
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogenation of furfural by noble metal-free nickel modified tungsten carbide catalysts†
Patrick Bretzler ab,
Michael Huberab,
Simon Nicklab and
Klaus Köhler*ab
ab,
Michael Huberab,
Simon Nicklab and
Klaus Köhler*ab
aDepartment of Chemistry, Inorganic Chemistry, Technical University of Munich, Lichtenbergstrasse 4, 85747 Garching, Germany. E-mail: klaus.koehler@tum.de; Tel: +49 89 289 13233
bCatalysis Research Center, Technical University of Munich, Ernst-Otto-Fischer-Strasse 1, 85747 Garching, Germany
First published on 22nd July 2020
Abstract
Nickel–tungsten carbide catalysts convert furfural to high value products in a liquid phase catalytic reaction. The product distribution depends on the solvent and the Ni–W-ratio of the catalyst. In isopropyl alcohol a combination of Ni and WxC enables the opening of the furan ring to yield 1,2-pentanediol. Nickel accelerates the tungsten oxide reduction in the tungsten carbide catalyst synthesis and facilitates the carbon insertion. Nickel modified tungsten carbide is a promising, noble metal-free catalyst system for the upgrading of furfural based renewable resources. Its preparation is facilitated compared to unmodified tungsten carbide catalysts.
Introduction
The valorization of renewable resource streams has become one of the most important ways to face declining availability of non-renewable resources. The production of high value chemicals for the chemical industry from biomass instead of petroleum-based resources is becoming increasingly important. Furfural is one of the platform chemicals available from biomass which is suitable for upgrading to high value products.1,2 Furfural is being produced on an industrial scale by the acidic hydrolysis of pentose sugars, especially arabinose and xylose. The structure of furfural lends itself to the production of several desirable products. Most of the products are formed via the intermediate furfuryl alcohol (FFA). From there three reaction pathways open up. The hydrodeoxygenation to 2-methylfuran (2-MF) and further to 2-methyltetrahydrofuran (2-MTHF), the hydrogenation to tetrahydrofurfuryl alcohol (THFA) and the ring opening to 1,5-pentanediol (1,5-PeD) and 1,2-pentanediol (1,2-PeD).3,4 A simplified network of these reaction products is depicted in Scheme 1. Several other products can be obtained from furfural as well.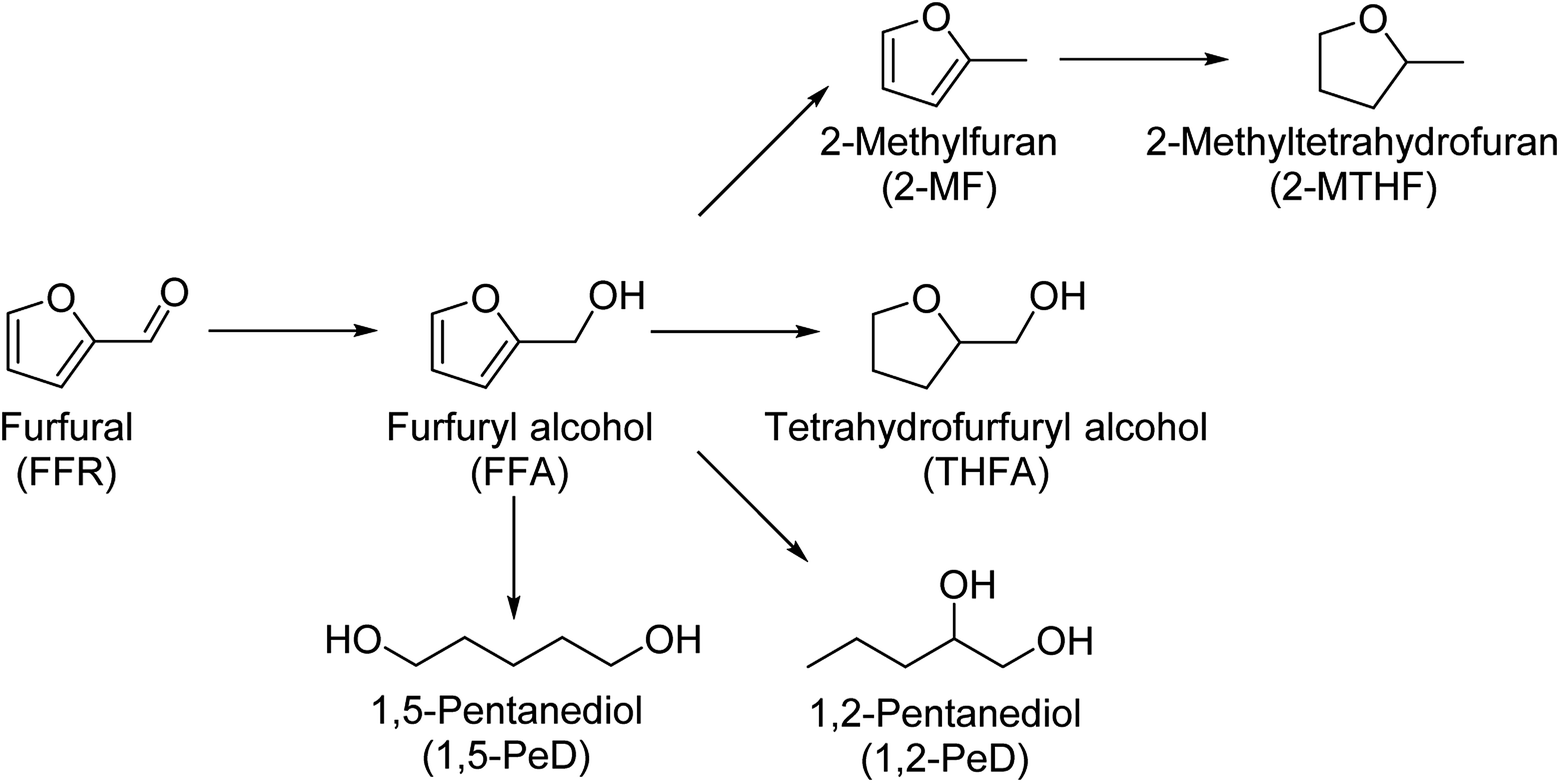 | ||
| Scheme 1 Reaction network of possible hydrogenation products starting from furfural.3,4 | ||
1,2-PeD can be used in the production of polyester,5,6 disinfectants,6 cosmetics7 and printing inks.8 Additionally, 1,2-PeD possesses interesting properties as an enhancer for the skin permeability of certain drugs.9,10 The conventional production of 1,2-PeD from non-renewable petroleum resources is performed in a multistep reaction. Pent-1-ene is oxidized, e.g. by the use of H2O2, to 1,2-epoxypentane which is further hydrolyzed to 1,2-PeD.5,11
While furfural has moved in to the limelight of research on biomass derived resource streams, only little research has been done on its ring opening to produce pentanediol and the majority of said research focuses on the production of 1,5-PeD.12 Most of the catalysts studied for the production of 1,2-PeD are based on noble metals like Ru,6 Rh,13–18 Pd19,20 and Pt.3,4,21–23 Most notable are two platinum-based catalysts. Mizugaki et al.4 prepared platinum nanoparticles supported on hydrotalcite and reached a selectivity of 73% to 1,2-PeD. Koch et al.23 used PtO2 supported on γ-Al2O3 and obtained a selectivity to 1,2-PeD of up to 80%.
Non-noble metal based catalysts suffer from low yield or limited industrial applicability due to the inherent toxicity of some of the catalysts components, especially Cr and Co. Adkins et al.24,25 prepared the first promising catalysts based on copper chromite already in 1931. More recent works have proposed the use of cobalt based catalysts for the ring opening of furfural.12,26,27 The product distribution of these catalysts favors 1,5-PeD over 1,2-PeD which was only obtained with relatively low yields. The most promising noble metal free catalysts are based on either Ni or Cu. The Ni catalysts typically show a preference to 1,5-PeD,28 while copper-based catalysts prepared by Liu et al.29,30 were able to reach a selectivity to 1,2-PeD of up to 51%.
Tungsten carbide has gained attention as a possible alternative to expensive noble metal hydrogenation catalysts. This interstitial alloy of tungsten and carbon shows similar catalytic properties as platinum in a number of reactions.31 The low price of tungsten and its widespread availability would make it a desirable catalyst material, if its performance can be tailored to be competitive to more established catalysts. Tungsten carbide has been successfully used in a number of reactions that aim to upgrade biomass-derived resources. Hollak et al.32 studied the deoxygenation of oleic acid. Bitter et al.33–35 converted several fatty acids and guaiacol over tungsten carbide and Guo et al.36 cracked the C–O bonds in lignin feedstock. The activity of tungsten carbide based catalysts can be improved by the addition of nickel. Both cellulose and sugars were successfully converted to ethylene glycol over nickel-modified tungsten carbide catalysts.37,38
Huang et al.39 investigated the hydrodeoxygenation of the furfural derivative 5-(hydroxymethyl)furfural to 2,5-dimethylfuran using a Ni-modified tungsten carbide catalyst. In the aprotic solvent tetrahydrofuran (THF) a high yield of 96% could be reached, while no ring opening was observed. The ring-opening to pentanediol is typically performed in protic solvents, like alcohol, or even with the addition of water.4,40 It stands to reason that a change in solvent could open up different reaction routes and products for the tungsten carbide catalyst system.
The tungsten carbide synthesis is performed at high temperatures, exceeding 700 °C. The carbide catalysts properties are strongly dependent on the synthesis conditions.41–43 The conversion of tungsten(VI) oxide to tungsten carbide has been studied intensively but the influence of a second metal on this conversion is not fully understood yet. It appears that nickel activates the methane decomposition and facilitates the carbide formation.44,45 At the same time, any carbon support present in the catalyst precursor is subject to methanation, resulting in extensive sintering.46
This work investigates the influence of different Ni-loadings on the isothermal tungsten carbide formation, illuminating the processes that occur during the high temperature carbide synthesis and the crystalline phase composition of the final catalysts. The catalysts are tested for their performance in the hydrogenation of furfural, comparing them to solely WxC, Ni or Ni17W3 based ones. A special focus is put on the change in product distribution, especially the yield of 1,2-pentanediol, dependent on solvent and Ni–W-ratio.
Experimental
Materials
Chemicals were obtained from Sigma-Aldrich Germany and used without further purification. The chemical's purities are as follows: ammonium metatungstate hydrate 99%, nickel nitrate hexahydrate 99%, furfural 99%, isopropyl alcohol for analysis, 1,4-dioxane 100%. Fumed silica Aerosil 200 was provided by Evonik. The gases are from Westfalen Germany with a purity of ≥99.999% (He, Ar, H2) or ≥99.5% (CH4). Argon was used as inert gas in all experiments, except for the temperature-programmed carburization in which He was used.Precursor
Silica was agglomerated, sieved to a particle size of 100–300 μm, dried at 200 °C for 1 h and calcined at 700 °C for 8 h. The agglomerated silica particles were loaded with 30 mol% (50.0 wt%) tungsten in an incipient wetness impregnation method using a 0.55 M aqueous solution of ammonium metatungstate. This material was dried for 2 h at 60 °C, for 6 h at 110 °C and calcined at 450 °C for 6 h. Ni was loaded on the WO3/SiO2 particles in a subsequent or on SiO2 in a direct incipient wetness impregnation using a nickel nitrate solution. A mixed Ni–W/SiO2 precursor was obtained in an incipient wetness co-impregnation using a mixed solution of nickel nitrate and ammonium metatungstate in a metal to metal molar ration of 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (Ni:W). The impregnation was followed by drying and calcination. Ni-loadings were between 0.5 and 9.0 wt% and 10.0 wt% for NiO/SiO2. For the Ni–W/SiO2-precursor the combined metal loading was 10.0 wt%.
:3 (Ni:W). The impregnation was followed by drying and calcination. Ni-loadings were between 0.5 and 9.0 wt% and 10.0 wt% for NiO/SiO2. For the Ni–W/SiO2-precursor the combined metal loading was 10.0 wt%.
Catalyst synthesis and activation
WxC/SiO2 and Ni/WxC/SiO2 catalysts were synthesized in a horizontal tube furnace under isothermal conditions. The sample was heated under inert gas flow to 700 °C before switching to reactive gas (20% CH4 in H2). The effluent gas stream was monitored using a ThermoStar mass spectrometer by Pfeiffer Vacuum. The reduction was determined to be complete once the CO-concentration dropped close to its original value. Afterwards, the catalyst was treated at synthesis temperature with pure H2 to remove any excess polymeric carbon from its surface.Ni/SiO2 was activated in a horizontal tube furnace by heating to 450 °C under inert gas and reduced under 50% H2 in Ar for 30 min and 100% H2 for additional 30 min. Ni17W3/SiO2 was prepared by heating the sample to 700 °C under inert gas and subsequent reduction in hydrogen. The reduction was deemed complete once the H2O evolution stopped. All catalysts were cooled under inert gas and transferred to a glove box without contact to air.
XRD
The crystalline phases were examined using an Empyrean X-ray diffractometer by Malvern Panalytical operating with Cu Kα radiation. Samples were measured after passivation in air in reflection geometry in the range from 5–70° 2θ.BET
The overall surface area of the catalysts was determined by isothermal nitrogen adsorption at −196 °C according to Brunauer–Emmet–Teller on a Quantachrome NovaTouch LX4. Before the measurement samples were degassed in vacuo at 350 °C for 4 h.Furfural hydrogenation
Catalysis reactions were performed in a 300 mL Parr mini reactor. The reaction mixture consisted of 3 mL furfural, 100 mL isopropyl alcohol and 1.5 mL of 1,4-dioxane as internal standard. The reaction mixture was flushed with Ar before inert addition of 0.50 g catalyst. The reactor was closed and pressurized with H2 to 65 bar. The reactor was heated to 200 °C before stirring was turned on. Aliquots of the liquid phase where taken through a dip tube several times during the reaction. Samples were analyzed with an Agilent 6980 gas chromatograph from Hewlett Packard equipped with an automated injector and FID. Samples were analyzed over a HP-1 MS column with the following temperature-program – holding at 50 °C for 3 min and heating to 270 °C with 30 K min−1. Retention times and response factors relative to the internal standard were determined using pure substances. The conversion and yield are calculated according to equations in the ESI.†Results and discussion
Synthesis of Ni-modified tungsten carbide catalysts and the role of nickel in the WxC synthesis
For tungsten carbide catalysts, the processes occurring during the carbide synthesis and in turn the properties of the final catalyst are highly dependent on the synthesis conditions. The addition of nickel is a substantial interference on the synthesis of the carbide-catalyst. Therefore, the reduction and carbon insertion occurring during the synthesis is explored in the first subsection. Silica is added to the catalyst to provide a non-reducible support and to stabilize a high tungsten carbide surface area. | ||
| Fig. 1 CO in the effluent gas stream during isothermal WxC synthesis in a horizontal tube furnace at 700 °C in reactive gas (20% CH4 in H2). The reaction is starting from WO3/SiO2 with different mass-loadings of Ni; no Ni: blue, dashed line; increasing amounts of Ni: darker shades of orange, solid lines. | ||
Without Ni the CO-evolution at 700 °C only starts after an initiation period of 25 min. The CO-concentration in the effluent gas stream slowly increases before gradually declining after passing through a maximum after 100 min and dropping back to the baseline after 130 min. The end of the CO-formation indicates the completion of the reduction and possibly the carbon insertion.
The addition of Ni cuts the duration of the initiation period in half and results in a steep increase of the CO-concentration in the exhaust gas stream. The CO-formation quickly passes through a maximum and gradually declines again. The CO-signal reaches the baseline typically after 75 min. The addition of Ni halves the time needed to complete the reduction, with only little influence of the total Ni-loading on this behavior. Only for the lowest Ni-loading of 0.5 wt% is this behavior slightly different. Here the CO-evolution is split into two maxima, a first one at 11 min and a second one at 28 min. Additionally, the complete reduction time is slightly longer with 95 min compared to 70 min for all higher Ni-loadings. The initiation period is shorter for low Ni-loadings and becomes longer when more Ni is added. It is possible that Ni has to be completely reduced to its metallic state before it can activate CH4, resulting in CO-formation. This Ni reduction takes more time when a larger amount of Ni is present.
The overall surface area of the catalysts, as measured by BET, gradually decreases from 97 m2 g−1 for no addition of Ni to 70 m2 g−1 for the addition of 10 wt% Ni. More details are shown in Table S1.† Some of this loss in surface area can be attributed to the second calcination step during the catalyst preparation when Ni is added. A second effect is likely the blocking of pores with increasing nickel loadings.
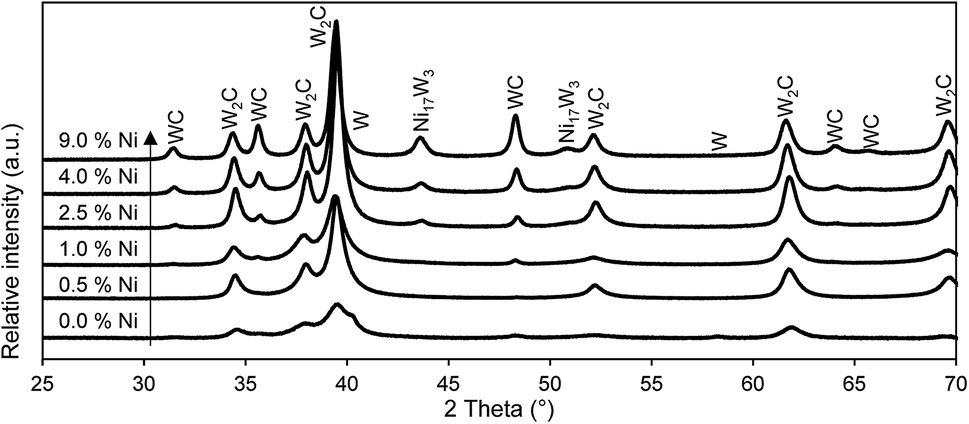 | ||
| Fig. 2 X-ray diffractograms of tungsten carbide catalysts with different Ni-loadings. The Ni-loading is indicated as weight% of the WO3/SiO2 precursor. Catalysts are prepared in an isothermal reaction at 700 °C. Reference patterns from ICDD database, WC: 00-025-1047,47 W2C: 00-35-0776,48 W: 00-001-120349 and Ni17W3: 03-065-4828.50 | ||
In summary, the addition of nickel to the WO3/SiO2 precursor alters the carbide formation in several ways. Nickel activates hydrogen and methane, resulting in altered reduction steps. Methane plays a more dominant role during the reduction. The isothermal reduction is substantially faster, even for the addition of only a small amount of nickel. The carbon insertion is assisted and the composition of the crystalline phases changes towards a higher amount of WC. Nickel itself forms a Ni17W3-alloy with tungsten. The synthesis of tungsten carbide catalysts is facilitated, forming the carbide more readily.
Catalytic conversion of furfural over Ni-modified tungsten carbide catalysts
The carbide synthesis resulted in tungsten carbide catalysts with Ni-loadings from 0.0 wt% to 9.0 wt%. These catalysts are investigated for the hydrogenation of furfural at 200 °C under 65 bar H2-pressure. The catalytic activity and the product distribution are the focus of this work. Both aspects are heavily influenced by the solvent and the Ni-loading, which are at the center of the next subsections.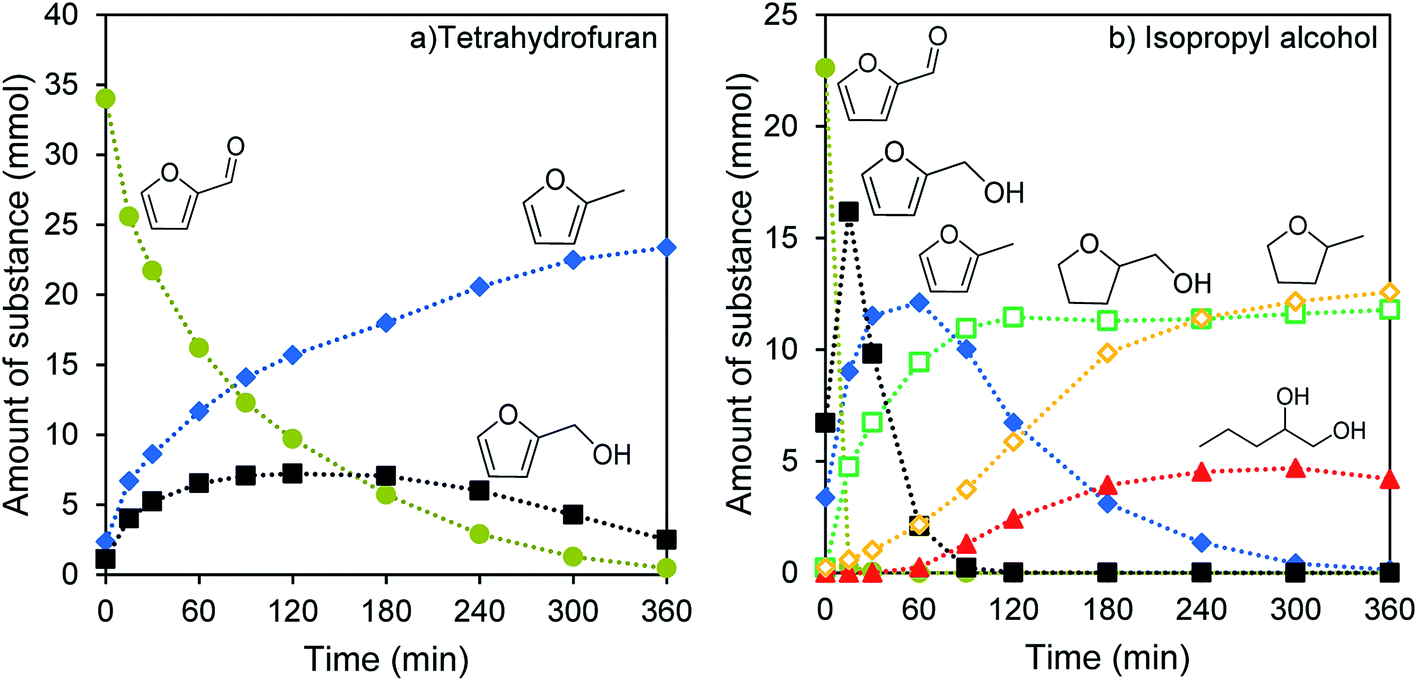 | ||
| Fig. 3 Reaction progress during the catalytic conversion of furfural in (a) tetrahydrofuran and (b) isopropyl alcohol as solvent. The reaction proceeds at 200 °C under 65 bar H2, with 0.50 g 4%-Ni/WxC/SiO2 and 3 mL furfural. | ||
Using isopropyl alcohol as a solvent results in a different product distribution and overall faster conversion of furfural. The reaction starts from furfural over furfuryl alcohol, where three competing reaction pathways open up. Furfural is hydrogenated much faster than in THF, with complete conversion to furfuryl alcohol after only 15 min instead of after 6 h. This secondary product is converted much faster too. All furfuryl alcohol is reacted after 90 min. Initially a mixture of three reaction products is formed: 2-methylfuran (2-MF), which is further hydrogenated to 2-methyltetrahydrofuran (2-MTHF), tetrahydrofurfuryl alcohol (THFA), and 1,2-pentanediol (1,2-PeD). In isopropyl alcohol, the alcohol functionality can be preserved to some extent and the opening of the furan ring is possible (Fig. 3b). The amount of unidentified products is similarly high for both solvents, with 23.8% for THF and 21.9% for isopropyl alcohol.
The two solvents differ in their polarity, their ability to form adducts with the reactants and intermediary products, and their hydrogen solubility. Isopropyl alcohol can form an acetal with furfural and ethers with all alcohol group bearing intermediary products. Together with the higher polarity of the protic solvent, this results in a preferential transformation to alcohol group containing products. The hydrogen solubility is much higher in isopropyl alcohol,51,52 explaining the lower reaction rate and incomplete hydrogenation of 2-MF in THF.
Unmodified tungsten carbide catalysts show poor catalytic activity and only reach full conversion after 14–24 h at 200 °C. Pure nickel catalysts on the other hand possess a very high catalytic activity, converting all furfuryl alcohol in only 30 min. Nickel modified tungsten carbide catalysts fall in between those two extremes, depending on their Ni-loading. A low Ni-loading of 0.5 wt% significantly improves the catalytic activity but still not all furfuryl alcohol is converted after 6 h. Further increasing the Ni-loading lowers the time until full conversion is reached, approaching the activity of pure Ni-catalysts at high Ni-loadings as shown in Fig. 4. The use of WxC does not improve the conversion rate compared to catalysts solely based on Ni but in the Ni-WxC-system activity can be traded for a change in product distribution, as will be shown in the next subsection.
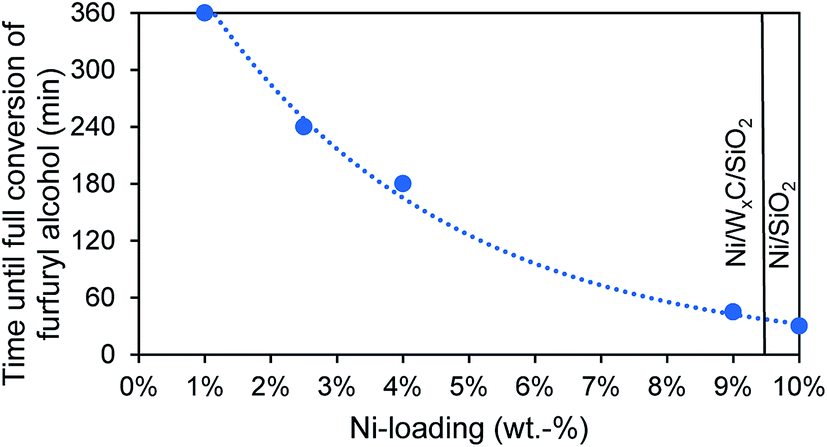 | ||
| Fig. 4 Time needed until full conversion of the intermediary product furfuryl alcohol for catalysts with different Ni-loadings. The catalyst with 10 wt% Ni consists only of Ni without a tungsten component. The reaction proceeds at 200 °C, 65 bar H2 with 0.50 g catalyst and 3 mL furfural in isopropyl alcohol as solvent. | ||
| Entry | Catalyst | Reaction time (h) | Yield% | ||||
|---|---|---|---|---|---|---|---|
| Hydrodeoxygenation | Hydrogenation | Ring opening | |||||
| 2-MF | 2-MTHF | THFA | 1,2-PeD | 1,5-PeD | |||
| 1 | WxC/SiO2 | 24 | 45 | — | — | — | 13 |
| 2 | 0.5%-Ni/WxC/SiO2 | 6 | 36 | 1 | 4 | 3 | — |
| 3 | 1.0%-Ni/WxC/SiO2 | 6 | 26 | 15 | 14 | 9 | — |
| 4 | 2.5%-Ni/WxC/SiO2 | 6 | 11 | 25 | 20 | 21 | — |
| 5 | 4.0%-Ni/WxC/SiO2 | 6 | — | 35 | 32 | 12 | — |
| 6 | 9.0%-Ni/WxC/SiO2 | 6 | 1 | 36 | 45 | 4 | — |
| 7 | 10.0%-Ni/SiO2 | 6 | — | 29 | 46 | — | — |
| 8 | 10.0%-Ni17W3/SiO2 | 6 | 10 | 39 | 35 | 2 | — |
Modifying the tungsten carbide catalyst with Ni changes the product distribution towards that of the Ni/SiO2-catalyst. The products of the hydrodeoxygenation pathway are more thoroughly hydrogenated to 2-MTHF with a decreasing total amount of substance following this pathway. THFA becomes more prevalent with increasing amounts of nickel. This change from the tungsten carbide to the nickel based system is very gradual and follows the changing ratio of Ni and W.
Interestingly, for medium Ni-loadings the ring opening of furfuryl alcohol to 1,2-PeD is observed. This product only appears for catalysts with a combination of nickel and tungsten carbide. The highest selectivity of 21% is obtained for 2.5 wt% Ni with a lower selectivity at either lower or higher Ni-loadings. The addition of W to Ni and the formation of Ni17W3 alone does only results in very miniscule amounts of this ring-opening product.
The combination of Ni17W3 and WxC gives rise to a different reaction pathway, which allows the opening of the furan ring while keeping both oxygen functionalities of the molecule. This reaction appears to be dependent on the simultaneous presence of tungsten carbide and nickel. The catalytic behavior of both tungsten carbide and nickel, for either the hydrodeoxygenation route or the formation of THFA, are mostly unaffected by this additional reactivity.
The furfural conversion is slower in the second catalytic run. Furfural is only consumed after 2 h instead of after 45 min. The intermediary furfuryl alcohol can be detected for 11 h instead of 5 h. The time needed until full conversion is doubled during the second catalysis test run (Fig. 5).
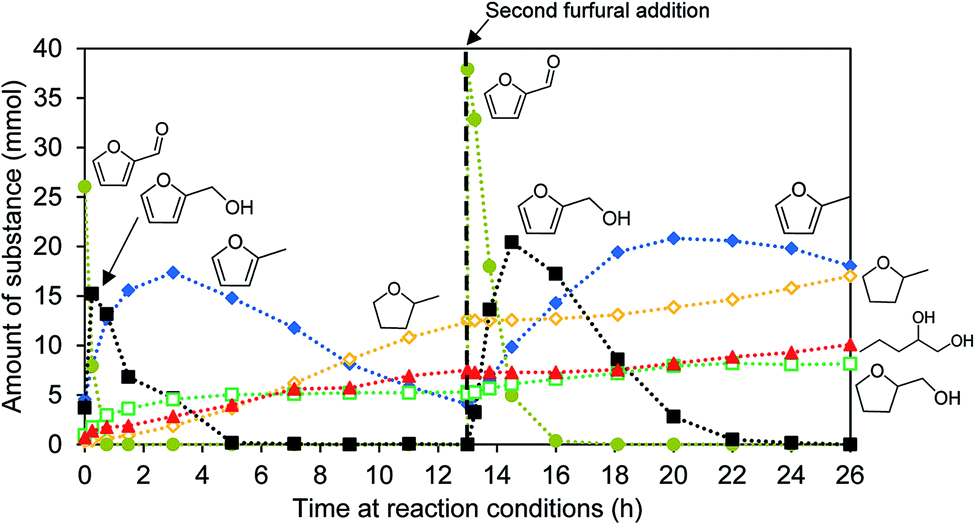 | ||
| Fig. 5 Reaction progress during catalytic conversion of furfural with 2.5 wt% Ni/WxC/SiO2. The reaction proceeds at 200 °C under 65 bar H2, with 0.5 g catalyst in isopropyl alcohol as solvent. Initially 3 mL furfural are added and after 13 h of reaction time the reactor is cooled down and depressurized to add further 3 mL of furfural before continuing the reaction. | ||
Not only the conversion rate changes, but the product distribution is modified as well (Table 2). The hydrodeoxygenation pathway is still prominent but the hydrogenation of the unsaturated bonds in the furan ring is hindered, resulting in more 2-MF. The hydrogenation of the double bonds and the ring opening without loss of the oxygen functionality is inhibited, producing less THFA and 1,2-PeD. The amount of unidentified products increases from 26.1% to 36.4%.
| Entry | Run | Yield% | ||||
|---|---|---|---|---|---|---|
| Hydrodeoxygenation | Hydrogenation | Ring opening | ||||
| 2-MF | 2-MTHF | THFA | 1,2-PeD | 1,5-PeD | ||
| 1 | First | 10.2 | 31.3 | 13.4 | 19.0 | — |
| 2 | Second | 31.1 | 15.4 | 7.9 | 9.2 | — |
The catalyst undergoes changes during the first catalysis run or the addition of the new substrate. These changes have an effect on the product distribution and conversion rate of the second run. It appears that several different active centers are available on the catalyst, which deactivate to a varying extent. The hydrodeoxygenation is less affected than the hydrogenation and ring opening. The reactions that are catalyzed by the Ni-component are more affected than the ones catalyzed by WxC. The X-ray diffraction pattern does not change during the catalysis test (Fig. S2†), therefore it is unlikely that one active component is lost during the reaction. A possible mechanism for this deactivation is the deposition of polymerized furfural derivatives and coke precursors blocking certain active sites.
Hypothesis on the nature of the active site for the 1,2-pentanediol formation
As the ring opening of furfural to produce 1,2-pentanediol belongs to the most interesting findings of this work, the possible nature of the active site responsible for this reaction is further discussed. The results obtained in this work can only give a first idea of a possible mechanism but to conclusively uncover the exact working of this catalyst more work will be needed. Only the mixture of nickel and tungsten carbide gives rise to the formation of 1,2-pentanediol. This valuable reaction product is obtained neither over tungsten carbide, nor over nickel catalysts on their own. The combination of the active materials Ni17W3 and WxC appears to enable the ring opening and the formation of this reaction product.Since the material is preferentially formed on a certain mixture of nickel and tungsten carbide, it can be expected that the active sites for the ring opening rely on a combination of both materials. The critical step for the determination of the product distribution is the reaction of the intermediary product furfuryl alcohol. Mizugaki et al.4 provided a possible reaction pathway of furfuryl alcohol to 1,2-pentanediol over Pt-nanoparticles supported on hydrotalcite, which places the active site at the boundary between platinum and support. In their proposed reaction pathway the alcohol group of FFA interacts with the basic surface sites of the hydrotalcite, orienting the furan ring towards the Pt-particle. The platinum then provides activated hydrogen and enables the ring opening at the C–O bond pointing away from the alcohol group.
A very similar reaction mechanism could be possible for Ni supported on WxC. In this case, the oxophilic tungsten carbide could attract the oxygen of the alcohol group and orient the furan ring towards the nickel-particle. The nickel then enables the actual ring opening. Scheme 2 depicts this process. Following this hypothetical reaction mechanism places the active site for the ring opening of furfural right at the boundary between WxC and Ni.
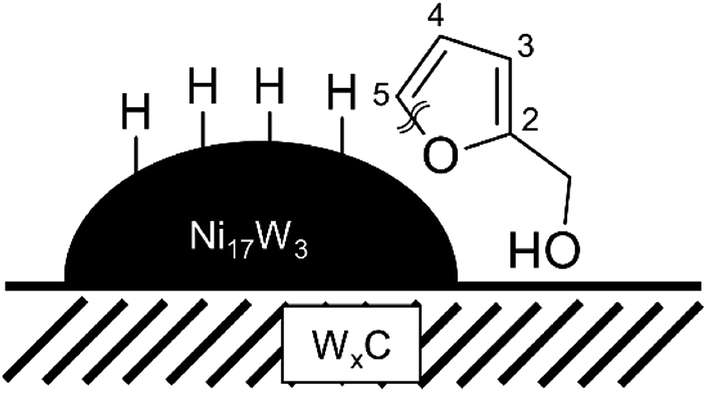 | ||
| Scheme 2 Possible orientation of furfuryl alcohol at the proposed active site for the ring opening, the boundary between Ni17W3 and WxC. Based on the proposed reaction path by Mizugaki et al.4 | ||
The reaction pathways occurring on solely Ni and WxC still dominate the overall product distribution for the tested catalysts. By improving the amount of active sites for the ring opening, the selectivity to 1,2-pentanediol could possibly be improved. At the same time, it might be possible to suppress selectively the hydrodeoxygenation and hydrogenation without ring opening by optimization of the synthesis conditions or a targeted deactivation of certain active sites. Investigations to improve the catalyst in this manner are already in progress.
Conclusion
The high temperature formation of the potential platinum substitute WxC is strongly influenced by the presence of Ni in the WO3-precursor. The added Ni activates the CH4 and H2 used in the carbide synthesis, facilitating the tungsten oxide reduction and carbon insertion. The time needed for complete reduction of WO3 is cut in half and the carbon insertion proceeds more completely, resulting in more WC and less W2C and W.The Ni–W-ratio and nature of the solvent play a crucial role in the formation of the high-value reaction product 1,2-pentanediol from furfural. No ring-opening occurs in THF, while in isopropyl alcohol, 1,2-pentanediol is formed.
The rate of conversion of furfural and furfuryl alcohol is strongly dependent on the Ni–W-ratio in the catalyst, with higher rates being achieved for high Ni-loadings. The combination of Ni and WxC gives rise to the formation of 1,2-pentanediol, which is fformed on neither a solely WxC- nor a Ni-based catalyst. By controlling the Ni–W-ratio the product distribution can be tailored to increase the yield of 1,2-pentanediol.
Future work must be focused to improve the selectivity and stability of the Ni/WxC-catalyst system. Nickel modified tungsten carbides provide a hopeful starting point for the development of non-noble metal catalysts for the upgrading of biomass derived furfural. Both problems of selectivity and stability can possibly be tackled by optimizing the precursor and tungsten carbide syntheses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. B. thanks the TUM Graduate School for financial support. We want to thank Hannah Augenstein for providing a nickel on silica catalyst.References
- N. S. Biradar, A. M. Hengne, S. N. Birajdar, P. S. Niphadkar, P. N. Joshi and C. V. Rode, ACS Sustainable Chem. Eng., 2014, 2, 272–281 CrossRef CAS.
- K. Yan, G. Wu, T. Lafleur and C. Jarvis, Renewable Sustainable Energy Rev., 2014, 38, 663–676 CrossRef CAS.
- W. Xu, H. Wang, X. Liu, J. Ren, Y. Wang and G. Lu, Chem. Commun., 2011, 47, 3924 RSC.
- T. Mizugaki, T. Yamakawa, Y. Nagatsu, Z. Maeno, T. Mitsudome, K. Jitsukawa and K. Kaneda, ACS Sustainable Chem. Eng., 2014, 2, 2243–2247 CrossRef CAS.
- R. Siegmeier, G. Prescher and H. Maurer, European Pat., 0,257,243, 1987.
- B. Zhang, Y. Zhu, G. Ding, H. Zheng and Y. Li, Green Chem., 2012, 14, 3402–3409 RSC.
- I. Agostini and S. Cupferman, US Pat., US 6,296,858 B1, 2001.
- K. Aoki and H. Mukai, Japan Pat., JP2012245721, 2012.
- L. Duracher, L. Blasco, J.-C. Hubaud, L. Vian and G. Marti-Mestres, Int. J. Pharm., 2009, 374, 39–45 CrossRef CAS PubMed.
- N. Li, W. Jia, Y. Zhang, M. C. Zhang, F. Tan and J. Zhang, AAPS PharmSciTech, 2014, 15, 354–363 CrossRef CAS PubMed.
- R. Siegmeier, G. Prescher, H. Maurer and G. Hering, US Pat., 4,605,795, 1986.
- T. P. Sulmonetti, B. Hu, S. Lee, P. K. Agrawal and C. W. Jones, ACS Sustainable Chem. Eng., 2017, 5, 8959–8969 CrossRef CAS.
- S. Liu, Y. Amada, M. Tamura, Y. Nakagawa and K. Tomishige, Catal. Sci. Technol., 2014, 4, 2535–2549 RSC.
- M. Chia, Y. J. Pagan-Torres, D. Hibbitts, Q. Tan, H. N. Pham, A. K. Datye, M. Neurock, R. J. Davis and J. A. Dumesic, J. Am. Chem. Soc., 2011, 133, 12675–12689 CrossRef CAS PubMed.
- J. Guan, G. Peng, Q. Cao and X. Mu, J. Phys. Chem. C, 2014, 118, 25555–25566 CrossRef.
- S. Koso, I. Furikado, A. Shimao, T. Miyazawa, K. Kunimori and K. Tomishige, Chem. Commun., 2009, 2035–2037 RSC.
- M. Chatterjee, H. Kawanami, T. Ishizaka, M. Sato, T. Suzuki and A. Suzuki, Catal. Sci. Technol., 2011, 1, 1466–1471 RSC.
- D. S. Pisal and G. D. Yadav, ACS Omega, 2019, 4, 1201–1214 CrossRef CAS PubMed.
- S. Liu, Y. Amada, M. Tamura, Y. Nakagawa and K. Tomishige, Green Chem., 2014, 16, 617–626 RSC.
- N. S. Date, R. C. Chikate, H.-S. Roh and C. V. Rode, Catal. Today, 2018, 309, 195–201 CrossRef CAS.
- W. E. Kaufmann and R. Adams, J. Am. Chem. Soc., 1923, 45, 3029–3044 CrossRef CAS.
- T. Tong, X. Liu, Y. Guo, M. Norouzi Banis, Y. Hu and Y. Wang, J. Catal., 2018, 365, 420–428 CrossRef CAS.
- O. Koch, A. Köckritz, M. Kant, A. Martin, A. Schöning, U. Armbruster, M. Bartoszek, S. Evert, B. Lange and R. Bienert, US Pat., US2014/0066666A1, 2014.
- H. Adkins and R. Connor, J. Am. Chem. Soc., 1931, 53, 1091–1095 CrossRef CAS.
- R. Connor and H. Adkins, J. Am. Chem. Soc., 1932, 54, 4678–4690 CrossRef CAS.
- F. Gao, H. Liu, X. Hu, J. Chen, Z. Huang and C. Xia, Chin. J. Catal., 2018, 39, 1711–1723 CrossRef CAS.
- J. Lee, S. P. Burt, C. A. Carrero, A. C. Alba-Rubio, I. Ro, B. J. O'Neill, H. J. Kim, D. H. K. Jackson, T. F. Kuech, I. Hermans, J. A. Dumesic and G. W. Huber, J. Catal., 2015, 330, 19–27 CrossRef CAS.
- H. W. Wijaya, T. Kojima, T. Hara, N. Ichikuni and S. Shimazu, ChemCatChem, 2017, 9, 2869–2874 CrossRef CAS.
- H. Liu, Z. Huang, H. Kang, C. Xia and J. Chen, Chin. J. Catal., 2016, 37, 700–710 CrossRef CAS.
- H. Liu, Z. Huang, F. Zhao, F. Cui, X. Li, C. Xia and J. Chen, Catal. Sci. Technol., 2016, 6, 668–671 RSC.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS PubMed.
- S. A. W. Hollak, R. W. Gosselink, D. S. van Es and J. H. Bitter, ACS Catal., 2013, 3, 2837–2844 CrossRef CAS.
- R. W. Gosselink, D. R. Stellwagen and J. H. Bitter, Angew. Chem., Int. Ed., 2013, 52, 5089–5092 CrossRef CAS PubMed.
- D. R. Stellwagen and J. H. Bitter, Green Chem., 2015, 17, 582–593 RSC.
- A. L. Jongerius, R. W. Gosselink, J. Dijkstra, J. H. Bitter, P. C. A. Bruijnincx and B. M. Weckhuysen, ChemCatChem, 2013, 5, 2964–2972 CrossRef.
- H. Guo, B. Zhang, C. Li, C. Peng, T. Dai, H. Xie, A. Wang and T. Zhang, ChemSusChem, 2016, 9, 3220–3229 CrossRef CAS PubMed.
- A. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef PubMed.
- R. Ooms, M. Dusselier, J. A. Geboers, B. Op de Beeck, R. Verhaeven, E. Gobechiya, J. A. Martens, A. Redl and B. F. Sels, Green Chem., 2014, 16, 695–707 RSC.
- Y.-B. Huang, M.-Y. Chen, L. Yan, Q.-X. Guo and Y. Fu, ChemSusChem, 2014, 7, 1068–1072 CrossRef PubMed.
- R. Ma, X.-P. Wu, T. Tong, Z.-J. Shao, Y. Wang, X. Liu, Q. Xia and X.-Q. Gong, ACS Catal., 2017, 7, 333–337 CrossRef.
- G. Leclercq, M. Kamal, J. M. Giraudon, P. Devassine, L. Feigenbaum, L. Leclercq, A. Frennet, J. M. Bastin, A. Löfberg, S. Decker and M. Dufour, J. Catal., 1996, 159, 142–169 CrossRef.
- A. Löfberg, A. Frennet, G. Leclercq, L. Leclercq and J. M. Giraudon, J. Catal., 2000, 189, 170–183 CrossRef.
- T. Xiao, A. Hanif, A. P. E. York, J. Sloan and M. L. H. Green, Phys. Chem. Chem. Phys., 2002, 4, 3522–3529 RSC.
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang, Y. Shu, A. L. Stottlemyer and J. G. Chen, Catal. Today, 2009, 147, 77–85 CrossRef CAS.
- N. Ji, T. Zhang, M. Zheng, A. Wang, H. Wang, X. Wang and J. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS.
- N. Ji, M. Zheng, A. Wang, T. Zhang and J. G. Chen, ChemSusChem, 2012, 5, 939–944 CrossRef CAS.
- L. N. Butorina, Sov. Phys. Crystallogr., 1960, 5, 216 Search PubMed.
- E. Rudy and S. Windisch, J. Am. Ceram. Soc., 1967, 50, 272 CrossRef CAS.
- W. P. Davey, Phys. Rev., 1925, 25, 753–761 CrossRef CAS.
- F. Abe and T. Tanabe, Z. Metallkd., 1985, 76, 420 Search PubMed.
- E. Brunner, Berichte der Bunsengesellschaft für physikalische Chemie, 1979, 83, 715–721 CrossRef.
- E. Brunner, J. Chem. Eng. Data, 1985, 30, 269–273 CrossRef CAS.
- A. Mehdad, R. E. Jentoft and F. C. Jentoft, J. Catal., 2017, 347, 89–101 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra02003f |
| This journal is © The Royal Society of Chemistry 2020 |