
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectron–phonon scattering and excitonic effects in T-carbon†
Xiangtian Bu and
Shudong Wang *
*
School of Physical Science and Technology, Inner Mongolia University, Hohhot 010021, China. E-mail: sdwang@imu.edu.cn
First published on 26th June 2020
Abstract
Through first-principles calculations combining many-body perturbation theory, we investigate electron–phonon scattering and optical properties including the excitonic effects of T-carbon. Our results reveal that optical and acoustic phonons dominate the scattering around the valence and the conduction band edges, respectively. In addition, the relaxation lifetimes of holes (0.5 ps) are longer than those of electrons (0.24 ps) around the band edges due to the weaker scattering. We also predict that mean free paths of hot holes are as high as 80 nm while only 15 nm for hot electrons, resulting in different hot carrier extraction ranges in T-carbon. Moreover, we demonstrate that there exist lowest energy dark excitons in T-carbon with radiative lifetime of about 3.4 s, which is revealed to be much longer than that of bright excitons and would lower the photoluminescence quantum yield of T-carbon.
1. Introduction
Electron–phonon (e–ph) interactions are a central topic in condensed matter physics and were first studied by Bloch in 1928.1–5 For example, e–ph interactions determine the carrier transport properties, phonon-assisted optical absorption,6,7 phonon transport and thermal conductivity,8 the thermoelectric properties of materials,9 Raman spectra,10 Peierls instability11,12 and the cooling process of hot carriers.13–15 In addition, the mobility of carriers and intrinsic carrier relaxation rates in materials are determined by e–ph scattering and electron–electron scattering.16 Consequently, e–ph scattering is crucial for applications in electronic and optoelectronic devices.To date, carbon has been a crucial and multifunctional element due to its possibility of forming numerous allotropes from zero-dimension to three-dimension17–21 and its outstanding physical and chemical properties. The most representative examples include zero dimensional (0D) hollow sphere fullerenes, one dimensional (1D) carbon nanotubes, and two dimensional (2D) one-atomic-thick graphene, and so on. With the increasing research on carbon allotropes, many novel carbon based allotropes have been discovered gradually, such as monoclinic M-carbon, body-centered tetragonal bct-C4 and orthorhombic W-carbon.22 All these carbon crystalline phases have been expected to be applied frontier domains on account of their abundant performance. For another, a novel carbon allotrope with mechanical and kinetical stabilities has been theoretically predicted by Sheng et al. via first-principles calculations.23,24 This carbon allotrope is consistent with diamond that a carbon tetrahedron substitute for each atom and it was defined as T-carbon in 2011.23 Very recently, an exciting news is that Zhang et al. successfully synthesized T-carbon nanowires (NWs) under picosecond laser irradiation.25
The T-carbon holds a much lower density and a much higher Vickers hardness with a direct band gap of about 3.0 eV.23 Previous theoretical results predict that the electron mobility in T-carbon is as high as 2.36 × 103 cm2 V−1 s−1, holding a potential efficient electron transport material.26 It is shown as well that T-carbon can be a potential candidate in energy fields applications.27 Molecular dynamics simulations show that T-carbon NWs have strong tensile properties and better ductility compared with other carbon phases, which determine the microstructure evolution and mechanical properties of T-carbon NWs under the action of tensile load.24 Furthermore, the optical properties of T-carbon were studied by adjusting its band gap with doping or strain engineering.28,29 Unfortunately, the primary mechanisms of intrinsic carrier scattering determined by the prominent processes of e–ph interaction have not yet been fully understood, as well as the excitonic effects related optical properties.
As a newly discovered 3D material, T-carbon has been found to be potential applications in energy storage, photocatalysis and so on. Considering its moderate direct band gap semiconductor, it is assumed that the carrier relaxation times are of great importance for the application in optoelectronic materials. In this study, we investigate the scattering mechanism30–34 based on density functional theory (DFT)35 combined with Wannier function interpolation method, including scattering rates, carrier lifetimes and mean free paths of T-carbon. By employing G0W0 approximation and Bethe–Salpeter equation formalism,36 we also obtain the optical properties of T-carbon including excitonic effects and the exciton radiative lifetimes.
2. Theories and computational details
The ground state calculations are performed within the local density approximation (LDA) implemented in the Quantum ESPPRESSO code.37,38 Norm-conserving pseudopotentials are taken to describe the core–valence interaction, and the kinetic energy cutoff we used is 80 Ry. We firstly compute the electronic and phonon properties on 12 × 12 × 12 k and 6 × 6 × 6 q point grids, and then interpolate to finer grids of 84 × 84 × 84 k and 84 × 84 × 84 q points based on the interpolation technique of Wannier functions implemented in the EPW code at 300 K.39,40The one-shot G0W0 approximation is adopted to deal with quasiparticle energies of T-carbon. The quasiparticle band gap is converged within 0.1 eV, with respect to the number of empty bands, the response block size and the k-point grids. Finally, the exciton related properties are calculated through solving the Bethe–Salpeter equation (BSE).41,42 250 unoccupied bands combined with the extrapolar correction scheme43 was taken to obtain the converged dielectric function under the random phase approximation (RPA). A 16 × 16 × 16 k-point grids, meanwhile, 3 valence bands and 4 conduction bands are considered to converge the optical spectra. The one-shot G0W0 and BSE calculations are performed by using the YAMBO code.44,45
Lattice vibrational properties are obtained by means of density-functional perturbation theory (DFPT). The imaginary part of the e–ph self-energy Im(Σe–phn,k)is defined as in the Brillouin zone (BZ)46
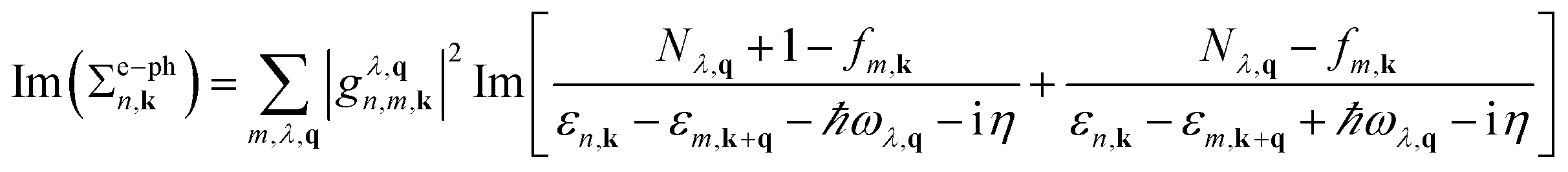 | (1) |
| gλ,qn,m,k = 〈Ψm,k+q|∂λ,qV|Ψn,k〉 | (2) |
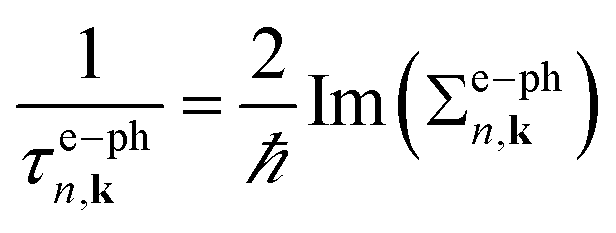 | (3) |
3. Results and discussion
The optimized primitive cell of T-carbon is shown in Fig. 1(a) where the carbon atoms in diamond are replaced by a carbon tetrahedron and its optimized lattice constant is 7.40 Å within the local density approximation (LDA) formalism, which agrees well to the value of 7.45 Å in ref. 23. The imaginary part of electron self-energy Im(Σe–phn,k)at 300 K along high-symmetry directions in the BZ combining with the electronic band structure of T-carbon within LDA is shown in Fig. 1(b). We choose finite bands in the energy windows, since relaxation times would shorten due to enhanced possible transitions with phonon emission. We observed the imaginary part of electron self-energy Im(Σe–phn,k) is strongly dependent on k and band for different electron energies in T-carbon. The absence of states near the band edges leads Im(Σe–phn,k) at the valence band maximum (VBM) and conduction band minimum (CBM) to be tiny. Since the scattering rates are proportional to Im(Σe–phn,k), they should be quite small aroud the band edges, while they will increase away from the band edges.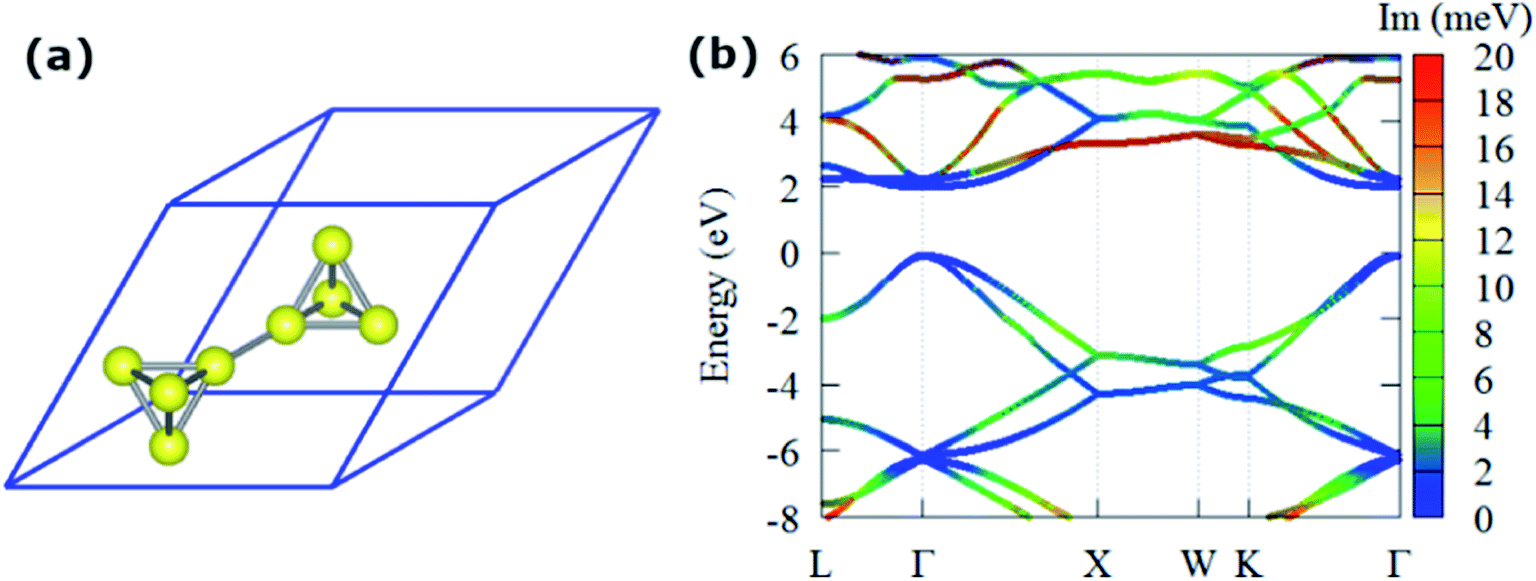 | ||
| Fig. 1 (a) Optimized primitive cell of T-carbon. (b) The LDA band structure with the Im(Σe–phn,k) (color mapped, at 300 K). | ||
The e–ph scattering rates and relaxation times (RTs) of T-carbon originating from acoustic and optical phonon scattering are shown in Fig. 2. There are multiple values at some points because only a few electrons and holes in the material near the Fermi energy level are scattered by phonons, and the valence bands of the T-carbon are degenerate near the Fermi level. The acoustic phonons scatter the electrons much more strongly than the optical phonons within the energy of 0–0.11 eV. However, the contribution of optical phonons is much larger than that of acoustic phonons above 0.22 eV. As can be seen in the band structure, the holes mainly come from the Γ-point within the energy range of 0–0.5 eV around the VBM. In case of electrons, there are several energy bands involved in electron–phonon scattering at ∼2 eV above the Fermi level. Generally, the scattering rates originated from optical phonons for holes are larger than the contribution from acoustic phonons within the energy range of 1.0 eV below VBM as shown in Fig. 2(a). For electrons in Fig. 2(b), acoustic phonons scatter electrons much more strongly than optical phonons at energies ranging from 0 to 0.11 eV. The scattering rate has a jump at the energy of 0.22 eV, meaning the L valley participates in the scattering. On the other hand, the RT from acoustic phonons scattering is as long as ∼14 ps around the VBM, as shown in Fig. 2(c). For the electrons, the optical phonon plays a dominant role around the CBM, which is much shorter than the RT around the VBM. This means holes and electrons closing to the band edges in T-carbon coupled with different phonon modes, and the optical phonon dominates the scattering around VBM while it is determined by acoustic phonon around CBM.
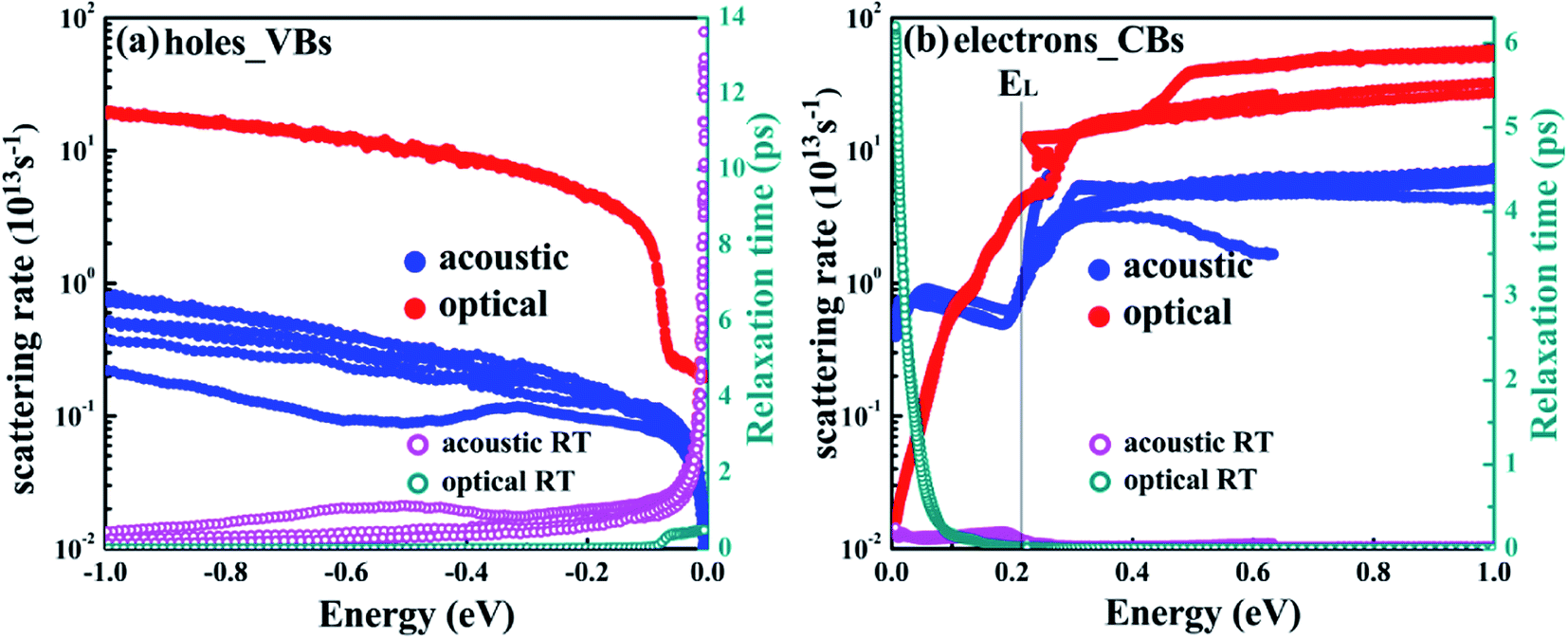 | ||
| Fig. 2 The e–ph scattering rates and relaxation times for holes (a) and electrons (b), respectively (including optical and acoustic phonon scattering). The VBM in (a) and CBM in (b) is set to the zero in the energy axis. | ||
In order to give an insight into the contribution by each phonon mode, we show the scattering rates according to the longitudinal acoustic (LA), transverse acoustic (TA), longitudinal optical (LO) and transverse optical (TO) phonon modes, respectively. Fig. 3(a) and (b) show the scattering rates contributed from different phonon branches. We observe that the TO mode dominates the scattering for both holes and electrons, exhibiting about an order of magnitude higher than any other modes generally except for the LO mode above 0.5 eV from CBM. Electrons and holes share a common jump at 0.25 eV, which is the emission onset of the LO phonon at Γ point.
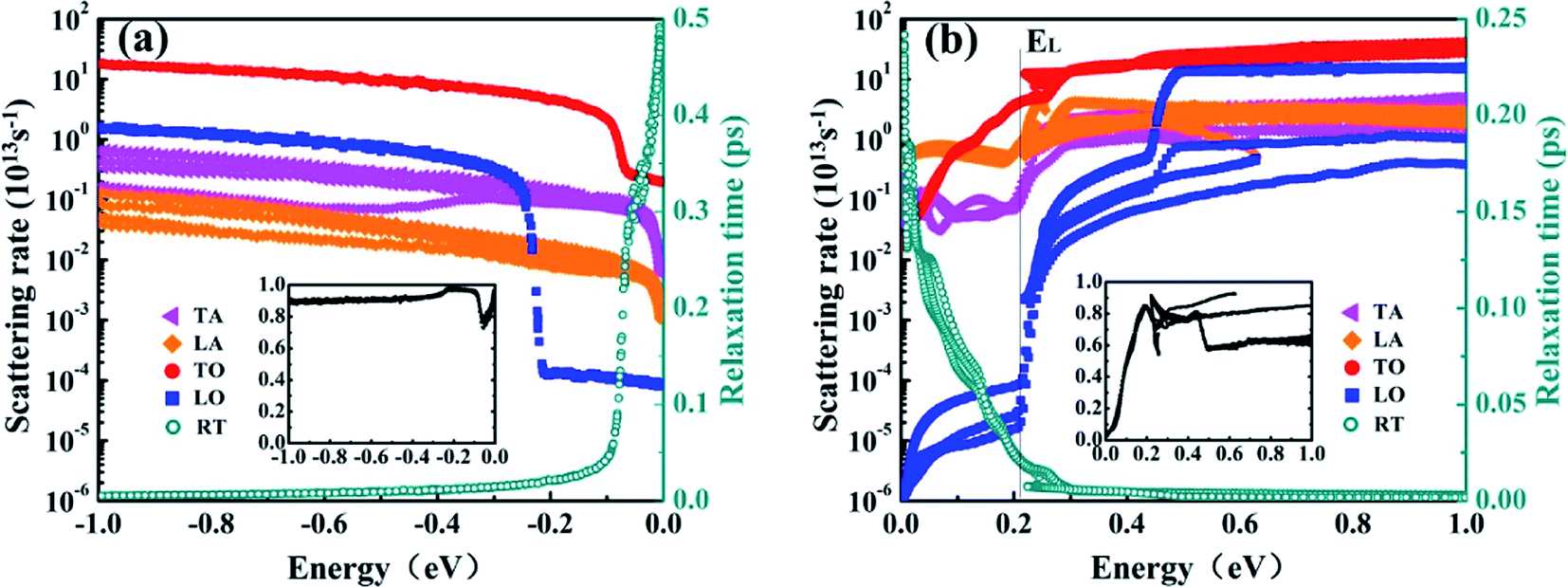 | ||
| Fig. 3 Mode-resolved scattering rates and total relaxation times of holes (a) and electrons (b). Inset: relative contribution from the TO branch. | ||
For holes with energy lower than 80 meV from VBM, the TO mode contributes about 90% to the total scattering, as revealed in the inset in Fig. 3(a). For electrons, the contribution from TO phonons is less than 55% at 0–120 meV, and it increases to more than 80% above 176 meV. Meanwhile, the maximum contributed about 93% at ∼220 meV and ∼620 meV, as shown in the inset in Fig. 3(b). The RTs are ∼0.5 ps and ∼0.25 ps at VBM and CBM, respectively. These results show that transverse optical phonons contribute mainly to electron–phonon scattering in T-carbon.
The conduction band of T-carbon shows two distinct valleys, with the Γ valley at CBM and the L valley at energy EL ≈ 0.22 eV above the CBM. Because the significance of e–ph scattering in the Γ and L valleys for charge transport in T-carbon, we further investigate the scattering rates of these two valleys. Fig. 4 shows our computed e–ph scattering rates and RTs for energies up to 0.5 eV at 300 K. We find Γ–L intervalley scattering occurs above energy EL and TO phonon scattering is dominant within this energy range, as shown in Fig. 4(a), when comparing the total scattering rates. The multi-branch feature indicates a strong k and band dependence of scattering for the anisotropy of the L valley. For electrons below EL, the scattering rate from Γ valley is only 6 × 1013 s−1 and it increases to more than 14 × 1013 s−1 above EL.
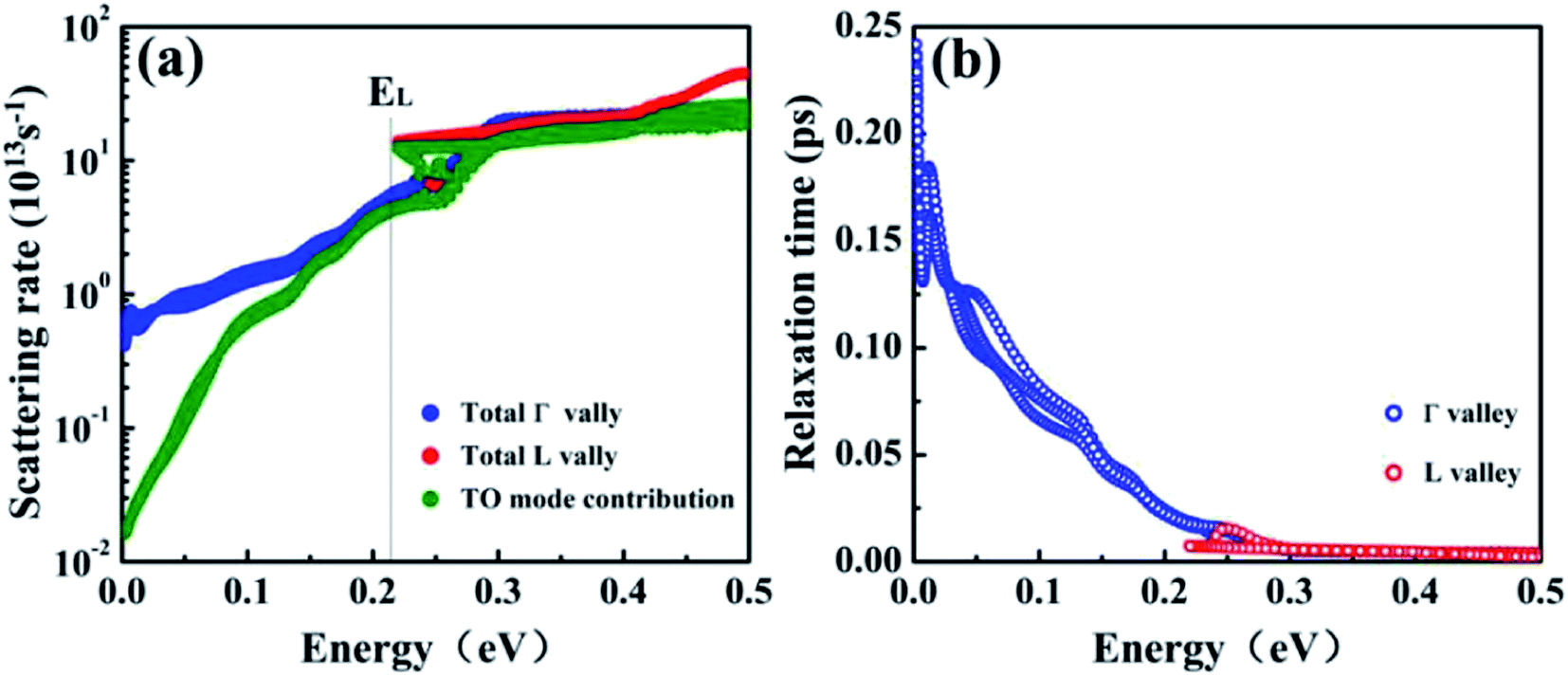 | ||
| Fig. 4 (a) The e–ph scattering rate and (b) relaxation times for electrons in T-carbon with energies within 0.5 eV from CBM. Blue and red data are for the states in the Γ and L valley, respectively. | ||
The e–ph scattering rates increase rapidly at 0–0.22 eV and it shows a strong energy dependence. Our results show that the RTs decrease with the increase of scattering rate until the intervalley scattering occurs, and the RTs tend to be constant as shown in Fig. 4(b).
Fig. 5 shows e–ph mean free paths (MFPs) for hot holes and hot electrons in T-carbon along the [100], [110], [111] directions. The values were obtained by computing vn,k and τn,k along the ΓX, ΓK, and ΓL directions in the BZ. The expression of mean free paths is defined as: Ln,k = vn,k·τn,k and the semiclassical velocities vn,k = ℏ−1∂En,k/∂k. For the hot holes, we find MFPs of up to 80 nm and distributed evenly in three directions at energy less than −0.1 eV. For hot electrons, MFPs are less than 5 nm at all directions at energies greater than 0.22 eV, and the longest MFPs are observed when energies are up to ∼73 meV above the CBM. As described above, the mean free paths is defined by group velocities and relaxation time. From the definition of group velocities, it can be found that the group velocities of holes is larger than that of electrons in the band structure. In addition, the RTs of the holes is about twice that of the electrons. These two factors cause the mean free paths of the hole to be much higher than those of electrons. Consequently, the extraction of hot holes along the [100] or [110] direction in T-carbon can be achieved within ∼88 nm range at 300 K. In addation, hot holes can be extracted from all directions of up to 80 nm less than 82 meV at VBM. The best extraction range for hot electrons is less than 15 nm along the [111] direction at 300 K.
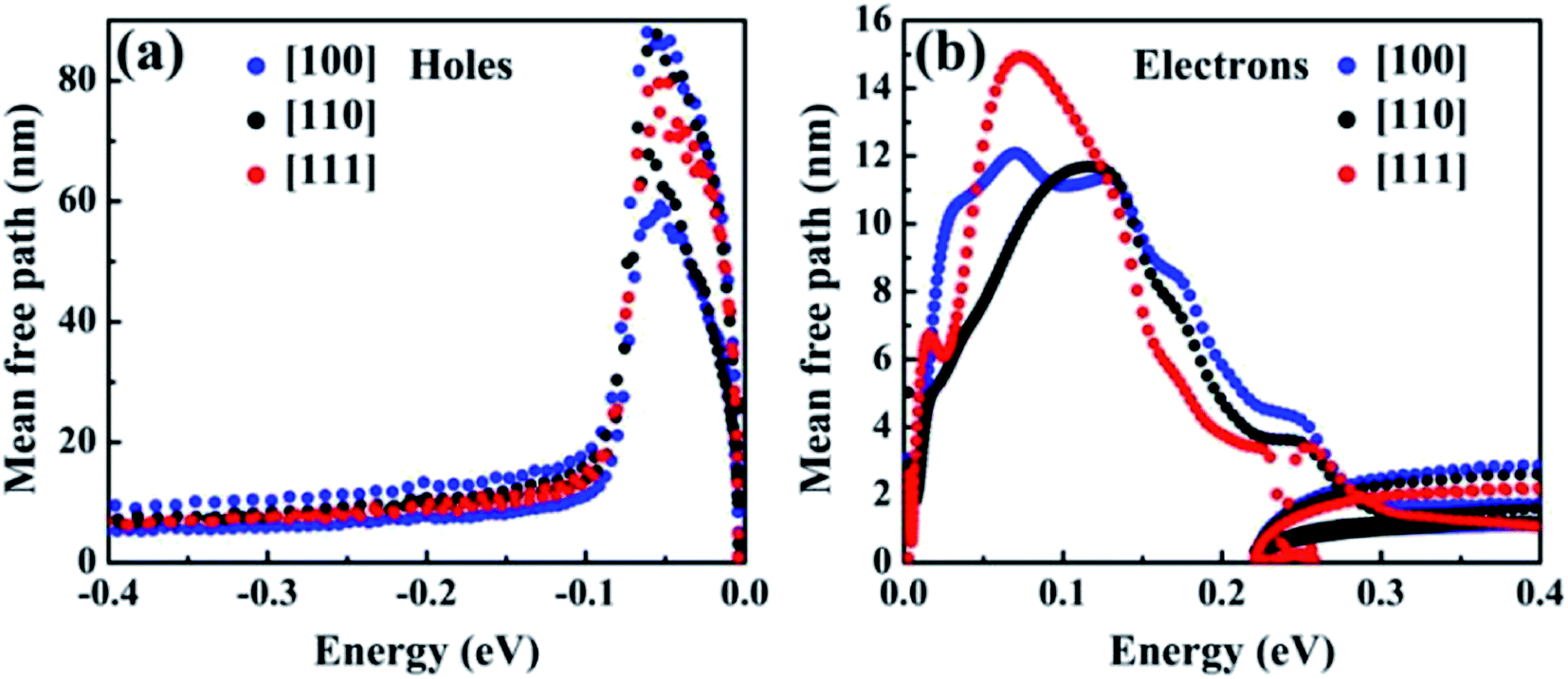 | ||
| Fig. 5 Mean free paths for hot holes (a) and hot electrons (b) in T-carbon along the three direction [100], [110], and [111]. The VBM and the CBM is set to zero in the energy axis. | ||
The e–ph coupling matrix elements determine the electron transfer probability between different states.47,48 We plot e–ph coupling matrix elements |g| of hot carriers at phonon dispersion with each branch along high-symmetry paths in Fig. 6(a) and (b), respectively. We find that |g| with LO modes at Γ point for holes are close to electrons and the strongest coupling comes from the LO phonon mode. As it can be easily seen from Fig. 6, the 18th |g| dispersion band and the corresponding highest TO phonon mode along ΓX are greater for holes than for electrons, while e–ph interactions are indeed weak among other phonon modes, especially acoustic phonons. Owing to the |g| depends strongly on the properties of the electronic orbitals,5 it is determined that the weak e–ph interactions is caused by non-bonding orbitals.49
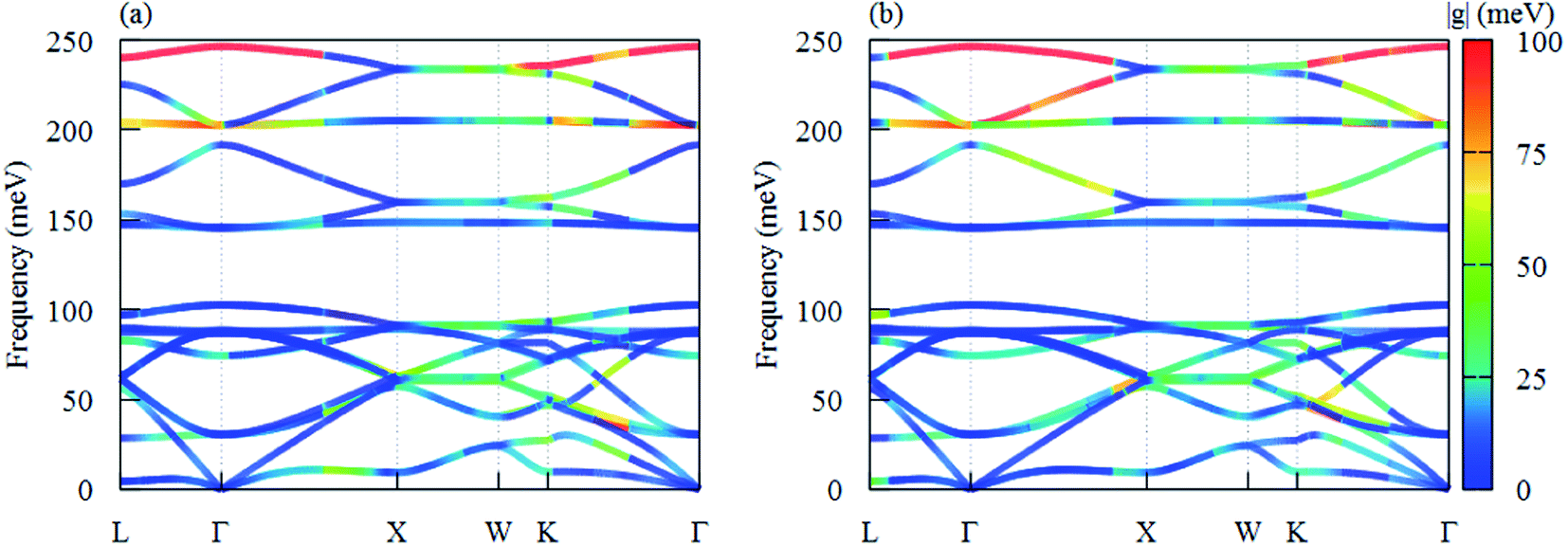 | ||
| Fig. 6 The phonon dispersion of T-carbon with color-mapped e–ph coupling matrix elements |gmk+qnk,qp| of (a) CBM and (b) VBM electron with different phonon branches in the first BZ. | ||
After obtaining the electron–phonon induced scattering, we here reveal the optical absorption process including excitonic effects in T-carbon. The electron–phonon scattering dominate the hot carrier lifetime, while the electron–hole interactions would affect the exciton lifetime. Since density functional theory deals with the ground state problem, we choose GW combined with Bethe–Salpeter equation formalism to deal with the excited state problem. The band gap calculated by one-shot G0W0 is about 3.4 eV, which is significantly higher than the band gap calculated by LDA (∼1.9 eV), the G0W0 band structures can be found in Fig. S1.† Compared to the independent-particle (IP) results with GW correction, the optical absorption spectrum has a red shift. The first absorption peak locates at 3.39 eV, corresponding to the first bright exciton denoted by A from the transition between the first valence band and the first conduction band around Γ point, as shown in Fig. 7. Below the bright exciton A, there are four dark excitons with the degenerate two locating at 3.23 eV, shown as D1 in Fig. 7. The first doubly degenerate dark excitons originate from the transitions between VBM/VBM-1 and CBM. The relative energy position of the dark excitons and the bright ones plays a key role for the photophysical properties of semiconductors.50 Here we evaluated the radiative lifetimes of the first dark and the first bright excitons of T-carbon at 300 K, as described by Bernardi and coworkers.51 Our computed results show that the lifetime of the first dark exciton is 3.4 s while it is 2.8 ns for the first bright exciton. The longer exciton lifetime of the dark exciton would trap much exciton population and further lower the photoluminescence quantum yield in T-carbon. One should note here we neglect the phonon-exciton related optical absorption here, which needs more computational resources and are beyond the current work.
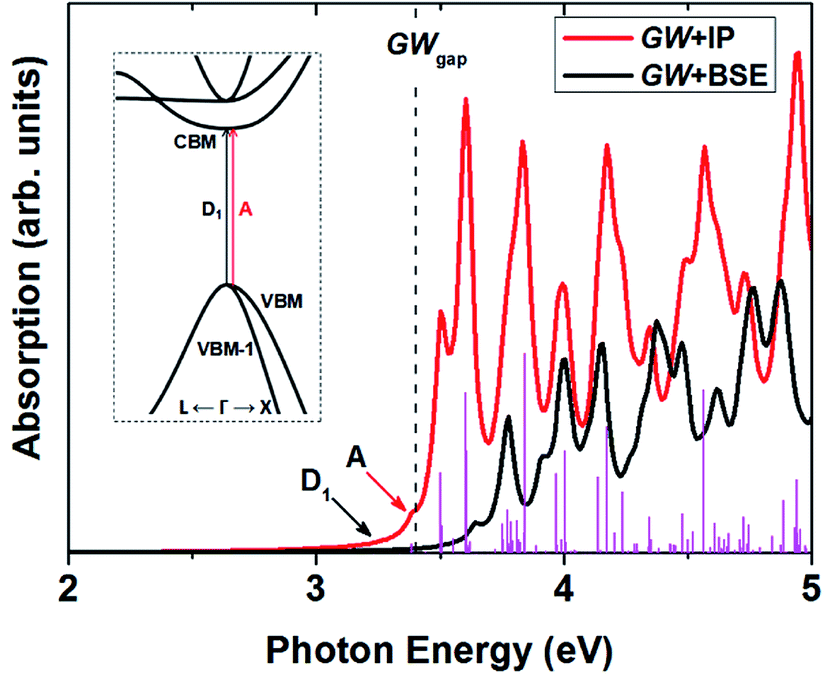 | ||
| Fig. 7 The optical absorption of T-carbon including excitonic effects. The first dark and the first bright exciton are denoted as D1 and A, respectively. The vertical lines are oscillator strength, and the inset is the absorption process of D1 and A. | ||
4. Conclusion
In summary, the electron–phonon scattering and excitonic effects of T-carbon is investigated by performing first-principles calculations. Our results indicate that the e–ph scattering rates are strongly dependent on and the electron energy. Particularly, the TO modes dominate the scatterings throughout the valence band energy region and the intervalley TO phonons dominate the scatterings of electrons above the energy of EL. The hot carrier relaxation times are up to ∼0.5 ps and ∼0.25 ps around the VBM and CBM, respectively. The mean free paths of hot holes are as high as 80 nm while only 15 nm for hot electrons, resulting in different hot carriers extraction ranges in T-carbon. We also demonstrate that there exist lowest energy dark excitons below the bright excitons, and thus lower the photoluminescence quantum yield in T-carbon.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the National Natural Science Foundation of China (Grant No. 11804173, 11547004, 11764031), and also acknowledge the computational resources at the IMU.References
- F. Bloch, Z. Phys., 1928, 52, 555 CrossRef CAS.
- M. Bernardi, Eur. Phys. J. B, 2016, 89, 239 CrossRef.
- F. Giustino, Rev. Mod. Phys., 2017, 89, 015003 CrossRef.
- T.-H. Liu, J. W. Zhou, B. L. Liao, D. J. Singh and G. Chen, Phys. Rev. B, 2017, 95, 075206 CrossRef.
- F. Meng, J. Ma, J. He and W. Li, Phys. Rev. B, 2019, 99, 045201 CrossRef CAS.
- M. D. Tillin and J. R. Sambles, J. Phys.: Condens. Matter, 1990, 2, 7055–7059 CrossRef CAS.
- J. Noffsinger, E. Kioupakis, C. G. Van de Walle, S. G. Louie and M. L. Cohen, Phys. Rev. Lett., 2012, 108, 167402 CrossRef PubMed.
- B. Liao, B. Qiu, J. Zhou, S. Huberman, K. Esfarjani and G. Chen, Phys. Rev. Lett., 2015, 114, 115901 CrossRef PubMed.
- B. Fu, G. Tang and Y. Li, Phys. Chem. Chem. Phys., 2017, 19, 28517–28526 RSC.
- A. H. Castro Neto and F. Guinea, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 045404 CrossRef.
- P. Sen, Chem. Phys. Lett., 2006, 428, 430–435 CrossRef CAS.
- S. Kivelson and D. Hone, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 4833–4835 CrossRef.
- J. Shah, A. Pinczuk, A. C. Gossard and W. Wiegmann, Phys. Rev. Lett., 1985, 54, 2045–2048 CrossRef CAS PubMed.
- G. Conibeer, et al., Sol. Energy Mater. Sol. Cells, 2009, 93, 713–719 CrossRef CAS.
- S. Mukherjee, et al., Nano Lett., 2013, 13, 240–247 CrossRef CAS PubMed.
- H. Haug and A.-P. Jauho, Quantum Kinetics in Transport and Optics of Semiconductors, Springer-Verlag, Berlin, Heidelberg, 2008 Search PubMed.
- A. Hirsch, Nat. Mater., 2010, 9, 868–871 CrossRef CAS PubMed.
- J. B. Howard, J. T. McKinnon, Y. Makarovsky, A. L. Lafleur and M. E. Johnson, Nature, 1991, 352, 139–141 CrossRef CAS PubMed.
- R. H. Baughman, A. A. Zakhidov and W. A. De Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
- Y. Liu, G. Wang, Q. Huang, L. Guo and X. Chen, Phys. Rev. Lett., 2012, 108, 225505 CrossRef PubMed.
- E. H. L. Falcao and F. Wudl, J. Chem. Technol. Biotechnol., 2007, 82, 524–531 CrossRef CAS.
- K. Xu, et al., Carbon, 2020, 157, 270–276 CrossRef CAS.
- X.-L. Sheng, Q. B. Yan, F. Ye, Q. R. Zheng and G. Su, Phys. Rev. Lett., 2011, 106, 155703 CrossRef PubMed.
- L. Bai, P.-P. Sun, B. Liu, Z. Liu and K. Zhou, Carbon, 2018, 138, 357–362 CrossRef CAS.
- J. Zhang, et al., Nat. Commun., 2017, 8, 683 CrossRef PubMed.
- P.-P. Sun, L. Bai, D. R. Kripalani and K. Zhou, npj Comput. Mater., 2019, 5, 9 CrossRef.
- G. Qin, K. R. Hao, Q. B. Yan, M. Hu and G. Su, Nanoscale, 2019, 11, 5798–5806 RSC.
- H. Ren, H. Chu, Z. Li, T. Yue and Z. Hu, Chem. Phys., 2019, 518, 69–73 CrossRef CAS.
- H. Alborznia, M. Naseri and N. Fatahi, Optik, 2019, 180, 125–133 CrossRef CAS.
- M. Bernardi, D. Vigil-Fowler, J. Lischner, J. B. Neaton and S. G. Louie, Phys. Rev. Lett., 2014, 112, 257402 CrossRef PubMed.
- M. Bernardi, D. Vigil-Fowler, C. S. Ong, J. B. Neaton and S. G. Louie, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 5291–5296 CrossRef CAS PubMed.
- M. Bernardi, J. Mustafa, J. B. Neaton and S. G. Louie, Nat. Commun., 2015, 6, 7044 CrossRef CAS PubMed.
- H. Tanimura, et al., Phys. Rev. B, 2016, 93, 161203 CrossRef.
- J.-J. Zhou and M. Bernardi, Phys. Rev. B, 2016, 94, 201201 CrossRef.
- R. M. Martin, Electronic Structure: Basic Theory and Practical Methods, Cambridge Univ. Press, Cambridge, United Kingdom, 2008 Search PubMed.
- M. S. Hybertsen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 5390 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, et al., J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli and M. Cococcioni, et al., J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- J. Noffsinger, F. Giustino, B. D. Malone, C.-H. Park, S. G. Louie and M. L. Cohen, Comput. Phys. Commun., 2010, 181, 2140–2148 CrossRef CAS.
- S. Poncé, E. R. Margine, C. Verdi and F. Giustino, Comput. Phys. Commun., 2016, 209, 116–133 CrossRef.
- E. E. Salpeter and H. A. Bethe, Phys. Rev., 1951, 84, 1232–1242 CrossRef.
- M. Rohlfing and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 4927–4944 CrossRef CAS.
- F. Bruneval and X. Gonze, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 085125 CrossRef.
- A. Marini, C. Hogan, M. Grüning and D. Varsano, Comput. Phys. Commun., 2009, 180, 1392–1403 CrossRef CAS.
- D. Sangalli, et al., J. Phys.: Condens. Matter, 2019, 31, 325902 CrossRef CAS PubMed.
- F. Giustino, M. L. Cohen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 165108 CrossRef.
- K. M. Borysenko, et al., Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 161402 CrossRef.
- J. Xi, D. Wang and Z. Shuai, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2015, 5, 215–227 CAS.
- J. Zhou, et al., Nat. Commun., 2018, 9, 1721 CrossRef PubMed.
- S. Kilina, E. Badaeva, A. Piryatinski, S. Tretiak, A. Saxena and A. R. Bishop, Phys. Chem. Chem. Phys., 2009, 11, 4113–4123 RSC.
- H.-Y. Chen, V. A. Jhalani, M. Palummo and M. Bernardi, Phys. Rev. B, 2019, 100, 075135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02343d |
| This journal is © The Royal Society of Chemistry 2020 |