
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Introducing chirality in halogenated 3-arylsydnones and their corresponding 1-arylpyrazoles obtained by 1,3-dipolar cycloaddition†
Marcel Mirel Popa a,
Sergiu Shovab,
Madalina Hrubarua,
Loredana Barbua,
Constantin Draghicia,
Florea Dumitrascu*a and
Denisa E. Dumitrescuc
a,
Sergiu Shovab,
Madalina Hrubarua,
Loredana Barbua,
Constantin Draghicia,
Florea Dumitrascu*a and
Denisa E. Dumitrescuc
aCenter of Organic Chemistry “C. D. Nenitzescu”, Romanian Academy, Spl Independentei 202B, 060023 Bucharest, Romania. E-mail: fdumitra@yahoo.com
b“Petru Poni” Institute of Macromolecular Chemistry, Aleea Gr. Ghica Voda 41A, Iasi 700487, Romania
cFaculty of Pharmacy, Ovidius University Constanta, Str. Cpt. Av. Al. Serbanescu 6, Campus Corp C, Constanta 900470, Romania
First published on 21st April 2020
Abstract
New 1-arylpyrazoles substituted with halogen atoms (Br, I) were synthesized from the corresponding sydnones by 1,3-dipolar cycloaddition. By introduction of a prochiral group such as isopropyl, in the ortho position of the benzene ring, in the starting phenylglycine 1 the rotamers caused by the hindered rotation between the phenyl and the heterocyclic ring were detected by NMR spectroscopy for 1-arylpyrazoles and for the first time for 3-arylsydnones. The N-nitrosophenylglycines present E–Z stereoisomerism due to the partial C–N double bond character. All the new compounds were structurally characterized by NMR spectroscopy and confirmed by X-ray crystallography. The crystal structures of N-nitrosophenylglycine 2c and of the sydnone 3c present similar Br⋯Br type II halogen contacts.
Introduction
Mesoionic compounds1–5 are versatile intermediates in the synthesis of bioactive pyrrole and pyrazole derivatives.6–11 Sydnones12,13 in particular, have been employed as 1,3-dipoles in obtaining N-arylpyrazoles by 1,3-dipolar cycloaddition reaction.14–28 N-Arylpyrazole derivatives have been shown to possess antithrombotic,29 anticancer,30 parathyroid dysfunction,31 antibiofilm,32 and antipain33 activity which makes them valuable targets for the pharmaceutical field.Chirality is important in drug-target recognition processes34 and atropisomerism could be a useful tool for generating chiral drugs34 by blocking the free rotation about a bond axis through steric factors. The most studied systems are biaryls.35 The structure of N-arylpyrazoles implies the existence of axial chirality when the right substituents are grafted on their framework. By fine tuning of the substituents on the aryl or pyrazole rings the free rotation about the bond axis between the two rings could be hindered and thus leading to the existence of the two rotational isomers. Depending on the value for the energy of the free rotation about the two rings, the enantiomers can be detected by NMR spectroscopy or even the physical separation of the conformational isomers could be achieved. However, even in the case the free energy of rotation is smaller and the atropisomers are not easily separable in normal conditions but instead rotamers are detectable, the conformational analysis is still an important thing to be investigated under de drug-target interaction paradigm, for example.36
Herein we present the synthesis of new halogenated 1-arylpyrazoles bearing in mind that halogen atoms could enhance their bioavailability37 due to halogen bonding38 formation with specific nucleophilic sites in proteins. The introduction of axial chirality in the new 1-arylpyrazoles was pursued and investigated during the synthesis steps starting with the N-nitrosoderivatives and sydnone intermediates. The structures of the compounds were confirmed by X-ray single crystal diffraction analysis.
Results and discussions
Synthesis and characterization of N-nitrosophenylglycines 2
The synthesis of the 3-arylsydnones was achieved from corresponding N-nitroso-N-(2-isopropylphenyl)glycines obtained according to literature procedures.16 The starting N-arylglycine 1a (Scheme 1), precursor of the N-nitrosoglycines, was obtained from 2-isopropylaniline by condensation with monochloroacetic acid. Compound 1 was further brominated with Br2 to yield the N-arylglycines 1b,c (Scheme 1).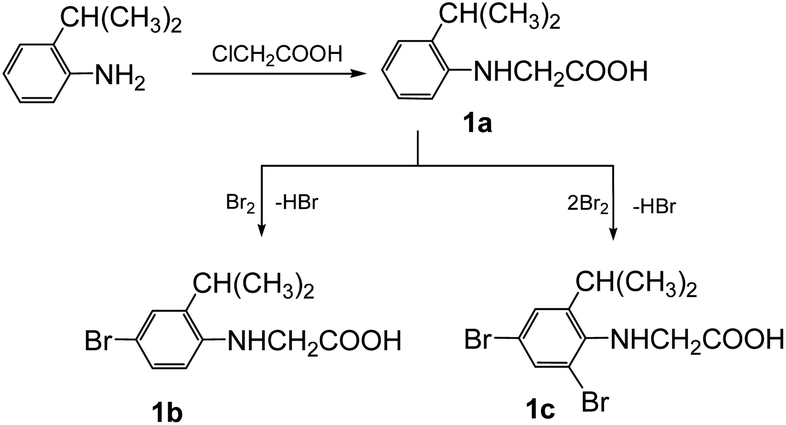 | ||
| Scheme 1 The synthesis of the starting N-arylglycines 1a–c. | ||
The 1H NMR spectra of the N-arylglycines 1a–c confirmed their structure. The isopropyl moiety appears as a doublet at 1.23–1.28 ppm with J = 6.8 Hz and a sextet at 2.96 ppm with the same value of the coupling constant. The aromatic region of the 1H NMR spectra of the compounds 1a–c presents the expected signals with the characteristic multiplicity (see Experimental section). The nitrosation of N-arylglycines 1a–c was made with NaNO2 in presence of hydrochloric acid according to Scheme 2. The nitroso derivatives 2a–c were obtained as crystalline solids in up to 90% yield.
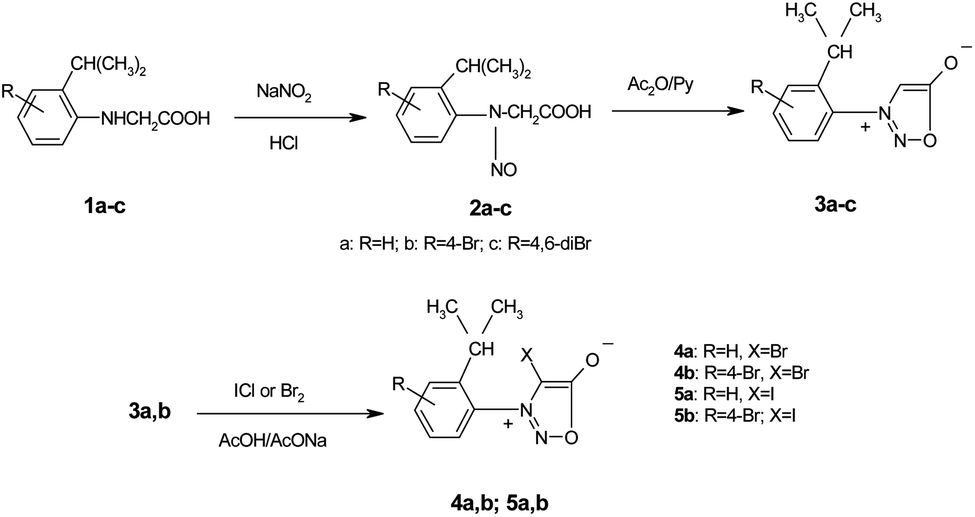 | ||
| Scheme 2 The synthesis of nitrosoaminoacids 2a–c and subsequent synthesis of the halogenated N-arylsydnones 3–5. | ||
From a careful examination of the NMR spectra of the nitrosoderivatives 2a–c we concluded that these compounds were obtained as mixtures of E–Z isomers which were unquestionably observed at low temperatures due to the fact that in normal conditions the reaction leads to the most stable isomer as will be discussed further. The E–Z isomerism appears in nitrogen containing compounds in which C–N (i.e. amides, oximes, etc) or N–N (i.e. nitroso) presents partial double bond character due to conjugation.39–44 However the E–Z isomerism in the case of nitrosoacids like 2a–c was less investigated. The activation parameters of the E–Z inter-conversion in the case of nitrosoamines were investigated by dynamic NMR techniques.45,46
Also the E–Z configuration was attributed on the rationale that the most voluminous substituent must be in cis position in respect to the oxygen atom in the nitroso group. The 1H NMR spectrum of the compound 2a shows that the signals of the protons in the isopropyl moiety and the protons in methylene attached to the nitrogen atom appear as two distinctive sets with the integral ratio of 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 (Fig. 1). For each distinctive sets of signals the multiplicities are different: the methylene attached to the N atom appears as a singlet (for the 95% isomer) and as two doublets forming an AB system with J = 17.8 Hz for the 5% isomer. The same is valid for the isopropyl moiety, Table 1 presenting the signals in the aliphatic region and their corresponding multiplicities. The presence of two conformers is observed for Z-2a (Fig. 1) due to the hindered rotation about the C–N bond induced by the bulky isopropyl group and the contribution of the nitroso moiety. The different chemical shifts of the analogous atoms in the two rotamers are slightly different due to the magnetic anisotropy of the nitroso group which is in a different spatial relation with the interacting hydrogen atoms.
:5 (Fig. 1). For each distinctive sets of signals the multiplicities are different: the methylene attached to the N atom appears as a singlet (for the 95% isomer) and as two doublets forming an AB system with J = 17.8 Hz for the 5% isomer. The same is valid for the isopropyl moiety, Table 1 presenting the signals in the aliphatic region and their corresponding multiplicities. The presence of two conformers is observed for Z-2a (Fig. 1) due to the hindered rotation about the C–N bond induced by the bulky isopropyl group and the contribution of the nitroso moiety. The different chemical shifts of the analogous atoms in the two rotamers are slightly different due to the magnetic anisotropy of the nitroso group which is in a different spatial relation with the interacting hydrogen atoms.
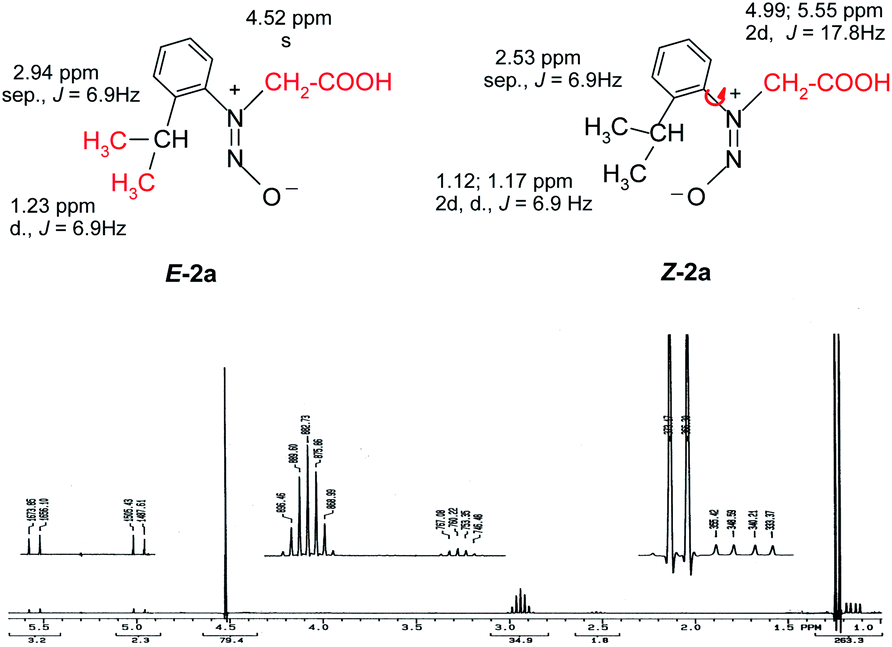 | ||
| Fig. 1 The 1H NMR spectrum of 2a showing the presence of both the Z–E isomers and the atropisomerism of the isomer Z-2a depicted by the signal multiplicities of the methyl and methylene protons. | ||
| Isomer | CH3, δ (ppm); J (Hz) | CH, δ (ppm); J (Hz) | CH2, δ (ppm); J (Hz) | H-6′ | % |
|---|---|---|---|---|---|
| E-2b | 1.22, d, 6.9 | 2.90, sep., 6.9 | 4.49, s | 7.28, s | 95 |
| Z-2b | 1.10, 1.16, 2d, 6.8 | 2.49, sep., 6.8 | 4.94, 5.53, 2d, 17.8 | 6.95, s | 5 |
| E-2c | 1.18, 1.21, 2d, 6.9 | 3.38, sep., 6.9 | 3.95, 4.94, 2d, 16.8 | — | 58 |
| Z-2c | 1.10, 2d, 6.8 | 2.82, sep., 6.8 | 4.91, 5.78, 2d, 17.8 | — | 42 |
The Z–E isomerism was also observed for compounds 2b and 2c. For 2b the protons in the aromatic ring, present two sets of signals corresponding to the two stereoisomers (Table 1) while H-6′ presents different chemical shifts also due to the spatial influence of the nitroso group.
The non-equivalence of the methyl protons of the isopropyl moiety suggests that Z-2a and Z-2b present axial isomerism which is induced by the hindered rotation about the C–N bond caused by the bulky isopropyl group. This is elegantly confirmed in the case of 2c, in which by introducing the bromine atom in the ortho position of the aryl ring with respect to the nitroso group gives the two E–Z stereoisomers in 58:42 ratio observed by 1H NMR spectroscopy (Fig. 1S†). Both Z-2c and E-2c present the coexistence of two stable conformers in solution (Table 1). The NMR studies indicated that Z-2c transforms in E-2c in solution similar to 2a and 2b. Thus one can conclude that E-2c is the thermodynamically stable stereoisomer and Z-2c is obtained by the nitrosation reaction of 1c to 2c which goes under kinetic control. We were able to separate E-2c from the mixture and to characterize it separately by NMR (Fig. 2S†) spectroscopy (see ESI†) and by single crystal X-ray diffraction analysis.
The structure of the N-nitrosophenylglycines 2 was investigated by X-ray single crystal diffraction of 2c as representative compound. Suitable crystals of 2c were grown by slow evaporation from acetonitrile. The X-ray diffraction analysis (Fig. 2) confirms the structure of 2c as being the E stereoisomer in solid state. The geometric parameters are listed in Table 2S (ESI).†
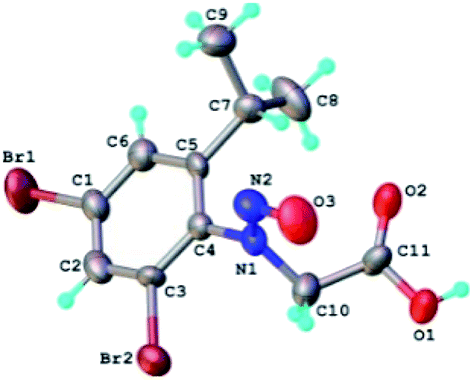 | ||
| Fig. 2 X-ray molecular structure of compound 2c with atom labeling and thermal ellipsoids at 50% level. | ||
The crystal structure of 2c can be characterized as the parallel packing of the discrete infinite ribbons running along a crystallographic axis (Fig. 3S†). The structure of one-dimensional ribbon-like supramolecular architecture is shown in Fig. 3 showing short Br⋯Br contacts. The value of the intermolecular Br⋯Br distance, 3.536(1) Å is under the sum of van der Waals radii (3.70 Å). Also the values of the angles C–Br⋯Br are 113.0° and 173.9°, respectively.
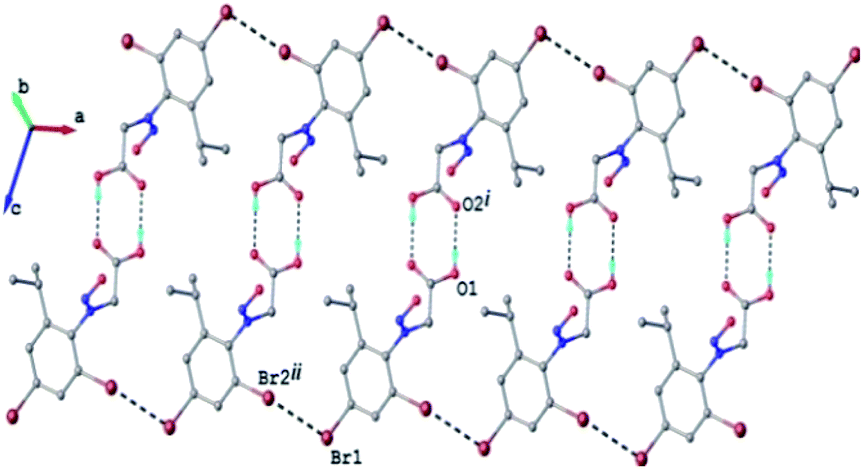 | ||
| Fig. 3 1D supramolecular network in the crystal structure 2c. H-atoms not involved in hydrogen bonding are omitted. H-bonds and Br⋯Br contacts are shown in dashed-black lines. | ||
The Br⋯Br contacts are by definition included in the class of the halogen bonds of type II.47
The energy of the cyclic dimer made by the hydrogen bond in the carboxylic acid was calculated at HF/3-21 level as implemented in Crystal Explorer 17.48,49 The calculated value of −73 kJ mol−1 place the O–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond strength as usual for such kind of compounds.50
C bond strength as usual for such kind of compounds.50
Synthesis and characterization of the 3-arylsydnones 3–5
The new nitrosophenylglycines 2a–c were used further in the synthesis of the N-arylsydnones 3a–c (Scheme 2). The new sydnones were obtained in good yields (see ESI†). The sydnones 3a,b were then halogenated in position 4 to give 4-bromosydnones 4a,b and 4-iodosydnones 5a,b by using iodine monochloride or molecular bromine. Unfortunately the halogenation in the case of 3c did not give satisfactory results owing to prolonged reaction time or required higher temperatures which led to the decomposition of the reaction products.The structure of the new compounds 3–5 (Table 2) was investigated and confirmed by NMR spectroscopy. The investigation of the NMR spectra showed that 1-arylsydnones 3c and 4b and 5a,b present also stable rotamers induced by the hindered rotation between the phenyl and sydnone ring given by the bulky isopropyl moiety and the voluminous halogen atom in position 4 of the sydnone ring in the case of 4 and 5. The diastereotopic isopropyl group has been used to probe the axial chirality which could be studied by NMR spectroscopy due to the magnetic non-equivalence of the methyl protons and carbon atoms caused by the hindered rotation around the C–N bond.
| No. | R | X | Yield (%) | Mp (°C) |
|---|---|---|---|---|
| 3a | H | H | 87 | 68–69 |
| 3b | 4-Br | H | 72 | 100–101 |
| 3c | 4,6-Dibromo | H | 54 | 169–171 |
| 4a | H | Br | 85 | 91–92 |
| 4b | 4-Br | Br | 82 | 142–144 |
| 5a | H | I | 82 | 179–180 |
| 5b | 4-Br | I | 82 | 195–197 |
In the case of the sydnones 3a–c, we could detect the presence of the two rotamers at room temperature, only in the case of 3c which possess a Br atom attached to the other ortho position of the phenyl ring in respect to the sydnone and contributes by its volume to the steric hindrance between the phenyl and sydnone rings. Thus, in the 1H NMR spectrum of 3c the methyl protons of the isopropyl appear as two doublets at room temperature (Fig. 4) while the 13C NMR spectrum presents two distinctive signals for the non-equivalent methyl carbon atoms.
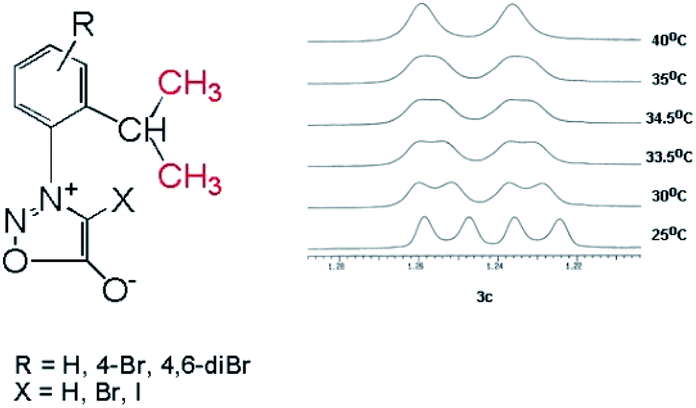 | ||
| Fig. 4 The two methyl protons in the isopropyl group become anisochronous due to the hindered rotation about the C–N group. The 1H NMR signals of the two methyl protons in 3c which coalesce at ∼35 °C. | ||
In the case of 4-bromosydnones 4a,b without a bromine in the position 6′ of the benzene ring the 1H NMR spectra does not provide relevant data regarding the presence of axial chirality at room temperature but the 13C NMR spectrum present the methyl groups C atoms as a broad singlet showing that at room temperature the energy barrier for the free rotation about the C–N bond is reached. Comparing the 1H and 13C NMR spectra of compounds 3c and 4a,b one can conclude that the presence of a Br atom on the ortho position of the phenyl ring has a greater influence upon the free rotation than in the case it is attached to the 4 position of the sydnone ring as the presence of the two rotamers is clearly distinguished in solution. For the 4-iodosydnone 5a,b the volume of iodine atom has an influence upon the signals of the methyl groups in the isopropyl moiety, the two methyl groups appearing as two distinct doublets in dilute solutions in 1H NMR. The 13C NMR spectra show also two distinct signals for the methyl groups. The free rotation energy barrier about the single bond must be therefore a little higher than in the analogous 4-brominated sydnones but still the free rotation has an energy sufficiently low to make to separate them in usual conditions.
The results of X-ray diffraction investigation for 3c is depicted in Fig. 5, while bond distances and angles are summarized in Table 2S.† This compound exhibits a molecular crystal structure resulting from the packing of two-dimensional supramolecular double-layer in parallel orientation (as shown in Fig. 4S†). The self-assembling of two-dimensional layer occurs through the weak H-bonds where C2–H group of the phenyl acts as donor towards carbonyl oxygen atom O2 of adjacent molecule as acceptor of proton (Fig. 4S†).
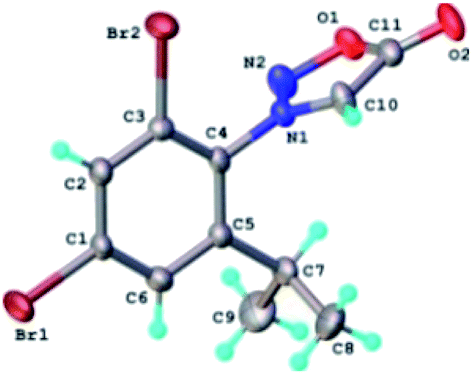 | ||
| Fig. 5 X-ray molecular structure of compound 3c with atom labeling and thermal ellipsoids at 50% level. | ||
Each double-layer architecture is built-up from two 2D networks (Fig. 6), which is consolidated due to the C–H⋯O hydrogen bonding and halogen⋯halogen interactions at 3.6020(8) Å for Br⋯Br′ distance (Fig. 6). In the sydnone 3c the intermolecular distance Br⋯Br 3.6020(8) is also under the value of vdW and values of the angles C–Br⋯Br of 119.57° and 169.39° respectively. The torsion angle between the plane of the phenyl ring and the plane of the sydnone ring has the value of 86.5°.
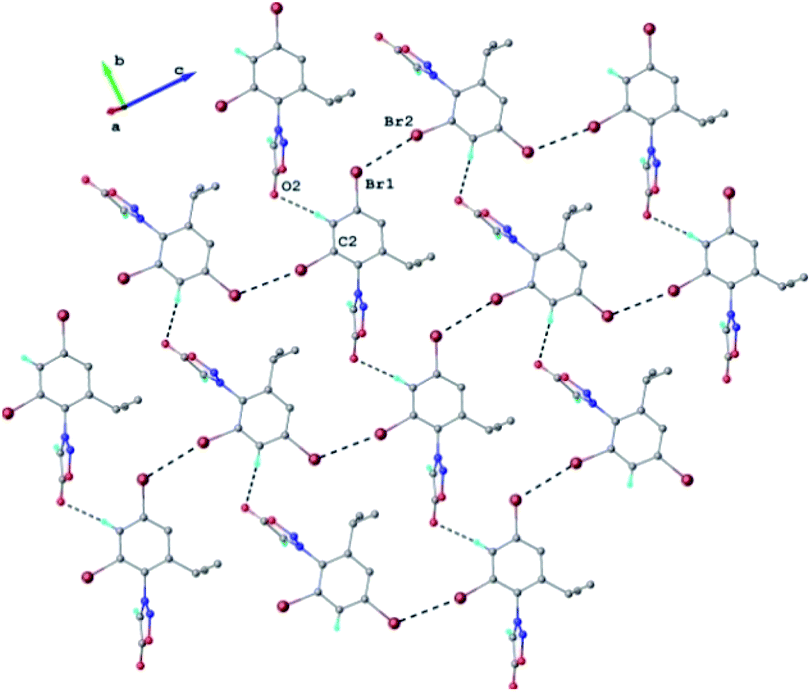 | ||
| Fig. 6 2D supramolecular network in the crystal structure 3c. H-atoms not-involved in hydrogen bonding are omitted. H-bonds and Br⋯Br contacts are shown in dashed-black lines. H-bond parameters: C2–H⋯O2 [C2–H 0.93 Å, H⋯O2 2.54 Å, C2⋯O2(x, 1 − y, z) 3.359(5) Å, ∠C2HO2 147.1°]. | ||
Beside the halogen Br⋯Br contacts in 3c we observed that 4-iodosydnones presented interesting halogen bonding affinity in solution51 in presence of different bases, and thus we envisaged that halogenated sydnones could provide ideal templates for studying the halogen bonding either in solution or solid state which will aim to investigate further.
Synthesis and characterization of the 1-arylpyrazoles 6a–g
The new halogenated 3-arylsydnones were employed in the final step in the synthesis of new halogenated 1-arylpyrazoles using the 1,3-dipolar cycloaddition reaction with activated acetylenic dipolarophiles. Thus, the new compounds 6a–g were obtained starting from the sydnones 3–5 at reflux temperature in toluene or xylene as solvent (Scheme 3). | ||
| Scheme 3 The synthesis of the halogenated 1-arylpyrazoles 6a–g. | ||
The mechanism of the [3 + 2] addition of the DMAD to sydnones was investigated28 and generally accepted as going through a formation of a bicyclic intermediate 7 as presented in the Scheme 4 which leads to the final pyrazole compounds by elimination of one CO2 molecule and further aromatization (Path A). Literature data52,53 suggest also another mechanism implying an acyclic intermediate of type 8 (Path B). In the case of the sydnones such intermediate was demonstrated to coexist to a lesser extent with the bicyclic one, giving pyrazoles with another substitution pattern, a 1,3-dipole structure being suggested.27
 | ||
| Scheme 4 Proposed mechanism pathways for the formation of the pyrazoles 6 by formal [3 + 2] cycloaddition of DMAD to sydnones. | ||
Table 3 presents the new synthesized pyrazoles 6a–g and their features.
| No. | R | X | Yield (%) | Mp (°C) |
|---|---|---|---|---|
| 6a | H | H | 87 | 62–63 |
| 6b | 4-Br | H | 94 | 139–140 |
| 6c | 2,4-DiBr | H | 67 | 113–115 |
| 6d | H | Br | 87 | 92–93 |
| 6e52 | H | I | 82 | 156–157 |
| 6f | 4-Br | Br | 86 | 103–104 |
| 6g | 4-Br | I | 80 | 128–131 |
Compounds 6a–g where characterized by NMR spectroscopy and their structure was confirmed (Experimental section). The singlet in the range 8.03–8.06 belongs to H-5 proton in the pyrazole moiety while the protons in the phenyl group are in accordance with the substitution pattern. In the compounds 6a,b,d the two methyl groups in the isopropyl moiety appear as doublets due to the coupling with the adjacent methine hydrogen. In compounds 6c the presence of the bromine atom in the ortho position the phenyl ring is sufficient to increase the rotation barrier of the two rings about the single bond. This is reflected in the 1H NMR spectrum by the magnetic non-equivalence of the two methyl groups which appear as two doublets at 1.12 and 1.19 ppm (Fig. 7). In the 13C NMR spectra the methyl groups in the isopropyl moiety appear as a singlet for 6a,b,d and for 6c the spectrum shows two different signals highlighting the magnetic non-equivalence of the two protons in discussion. For the 5-bromopyrazoles 6d,f the most important proof of the hindered rotation is the 13C NMR which shows two signals for the methyl groups while the proton NMR being more sensible to dilution shows only one doublet. For the 5-iodopyrazoles 6e,g the magnetic non-equivalence of the two methyl groups is clear in both 1H and 13C NMR spectra. The NMR data suggests that the most effective hindrance of the free rotation about Ar–N bond is when the halogen atom and the isopropyl group are both grafted on the same ring, the phenyl ring in particular.
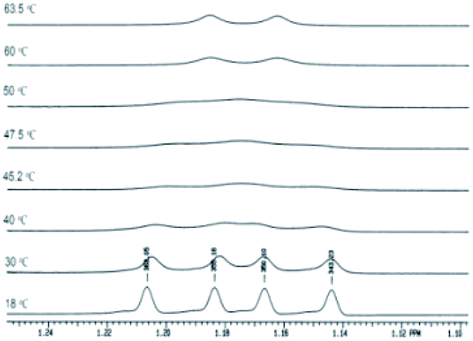 | ||
| Fig. 7 The 1H NMR signals of the methyl groups in the isopropyl moiety for compound 6e. | ||
The compound 6e was studied by dynamic NMR and the resulted energy barrier of rotation was calculated at a coalescence temperature of 50 °C using the Gutowski relation. The energy was calculated to be 70.5 kJ mol, which is high enough to try the separation of the two enantiomers.
The structure of the highly substituted pyrazoles has been also confirmed by single crystal X-ray diffraction analysis. The molecular structure of the representative compound 6b is shown in Fig. 8 and bond distances and angles in Table 1S.†
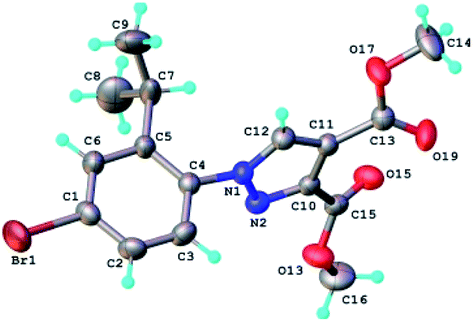 | ||
| Fig. 8 X-ray molecular structure of compound 6b with atom labeling and thermal ellipsoids at 50% level. | ||
The analysis of the crystal packing shows the presence of two-dimensional supramolecular layers, which are formed by a system of intermolecular C–H⋯O hydrogen bonding. A view of this layer is depicted in Fig. 5S.† The crystal structure essentially results from the packing of these layers parallel to 011 plane, as shown in Fig. 6S.† The torsion angle between the plane of the phenyl ring and the plane of the pyrazole ring has the value of 75.57°.
We have investigated recently the propensity of halogen bonding in halogenated 1-arylpyrazoles and we found them as versatile structures for study of this interesting interaction.54
Conclusions
In conclusion, new halogenated 3-(2-isopropylphenyl)sydnones and 1-(2-isopropylphenyl)pyrazoles were obtained from the corresponding N-nitrosoderivatives. The bulky halogen atoms on the phenyl or the five membered rings (sydnone and pyrazole) and the presence of the isopropyl on the ortho position of the phenyl ring induced a hindered rotation about the C–N bond, two stable rotamers being confirmed by NMR spectroscopy in solution. The X-ray single crystal analysis confirmed the structure of the new compounds and highlighted their conformation in solid state. The X-ray diffraction confirmed the structure of the E-2c N-nitrosophenylglycine and showed similar Br⋯Br short contacts with sydnone 3c. All the halogenated sydnones and pyrazoles are to be further investigated regarding their propensity for halogen bonding as the solution studies and X-ray diffraction analysis showed promising results.Experimental
The synthesis of N-(2-isopropyl)phenylglycine (1a)
Mp 139–141 °C. Yield 46%. 30 mL (0.21 mol) of 2-isopropylaniline are mixed with 17 g (0.18 mol) of monochloroacetic acid in 200 mL water and 10 mL ethanol. The reaction mixture is kept under reflux for 3 h. The reaction mass is cooled with water and then stirred for 1 h on an ice bath. The formed precipitate is removed by filtration and then washed with water and with benzene after drying to remove any unreacted 2-isopropylaniline. Anal. for C11H15NO2 (193.2): found: C 68.67; H 8.04; N 7.52. Calcd: C 68.37; H 7.82; N 7.25. 1H NMR (CDCl3; δ, ppm; J, Hz): 1.28 (d, 6H, 6.8; Me); 2.96 (sep., 1H, 6.8; CHMe2); 4.03 (s, 2H, CH2); 6.15 (sl, 2H, COOH, NH); 6.54 (dd, 1H, 7.6; 1.5; H-6′); 6.83 (td, 7.6; 1.7; H-4′); 7.13 (td, 7.6; 1.5; H-5′); 7.19 (dd, 7.6; 1.7; H-3′). 13C NMR (CDCl3; δ, ppm): 22.3 (Me); 27.3 (CHMe2); 46.0 (CH2); 110.8 (C-6′); 118.9 (C-4′); 125.3 (C-3′); 126.8 (C-5′); 133.0 (C-2′); 143.3 (C-1′); 176.1 (CO).The synthesis of N-phenylglycines 1b,c
N-(2-Isopropyl)phenylglycine 3.9 g (20 mmol) are suspended in 20 mL glacial acetic acid. Over this solution is added by stirring 20 mmol Br2 for 1b or 40 mmol Br2 for 1c dissolved in 5 mL acetic acid. The reaction is kept under stirring for cca. 15–20 min and then the reaction mass is poured over cold water under continuous stirring. The formed precipitate is filtered and washed with water on the filter paper, and then the compound is dried.Procedures for obtaining N-nitroso-N-phenylglycines
N-(2-Isopropylphenyl)glycine 20 mmol are dissolved in 40 mL solution of 5% NaOH and 21.5 mmol of NaNO2 are added. Over this mixture 10 mL conc. HCl are added under vigorous stirring and cooling on an ice bath, until the pH will reach a value of 1.5–2. The stirring continues for another 15 min while the nitrosoderivative precipitates. The N-nitrosophenylglycine is filtered washed with water on the filter and let to dry in air.General procedure for the synthesis of 3-(2-isopropylphenyl)sydnones 3–5
N-nitroso-N-(2-isopropylphenyl)glycine 20 mmol was dissolved in 30 mL acetic anhydride. On this solution is added 1 mL of dried pyridine. The reaction mixture is gently heated on a water bath at reduced pressure in order to evaporate the excess acetic anhydride and the acetic acid resulted from the reaction. The sydnone precipitates as oil which crystallizes upon cooling.General procedure for obtaining 4-brom-3-(2-isopropylphenyl)sydnones (4a,b)
3-(2-Isopropylphenyl)sydnone 10 mmol and 12 mmol sodium acetate anhydrous are dissolved in 15 mL glacial acetic acid. Over the reaction mixture are added under stirring and on an ice bath 11 mmol Br2 dissolved in 15 mL glacial acetic acid. The stirring is kept 10–15 minutes at room temperature and then the reaction mixture is poured over 150 mL of cold water. The formed precipitate is removed by filtration and washed with water on the filter paper.General procedure for the synthesis of the 1-arylpyrazoles 6
Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Ollis and A. Ramsden, Meso-ionic Compounds, Adv. Heterocycl. Chem., 1976, 19, 1 CrossRef.
- F. Dumitrascu, M. R. Caira, B. Draghici, M. T. Caproiu and D. G. Dumitrescu, A novel approach for the synthesis of highly fluorescent pyrrolo[1,2-b]pyridazines, Synlett, 2008, 813–816 CAS.
- K. Sugimoto, Y. Miyakawa and H. Tokuyama, Total synthesis of (–)-rhazinilam using 1,3-dipolar cycloaddition of optically active münchnone intermediate, Tetrahedron, 2015, 71, 3619–3624 CrossRef CAS.
- Y. Fang, R. C. Larock and F. Shi, Aryne Cycloaddition with Stable Münchnones: Synthesis of 9,10-Dihydro-9,10-epiminoanthracenes and Isoindoles, Asian J. Org. Chem., 2014, 3, 55–57 CrossRef CAS.
- C. M. Torres, J. S. Quesnel, D. B. Bijou and A. Arnstden, From Aryl Iodides to 1,3-Dipoles: Design and Mechanism of a Palladium Catalyzed Multicomponent Synthesis of Pyrroles, J. Am. Chem. Soc., 2016, 138, 7315–7324 CrossRef PubMed.
- F. Bellina and R. Rossi, Synthesis and biological activity of pyrrole, pyrroline and pyrrolidine derivatives with two aryl groups on adjacent positions, Tetrahedron, 2006, 62, 7213–7256 CrossRef CAS.
- A. Ansari, A. Ali, M. Asif and S. Shamsuzzaman, Review: biologically active pyrazole derivatives, New J. Chem., 2017, 41, 16–41 RSC.
- S. Fustero, M. Sanchez-Rosello, P. Barrio and A. Simon-Fuentes, From 2000 to Mid-2010: A Fruitful Decade for the Synthesis of Pyrazoles, Chem. Rev., 2011, 111, 6984–7034 CrossRef CAS PubMed.
- D. Vuluga, J. Legros, B. Crousse and D. Bonnet-Delpon, Synthesis of pyrazoles through catalyst-free cycloaddition of diazo compounds to alkynes, Green Chem., 2009, 11, 156–159 RSC.
- Z.-X. Wang and H.-L. Qin, Solventless syntheses of pyrazole derivatives, Green Chem., 2004, 6, 90–92 RSC.
- J. Jansa, R. Schmidt, A. D. Mamuye, L. Castoldi, A. Roller, V. Pace and W. Holzer, Synthesis of tetrasubstituted pyrazoles containing pyridinyl substituents, Beilstein J. Org. Chem., 2017, 13, 895–902 CrossRef CAS PubMed.
- A. M. Abdualkader, M. Taher and N. I. N. Yusoff, Mesoionic sydnone: A review in their chemical and biological properties, Int. J. Pharm. Pharmaceut. Sci., 2017, 9, 1–9 CrossRef CAS.
- F. Albota, M. R. Caira, C. Draghici, F. Dumitrascu and D. E. Dumitrescu, Sydnone C-4 heteroarylation with an indolizine ring via Chichibabin indolizine synthesis, Beilstein J. Org. Chem., 2016, 12, 2503–2510 CrossRef CAS PubMed.
- F. Albota, C. Draghici, M. R. Caira and F. Dumitrascu, 1,3-Dipolar cycloaddition between acetylenic dipolarophiles and sydnone-N-ylides as bis(1,3-dipoles), Tetrahedron, 2015, 71, 9095–9100 CrossRef CAS.
- N. Pétry, T. Vanderbeeken, A. Malher, Y. Bringer, P. Retailleau, X. Bantreil and F. Lamaty, Mechanosynthesis of sydnone-containing coordination complexes, Chem. Commun., 2019, 55, 9495–9498 RSC.
- F. Dumitrascu, C. Draghici, D. Dumitrescu, L. Tarko and D. Raileanu, Direct iodination of sydnones and their cycloadditions to form 5-iodopyrazoles, Liebigs Ann./Recl., 1997, 2613–2616 CrossRef CAS.
- F. Dumitrascu, C. I. Mitan, D. Dumitrescu, C. Draghici and M. T. Caproiu, Steric effects on the sydnones reactivity. New sydnones and pyrazoles, Arkivoc, 2002, 80–86 Search PubMed.
- F. Dumitrascu, C. Draghici, D. Vuluga and M. T. Caproiu, New pyrazoles by 1,3-dipolar cycloaddition reactions between sydnones and activated alkynes, Rev. Roum. Chim., 2006, 51, 255–260 Search PubMed.
- W.-J. Hung and H.-J. Tien, Dehalogenation and Halogenation of Sydnone Compounds with Duet Electrosynthesis, J. Chin. Chem. Soc., 1993, 40, 637–639 CrossRef CAS.
- T. Delaunay, P. Genix, M. Es-Sayed, J.-P. Vors, N. Monteiro and G. Balme, A Modular Sydnone Cycloaddition/Suzuki–Miyaura Cross-Coupling Strategy to Unsymmetrical 3,5-Bis(hetero)aromatic Pyrazoles, Org. Lett., 2010, 12, 3328–3331 CrossRef CAS PubMed.
- T. Delaunay, M. Es-Sayed, J.-P. Vors, N. Monteiro and G. Balme, Facile Access to 3,5-Dihalogenated Pyrazoles by Sydnone Cycloaddition and their Versatile Functionalization by Pd-Catalyzed Cross-Coupling Processes, Eur. J. Org. Chem., 2011, 3837–3848 CrossRef CAS.
- D. C. Brown and K. Turnbull, Improved Method for the Iodination of Sydnones, Synth. Commun., 2013, 43, 3233–3237 CrossRef CAS.
- I. A. Cherepanov and S. K. Moiseev, Recent developments in the chemistry of sydnones and sydnone imines, Adv. Heterocycl. Chem., 2020, 131, 49–164 CrossRef.
- I. F. Nashashibi, J. M. Tumey, B. L. Owens and K. Turnbull, Chlorination of 3-Arylsydnones with Iodine Monochloride, Org. Prep. Proced. Int., 2017, 49, 59–63 CrossRef CAS.
- E. Decuypere, S. Specklin, S. Gabillet, D. Audisio, H. Liu, L. Plougastel, S. Kolodych and F. Taran, Copper(I)-Catalyzed Cycloaddition of 4-Bromosydnones and Alkynes for the Regioselective Synthesis of 1,4,5-Trisubstituted Pyrazoles, Org. Lett., 2015, 17, 362–365 CrossRef CAS PubMed.
- H. Liu, D. Audisio, L. Plougastel, E. Decuypere, D.-A. Buisson, O. Koniev, S. Kolodych, A. Wagner, M. Elhabiri, A. Krzyczmonik, S. Forsback, O. Solin, V. Gouverneur and F. Taran, Ultrafast Click Chemistry with Fluorosydnones, Angew. Chem., Int. Ed., 2016, 55, 12073–12077 CrossRef CAS PubMed.
- R. S. Foster, H. Adams, H. Jakobi and J. P. Harrity, Synthesis of 4-Fluoromethylsydnones and their Participation in Alkyne Cycloaddition Reactions, J. Org. Chem., 2013, 78, 4049–4064 CrossRef CAS PubMed.
- V. Hladikova, J. Vana and J. Hanusek, [3 + 2]-Cycloaddition reaction of sydnones with alkynes, Beilstein J. Org. Chem., 2018, 14, 1317–1348 CrossRef CAS PubMed.
- G. Zech, G. Hessler, A. Evers, T. Weiss, P. Florian, M. Just, J. Czech, W. Czechtizky, J. Gorlitzer, M. Kohlmann and M. Nazare, Identification of High-Affinity P2Y12 Antagonists Based on a Phenylpyrazole Glutamic Acid Piperazine Backbone, J. Med. Chem., 2012, 55, 8615–8629 CrossRef CAS PubMed.
- M. Ito, T. Tanaka, D. R. Cary, M. Iwatani-yoshihara, Y. Kamada, T. Kawamoto, S. Aparicio, A. Nakanishi and Y. Imaeda, Discovery of Novel 1,4-Diacylpiperazines as Selective and Cell-Active eIF4A3 Inhibitors, J. Med. Chem., 2017, 60, 3335–3351 CrossRef CAS PubMed.
- S. Poon, D. J. St. Jeana, P. E. Harrington, C. Henley III, J. Davis, S. Morony, F. D. Lott, J. D. Reagan, J. Y.-L. Lu, Y. Yang and C. Fotsch, Discovery and Optimization of Substituted 1-(1-Phenyl-1H-pyrazol-3-yl)methanamines as Potent and Efficacious Type II Calcimimetics, J. Med. Chem., 2009, 52, 6535–6538 CrossRef CAS PubMed.
- S. Cascioferro, B. Maggio, D. Raffa, M. V. Raimondi, M. G. Cusimano, D. Schillaci, B. Manachini, F. Plescia and G. Daidone, Synthesis and biofilm formation reduction of pyrazole-4-carboxamide derivatives in some Staphylococcus aureus strains, Eur. J. Med. Chem., 2016, 123, 58–68 CrossRef CAS PubMed.
- J. H. Kim, G. Keum, H. Chung and G. Nam, Synthesis and T-type calcium channel-blocking effects of aryl(1,5-disubstituted-pyrazol-3-yl)methyl sulfonamides for neuropathic pain treatment, Eur. J. Med. Chem., 2016, 123, 665–672 CrossRef CAS PubMed.
- S. R. LaPlante, L. D. Fader, K. R. Fandrick, O. Hucke, R. Kemper, S. P. F. Miller and P. J. Edwards, Assessing Atropisomer Axial Chirality in Drug Discovery and Development, J. Med. Chem., 2011, 54, 7005–7022 CrossRef CAS PubMed.
- J. Wencel-Delord, F. R. Panossian and F. Colobert, Recent advances and new concepts for the synthesis of axially stereoenriched biaryls, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC.
- K. A. Brameld, B. Kuhn, D. C. Reuter and M. Stahl, Small Molecule Conformational Preferences Derived from Crystal Structure Data. A Medicinal Chemistry Focused Analysis, J. Chem. Inf. Model., 2008, 48, 1–24 CrossRef CAS PubMed.
- M. Z. Hernandes, S. M. T. Cavalcanti, D. R. M. Moreira, W. F. de Azevedo Jr. and A. C. Lima-Leite, Halogen atoms in the modern medicinal chemistry: hints for the drug design, Curr. Drug Targets, 2010, 11, 303–314 CrossRef CAS PubMed.
- Y. Lu, Y. Wang and W. Zhu, Nonbonding interactions of organic halogens in biological systems: implications for drug discovery and biomolecular design, Phys. Chem. Chem. Phys., 2010, 12, 4543–4551 RSC.
- D. S. Bohle, Z. Chua, I. Perepichka and K. Rosadiuk, E/Z Oxime Isomerism in PhC(NOH)CN, Chem.–Eur. J., 2013, 19, 4223–4229 CrossRef CAS PubMed.
- G. Szalontai, P. Sandor, F. Bangerter and L. Kollar, 1H and 13C dynamic NMR study of the E, Z isomers of vinylogous diamides. Effect of conjugation and electrostatic and steric repulsion on the C(O)-N rotation barriers and low-temperature conformation, Magn. Reson. Chem., 1989, 27, 216–222 CrossRef CAS.
- A. B. Lopes, E. Miguez, A. E. Kummerle, V. M. Rumjanek, C. A. M. Fraga and E. J. Barreiro, Characterization of Amide Bond Conformers for a Novel Heterocyclic Template of N-acylhydrazone Derivatives, Molecules, 2013, 18, 11683–11704 CrossRef PubMed.
- J. S. Laursen, J. Engel-Adreasen, P. Fristrup, P. Harris and C. A. Olsen, Cis–Trans Amide Bond Rotamers in β-Peptoids and Peptoids: Evaluation of Stereoelectronic Effects in Backbone and Side Chains, J. Am. Chem. Soc., 2013, 135, 2835–2844 CrossRef CAS PubMed.
- P. Subramaniam, C. Ramasubbu and S. Athiramu, Exploiting intramolecular hydrogen bonding for the highly (Z)-selective & metal free synthesis of amide substituted β-aminoenones, Green Chem., 2017, 19, 2541–2545 RSC.
- S. Ianelli, G. Pelosi, G. Ponticelli, M. T. Cocco and V. J. Onnis, Crystal and molecular structure of acetamidrazone derivatives, J. Chem. Crystallogr., 2001, 31, 149–154 CrossRef CAS.
- M. Oki, Applications of Dynamic NMR Spectroscopy to Organic Chemistry, VCH, New York, 1985 Search PubMed.
- M. Oki, The chemistry of Rotational isomers, Springer, Berlin, 1993 Search PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The Halogen Bond, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Energy frameworks: insights into interaction anisotropy and the mechanical properties of molecular crystals, Chem. Commun., 2015, 51, 3735–3738 RSC.
- M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer17, University of Western Australia, 2017 Search PubMed.
- A. Gavezzoti, Hydrogen bond strength and bond geometry in cyclic dimers of crystalline carboxylic acids, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 401–403 CrossRef PubMed.
- C. Draghici, M. R. Caira, D. E. Dumitrescu and F. Dumitrascu, Halogen Bonds of 4-Iodosydnones in Solution Deduced from 13C-NMR Spectra, Rev. Chem., 2018, 69, 843–845 CAS.
- G. Mloston, M. Celeda, R. Jasinski and H. Heimgartner, Experimental and computational studies on stepwise [3+2]-cycloadditions of diaryldiazomethanes with electron-deficient dimethyl (E)- and (Z)-2,3-dicyanobutenedioates, Eur. J. Org. Chem., 2019, 422–431 CrossRef CAS.
- R. Jasinski, K. Kula, A. Kacka and B. Miroslaw, Unexpected course of reaction between (E)-2-aryl-1-cyano-1-nitroethenes and diazafluorene: why is there no 1,3-dipolar cycloaddition?, Monatsh. Chem., 2017, 148, 909–915 CrossRef CAS PubMed.
- M. M. Popa, I. C. Man, C. Draghici, S. Shova, M. R. Caira, F. Dumitrascu and D. Dumitrescu, Halogen bonding in 5-iodo-1-arylpyrazoles investigated in the solid state and predicted by solution 13C-NMR spectroscopy, CrystEngComm, 2019, 21, 7085–7093 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1849326, 1849327 and 1849329. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra02368j |
| This journal is © The Royal Society of Chemistry 2020 |