solution is presented. Inkjet printed oxide layers produced from the solution after xenon flash lamp curing shows that the photosensitive complexes break down and that the carbon content is reduced to form tantalum oxide films, which potentially could be used in various applications. Electrical characterization demonstrates that the capacitance and the conductivity of the layers change after photocuring on interdigitated electrodes, which we believe will inspire further research on metal oxide films deposited in this manner.Experimental
Proposed synthesis – acetylacetone
Anhydrous ethanol (42.33 mmol, 1950 mg [AcrosOrganics, 99.5%, ExtraDry, absolute]), acetylacetone (6.15 mmol, 616 mg, [Alfa-Aesar, 95%]), and tantalum(V) ethoxide (6.15 mmol, 2499 mg [Alfa Aesar, 99+%]) were mixed together via magnetic stirring in a dry Schlenk flask in an argon filled glovebox, and were subsequently removed from the glovebox and connected to a Schlenk line. The Schlenk flask containing the reagents (reaction flask) was immersed in silicone oil, with the oil temperature being held and maintained at 80 °C to promote chelation of acetylacetone. The reaction was carried out in argon, and reflux was conducted for 8 hours with mixing to allow enough time for complexation of acetylacetone to tantalum;48,55,63 the reaction time was minimized as much as possible in order to prevent decomposition of the complex.59After 8 hours, the flask was cooled to 23 °C over a 2 hour period. 1,3-Propanediol (42.04 mmol, 3199 mg [Sigma-Aldrich, for synthesis]) was then added via syringe through the Schlenk flask valve and mixed, where bubbles appeared for a short time but eventually disappeared. After 12 hours of mixing at a reduced temperature, the Schlenk flask was removed from the Schlenk line. The product was subsequently transferred from the Schlenk flask to a round bottom flask, and the round bottom flask was then connected to a rotary evaporator. The rotation speed was set to 95 rpm, the pressure was set to 30 mbar, and the water bath temperature was set to 40 °C. Vigorous bubbling was present at the beginning, but the bubbling eventually dissipated. The flask remained attached for 8 hours, where afterward it was found that the mass loss (3236 mg) was close to the calculated ethanol loss (3367 mg). The product was prepared for NMR, but not enough distillate could be collected for NMR analysis.
Proposed synthesis – benzoylacetone
The synthesis is the same as that proposed for acetylacetone, except with the following reagent quantities: anhydrous ethanol (54.24 mmol, 2499 mg [AcrosOrganics, 99.5%, ExtraDry, absolute]), benzoylacetone (5.99 mmol, 972 mg [Sigma-Alrich, 99%]), tantalum(V) ethoxide (6.12 mmol, 2486 mg [Alfa Aesar, 99+%]), and 1,3-propanediol (42.16 mmol, 3208 mg [Sigma-Aldrich, for synthesis]). For ethanol removal, the mass loss (3681 mg) was close to the calculated ethanol loss (3909 mg). The product was prepared for NMR, but not enough distillate could be collected for NMR analysis.NMR measurements
All NMR spectra were recorded at 25 °C using a 600 MHz AVANCE NMR spectrometer (Bruker Corporation, USA). Chemical shifts for the reagents and products are relative to the residual proton signal of the external deuterated solvent, D2O, used as a reference; one exception is benzoylacetone, which was dissolved in a CDCl3 reference solvent.Inkjet printing and photonic curing
Dilutions for ink production from the proposed methods were performed by vortexing the product with diethylene glycol monoethyl ether (DEGEE) (Sigma-Aldrich, ReagentPlus 99%) in order to achieve the proper viscosity and surface tension for inkjet printing.72 Viscosity was measured with a DV3T rheometer (AMETEK Brookfield, USA), and surface tension with a surface tensiometer model A3 (KINO Industry Co., Ltd., USA). All inkjet printing was performed with an OmniJet 300 printer (Unijet Co., Ltd., South Korea), using 10 pL Dimatix cartridges (FUJIFILM Dimatix, Inc., USA) where the ink was filtered using a Whatman Puradisc 13, GF/F, 0.7 μm glass syringe filter (GE Healthcare, USA). The printing waveform was set to 1 μs–10 μs–1 μs at 25 V, with a frequency of 1000 Hz.After deposition, all samples were treated with thermal annealing, which entailed a 1 hour ramp to 125 °C and a 2 hour hold on a hot plate. This was followed by xenon flash lamp exposure for photocured samples, which was carried out with the PulseForge 1200 (Novacentrix, USA) with an output spectrum from roughly 230 nm–1000 nm at the set voltage.84 Multiple, low power “shots” were utilized in order to protect the fragile gold foil structures for electrical characterization (described below), with each shot having the following parameters: 400 V, 900 μs envelope containing 3 pulses with a 100 μs rise time and a 200 μs fall time, with a firing frequency of 0.33 Hz and a working distance of 25 mm. The estimated energy for each shot was 0.623 J cm−2. The SimPulse 2.5 software provided by the manufacturer estimates a plateau in surface temperature during the 1000 shot treatment at 0.33 Hz and an operating temperature of 25 °C: 500 μm thick a-SiO2, ∼25 °C; 500 μm thick c-Si ∼60 °C; 80 μm thick PET, ∼25 °C; 80 μm thick PET with 5 nm Ti adhesion layer and 50 nm Au layer, ∼300 °C.
Deposition on a-SiO2 for UV/Vis spectrophotometry
0.5 mm thick a-SiO2 was chosen as the best substrate for transparency in the near UV range. Before printing, the substrate was subjected to an oxygen plasma treatment for 5 minutes (120 W, 0.6 mbar) using the NANO plasma oven (Diener electronic GmbH + Co. KG, Germany).Product from the proposed method with acetylacetone was diluted with DEGEE, with the dilution containing 706 mg of DEGEE (70 wt%) and 308 mg of product (30 wt%). The ink was then printed on the substrate with a 150 μm drop pitch having dimensions of 250 × 120 drops to create a uniform layer.
For benzoylacetone, the dilution contained 732 mg DEGEE (69 wt%) and 328 mg of product (31 wt%). The ink was then printed with a smaller drop pitch of 75 μm to create uniform layers (the benzoylacetone ink did not spread as well as the acetylacetone ink), having dimensions of 400 × 220 drops to create a uniform layer.
UV/Vis spectrophotometry
UV/Vis spectra of the films on a-SiO2 were measured versus a 0.5 mm thick a-SiO2 reference, with a 1 nm step in the 200 nm to 400 nm range. The device used was a Lambda 900 spectrometer (PerkinElmer, USA).Deposition on a-SiO2 for Raman/XRD
a-SiO2 was used to avoid interfering peaks in both Raman spectroscopy and X-ray diffraction (XRD) measurements. Before printing, the substrate was subjected to an oxygen plasma treatment for 5 minutes (150 W, 0.6 mbar). The power was set slightly higher to increase spreading as the ink contained less DEGEE.Product from the proposed method with acetylacetone was diluted with DEGEE, with the dilution containing 611 mg of DEGEE (60 wt%) and 404 mg of product (40 wt%). The ink was then printed on 0.5 mm thick a-SiO2, with a 70 μm drop pitch having dimensions of 450 × 275 drops to create a uniform layer. The ink was printed with a smaller pitch and diluted with less DEGEE than for the UV/Vis samples, in order to obtain higher quality Raman spectra.
Raman spectroscopy
Confocal Raman microscopy was performed using an alpha300 R setup (WITec, Germany). Illumination of the sample was performed using a 532 nm excitation line from a single-mode frequency doubled Nd:YAG laser via a 100 μm single-mode glass fiber. A LD EC Epiplan-Neofluar 50×/0.55 objective (Carl Zeiss AG, Germany) was used and the laser power at the sample behind the objective was 12 mW.An edge filter was used to separate the Raman signal from the excitation line. Confocality of the Raman signal was achieved via a 50 μm multi-mode fiber glass between the microscope and the Raman spectrometer, where the fiber serves as a pin-hole. The Raman spectrometer was equipped with a holographic grating of 600 lines per mm. For the detector, a Newton 970 EMCCD camera (Andor Technology Ltd, United Kingdom) with 1600 × 200 pixels was used, where this configuration allows a spectral resolution of about 2 cm−1 to be obtained.
An integration time of roughly 0.1 s per spectrum and pixel was used to improve the signal-to-noise ratio (S/N). For Raman depth scans, 100 pixel × 80 pixel scans were used for covering an area of 50 μm × 40 μm. All data sets were analyzed using cluster analysis and non-negative matrix factorization.
XRD
X-ray measurements were accomplished with a high-resolution D8 diffractometer (Bruker Corporation, USA), using the CuKα1 wavelength (λ = 1.54016 Å) collimated and filtered by means of a Göbel mirror and a Ge-220 four-crystal monochromator. On the detector side, a scintillator with a mechanical slit of 3 mm was employed. The 2θ/θ scans were carried out in a range from 2θ = 3–120° with angular resolutions of 0.1°. Each data point was measured for 10 s.Deposition on c-Si for RBS/NRA
The ink was deposited on c-Si in order to determine whether PCSD is possible on this standard substrate, and used Rutherford backscattering spectrometry (RBS) and nuclear reaction analysis (NRA) to measure tantalum, carbon, and oxygen content after treatment. Before printing, the substrate was subjected to an argon plasma treatment for 5 minutes (150 W, 0.6 mbar).Product from the proposed method with acetylacetone was diluted with DEGEE, with the dilution containing 622 mg of DEGEE (60 wt%) and 414 mg of product (40 wt%). The ink was then printed on 0.5 mm thick c-Si, with a 70 μm drop pitch having dimensions of 75 × 75 drops to create a uniform layer.
RBS/NRA
RBS and NRA were performed using 1.43 MeV deuterons. A beam spot diameter of about 200 μm was set up using a triple-focus. A 20 μm resolution imaging camera together with a piezo manipulator enabled positioning of the beam spot on the regions of interest on each sample. Each sample spot was integrated for about 2000 s to about 3 μC of ion charge for sufficient counting statistics. Signals were acquired using a 100 μm thick PIPS detector with 11 keV FWHM resolution at 150° reaction angle.RBS and NRA data were analysed using SimNRA 7.02 with Rutherford cross-sections for all elements except for C and O which were analyzed using 16O(d,p0)17O and the 12C(d,p0)13C reactions and Sigmacalc 2.0 cross-sections. The measurement achieves a total uncertainty of 10% originating from the Particle*Sr value determined from the substrate RBS and NRA counting statistics. The good counting statistics of tantalum results in negligible uncertainties for this element.
Deposition on gold foil for XPS and LCR meter measurements
The ink was deposited on gold/titanium/PET foil (metallization via electron-beam physical vapor deposition of less than 50 nm of gold with ∼5 nm titanium as adhesion layer on 80 μm DuPont PCS) to see if the ink could be cured on a low melting point substrate. The gold foils also allowed for electrical characterization and for higher quality X-ray photoelectron spectroscopy (XPS) spectra, which were wet-etched to create interdigitated structures consisting of 250 digits in a 4 mm × 4 mm area (digit length of ∼3992 μm, width and gap length of ∼8 μm); the structures were connected to feedlines to allow for LCR meter measurements. Before printing, the substrate was subjected to an oxygen plasma treatment for 2 minutes (150 W, 0.6 mbar).Product from the proposed method with acetylacetone was diluted with DEGEE, with the dilution containing 622 mg of DEGEE (60 wt%) and 414 mg of product (40 wt%). The ink was then printed on the gold PET foil with interdigitated structures, with a 150 μm drop pitch having dimensions of 35 × 31 drops to create a uniform layer.
XPS
Spectra were obtained using a Phi5000 VersaProbe II (ULVAC-Phi Inc., USA). The source was AlKα, monochromatic (1.486 keV) with X-ray settings of 50 W, 15 kV in a 200 μm spot. Survey spectra were obtained with a 187.5 eV pass energy, with a 0.8 eV step and 100 ms per step. Detailed spectra used a 23.5 eV pass energy, with a 0.1 eV step and 100 ms per step.Atomic quantification assumed a homogenous volume, using atomic% (at%) with a 15% relative error. Quantification was normalized to 100 at%. The Shirley-background was subtracted and empirical relative sensitivity factors were used. Charge compensation was performed by setting the main peak of the C 1s signal to 285 eV.
LCR meter measurements
After the 125 °C preliminary anneal, the layers were exposed to the xenon flash lamp, with the parallel capacitance (with parasitic capacitance subtracted) and the parallel resistance measured at 1000 Hz every 50 shots, using a DE-5000 LCR meter with TL-22 probes (DER-EE, Taiwan). Plotted values were an average from 5 structures, with the error bar reporting the standard deviation.FIB and SEM
Focused ion beam (FIB) sectioning and subsequent scanning electron microscopy (SEM) imaging was carried out with a HELIOS 600i NanoLab (FEI, Thermo Fisher Scientific, USA). First the region of interest on the sample was covered in situ with protective Pt/C layers. Rough milling was processed with Ga+ ions at 0.43 or 0.79 nA and 30 kV followed by a fine polishing step at 40 pA and 30 kV. SEM imaging was performed at 21 pA and 3 kV using the TL detector for secondary electrons.Conflicts of interest
We declare that Christopher Beale, Stefanie Hamacher, Dirk Mayer, and Andreas Offenhäusser have filed a German patent application (Date: Oct 08, 2019; application number: DE 10 2019 006 976.5) on behalf of Forschungszentrum Jülich GmbH. The patent application includes the synthesis methods and their products described in this publication, with the intended use being the deposition of metal oxide material. All other authors have no conflicts to declare.Acknowledgements
The authors would like to thank Michael Prömpers for preparing the wet-etched, interdigitated gold structures on PET foil, as well as the Helmholtz Nano Facility staff for preparing the other substrates. The authors would also like to thank Jekaterina Viktorova for sol–gel and inkjet chemistry advice. Funding from “Europäischer Fonds für regionale Entwicklung”, for the project “EFRE-0800361 Packsense”, was used for this study.References
- O. H. Fulcher, J. Am. Med. Assoc., 1943, 121, 931 CrossRef.
- G. L. Burke, Can. Med. Assoc. J., 1940, 43, 125–128 CAS.
- D. Fraser, G. Mendonca, E. Sartori, P. Funkenbusch, C. Ercoli and L. Meirelles, Clin. Oral Implants Res., 2019, 30, 156–168 CrossRef PubMed.
- M. Papakyriacou, Int. J. Fatigue, 2000, 22, 873–886 CrossRef CAS.
- Y. Freeman, Tantalum and Niobium-Based Capacitors, Springer International Publishing, Cham, 2018 Search PubMed.
- J. Ma, X. Guo, H. Xue, K. Pan, C. Liu and H. Pang, Chem. Eng. J., 2020, 380, 122428 CrossRef CAS.
- S. Uhlenbruck, J. Dornseiffer, S. Lobe, C. Dellen, C.-L. Tsai, B. Gotzen, D. Sebold, M. Finsterbusch and O. Guillon, J. Electroceram., 2017, 38, 197–206 CrossRef CAS.
- N. Özer and C. M. Lampert, Windows Innovations 95 Conference Proceedings, Berkeley Laboratory, Livermore, 1995, LBL-38526, http://https://windows.lbl.gov/publications/sol-gel-deposited-amorphous-tantalum-oxide-and-niobium-oxide-films-protonic-conductors Search PubMed.
- R. E. G. van Hal, J. C. T. Eijkel and P. Bergveld, Sens. Actuators, B, 1995, 24, 201–205 CrossRef CAS.
- W. Olthuis, W. Streekstra and P. Bergveld, Sens. Actuators, B, 1995, 24, 252–256 CrossRef CAS.
- M. J. Schöning, D. Brinkmann, D. Rolka, C. Demuth and A. Poghossian, Sens. Actuators, B, 2005, 111–112, 423–429 CrossRef.
- C. H. Kao, H. Chen, L. T. Kuo, J. C. Wang, Y. T. Chen, Y. C. Chu, C. Y. Chen, C. S. Lai, S. W. Chang and C. W. Chang, Sens. Actuators, B, 2014, 194, 419–426 CrossRef CAS.
- L. Manjakkal, K. Cvejin, B. Bajac, J. Kulawik, K. Zaraska and D. Szwagierczak, Electroanalysis, 2015, 27, 770–781 CrossRef CAS.
- M. Biednov, T. Lebyedyeva and P. Shpylovyy, Proc. of SPIE Optical Sensors, 2015, vol. 9506, pp. 95061P Search PubMed.
- D. Li, W. Zhang, H. Liu, J. Hu and G. Zhou, IEEE Photonics J., 2017, 9, 1–8 CAS.
- M. R. Hasan, S. Akter, K. Ahmed and D. Abbott, IEEE Photonics Technol. Lett., 2018, 30, 315–318 CAS.
- A. González García, Láminas delgadas y ultradelgadas de tantalato de estroncio y bismuto obtenidas a partir de derivados de glicolato de tántalo para su uso en memorias ferroeléctricas no volátiles, PhD thesis, Autonomous University of Madrid, Spain, November 2002.
- M. L. Calzada and A. González, J. Am. Ceram. Soc., 2005, 88, 2702–2708 CrossRef CAS.
- M. L. Calzada, M. Algueró, J. Ricote, A. Santos and L. Pardo, J. Sol-Gel Sci. Technol., 2007, 42, 331–336 CrossRef CAS.
- M. L. Calzada, M. Algueró, A. Santos, M. Stewart, M. G. Cain and L. Pardo, J. Mater. Res., 2009, 24, 526–533 CrossRef CAS.
- M. D. Anderson, B. Aitchison and D. C. Johnson, ACS Appl. Mater. Interfaces, 2016, 8, 30644–30648 CrossRef CAS PubMed.
- T. Damart, E. Coillet, A. Tanguy and D. Rodney, J. Appl. Phys., 2016, 119, 175106 CrossRef.
- N. A. Mancheri, B. Sprecher, S. Deetman, S. B. Young, R. Bleischwitz, L. Dong, R. Kleijn and A. Tukker, Resour., Conserv. Recycl., 2018, 129, 56–69 CrossRef.
- T. Nakajima, K. Shinoda and T. Tsuchiya, Chem. Soc. Rev., 2014, 43, 2027–2041 RSC.
- A. Matavž and B. Malič, J. Sol-Gel Sci. Technol., 2018, 87, 1–21 CrossRef.
- I. Bretos, R. Jiménez, J. Ricote and M. L. Calzada, Chem. Soc. Rev., 2018, 47, 291–308 RSC.
- D. Grosso, C. Boissière and M. Faustini, in The Sol-Gel Handbook, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015, pp. 277–316 Search PubMed.
- M. L. Calzada, in The Sol-Gel Handbook, ed. D. Levy and M. Zayat, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015, pp. 841–882 Search PubMed.
- R. Deshmukh and M. Niederberger, in The Sol-Gel Handbook, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2015, p. 34 Search PubMed.
- G. A. Seisenbaeva and V. G. Kessler, Nanoscale, 2014, 6, 6229–6244 RSC.
- H. C. Ling, M. F. Yan and W. W. Rhodes, in Science of Ceramic Chemical Processing, ed. L. L. Hench and D. R. Ulrich, John Wiley & Sons, Inc., New York, 1986, pp. 285–303 Search PubMed.
- L. A. Silverman, G. Teowee and D. R. Uhlmann, MRS Proceedings, 1986, 72, 331 CrossRef CAS.
- L. A. Silverman, Sol-gel derived tantalum oxide thin films, PhD thesis, Massachusetts Institute of Technology, United States, June 1987.
- P. Griesmar, G. Papin, C. Sanchez and J. Livage, Chem. Mater., 1991, 3, 335–339 CrossRef CAS.
- I. Bretos and M. L. Calzada, in Multifunctional Polycrystalline Ferroelectric Materials - Processing and Properties, ed. L. Pardo and J. Ricote, Springer, Dordrecht, 2011, vol. 152–155, pp. 182–186 Search PubMed.
- J. F. Scott, in Ferroelectric Memories, Springer-Verlag, Berlin, 2000, p. 166 Search PubMed.
- R. C. Mehrotra and P. N. Kapoor, J. Less-Common Met., 1965, 8, 419–427 CrossRef CAS.
- R. C. Mehrotra and P. N. Kapoor, J. Less-Common Met., 1965, 10, 237–245 CrossRef.
- T. Ohishi, S. Maekawa and A. Katoh, J. Non-Cryst. Solids, 1992, 147–148, 493–498 CrossRef CAS.
- I. W. Boyd and J.-Y. Zhang, Solid-State Electron., 2001, 45, 1413–1431 CrossRef CAS.
- N. Tohge, K. Shinmou and T. Minami, J. Sol-Gel Sci. Technol., 1994, 2, 581–585 CrossRef CAS.
- K. Suzuki, W. Sakamoto, T. Yogo and S. Hirano, J. Ceram. Soc. Jpn., 1999, 107, 1032–1036 CrossRef CAS.
- T. H. Park, K. H. Yang, D. K. Kang, T. Y. Lim, K. H. Auh and B. H. Kim, J. Mater. Sci., 2003, 38, 1295–1300 CrossRef CAS.
- T. Y. Lim, K. H. Yang, B. H. Kim and K. H. Auh, Thin Solid Films, 2005, 471, 12–18 CrossRef CAS.
- N. Noma and M. Hamano, J. Sol-Gel Sci. Technol., 2012, 64, 297–303 CrossRef CAS.
- H. Segawa, S. Inoue, K. Watanabe, R. Ohashi, H. Nitani and M. Nomura, J. Ceram. Soc. Jpn., 2015, 123, 793–799 CrossRef CAS.
- R. J. Squibb, M. Sapunar, A. Ponzi, R. Richter, A. Kivimäki, O. Plekan, P. Finetti, N. Sisourat, V. Zhaunerchyk, T. Marchenko, L. Journel, R. Guillemin, R. Cucini, M. Coreno, C. Grazioli, M. Di Fraia, C. Callegari, K. C. Prince, P. Decleva, M. Simon, J. H. D. Eland, N. Došlić, R. Feifel and M. N. Piancastelli, Nat. Commun., 2018, 9, 1–7 CrossRef CAS PubMed.
- A. Whitley, New metal alkoxides, PhD thesis, Birkbeck College, University of London, United Kingdom, November 1954.
- D. C. Bradley, W. Wardlaw and A. Whitley, J. Chem. Soc., 1955, 726–728 RSC.
- H. Funk and K. Niederländer, Ber. Dtsch. Chem. Ges., 1929, 62, 1688–1691 CrossRef.
- H. Funk, Ber. Dtsch. Chem. Ges., 1934, 67, 1801–1804 CrossRef.
- D. C. Bradley, R. C. Mehrotra, I. P. Rothwell and A. Singh, in Alkoxo and Aryloxo Derivatives of Metals, Academic Press, London, 2001, vol. 121 Search PubMed.
- R. C. Mehrotra and P. N. Kapoor, J. Less-Common Met., 1964, 7, 453–457 CrossRef CAS.
- R. C. Mehrotra and P. N. Kapoor, J. Less-Common Met., 1964, 7, 176–179 CrossRef CAS.
- P. N. Kapoor and R. C. Mehrotra, J. Less-Common Met., 1965, 8, 339–346 CrossRef CAS.
- R. N. Kapoor, S. Prakash and P. N. Kapoor, Bull. Chem. Soc. Jpn., 1967, 40, 1384–1386 CrossRef CAS.
- M. L. Calzada, R. Sirera, F. Carmona and B. Jiménez, J. Am. Ceram. Soc., 1995, 78, 1802–1808 CrossRef CAS.
- M. L. Calzada, A. González, R. Poyato and L. Pardo, J. Mater. Chem., 2003, 13, 1451–1457 RSC.
- T. Kemmitt and M. Daglish, Inorg. Chem., 1998, 37, 2063–2065 CrossRef CAS.
- G. Ambrožič, S. D. Škapin, M. Žigon and Z. C. Orel, J. Colloid Interface Sci., 2010, 346, 317–323 CrossRef PubMed.
- M. Staniuk, D. Zindel, W. van Beek, O. Hirsch, N. Kränzlin, M. Niederberger and D. Koziej, CrystEngComm, 2015, 17, 6962–6971 RSC.
- R. K. Sodhi and S. Paul, Catal. Surv. Asia, 2018, 22, 31–62 CrossRef CAS.
- K. D. Pollard and R. J. Puddephatt, Chem. Mater., 1999, 11, 1069–1074 CrossRef CAS.
- D. Ambrose and C. H. S. Sprake, J. Chem. Thermodyn., 1970, 2, 631–645 CrossRef CAS.
- “Ethanol” National Institute of Standards and Technology Chemistry WebBook, United States Department of Commerce, 2018 Search PubMed.
- D. R. Stull, Ind. Eng. Chem., 1947, 39, 517–540 CrossRef CAS.
- “1,3-propanediol,” National Institute of Standards and Technology Chemistry WebBook, United States Department of Commerce, 2018 Search PubMed.
- S. S. Likhodii and W. M. Burnham, Med. Sci. Monit., 2002, 8, HY19–HY24 CAS.
- J. B. Lambert, E. P. Mazzola and C. D. Ridge, Nuclear Magnetic Resonance Spectroscopy: An Introduction to Principles, Applications, and Experimental Methods, John Wiley & Sons, Inc., Hoboken, 2nd edn, 2019, vol. 86, p. 99 Search PubMed.
- Y. Zhao, J. Q. L. Ang, A. W. T. Ng and Y.-Y. Yeung, RSC Adv., 2013, 3, 19765 RSC.
- Y. Takebayashi, S. Yoda, T. Sugeta, K. Otake, T. Sako and M. Nakahara, J. Chem. Phys., 2004, 120, 6100–6110 CrossRef CAS PubMed.
- A. Matavž, R. C. Frunza, A. Drnovšek, V. Bobnar and B. Malič, J. Mater. Chem. C, 2016, 4, 5634–5641 RSC.
- P. K. Verma, A. Steinbacher, F. Koch, P. Nuernberger and T. Brixner, Phys. Chem. Chem. Phys., 2015, 17, 8459–8466 RSC.
- C. Joseph, P. Bourson and M. D. Fontana, J. Raman Spectrosc., 2012, 43, 1146–1150 CrossRef CAS.
- Y. Zhu, F. Yu, Y. Man, Q. Tian, Y. He and N. Wu, J. Solid State Chem., 2005, 178, 224–229 CrossRef CAS.
- G. Amsel, J. P. Nadai, C. Ortega, S. Rigo and J. Siejka, Nucl. Instrum. Methods, 1978, 149, 705–712 CrossRef CAS.
- G. Amsel, J. P. Nadai, C. Ortega and J. Siejka, Nucl. Instrum. Methods, 1978, 149, 713–720 CrossRef CAS.
- G. Amsel and J. A. Davies, Nucl. Instrum. Methods Phys. Res., 1983, 218, 177–182 CrossRef.
- R. Simpson, R. G. White, J. F. Watts and M. A. Baker, Appl. Surf. Sci., 2017, 405, 79–87 CrossRef CAS.
- J.-H. Zhou, Z.-J. Sui, J. Zhu, P. Li, D. Chen, Y.-C. Dai and W.-K. Yuan, Carbon, 2007, 45, 785–796 CrossRef CAS.
- L. Chen and R. W. Hoffman, J. Vac. Sci. Technol., A, 1993, 11, 2303–2307 CrossRef CAS.
- K. Tetzner, K. A. Schroder and K. Bock, Ceram. Int., 2014, 40, 15753–15761 CrossRef CAS.
- K. Tetzner, Y. H. Lin, A. Regoutz, A. Seitkhan, D. J. Payne and T. D. Anthopoulos, J. Mater. Chem. C, 2017, 5, 11724–11732 RSC.
- S. Luo, S. Zhang, B. B. Bourgeois, B. C. Riggs, K. A. Schroder, Y. Zhang, J. He, S. Adireddy, K. Sun, J. T. Shipman, M. M. Oguntoye, V. Puli, W. Liu, R. Tu, L. Zhang, S. Farnsworth and D. B. Chrisey, J. Mater. Res., 2017, 32, 1701–1709 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02558e |
| This journal is © The Royal Society of Chemistry 2020 |