
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA polycarboxylic chelating ligand for efficient resin purification of His-tagged proteins expressed in mammalian systems†
Codruţa C. Popescu‡
a,
Marius C. Stoian‡ab,
Lia-Maria Cucos‡ c,
Anca G. Comana,
Antonio Radoib,
Anca Pauna,
Niculina D. Hădaded,
Arnaud Gautiere,
Costin-Ioan Popescu*c and
Mihaela Matache*a
c,
Anca G. Comana,
Antonio Radoib,
Anca Pauna,
Niculina D. Hădaded,
Arnaud Gautiere,
Costin-Ioan Popescu*c and
Mihaela Matache*a
aUniversity of Bucharest, Faculty of Chemistry, Department of Organic Chemistry, Biochemistry and Catalysis, Research Centre of Applied Organic Chemistry, 90-92 Panduri Street, RO-050663 Bucharest, Romania. E-mail: mihaela.matache@g.unibuc.ro
bNational Institute for Research and Development in Microtechnology – IMT Bucharest, 126A Erou Iancu Nicolae Street, 077190, Voluntari, Romania
cInstitute of Biochemistry of the Romanian Academy, 296 Spl. Independentei, 060031, Bucharest, Romania. E-mail: pop@biochim.ro
dFaculty of Chemistry and Chemical Engineering, Supramolecular Organic and Organometallic Chemistry Centre, ‘‘Babes--Bolyai’’ University, 11 Arany Janos Str., RO-400028-Cluj-Napoca, Romania
eUniversité Clermont Auvergne, CNRS, Sigma Clermont, ICCF, F-63000 Clermont-Ferrand, France
First published on 23rd June 2020
Abstract
We describe the synthesis of a novel polyamino polycarboxylic ligand, its ability to coordinate metal-ions and attachment to a solid support designed for protein purification through Immobilised Metal-ion Affinity Chromatography (IMAC). The resin was found to be highly efficient for purification of His-tagged HCV E2 glycoproteins expressed in 293T mammalian cells.
Protein purification has become a major research concern due to the increasing market of recombinant proteins and the necessity to provide highly pure proteins in good yields.1 Immobilised Metal-ion Affinity Chromatography (IMAC) represents one of the most attractive choices due to several advantages including competitive costs, simplicity of the experimental procedures and small tag protein modifications that do not usually interfere with protein activity.2 IMAC consists of immobilising transition metal ions (Co2+, Ni2+, Cu2+, Zn2+) on a solid matrix considering the ability of metal-ion complexes to interact with protein tags containing donor atoms such as oxygen, nitrogen and sulfur.3 A common tag is polyhistidine i.e. four to eight histidine residues, which can be easily incorporated into proteins. This small tag yields a very strong interaction with metal ions and, in most cases, does not significantly affect protein functions and can be conveniently removed by proteolysis.4 The solid matrices used for such applications are usually functionalised-saccharide polymers on which organic chelators are grafted, such as iminodiacetic acid (IDA, I) or nitrilotriacetic acid (NTA, II) (Fig. 1). The most known used system in IMAC technique is the one formed by Ni–NTA complexes and His-tagged proteins.4,5 This interaction is reversible, therefore captured proteins are eluted by adjusting pH or by using an imidazole containing buffer. Depending on the composition of the buffer, the nature of the column-bound molecules changes with the coordination sphere of the immobilized metal ions. Thus, IMAC is highly suitable for the extensive purification of His-tagged proteins, achieving purities of up to 95%, in high yields. Chelating ligands covalently attached to solid matrices are very important and are usually multidentate organic ligands that bind to metal ions. The most encountered ligands are tridentate (i.e. IDA, I) and tetradentate (i.e. NTA, II); nevertheless, intensive studies for discovering new ligands that could generate proper matrices for maximizing protein purification yields are present in the literature.3b,6 Most of these ligands take the form of acyclic or cyclic polyamino polycarboxylic acids, which have been often called bifunctional chelating agents, also useful for a wide range of applications involving biomolecules labelling.7 A new type of chelating agents holding numerous coordinating sites, prone to lanthanide coordination and formation of luminescent stable complexes, such as compounds of type III (Fig. 1), has been increasingly developed for MRI applications or for naked eye visualization of labelled proteins.8
In this context, we describe herein synthesis of a new functional chelating agent 1 (Fig. 1), its ability to form metal complexes and utility for preparation of a new solid matrix to be employed in affinity chromatography and proteins purification. Similar compounds with 1 have been previously investigated as ligands for metal-ion complexes, bearing various substituents in para-position relative to the phenol group and information is readily available regarding stoichiometry, electronic and magnetic properties of complexes with Cu(II)9 and lanthanides.10
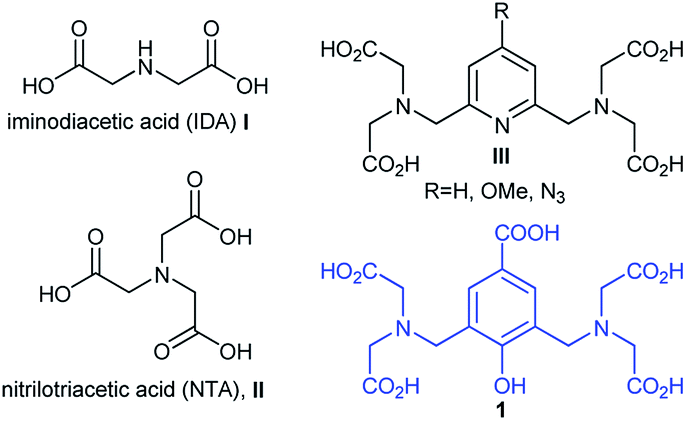 | ||
| Fig. 1 Structures of various polyamino polycarboxylic chelating agents and structure of compound 1 used in this work. | ||
Synthesis of the key intermediate 3 (Scheme 1) was achieved in a two steps sequence starting from p-hydroxybenzoic acid by bromomethylation and subsequent alkylation with tert-butyl iminodiacetate, in 50% global yield. Previously reported11 synthetic approaches of such compounds refer to Mannich reactions11a–c of the corresponding phenols using paraformaldehyde and secondary amines or hydroxymethylation of the phenols followed by bromination with hydrogen bromide.11d However, in most cases, the Mannich monosubstituted product predominates, while the hydroxymethylation reactions in basic medium tend to provide polymerisation products, especially in presence of electron-withdrawing substituents. Thus, direct bromomethylation in acidic conditions was found to be the best option, due to the cleanliness of the reaction procedure, as it occurred free of by-products (i.e. polymers). N-Alkylation proceeded smoothly and the esters was conveniently obtained. With compound 3 in hand, we performed deprotection of the tert-butyl ester using 25% TFA in dichloromethane to afford compound 1. We monitored the reaction progress by RP-HPLC (ESI, Fig. S1†) and observed that complete deprotection occurred overnight. Compound 1 is water soluble and the recorded UV-vis spectra indicated an absorption maximum at λmax = 253 nm (ε = 7591.6 L mol−1 cm−1, ESI, Fig. S2†).
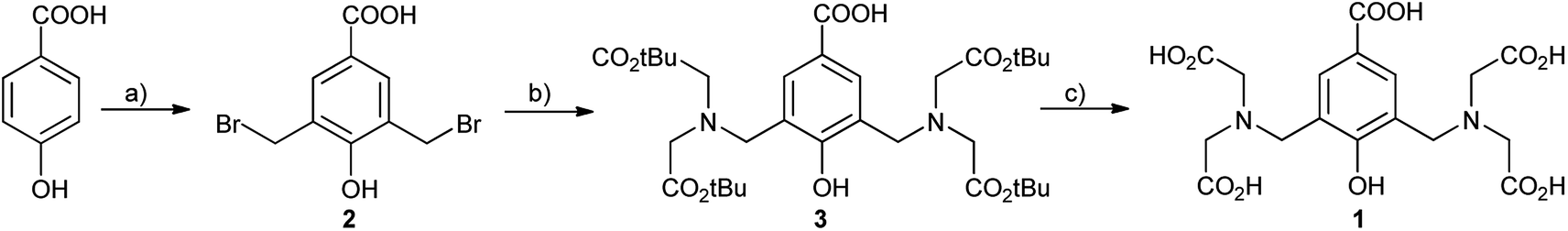 | ||
| Scheme 1 Synthesis of the compounds: (a) (CH2O)n, 48% aq HBr, 60–80 °C, 6 days, 76%; (b) NH(CH2CO2tBu)2, NaHCO3, MeCN, rt, overnight, 70%; (c) 25% TFA in CH2Cl2, overnight, 94%. | ||
Ligand 1 was investigated for the complexation properties with various metal ions and we performed a screening among Ni(II), Zn(II), Cu(II), Fe(II), Yb(III), Er(III), Nd(III) (ESI, Fig. S3†). The absorption spectra of the mixtures between the ligand and the metal ions suffered a general shifting of the absorption maxima towards higher wavelengths indicating formation of metal-ion complexes.
We were particularly interested in formation of the complexes with Zn(II) and Ni(II) ions that were studied by UV-vis in order to establish the ligand to metal stoichiometry. Formation of the complex with Zn(II) was followed by addition of increasing amounts of the metal ions in aqueous solutions of compound 1, in basic medium ensured by triethylamine (TEA) (see ESI, Fig. S4†). The experiments revealed a 1:2 ligand to metal stoichiometry and the association constants calculated using the on-line tool "supramolecular.org"12 were K11 = 3.66 × 105 ± 243 M−1 and K12 = 4.19 × 106 ± 233 M−1.
The UV-vis absorption spectra of the complex resulted between compound 1 and Ni(II) ions revealed multiple bands, specific to green complex formation, both in solution (Fig. 2) and solid state (Fig. 4). Thus, the absorption band in the visible region, assigned to phenoxo-to-Ni(II) ligand-to-metal charge-transfer (LMCT) band has λmax = 381 nm and the large band between 600–800 nm corresponds to Ni(II) d–d transitions, with maximum at λmax = 651 nm.9
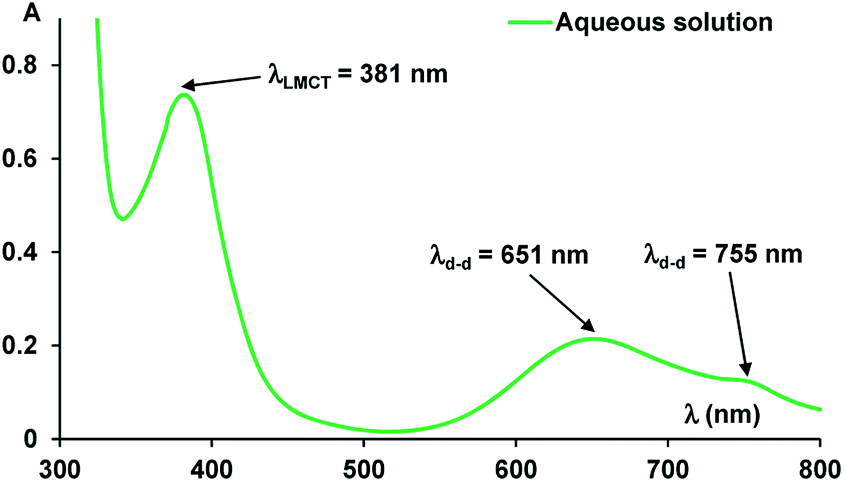 | ||
| Fig. 2 The absorption spectra of the Ni(II) complex in aqueous solution. | ||
UV-vis titration experiments (Fig. 3) of compound 1 with Ni(II) revealed absorbance modification at λmax = 282 nm. Data processing (ESI, Fig. S6†)12 confirmed a ligand to metal ions stoichiometry of 1:2, yielding high values for the association constant (K11 = 1.33 × 109 ± 1000 M−1 and K12 = 2.7 × 106 ± 127 M−1) only ten times smaller than the value of Ni–NTA complex (KD = 1.8 × 10−11 M).6g The values of the association constants are consistent with previous data for similar polyamino polycarboxylic acids13 with the corresponding metal-ions.
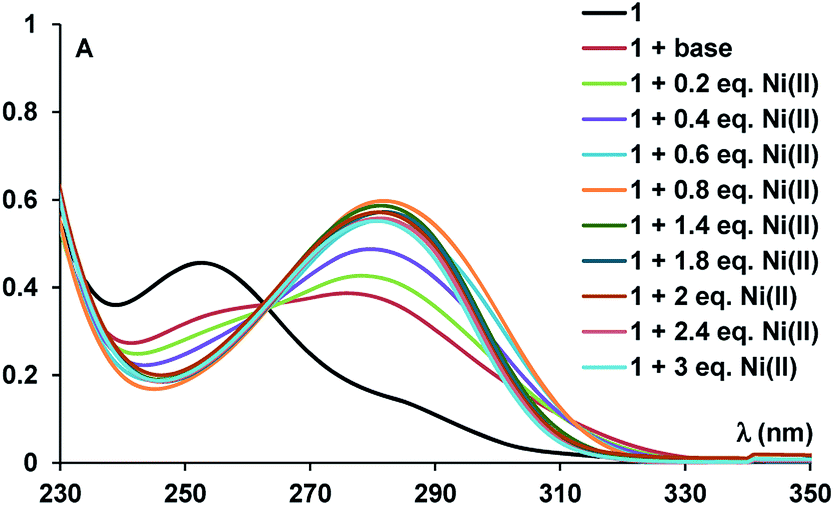 | ||
| Fig. 3 UV-vis spectra during Ni2+ titration of compound 1. | ||
Once compound 1 characterised, we moved further to prepare a solid matrix able to afford His-tagged proteins purification. For this, we used commercial Thermo Scientific™ CarboxyLink™ Coupling Gel (Immobilized Diaminodipropylamine) which is a crosslinked beaded agarose support functionalised with diaminodipropylamine residues, able to react with carboxyl groups. Attachment of compound 3 was performed using peptide coupling conditions after solvent exchange with DMF (N,N-dimethylformamide), using PYBOP (benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluoro phosphate) as coupling reagent, DIPEA (N,N-diisopropylethylamine) as base and two equivalents of 1 relative to the resin loading. Qualitative check of the coupling was performed using ninhydrin test. i.e. colourless solution of the mixture of resin with ninhydrin, indicating complete block of the amino groups (see ESI† for details regarding experimental procedure). Loading of Ni(II) ions was further performed by prior solvent exchange with water and treatment of the resin with lithium hydroxide to ensure deprotection of the tert-butyl esters. Finally, the resin was thoroughly washed with water in order to remove unbounded metal-ions.
We analysed the functionalised solid support by UV-vis (Fig. 4), Fourier-transform infrared spectroscopy (FTIR) and RAMAN spectroscopy. Thus, the UV-vis spectrum of a sample containing a mixture of sepharose and Ni(II) ions displayed the d–d transition band, specific to the Ni(II) ions, while the UV-vis spectrum of the resin containing the complex had a similar profile with the spectrum of the isolated complex, displaying a band at λmax = 350 nm and indicating the presence of the nickel complex, due to the band at λmax = 665 nm (Fig. 4).
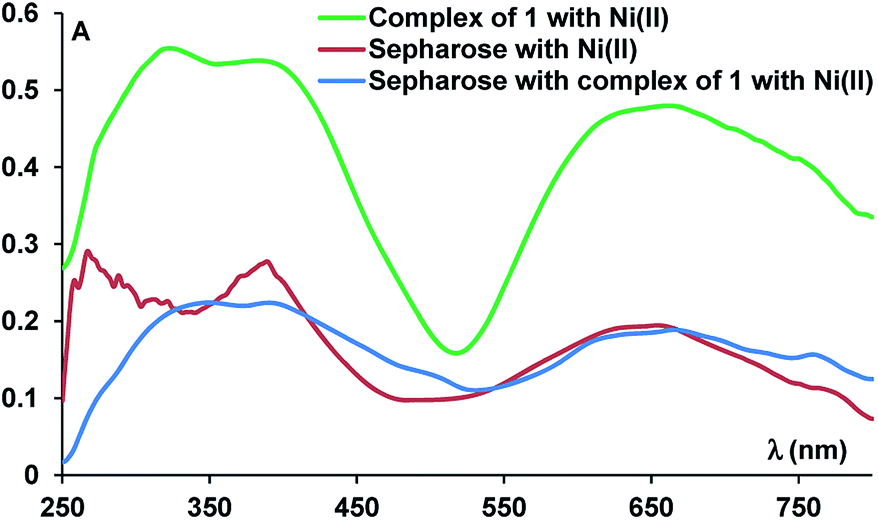 | ||
| Fig. 4 Solid UV-vis spectra of Ni(II) complex derived sepharose. | ||
The FTIR of the Sepharose grafted with 3 showed specific bands assigned to stretching of the carboxyl and amide groups at 1719 and 1640 cm−1, respectively, suggesting successful modification of the sepharose resin. Presence of the Ni(II) complex of 1 was confirmed by the two bands with maxima at 1602 and 1397 cm−1 that were correlated with the carboxylate asymmetric and symmetric vibrations, concomitant with decrease of the initial carbonyl stretching band; moreover, the difference of 205 cm−1 between the bands of the carboxylates proved coordination of Ni(II) to ligand 1.14 Additional bands at 532 and 439 cm−1 were attributed to Ni–N and Ni–O respectively, confirming presence of the metal ion complex.15 The acquired spectra and FTIR assignment table are presented in ESI (see Fig. S6†). RAMAN spectra also confirmed formation of the Ni(II) complex (see Fig. S6†).
Next, the retention capacity of the resin for His-tagged proteins was evaluated. We started testing a pure recombinant protein expressed in E. coli, a frequently used prokaryotic expression system.16 The protein to be expressed, namely S100 calcium binding protein B (S100B), was fused in the N-terminus with a hexahistidine tag having a molecular mass of 13.8 kDa. An IMAC experiment was performed using equal amounts of the following resins: commercial amino-sepharose (S), sepharose resin derivatized with 1 (S–1), commercial Ni-NTA resin (NiNTA), sepharose derivatized with Ni(II) complex of 1 (S–1–Ni). Resins were incubated with approximately 400 μg of HisS100B protein. After washing and performing a specific elution with imidazole, bound (B) and unbound (UN) protein fractions were resolved by SDS-PAGE and visualized by Coomassie Blue staining (Fig. 5). Binding of HisS100B was specific for S–1–Ni and NiNTA resins with no visible non-specific binding to the control resins. Binding efficacy was comparable for S–1–Ni and NiNTA since the bound and unbound fractions of HisS100B are similar for the two resins.
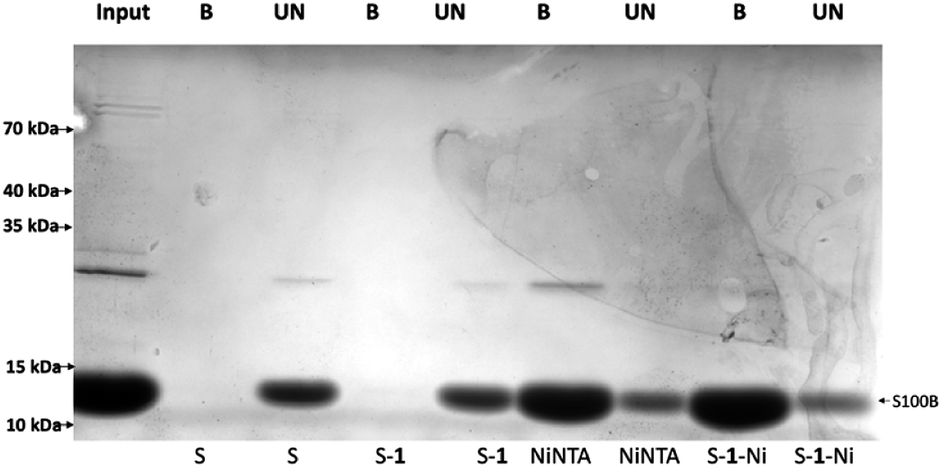 | ||
| Fig. 5 Pulldown of recombinant HisSB100. IMAC was performed using 400 μg of HisS100B purified protein on commercial amino-sepharose (S), sepharose resin derivatized with 1 (S–1), commercial Ni–NTA resin (NiNTA), sepharose derivatized with Ni(II) complex of 1 (S–1–Ni). The eluates (B) and 1/30 of the supernatants (UN) were separated by SDS-PAGE and stained with Coomassie Blue. 60 μg of purified HisS100B were loaded as the reference input. | ||
Then, the resin binding efficacy and specificity was tested in a complex lysate of E. coli expressing HisS100B. After protein expression induction, the cell pellet was sonicated in lysis buffer and the soluble protein fraction was further processed. A pulldown assay was further performed for the soluble fraction similarly to the pure protein experiment (Fig. 6). HisS100B interacted minimally with the control resin S–1. Both S–1–Ni and NiNTA pulled down HisS100B, while S–1–Ni interacted non-specifically with more prokaryotic proteins compared to NiNTA. A higher affinity of the interaction between S–1–Ni and histidine or clusters of histidine residues on specific prokaryotic proteins could explain the difference between resins specificity.17 To assess the stability of the resin we repeated the pull-down for three times without an obvious loss of the binding efficacy as estimated by Coomassie Blue staining of SDS-PAGE gels in Fig. S10.† The binding capacities of the two resins were determined as described in the ESI.† The binding capacities of S–1–Ni (409 ± 42 μg ml−1 resin) and NiNTA (412 ± 78 μg ml−1 resin) were not significantly different. Next, we tested the affinity purification of HisRAGE, a protein with a higher molecular mass (40k Da apparent MM). As shown in Fig. S11,† the S–1–Ni pulled down HisRAGE as efficiently as NiNTA, along with non-specific prokaryotic proteins.
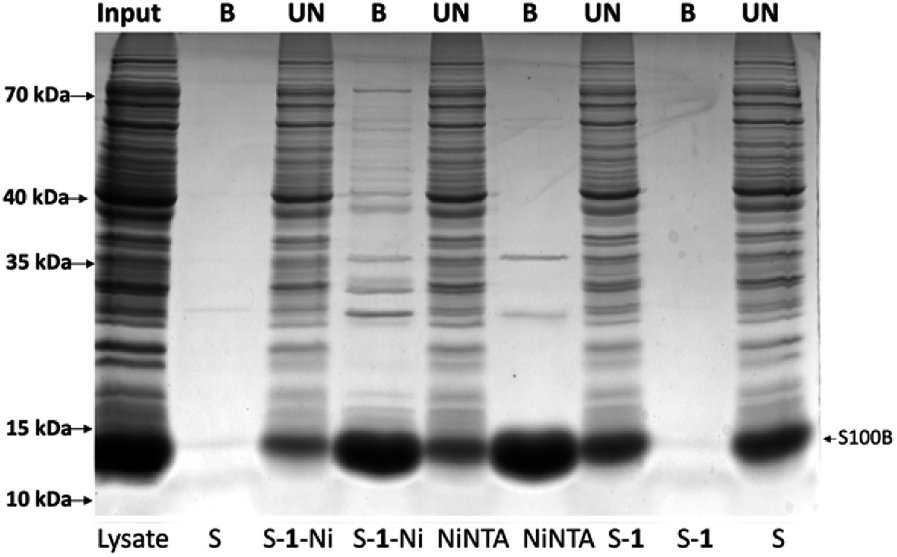 | ||
| Fig. 6 Purification of recombinant HisS100B from E. coli BL21 cell lysate using commercial amino-sepharose (S), sepharose resin derivatized with 1 (S–1), commercial Ni–NTA resin (NiNTA), sepharose derivatized with Ni(II) complex of 1 (S–1–Ni). 500 μg of clarified lysate was used for HisS100B pull down. Clarified lysate (input), the eluates (B), and 1/30 of the supernatants (UN) were separated by SDS-PAGE and visualized with Coomassie Blue staining. | ||
Proteins with higher molecular masses and post-translational modifications (PMTs) are difficult and sometimes impossible to express in prokaryotic expression systems. Thus, eukaryotic expression systems (yeast, insect, mammalian) remain the main choice for producing proteins with high molecular mass, disulphidic bonds, glycosylation or phosphorylation.18 Mammalian cell lines such as CHO and 293T are used extensively for recombinant protein purification for industrial or academic purposes.19 IMAC is a popular protein purification technique with reasonable costs, yields and protein purity. However, if secreted proteins are to be purified from mammalian cell culture media, animal serum proteins compete with secreted recombinant proteins during IMAC resin binding.20 Thus, chemically defined, serum-free media are used which are often much more expensive and incompatible with classic production adherent cell lines. Therefore, we decided to test the capacity of S–1–Ni in the context of a soluble glycoprotein secreted in a serum rich cell culture media. We chose Hepatitis C Virus E2 glycoprotein which is a type I transmembrane glycoprotein with 11 glycans.21 HCV E2 ectodomain (50 kDa) was expressed in 293T cells cultured in media with 2% or 10% fetal bovine serum (FBS). Following the pulldown procedure, the protein was eluted, resolved in an SDS-PAGE gel and detected by western blot. As shown in Fig. 7, the protein was abundantly expressed in the cell lysate. In the cell culture media, a higher molecular mass species was secreted suggesting glycan processing to complex structures, as previously reported.22
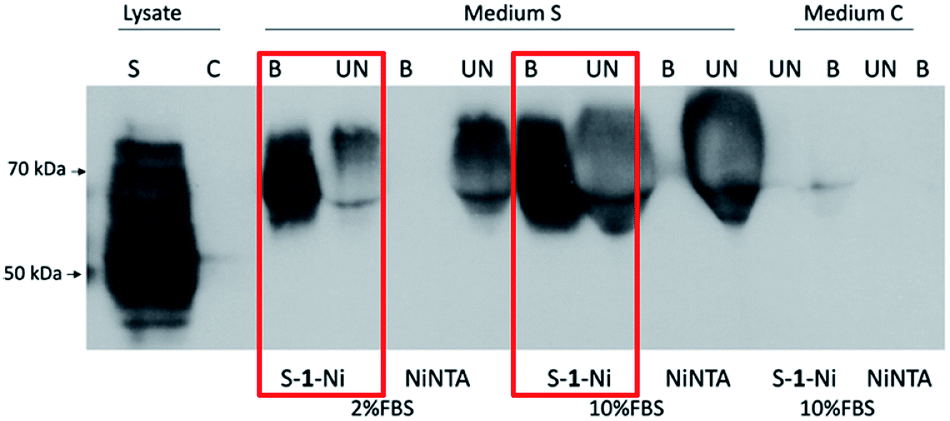 | ||
| Fig. 7 Purification of recombinant HCV E2s protein. 293T cells were transfected with a plasmid coding for HCV E2 ectodomain (AB154191_E2_C1b, aa 384_656) (S) or pcDNA3.1 empty vector (C). 48 h post-transfection, the clarified media was subjected to pulldown using either NiNTA or S–1–Ni resin. Cell lysates, eluates (B), and 1/30 of the supernatants (UN) were separated by SDS-PAGE and HCV E2s was detected by western blot. | ||
Strikingly, S–1–Ni was able to pull down E2s both in presence of 2% and 10% FBS while there was no protein detected in NiNTA eluates. A possible explanation is a difference in the binding affinity of the His tag to the two resins in the context of E2s and high serum protein concentration. As noticed for the prokaryotic proteins purification, the S–1–Ni interacted non-specifically with the bovine serum albumin present in high concentrations in the input media (3.7 mg ml−1) (Fig. S9†). In these pulldown conditions the specific binding capacity was estimated at 200–300 ng ml−1 of S–1–Ni and 10–20 ng ml−1 of NiNTA for the two serum concentrations. Although further work is required to evaluate the general use of the new resin for protein purification in the presence of high concentrations of serum, our work paves the way for designing new chelators which could enable a cost efficient purification of proteins with complex structural elements produced in mammalian expression systems.
Conclusions
In conclusion, we presented synthesis of a new bifunctional chelating ligand with iminodiacetate moieties and investigation of the behaviour towards metal-ions, indicating excellent association constant with nickel(II) ions. Further, we used these properties to create a novel solid matrix derived from Sepharose which was able to act as an Immobilised Metal Ions Affinity Chromatography support for purification of recombinant proteins expressed in prokaryotic and eukaryotic expression systems. The structure of the new ligand may allow a stronger affinity to His-tagged proteins, but also towards untagged matrix proteins. Thus, it showed a higher purification efficiency of a His-tagged glycoprotein both in the presence of 2% and 10% FBS compared to commercial NiNTA resin. However, further protein purification polishing steps are required along with the affinity chromatography conditions optimization for each particular protein expressed in either prokaryotic or mammalian systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by a grant of the Romanian National Authority for Scientific Research and Innovation, CNCS UEFISCDI, project number PN-II-RU-TE-2014-4-0808 and the University of Bucharest. We thank Dr Mihaela Florea for RAMAN spectroscopic investigations. The research leading to these results has received funding from the EEA Grants 2014-2021, the SmartVac Project, Contract No 1/2019. We thank Dr Carmen Tanase and Dr Ioana Popa for kindly providing the HisS100B and His-sRAGE prokaryotic expression plasmids and advice regarding protein purification protocols. We also thank Dr Horia Szedlacsek for technical assistance with HisS100B protein purification.Notes and references
- C. Oliveira and L. Domingues, Appl. Microbiol. Biotechnol., 2018, 102, 81–92 CrossRef CAS PubMed.
- W.-H. K. Kuo and H. A. Chase, Biotechnol. Lett., 2011, 33, 1075–1084 CrossRef CAS PubMed.
- (a) J. Porath, Protein Expression Purif., 1992, 3, 263 CrossRef CAS; (b) R. Gutiérrez, E. M. Martín del Valle and M. A. Galán, Sep. Purif. Rev., 2007, 36, 71–111 CrossRef.
- (a) V. Gaberc-Porekar and V. Menart, Chem. Eng. Technol., 2005, 28, 1306–1314 CrossRef CAS; (b) S. Uchinomiya, A. Ojida and I. Hamachi, Inorg. Chem., 2014, 53, 1816–1823 CrossRef CAS PubMed; (c) J. A. Bornhorst and J. J. Falke, Methods Enzymol., 2000, 326, 245–254 CAS.
- J. Crowe, H. Döbeli, R. Gentz, E. Hochuli, D. Stüber and K. Henco, Methods Mol. Biol., 1994, 31, 371–387 CAS.
- (a) C. You and J. Piehler, Anal. Bioanal. Chem., 2014, 406, 3345–3357 CrossRef CAS PubMed; (b) Z. Huang, J. I. Park, D. S. Watson, P. Hwang and F. C. Szoka Jr, Bioconjugate Chem., 2006, 17, 1592–1600 CrossRef CAS PubMed; (c) I. Takahira, H. Fuchida, S. Tabata, N. Shindo, S. Uchinomiya, I. Hamachi and A. Ojida, Bioorg. Med. Chem. Lett., 2014, 24, 2855–2858 CrossRef CAS PubMed; (d) S.-h. Fujishima, H. Nonaka, S.-h. Uchinomiya, Y. A. Kawase, A. Ojidab and I. Hamachi, Chem. Commun., 2012, 48, 594–596 RSC; (e) D. Keller, A. Beloqui, M. Martínez-Martínez, M. Ferrer and G. Delaittre, Biomacromolecules, 2017, 18, 2777–2788 CrossRef CAS PubMed; (f) X.-M. He, B.-F. Yuan and Y.-Q. Feng, J. Chromatogr. B, 2017, 1043, 122–127 CrossRef CAS PubMed; (g) S. Knecht, D. Ricklin, A. N. Eberle and B. Ernst, J. Mol. Recognit., 2009, 22, 270–279 CrossRef CAS PubMed.
- L. Lattuada, A. Barge, G. Cravotto, G. B. Giovenzanac and L. Teid, Chem. Soc. Rev., 2011, 40, 3019–3049 RSC.
- (a) L. Pellegatti, J. Zhang, B. Drahos, S. Villette, F. Suzenet, G. Guillaumet, S. Petoud and E. Tóth, Chem. Commun., 2008, 48, 6591–6593 RSC; (b) N. Candelon, N. D. Hădade, M. Matache, J. L. Canet, F. Cisnetti, D. P. Funeriu, L. Nauton and A. Gautier, Chem. Commun., 2013, 49, 9206–9208 RSC; (c) G. B. Giovenzana, L. Lattuada and R. Negri, Isr. J. Chem., 2017, 57, 825–832 CrossRef CAS.
- (a) S. Feng, L. Mab, G. Feng, Y. Jiao and M. Zhu, J. Supramol. Struct., 2014, 1059, 27–32 CrossRef CAS; (b) L. Ma, L. Lu, M. Zhu, Q. Wang, F. Gao, C. Yuan, Y. Wu, S. Xing, X. Fu, Y. Mei and X. Gao, J. Inorg. Biochem., 2011, 105, 1138–1147 CrossRef CAS PubMed; (c) R. C. Holz, J. M. Bradshaw and B. Bennett, Inorg. Chem., 1998, 37, 1219–1225 CrossRef CAS PubMed.
- L. S. Natrajan, P. L. Timmins, M. Lunn and S. L. Heath, Inorg. Chem., 2007, 46, 10877–10886 CrossRef CAS PubMed.
- (a) F. F. Blicke and F. I. McCarty, J. Org. Chem., 1959, 24, 1061–1069 CrossRef CAS; (b) J. Kankare, A. Karppi and H. Takalo, Anal. Chim. Acta, 1994, 295, 27–35 CrossRef CAS; (c) A. Du Moulinet d'Hardemare, O. Jarjayes and F. Mortini, Synth. Commun., 2004, 34, 3975–3988 CrossRef; (d) B. M. Trost, V. S. C. Yeh, H. Ito and N. Bremeyer, Org. Lett., 2002, 4, 2621–2623 CrossRef CAS PubMed.
- D. B. Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- (a) I. Svanedal, S. Boija, A. Almesaker, G. Persson, F. Andersson, E. Hedenstrom, D. Bylund, M. Norgren and H. Edlund, Langmuir, 2014, 30, 4605–4612 CrossRef CAS PubMed; (b) G. Anderegg, F. Arnaud-Neu, R. Delgado, J. Felcman and K. Popov, Pure Appl. Chem., 2005, 77, 1445–1495 CAS.
- G. B. Deacon and R. I. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- D. A. Köse and H. Necefoglu, J. Therm. Anal. Calorim., 2008, 93, 509–514 CrossRef.
- J. V. Pham, M. A. Yilma, A. Feliz, M. T. Majid, N. Maffetone, J. R. Walker, E. Kim, H. J. Cho, J. M. Reynolds, M. C. Song, S. R. Park and Y. J. Yoon, Front. Microbiol., 2019, 10, 1404 CrossRef PubMed.
- J. Schmitt, H. Hess and H. G. Stunnenberg, Mol. Biol. Rep., 1993, 18, 223–230 CrossRef CAS PubMed.
- (a) S. Gräslund, P. Nordlund and J. Weigelt, et al., Nat. Methods, 2008, 5, 135–146 CrossRef PubMed; (b) P. T. Wingfield, Curr. Protoc. Protein Sci., 2015, 80, 6.1.1–6.1.35 Search PubMed.
- R. Adiga, M. Al-adhami and A. Andar, et al., Nat. Biomed. Eng., 2018, 2, 675–686 CrossRef CAS PubMed.
- J. Porath, J. Carlsson, I. Olsson and G. Belfrage, Nature, 1975, 258, 598–599 CrossRef CAS PubMed.
- M. Lavie, A. Goffard and J. Dubuisson, Curr. Issues Mol. Biol., 2007, 9, 71–86 CAS.
- M. Flint, J. Dubuisson, C. Maidens, R. Harrop, G. R. Guile, P. Borrow and J. A. McKeating, J. Virol., 2000, 74, 702–709 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02382e |
| ‡ These authors equally contributed. |
| This journal is © The Royal Society of Chemistry 2020 |