
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in catalytic chain transfer polymerization of isobutylene: a review
Tota Rajasekhar *,
Gurmeet Singh,
Gurpreet Singh Kapur* and
S. S. V. Ramakumar
*,
Gurmeet Singh,
Gurpreet Singh Kapur* and
S. S. V. Ramakumar
R&D Center, Indian Oil Corporation Limited, Sector-13, Faridabad-121007, Haryana, India. E-mail: rajasekhart@indianoil.in; kapurgs@indianoil.in
First published on 12th May 2020
Abstract
This review presents the development of highly reactive polyisobutylene (HRPIB), a major commercial intermediate toward fuel and lubricant additives. Recent years have witnessed very substantial advances in the catalytic chain transfer polymerization (CCTP) of isobutylene/industrial Raffinate-1 (C4 Raffinate) to produce HRPIB, particularly in nonpolar solvents at elevated temperatures. The main subjects of this review are cationic polymerization of isobutylene, progress in HRPIB research and existing challenges, and recent advances of CCTP. New initiating/catalyst systems based on ionic liquids with Lewis acids are detailed, and this approach may open new views in the synthesis of HRPIB. Some current developments in CCTP of industrial Raffinate-1 and mechanistic studies are also described. This review strongly supports that the hydrocarbon soluble Lewis acid·ether (LA·ether) complex catalyzed CCTP will become the most popular technique for preparing HRPIB and could replace the traditional BF3 catalyzed industrial method.
Introduction
In general, sludge, soot, oxidation products, and other deposit precursors are generated in engine oil during operation of an engine, which could be due to the incomplete oxidation/combustion of fuel. These incomplete combustion by-products cause corrosion, deposit formation, and wear within the engine over time, which directly affects engine performance. To avoid this, additives are typically added to lubricating motor oils.1 Furthermore, the engine performance could be increased by formulating engine oil with dispersants, antioxidants, viscosity modifiers and pour point depressants.2 Usually, formulated engine oil consists of ∼15 wt% of additives with major composition of dispersants, which are ∼50 wt% of the total additive percentage. Since 1950s, dispersants have been used in engine oils. Dispersants are composed of a polar head group and an oil soluble non-polar tail parts. The dispersant is adsorbed on to the surface of ultra-fine particle sized soot surface due to polar interactions, which avoids agglomeration of the particles by suspending in the form of micelles in the oil.1,2 This has been schematically represented in Fig. 1.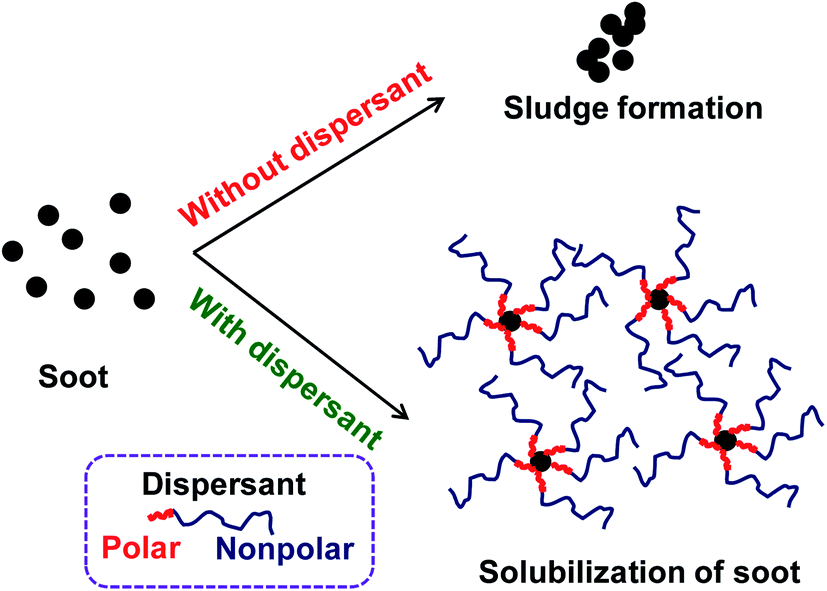 | ||
| Fig. 1 Schematic representation of role of dispersants in engine oil. | ||
Dispersants are amphiphilic polymers possessing nonpolar, hydrocarbon chain and a polar moiety/chain at the chain end. Different types of dispersants are being typically used by the oil additive industry; for instance polyalphaolefins; polyisobutylene (PIB) and polypropylene, and acrylate-based polymers. PIB exhibits low ceiling temperature, 175 °C, which is an important characteristic of PIB that makes it well appropriate for the use of as ashless dispersants. Moreover, cationic polymerization is used to synthesize PIB component of dispersants. Polyisobutylene succinimide (PIBSI) dispersants are of the most common ashless dispersants used in engine oil, which have been initially developed by Le Suer and Stuart.3,4 The PIBSI dispersants are prepared in two steps. In the first step, PIB chain terminated with succinic anhydride (PIBSA) is generated mostly via Alder-ene reaction. Then, PIBSA is reacted with a polyamine to produce PIBSI dispersants (Scheme 1). In comparison to internal olefin ends (tri and tetra substituted) containing PIBs (conventional PIBs), PIB with terminal vinylidene functionality (exo group) is highly reactive toward maleic anhydride5–9 to give PIBSA. Because of the higher reactivity for post-polymerization reaction, exo group containing PIB is termed as highly reactive polyisobutylene (HRPIB).
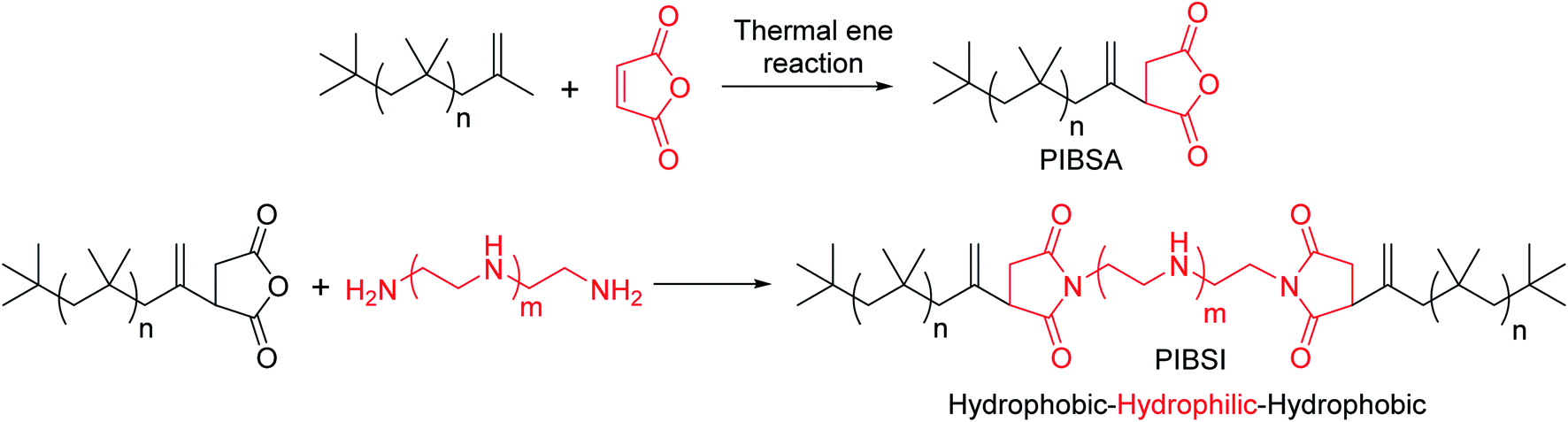 | ||
| Scheme 1 Preparation of PIBSI from precursor, HRPIB. | ||
Conventional PIBs have low reactivity for further post-polymerization functionalization due to steric crowding of the internal double bond.10–13 Earlier, the conventional PIBs required to chlorination followed by dehydrochlorination to react with maleic anhydride to give PIBSA, which was further reacted with oligo-alkylenimines to form PIBSI dispersants (Scheme 2).14–17 The main disadvantage of this method was that resulting dispersants contain residual chlorine as organic chlorides and also produces large quantities of chlorine containing waste water, which is the major environmental concern. Thus, HRPIB is the most preferred precursor for the preparation of ashless dispersants.
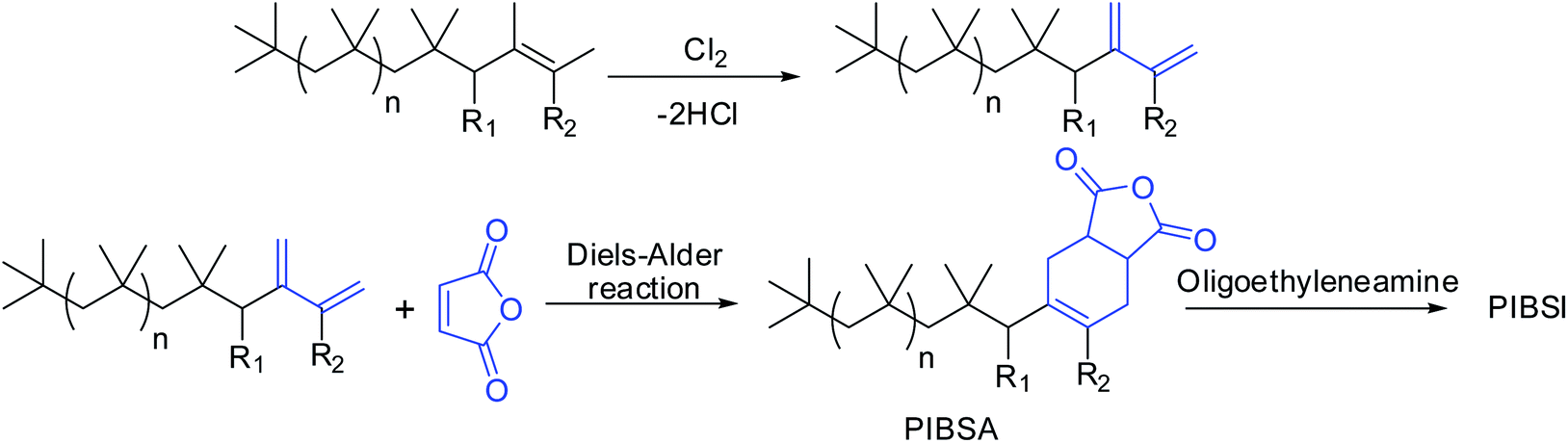 | ||
| Scheme 2 Preparation of PIBSI by using conventional PIB. | ||
More than 750![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tons of HRPIB, PIBs with terminal double bonds (exo bonds) are produced each year. Exxon, Amoco, BP, and BASF are globally major manufacturers of HRPIB. Dispersant properties are significantly affected by the molecular weight of polymer. Based on the molecular weight, PIB market size is divided into low molecular weight PIB (Mn < 5000 g mol−1), medium molecular weight PIB (Mn = 10000–100000 g mol−1) and high molecular weight PIB (Mn > 100000 g mol−1). Viscosity is an important physical parameter to differentiate low, medium and high molecular weight HRPIBs. Viscosity of the low molecular weight HRPIB for Mn = 1000 g mol−1 is approximately 200 centistokes (cSt) at 100 °C, which is significantly lower than the viscosity of medium and high molecular weight HRPIBs. Amongst them, low molecular weight HRPIB is the most important class because of their demanding as lubricant and fuel additives. Low molecular weight HRPIB, 1000–2500 g mol−1 with polydispersity index of 2–3 are used as the additives.18,19
000 metric tons of HRPIB, PIBs with terminal double bonds (exo bonds) are produced each year. Exxon, Amoco, BP, and BASF are globally major manufacturers of HRPIB. Dispersant properties are significantly affected by the molecular weight of polymer. Based on the molecular weight, PIB market size is divided into low molecular weight PIB (Mn < 5000 g mol−1), medium molecular weight PIB (Mn = 10000–100000 g mol−1) and high molecular weight PIB (Mn > 100000 g mol−1). Viscosity is an important physical parameter to differentiate low, medium and high molecular weight HRPIBs. Viscosity of the low molecular weight HRPIB for Mn = 1000 g mol−1 is approximately 200 centistokes (cSt) at 100 °C, which is significantly lower than the viscosity of medium and high molecular weight HRPIBs. Amongst them, low molecular weight HRPIB is the most important class because of their demanding as lubricant and fuel additives. Low molecular weight HRPIB, 1000–2500 g mol−1 with polydispersity index of 2–3 are used as the additives.18,19
In the past 10 years, substantial advances in synthesis of commercially valued, HRPIB via different cationic polymerization approaches have been achieved.20–50 In particular, the catalytic chain transfer polymerization (CCTP) in nonpolar solvents using Lewis acid·ether complexes (LA·ether), has recently recognized much interest in the view of their industrial relevance.27–50 Because the recent CCTP embodies attractive features compared to the traditional BF3 catalyzed polymerization method,51,52 such as the simple experimental operation and cost, tolerance of ambient reaction conditions, and applicability of various kinds of reactor systems.19,53,54 To date, low molecular weight HRPIB is the most important industrial precursor material in the preparation of motor oil and fuel auxiliaries.
The endeavour of this perspective is clearly illustrated in Fig. 1 and Scheme 1, which presents importance and motivation on behind in the development of HRPIB. We will first briefly discuss the existing synthetic methods of engine oil/lubricant dispersant precursor and challenges. Subsequently, we will also review the basics of isobutylene cationic polymerization, and conventional & living polymerization of isobutylene approaches towards in development of HRPIB. This review finally covers the recent progress in CCTP of isobutylene/industrial Raffinate-1 feed for HRPIB synthesis and shows advantages of this method in comparison to conventional polymerization method. Furthermore, fundamental mechanistic aspects of chain transfer agent involved cationic polymerization will also be focused. Development of new initiating system and cost-effective processes for the production of HRPIB is currently an active area of research in academics and industry. An interested reader can consult previous reports on various developments in cationic polymerization of isobutylene.55–60
Synthesis of engine oil/lubricant dispersant precursor and challenges
Oil additive industry typically use PIB based dispersants, due to their unique properties of cleaner burning, low toxicity, good thickening properties, high shear stability, water and oxidation resistance, tackiness, and cohesive strength. Moreover, PIB based dispersants possess various other value-added advantage including less char formation at high temperature performance conditions and ability to improve final lubricant and fuel performance. Basically the precursors of PIB dispersants consist of olefinic group/double bond. Based on the double bond position, the PIB precursors are broadly classified in to two categories: (i) conventional PIB consists of internal double bonds (tri and tetra substituted olefinic groups), and (ii) HRPIB consists of external double bond (exo olefinic group).The internal double bonds containing PIB preparation with low molecular weight has been well known as conventional process/Exxon process. In this process, isobutylene polymerization in the presence of cationogen and Lewis acid, AlCl3 or EtAlCl2 as coinitiator produces the low molecular weight conventional PIB.61–64 Importantly, the reaction mechanism of conventional PIB preparation is associated with a complex set of isomerisation and chain scission reactions. Faust et al., clearly demonstrated that the formation of ‘‘conventional’’ PIBs with internal tri- and tetra-substituted olefinic groups (Schemes 3 and 4) through complex mechanism of the carbocationic rearrangements.65 Here, the internal olefins formation in the polymerization of IB has been understood by ionization of PIB-Cl to PIB+ using EtAlCl2 in the presence of a proton trap.
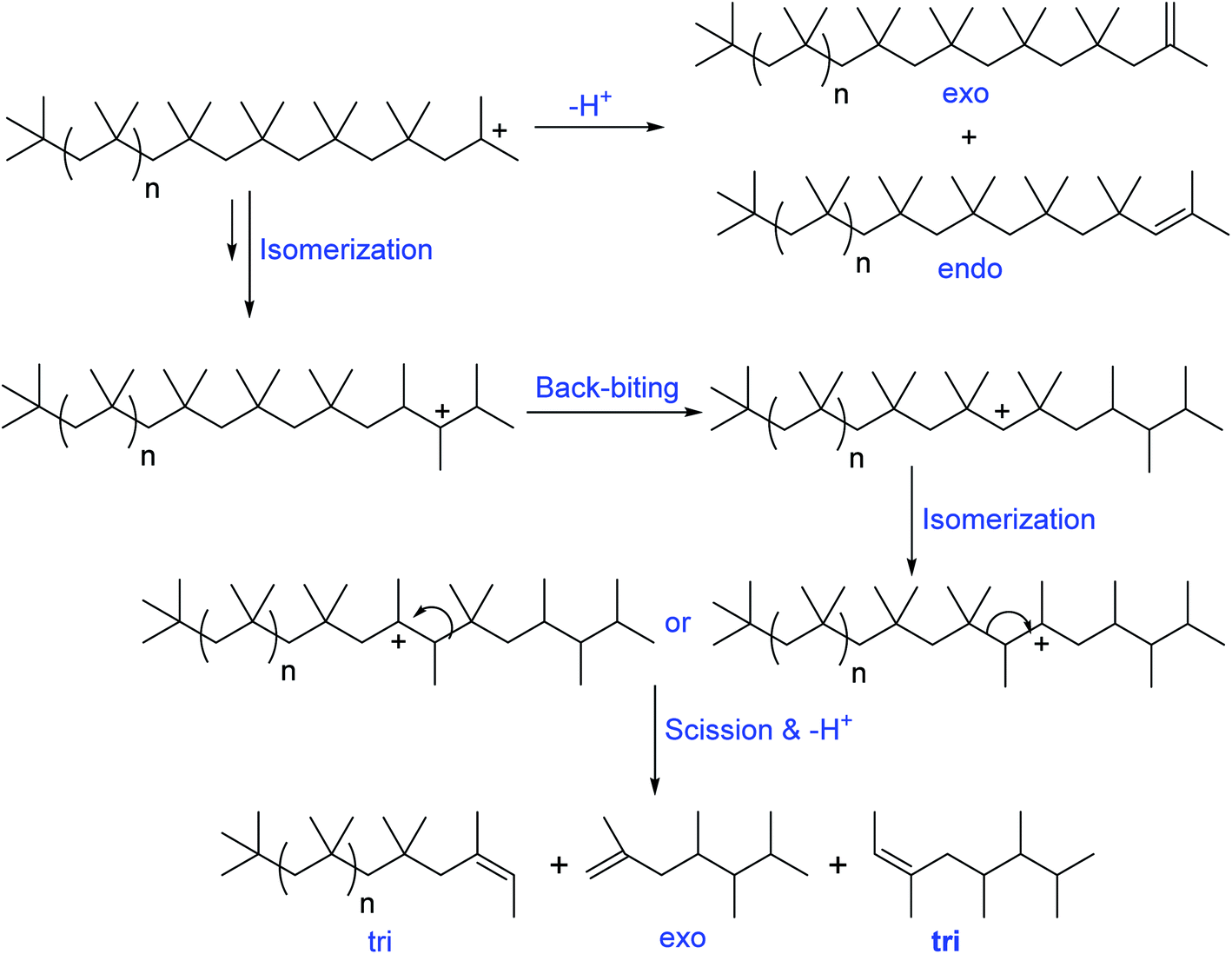 | ||
| Scheme 3 Mechanism of formation of different olefinic groups in the conventional cationic polymerization of isobutylene (proposed by Faust et al.65). | ||
 | ||
| Scheme 4 Mechanism of formation of internal olefinic group, tetra substituted olefin in the conventional cationic polymerization of isobutylene (proposed by Faust et al.65). | ||
A possible mechanism has been proposed by Faust et al., to explain the olefin structures. The mechanism involving a sterically hindered cation arising via hydride and methyde shifts from the chain growing PIB+. Tri-substituted olefins form through a distant hydride shift by backbiting followed by a methyde shift and chain scission of the PIB+. Moreover, the tri-substituted olefins possess of irregular carbon numbers, which clearly supports the chain scission pathway in the mechanism (Scheme 3). But the exo- and endo olefins are formed by simple β-proton elimination from the PIB+.
The formation of exo-, endo-, and tetra-substituted olefins was found accidentally, which form under milder conditions. The ionization of PIB-Cl at room temperature in the absence of a proton trap resulted to the formation of these three olefins. As shown in Scheme 4, PIB+ undergoes to hydride and methyde shifts followed by proton elimination, which results to tetra-substituted olefins.65
The external double bond containing PIBs (HRPIB) readily react with maleic anhydride,5 which clearly indicates that chlorination followed by dehydrochlorination is not essential to prepare PIBSA from HRPIB. Importantly, HRPIB route is environmental friendly one for the manufacture of ashless dispersants. Cationic polymerization of IB initiated by BF3 complex with either alcohol or ethers as catalyst system is the traditional process, to prepare commercial low molecular weight HRPIBs with more than 80 mol% of exo-olefin content. The process was first developed by BASF and is more commonly known as BASF process.66,67 Major drawback of this process is the critical low polymerization temperature, because of the requirement of low temperatures and highly purified feed making the process more expensive. Another disadvantage of this process is that usage of BF3 catalyst system produces fluorine based byproducts (Scheme 5). These fluorine based products, under the thermal stress, get converted to highly corrosive hydrogen fluoride. Moreover, in this process the fluorine content in HRPIB is noticed up to 200 ppm.54 Furthermore, researchers have been trying different ways to produce HRPIB with the aim to replace BF3 catalyst used in the traditional process.
 | ||
| Scheme 5 Schematic representation of formation of byproduct in traditional HRPIB synthesis process. | ||
In the recent years, significant contributions have been made by industries and academic-industrial collaborations for developing new catalyst systems and improvising process for producing HRPIB.20–54 Before going to discuss about the insights in development of HRPIB research, a summary is given below on historical developments in cationic polymerization of isobutylene. These past findings have been actually shown the route map to commercial valued HRPIB developments.
Cationic polymerization of isobutylene
Cationic polymerization offers the best existing methodology for the homopolymerization and copolymerization of isobutylene, which cannot be polymerized by any another polymerization method.68–70 Free radical polymerization of inactive double containing monomers (ethylene, propylene, isobutylene, etc.) requires extremely high pressure (>1000 bar) and temperatures (>150 °C). This is because of high activation energy of the free radical polymerization of the inactive monomers.71,72 However, under the certain polymerization conditions, the formed low molecular weight PIB chains can undergo depolymerization and regenerates isobutylene by backward reaction. Thus, scientific community does not show interest in free radical polymerization of isobutylene. Anionic polymerization of isobutylene can't be observed, due to the low reactivity of isobutylene with anionic species/initiator. The cationic polymerization is initiated by a cationic initiator (for example t-butyl chloride) in conjunction with a Lewis acid (LA), where heterolytic bond cleavage of the initiator leads to reactive cation species and counter anion. In the subsequent step, the cation species reacts with isobutylene to yield a cationic-adduct. Propagation involves the repetitive addition of isobutylene to the growing carbenium ion until a chain-breaking reaction via chain transfer or termination occurs.73–81 In the chain transfer step, exo, endo, tri and tetra substituted double bond containing PIBs are formed by a complex set of isomerisation and chain scission reactions followed by proton elimination of the propagating PIB+. In the presence of a chain transfer agent, chain transfer reactions predominantly involve β-proton elimination to form exo end groups. Adding ethers as the chain transfer agent has been found to be effective to improve the controllability to form exo-olefin end groups.38–50 These developments are clearly discussed in the next section: development of HRPIB synthesis. Additionally, the molecular weight of the polymerization can be limited by chain transfer. In the termination step PIB+ cations usually undergo ion collapse by transferring of an anionic fragment from the complex counterion. In the present illustration (Scheme 6), PIB+ cation is converted to PIB-Cl by ion collapse in the termination step.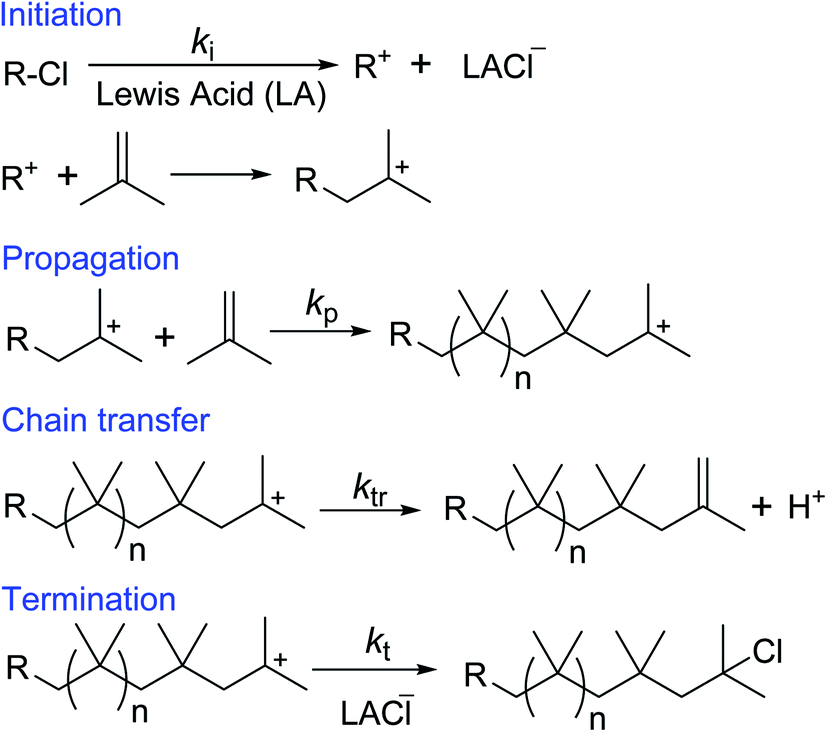 | ||
| Scheme 6 Schematic representation of steps involved in cationic polymerization of isobutylene. | ||
Interestingly, overall activation energy of the isobutylene polymerization is negative, and increase in temperature causes decrease in the rate of propagation of polymerization.34,74 The activation energy can be expressed by the following equation.
| Activation energy of polymerization = (sum of activation energies of initiation and propagation) − (activation energy of termination or chain transfer) |
The above equation is a fundamental statement for activation energy of polymerization. In the case of chain transfer polymerization, activation energy of termination could be replaced by activation energy of chain transfer.71 In the above equation, the sum of activation energies of initiation and propagation is always smaller than activation energy of chain transfer. Increase in polymerization temperature leads to shorter polymer chains, which is due to overcoming the energy barrier for the chain transfer reaction.34,76–81 This fundamental aspect helps in controlling the molecular weight of oil and lubricant additives during their synthesis. Moreover, chain transfer step plays a significant role in HRPIB synthesis,20–50 which could be discussed on detail in the following sections.
Cationic polymerization of isobutylene has a long story, the first report of the isobutylene polymerization by acid initiation at room temperature was revealed in 1873, where the polymerization was restricted to formation of low molecular weight PIB oligomers. Since then, polymerization reactions of various conditions were examined,82–84 for which several types of catalyst systems have been developed to prepare high-molecular weight PIB.85,86 The discovery of butyl rubber/synthetic rubber in early 1900s brings cationic polymerization into light. In the initial reports recognized that low temperatures and AlCl3 as catalyst have been required to obtain high molecular weight PIBs. Even though achieving of the molecular weight is more than 100000 g mol−1, the polymer was not used for practical end use application. Later, the issue was overcome by copolymerization of isobutylene with isoprene. Here, the unsaturation unit of isoprene could be used for vulcanization of copolymer, butyl rubber in the formulation process. The discovery of butyl rubber led to in detail understanding of cationic polymerization.87 Control in cationic polymerization was lagging because of thermodynamically favoured chain transfer reaction. It was well known that chain transfer could be suppressed by lower temperatures, which favours higher molecular weight polymer. Based on these Kennedy et al., the conditions for the “quasiliving” polymerization of isobutylene have been realized.88 Moreover, the polymerizations having controlled initiation and the absence of termination or chain transfer during the propagation. These understandings gave direction for the controlled synthesis of low molecular weight PIBs having distinct end groups with predictable molecular weight. PIB of low molecular weights are valuable viscosity modifiers, fuel and lubricating oil additives.89–91 Relevant for this review are the currently practiced “conventional” and “living” cationic polymerizations of isobutylene as well as end group functionalities derived there from. The present review focused on chain end olefinic functionality of low molecular weight (<5000 g mol−1) isobutylene-based polymers.
Development of HRPIB synthesis
In recent decades, numerous new methods for the synthesis of HRPIB have been reported. Broadly three approaches on independently for the synthesis of HRPIB are being developed: (i) through living cationic polymerization (ii) using solvent-ligated complexes and (iii) modified conventional cationic polymerization or CCTP. The basic developments in the three approaches are shorted out here.:1 complexation. Here, the added nucleophilic agent is more likely to be rapidly consumed by interaction with the Lewis acid rather than reacting with the carbenium ion chain ends. The result is ion-pair collapse and production of unmodified PIB-Cl. While in the case of sterically hindered amines, uncomplexed base with TiCl4 is responsible for the exo olefin formation at the carbenium ion chain end. Later on found that the most effective classes of quenchers are sulfides and ethers.20 It has been projected to use sulfides or ethers as quenchers instead of hindered bases. In this case, the quenching proceed into two steps through the formation of stable sulfonium or oxonium PIB ions, which followed by decomposition of resulting onium ions by addition of excess of nucleophile to yield HRPIB.20–22 Main disadvantages with these imitating systems are that the reactions have been conducted at low temperatures in chlorinated solvent and expensive reagents have been used for the synthesis.117–119Recent developments in CCTP of isobutylene for HRPIB
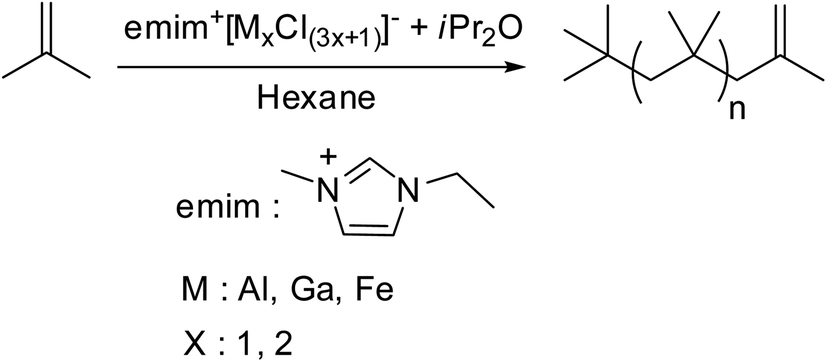 | ||
| Scheme 7 Ionic liquid catalyzed cationic polymerization of isobutylene. | ||
Later on Kostjuk et al. in their successive work, to solve the problem of slow polymerization with emimCl–AlCl3 imitating system, they have studied the influence of ionic liquid acidity, nature of Lewis acid and imidazolium salt on the reaction rate.49 In this work, various Lewis acids (AlCl3, AlBr3, iBuAlCl2, FeCl3, GaCl3 and BBr3) have been used for the preparation of acidic ionic liquids by the reaction with imidazole chloride, and their activity in isobutylene polymerization were examined. High exo content and relatively narrow PDI have been obtained only with emimCl–AlCl3, emimCl–FeCl3 and emimCl–GaCl3. Moreover, the polymerization rate depended on the rate of partial hydrolysis of ionic liquid and increased in the following order: emimCl–FeCl3 < emimCl–GaCl3 < emimCl–AlCl3. For efficient synthesis of HRPIB, the ionic liquid based catalyst needs to be soluble at polymerization temperature, and possess high Lewis acidity. HRPIB with targeted molecular weight, PDI and high exo content was achieved emimCl–FeCl3, due to low selectivity to emimCl–FeCl3:ether ratio. But sonication of polymerization mixture has been required to get high polymerization rate, due to poor dispersion of emimCl–FeCl3 in n-hexane. The main disadvantage of previously developed initiating systems of CCTP is relatively high PDI of the synthesized HRPIB, which is over come with this new system.
Subsequently, titanium tetrachloride, boron trichloride, and ethylaluminum sesquichloride initiating systems in 1-butyl-3-methylimidazolium hexafluorophosphate ([Bmim][PF6]) ionic liquid have been studied by Wu and co-workers.133 Initially in this study, [PF6]−, [BF4]−, and [NTf2]− based ionic liquids were screened for their potential in the synthesis of HRPIB. It was shown that [Bmim][PF6] is the best ionic liquid for polymerization of isobutylene at −10 °C using an H2O/TiCl4 initiating system, and it was a suspension polymerization. In this study, HRPIB with a high exo-olefin end group content (>80%) was obtained. Furthermore, the exo content is relatively higher in the ionic liquid ([Bmim][PF6]) than in a polar solvent, dichloromethane, whereas lower Mn of HRPIB with a relatively narrow MWD were obtained in the ionic liquid as compared to polymers obtained in dichloromethane. Density functional theory was used to understand the effects of solvent polarity and ionic structure on the initiating systems and active centre of the polymerization system, and proposed that the polymerization proceeds at the interface of ionic liquid particles. This innovative ionic liquid based catalyst system approach can lead to prospect of new HRPIB technologies.133
Rajasekhar et al., thoroughly studied copolymerization of isobutylene in the presence of other C4 olefins (1B, C2B and BD) using EADC·CEE initiating system.50 To understand the effect of each of the C4 olefin on the isobutylene polymerization rate and olefin distribution in the product, the copolymerization of isobutylene with each of the C4 olefins was studied individually. Polymerization of isobutylene in the presence of 1B using EADC·CEE in conjunction with t-BuCl was studied at 0 °C with different ratios of [isobutylene]/[1B] = [95/05, 90/10, 80/20, 70/30, 60/40 and 10/90]. The first order plots with [isobutylene]/[1B] > 80/20 are linear starting at the origin suggesting a constant steady state concentration of the active species. However, the first-order plots are curved downward with [isobutylene]/[1B] ≤ 80/20, suggesting decreasing active species concentration. The 1H NMR spectrum of obtained HRPIB in this study suggested that capping of PIB+ with 1B gives PIB-1B-Cl due to termination by ion collapse. Noticeable decrease in termination was observed for the polymerization of isobutylene with [isobutylene + 1B] ≥ 2 M up to 8 M. The first order plots are moving downward with decreasing [isobutylene]. This suggests that termination has been suppressed at [isobutylene + 1B] ≥ 2 M relative to propagation. Importantly, determined the reactivity ratio of isobutylene is risobutylene = 1100, which means that isobutylene is 1100 times more reactive than 1B.
Similarly, polymerization of isobutylene in the presence of C2B with [isobutylene]/[C2B] = 92/08, and in the presence of BD with [isobutylene]/[BD] = 99.5/0.5 and 97.5/2.5 were conducted at different [isobutylene + C2B] = 1, 2, 4 and 6 M. In addition, the first-order plots are linear when the copolymerization was carried out with [isobutylene + C2B or BD] > 2 M.
Polymerization of C4 olefin mixture, IB:1B:C2B:BD = 43:28:4:0.1, at [C4 olefin mixture] = 6 M was also conducted at 0 °C. As similar to isobutylene polymerization with individual other C4 olefin, here the first-order plot is curved downward. Moreover, molecular weights and exo-olefin contents are comparable to those obtained with pure isobutylene.
It has to note that 1H NMR spectrum of HRPIB obtained by polymerization of C4 feed is more complicated in comparison to synthesized using pure isobutylene. Fig. 2 represents a typical 1H NMR spectrum of a representative HRPIB sample prepared by the copolymerization isobutylene. The two protons characteristic of the exo-olefin end group (protons a1 and a2) appeared as two well resolved peaks at 4.85 and 4.64 ppm, respectively, while the endo olefin end group (proton d) appeared as a single peak at 5.15 ppm. Small amounts of the E and Z configurations of another tri-substituted olefin end group (proton e1 represents Z configuration and proton e2 represents E configuration) were also observed in some samples at 5.37 and 5.17 ppm. The signal corresponding to the tetra-substituted olefin end group (proton f) appeared as a broad multiplet at 2.85 ppm. The methine protons in the PIB-1B-Cl group (protons –CHCl–) which appear at 3.95 ppm. Resonances for BD coupled PIB chains (protons BD1 and BD2) appeared at 4.82 ppm. The methylene and methyl protons of the isobutylene repeat unit (protons b and c, respectively) usually appeared at 1.42 and 1.11 ppm, respectively.
 | ||
| Fig. 2 Typical 1H NMR spectrum of HRPIB obtained by copolymerization of isobutylene. | ||
It has been shown that a complex set of isomerization is basis for the formation of different types of PIB terminal ends with 1B, C2B and BD units (Scheme 8). To explain the complex mechanism, low-temperature 1H NMR studies were carried. As part of this the sec-alkyloxoniums were formed individually from the reaction of TMPCl with B1 or C2B or BD in the presence of EADC·CEE at 0 °C. The NMR studies clearly support the proposed possible mechanistic pathways.
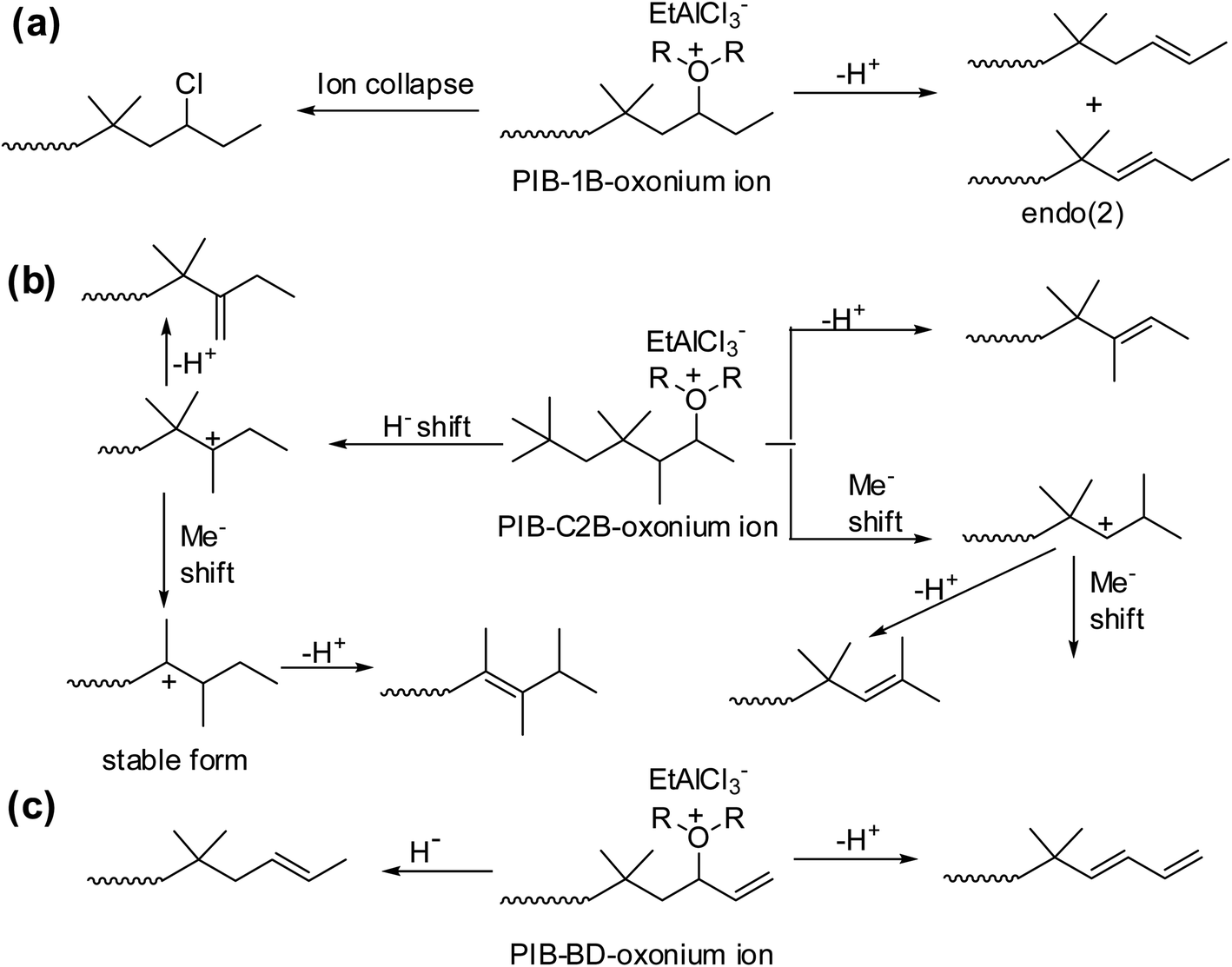 | ||
| Scheme 8 Schematic representation of the sec-alkyloxonium ions, (a) PIB-1B-oxonium ion, (b) PIB-C2B-oxonium ion and (c) PIB-BD-oxonium ion, rearrangement on copolymerization of isobutylene using EtAlCl2·ROR initiating system (proposed by Rajasekhar et al.50). | ||
Very recently, the copolymerization of isobutylene in the presence of alcohol as an exo-enhancer with t-BuCl/EADC.CEE initiating system under simulated CSTR conditions has been investigated by Rajasekhar et al.54 The desired Mn (∼2200 g mol−1) and high exo content of HRPIB can be prepared only in the presence of exo enhancer under the CSTR conditions. In the absence of exo enhancer, the desired molecular weight have been achieved only at 70–80% CSTR conversions at 0 °C with [CEE]/[EADC] = 1.5 with exo content ∼80%. But the desired molecular weight was achieved with high CSTR conversion (90–95%) at [CEE]/[EADC] = 1. However, low [CEE] caused to decrease in exo content to ∼70%. It means that [CEE]/[EADC] > 1 is necessary to maintain high exo content. In this report found that alcohols could be used as exo-enhancers, where the content of exo-olefin end groups increased with increasing bulkiness of the alkyl group of the alcohol in the following series: primary alcohol < sec-alcohol < tert-alcohol. Molecular weights were virtually unaffected in the presence of exo enhancer with [CEE]/[EADC] = 1. The real exo-enhancer in these polymerizations is alkoxyaluminum dichloride (ROAlCl2), which could be formed in the polymerization mixture by the reaction of EADC·CEE with alcohol. Moreover, molecular weights have been almost unaffected in the presence exo enhancer, ROAlCl2 with [ROAlCl2]:[EADC·CEE] < 0.5, and exo-olefin content improved 15% in comparison to polymerization in the absence of exo-enhancer. The systematic experimental results discussed in this study suggest that t-BuOAlCl2 suppressing the isomerization of PIB+, but does not act as a chain transfer agent, for the proposed mechanism (see Scheme 9). The increase of the exo content in the presence of exo-enhancer is attributed to the stabilization of PIB+ by the enhancer. The stabilization of PIB+ has been illustrated by a model study. In the model study, interaction of carbenium ion, PhCH2+ and t-BuOAlCl2 was studied using low-temperature NMR spectroscopy. This is the original report that stabilization of carbocations in the cationic polymerization has also been proven, which opens new vistas in this area of research. From this report understood that RAlCl2·ether initiating systems in the presence of tert-alcohol are the best candidates to synthesize desired low molecular weight HRPIB with high exo-olefin end group content in non-polar solvent conditions.
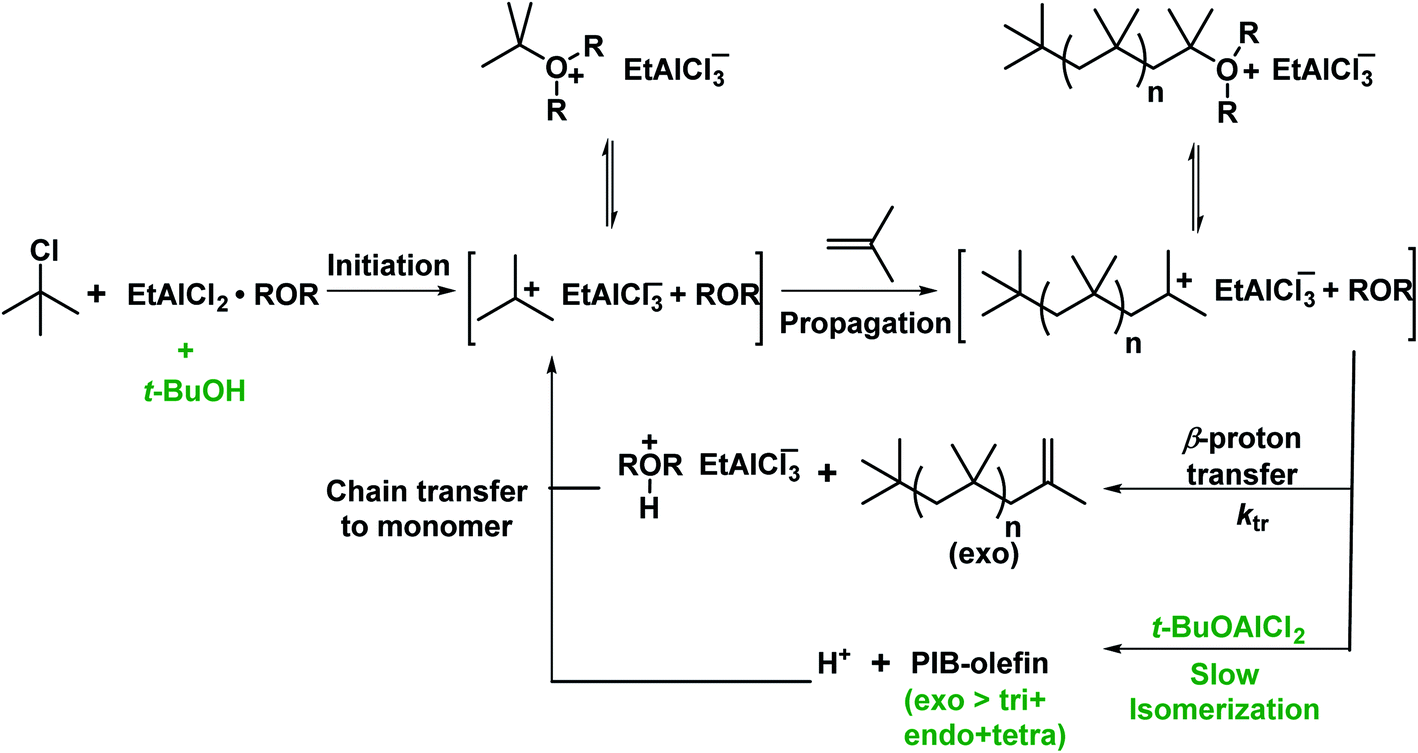 | ||
| Scheme 9 Possible mechanism for polymerization of IB with t-BuCl/EADC·CEE in the presence of t-BuOH (proposed by Rajasekhar et al.54). | ||
Later on, Kostjuk et al., extended the study to cationic polymerization of isobutylene to produce HRPIB of low molecular weight (∼1000 g mol−1) to medium molecular weight (up to 50000 g mol−1) under mild experimental conditions using alkoxy aluminum chlorides as catalyst. From experimental observations, authors conclude that alkoxy aluminum chloride acts concurrently as co-initiator, carbocation stabilizer, and chain-transfer agent.134
Overall, the specific merits of the recent developments in CCTP and their advantages over the traditional polymerization system show industrial interest. Recently developed approaches coupled with existed industrial approaches will give out to intensify the mechanistic understanding of CCTP and thus eventually help in the development of new ones.
Conclusion
In this review, we have discussed advances in the preparation of HRPIB using cationic polymerization of isobutylene. Following the development of CCTP about last few years, tremendous efforts have been made to explore new approaches for HRPIB. The CCTP has emerged as an extremely influential method, which is of vast industrial and fundamental importance. The recent rapid development in this area of research has been explored to new initiating systems in nonpolar solvents, LA·ether complexes in conjunction with cationogen, which are the best alternative systems in the place of traditional BF3 catalyzed process. Compare to the traditional catalyst system, LA·ether complexes significant advantage of highly selective for the more useful exo olefin end groups and the most suitable ambient reaction conditions. Moreover, this new initiating systems demonstrate significant efficiency for isobutylene/industrial Raffinate-1 polymerization, which is also endowed with conceivable industrial importance for the synthesis of low molecular weight HRPIB. The kinetic and mechanistic aspects can attribute to make the way for creating rules for the reasonable selection of reaction conditions to achieve the synthesis of the targeted PIB precursors, HRPIB. Furthermore, due to these recent accomplishments in economic way of synthesis of HRPIB, advance industrial developments are expected.Conflicts of interest
There are no conflicts to declare.References
- R. M. Mortier, F. F. Malcolm and S. T. Orszulik, Chemistry and Technology of Lubricants, Springer, London, 2009 Search PubMed.
- S. Q. A. Rizvi, Additives and additive chemistry, fuels and lubricants: technology, properties, performance, and testing, ASTM International, West Conshohocken, PA, 2003, p. 199 Search PubMed.
- W. M. Le Suer and G. R. Norman, US Pat., US3172892A, 1965.
- A. F. Stuart, R. G. Orinda, N. Anderson and A. Y. Drummond, US Pat., US3202678, 1965.
- H. Mach and P. Rath, Lubr. Sci., 1999, 175, 11 Search PubMed.
- J. J. Harrison, C. M. Mijares, M. T. Cheng and J. Hudson, Macromolecules, 2002, 35, 2494 CrossRef CAS.
- I. Puskas and S. Meyerson, J. Org. Chem., 1984, 49, 258 CrossRef CAS.
- F. Balzano, A. Pucci, R. Rausa and G. Uccello-Barretta, Polym. Int., 2012, 61, 1256–1262 CrossRef CAS.
- I. Puskas, E. M. Banas and A. G. Nerheim, J. Polym. Sci., Polym. Symp., 1976, 56, 191–202 CrossRef CAS.
- F. Balzano, A. Pucci, R. Rausa and G. Uccello-Barretta, Polym. Int., 2012, 61, 1256–1262 CrossRef CAS.
- I. Puskas, E. M. Banas and A. G. Nerheim, J. Polym. Sci., Polym. Symp., 1976, 56, 91–202 Search PubMed.
- I. Puskas and S. J. Meyerson, Org. Chem., 1984, 49, 258 CrossRef CAS.
- J. J. Harrison, C. M. Mijares, M. T. Cheng and J. Hudson, Macromolecules, 2002, 35, 2494 CrossRef CAS.
- J. P. Kennedy, V. S. C. Chang, R. A. Smith and B. Ivan, Polym. Bull., 1979, 1, 575 CrossRef CAS.
- J. P. Kennedy and R. A. Smith, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 1523 CrossRef CAS.
- B. Ivan and J. P. Kennedy, Macromolecules, 1990, 23, 2880 CrossRef CAS.
- M. K. Mishra, B. Sar-Mishra and J. P. Kennedy, Polym. Bull., 1985, 13, 435 CrossRef CAS.
- S. Zhu, Y. C. Lu, K. Wang and G. S. Luo, Eur. Polym. J., 2016, 80, 219 CrossRef CAS.
- S. Zhu, Y. C. Lu, K. Wang and G. S. Luo, RSC Adv., 2016, 6, 97983 RSC.
- S. Ummadisetty, D. L. Morgan, C. D. Stokes and R. F. Storey, Macromolecules, 2011, 44, 7901 CrossRef CAS.
- S. Ummadisetty, D. L. Morgan, C. D. Stokes, J. J. Harrison, C. G. Campbell and R. F. Storey, Macromol. Symp., 2013, 323, 6 CrossRef CAS.
- S. Ummadisetty and R. F. Storey, Macromolecules, 2013, 46, 2049 CrossRef CAS.
- M. R. Lichtenthaler, A. Higelin, A. Kraft, S. Hughes, A. Steffani, D. A. Plattner, J. M. Slattery and I. Krossing, Organometallics, 2013, 32, 6725 CrossRef CAS.
- A. K. Hijazi, N. Radhakrishnan, K. R. Jain, E. Herdtweck, O. Nuyken, H.-M. Walter, P. Hanefeld, B. Voit and F. E. Kühn, Angew. Chem., Int. Ed., 2007, 46, 7290 CrossRef CAS PubMed.
- N. Radhakrishnan, A. K. Hijazi, H. Komber, B. Voit, S. Zschoche, F. E. Kühn, O. Nuyken, M. Walter and P. Hanefeld, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5636 CrossRef CAS.
- H. Y. Yeong, Y. Li, F. E. Kühn and B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 158 CrossRef CAS.
- R. Kumar, B. Zheng, K.-W. Huang, J. Emert and R. Faust, Macromolecules, 2014, 47, 1959 CrossRef CAS.
- I. V. Vasilenko, D. I. Shiman and S. V. Kostjuk, Polym. Chem., 2014, 5, 3855 RSC.
- D. I. Shiman, I. V. Vasilenko and S. V. Kostjuk, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2386 CrossRef CAS.
- S. Banerjee, J. Emert, P. Wright, T. Skourlis, R. Severt and R. Faust, Polym. Chem., 2015, 6, 4902 RSC.
- S. Banerjee, B. N. Jha, P. De, J. Emert and R. Faust, Macromolecules, 2015, 48, 5474 CrossRef CAS.
- D. I. Shiman, I. V. Vasilenko and S. V. Kostjuk, Polymer, 2016, 99, 633 CrossRef CAS.
- I. V. Vasilenko, P. A. Nikishev, D. I. Shiman and S. V. Kostjuk, Polym. Chem., 2017, 8, 1417 RSC.
- T. Rajasekhar, J. Emert and R. Faust, Polym. Chem., 2017, 8, 2852 RSC.
- S. Zhu, Y. Lu and R. Faust, RSC Adv., 2017, 7, 27629 RSC.
- Q. Liu, Y.-X. Wu, Y. Zhang, P.-F. Yan and R.-W. Xu, Polymer, 2010, 51, 5960 CrossRef CAS.
- Q. Liu, Y. Wu, P. Yan, Y. Zhang and R. Xu, Macromolecules, 2011, 44, 1866 CrossRef CAS.
- I. V. Vasilenko, A. N. Frolov and S. V. Kostjuk, Macromolecules, 2010, 43, 5503 CrossRef CAS.
- I. V. Vasilenko, D. I. Shiman and S. V. Kostjuk, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 750 CrossRef CAS.
- P. Dimitrov, J. Emert and R. Faust, Macromolecules, 2012, 45, 3318 CrossRef CAS.
- R. Kumar, P. Dimitrov, K. J. Bartelson, J. Emert and R. Faust, Macromolecules, 2012, 45, 8598 CrossRef CAS.
- K. J. Bartelson, P. De, R. Kumar, J. Emert and R. Faust, Polymer, 2013, 54, 4858 CrossRef CAS.
- D. I. Shiman, I. V. Vasilenko and S. V. Kostjuk, Polymer, 2013, 54, 2235 CrossRef CAS.
- R. Kumar, P. De, B. Zheng, K.-W. Huang, J. Emert and R. Faust, Polym. Chem., 2015, 6, 322 RSC.
- R. Kumar, J. Emert and R. Faust, Polym. Bull., 2015, 72, 49 CrossRef CAS.
- S. Zhu, Y. C. Lu, K. Wang and G. S. Luo, RSC Adv., 2016, 6, 9827 RSC.
- T. Rajasekhar, U. Haldar, J. Emert, P. Dimitrov, R. Severt and R. Faust, J. Polym. Sci., Part A: Polym. Chem., 2017, 22, 3697 CrossRef.
- I. V. Vasilenko, I. A. Berezianko, D. I. Shiman and S. V. Kostjuk, Polym. Chem., 2016, 7, 5615–5619 RSC.
- I. A. Berezianko, I. V. Vasilenko, D. I. Shiman and S. V. Kostjuk, Polymer, 2018, 145, 382 CrossRef CAS.
- T. Rajasekhar, U. Haldar, P. De, J. Emert and R. Faust, Macromolecules, 2017, 50, 8325 CrossRef CAS.
- H. P. Rath, U. Kanne and F. V. Deyck, US Pat., US6407186B1, 2002.
- H. P. Rath, US Pat., US5962604A, 1999.
- S. Zhu, Y. Lu and R. Faust, RSC Adv., 2017, 7, 27629 RSC.
- T. Rajasekhar, J. Emert, L. M. Wolf and R. Faust, Macromolecules, 2018, 51, 3041 CrossRef CAS.
- S. V. Kostjuk, RSC Adv., 2015, 5, 13125 RSC.
- S. V. Kostjuk, I. V. Vasilenko, D. I. Shiman, A. N. Frolov and L. V. Gaponik, Macromol. Symp., 2015, 349, 94 CrossRef CAS.
- Y. Li, M. Cokoja and F. E. Kühn, Coord. Chem. Rev., 2011, 255, 1541 CrossRef CAS.
- S. V. Kostjuk, H. Y. Yeong and B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 471 CrossRef CAS.
- J. E. Puskas and G. Kaszas, Prog. Polym. Sci., 2000, 25, 403 CrossRef CAS.
- R. Faust, Cationic polymerization of nonpolar vinyl monomers, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski and M. Möller, Elsevier, 2012, vol. 3, pp. 501–526 Search PubMed.
- J. P. Kennedy and E. Marechal, Carbocationic Polymerization, Wiley, New York, 1982 Search PubMed.
- I. Puskas and S. Meyerson, J. Org. Chem., 1984, 49, 258 CrossRef CAS.
- J. J. Harrison, C. M. Mijares, M. T. Cheng and J. Hudson, Macromolecules, 2002, 35, 2494 CrossRef CAS.
- J. J. Harrison, D. C. Young and C. L. Mayne, J. Org. Chem., 1997, 62, 693 CrossRef CAS PubMed.
- P. Dimitrov, J. Emert, J. Hua, S. Keki and R. Faust, Macromolecules, 2011, 44, 1831 CrossRef CAS.
- H. P. Rath, US Pat., 5286823, BASF AG, 1994.
- H. P. Rath, F. Hoffmann, P. Reuter and H. Mach, US Pat., US5191044, 1993.
- S. Aoshima and S. Kanaoka, Chem. Rev., 2009, 109, 5245 CrossRef CAS PubMed.
- D. C. Pepper, in Friedel-Crafts and Related Reactions, ed. G. A. Olah, Interscience, New York, 1964, ch. 30 Search PubMed.
- J. P. Kennedy, Cationic Polymerization of Olefins: A Critical InVentory, John Wiley and Sons, New York, 1975 Search PubMed.
- G. Odian, Principles of Polymerization, Wiley Interscience, 2004, ISBN: 978-0-471-27400-1 Search PubMed.
- G. A. Mortimer and L. C. Arnold, J. Polym. Sci., Part A: Gen. Pap., 1964, 2, 4247 CrossRef.
- R. Faust and J. P. Kennedy, Polym. Bull., 1986, 15, 317 CrossRef CAS.
- M. Miyamoto, M. Sawamoto and T. Higashimura, Macromolecules, 1984, 17, 265 CrossRef CAS.
- M. Miyamoto, M. Sawamoto and T. Higashimura, Macromolecules, 1984, 17, 2228 CrossRef CAS.
- Cationic Polymerizations: Mechanism, Synthesis, and Applications, ed. K. Matyjaszewski, Marcel Dekker, New York, 1996 Search PubMed.
- R. Faust and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 1847 CrossRef CAS.
- M. Sawamoto, Prog. Polym. Sci., 1991, 16, 111 CrossRef CAS.
- J. P. Kennedy and B. Iván, Designed Polymers by Carbocationic Macromolecular Engineering: Theory and Practice, Oxford University Press, New York, 1992 Search PubMed.
- P. Sigwalt, Some still unsolved problems in carbocationic polymerization, Macromol. Symp., 1998, 132, 127 CrossRef CAS.
- H. Mayr, in Ionic Polymerizations and Related Processes; NATO Science Series E, ed. J. E. Puskas, Kluwer Academic Publishers, Dordrecht/Boston/London, 1999, vol. 359, p. 99 Search PubMed.
- D. Cook, Infrared spectra of xanthone: Lewis acid complexes, Can. J. Chem., 1963, 41, 522 CrossRef CAS.
- J. P. Kennedy and B. Iván, Designed Polymers by Carbocationic Macromolecular Engineering, Hanser Publishers, Munich, Vienna, New York, Barcelona, 1991 Search PubMed.
- Z. S. Fodor and R. Faust, J. Macromol. Sci., Part A: Pure Appl.Chem., 1998, 35(2), 375 CrossRef.
- R. M. Thomas, I. E. Lightbown, W. J. Sparks, K. P. Frolich and E. L. Murphree, Ind. Eng. Chem., 1940, 32, 1283 CrossRef CAS.
- R. M. Thomas, W. J. Sparks, P. K. Frolich and M. Otto, J. Am. Chem. Soc., 1940, 62, 276 CrossRef CAS.
- R. M. Thomas and W. J. Sparks, US Pat., US2356128, 1944.
- R. Faust and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 1986, 25, 1847 CrossRef.
- S. Ouardad, A. Deffieux and F. Peruch, Pure Appl. Chem., 2012, 84, 2065 CAS.
- V. A. Rozentsvet, V. G. Kozlov and Y. B. Monakov, Cationic Polymerization of conjugated dienes, Nauka, Moscow, 2011 Search PubMed.
- S. Ouardad, M.-E. Bakleh, S. V. Kostjuk, F. Ganachaud, J. E. Puskas, A. Deffieux and F. Peruch, Polym. Int., 2012, 61, 149 CrossRef CAS.
- J. J. Harrison, D. C. Young and C. L. Mayne, J. Org. Chem., 1997, 62, 693 CrossRef CAS PubMed.
- M. Mueller-Cunradi and M. Otto, US Pat., US2203873, 1940.
- E. J. Goethals and F. Du Prez, Prog. Polym. Sci., 2007, 32, 220 CrossRef CAS.
- S. Penczek, Makromol. Chem., Rapid Commun., 1992, 13, 147 CrossRef CAS.
- T. Pernecker, J. P. Kennedy and B. Iván, Macromolecules, 1992, 25, 1642 CrossRef CAS.
- T. Pernecker and J. P. Kennedy, Polym. Bull., 1992, 29, 27 CrossRef CAS.
- M. Kamigaito, Y. Maeda, M. Sawamoto and T. Higashimura, Macromolecules, 1993, 26, 1643 CrossRef CAS.
- J. V. Crivello and K. Dietliker, Photoinitiators for Free Radical Cationic and Anionic Photopolymerization, ed. G. Bradley, Wiley, New York, 1998, p. 53 Search PubMed.
- J. V. Crivello, Des. Monomers Polym., 2002, 5, 141 CrossRef CAS.
- C. C. Chen, J. Si and J. P. Kennedy, J. Macromol. Sci., Part A: Pure Appl.Chem., 1992, 29, 669 CrossRef.
- J. E. Puskas and M. Grassmuller, Makromol. Chem., Macromol. Symp., 1998, 132, 117 CrossRef CAS.
- B. Rajabalitar, H. A. Nguyen and H. Cheradame, Macromolecules, 1996, 29, 514 CrossRef.
- M. Bahadur, T. D. Shaffer and J. R. Ashbaugh, Macromolecules, 2000, 33, 9548 CrossRef CAS.
- S. Hadjikyriacou, M. Acar and R. Faust, Macromolecules, 2004, 37, 7543 CrossRef CAS.
- J. P. Kennedy, V. S. C. Chang, R. A. Smith and B. Ivan, Polym. Bull., 1979, 1, 575 CrossRef CAS.
- J. P. Kennedy and R. A. Smith, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 1523 CrossRef CAS.
- M. K. Mishra, B. Sar-Mishra and J. P. Kennedy, Polym. Bull., 1985, 13, 435 CrossRef CAS.
- B. Ivan and J. P. Kennedy, Macromolecules, 1990, 23, 2880 CrossRef CAS.
- L. Wilczek and J. P. Kennedy, Polym. Bull., 1987, 17, 37 CrossRef CAS.
- L. Wilczek and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 3255 CrossRef CAS.
- B. Ivan and J. P. Kennedy, Polym. Mater. Sci. Eng., 1988, 58, 869 CAS.
- B. Ivan and J. P. Kennedy, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 89 CrossRef CAS.
- L. V. Nielsen, R. R. Nielsen, B. Gao, J. Kops and B. Ivan, Polymer, 1997, 38, 2529 CrossRef CAS.
- M. Roth and H. Mayr, Macromolecules, 1996, 29, 6104 CrossRef CAS.
- L. K. Kemp, J. E. Poelma, T. R. Cooper and R. F. Storey, J. Macromol. Sci., Part A: Pure Appl.Chem., 2008, 45, 137 CrossRef CAS.
- K. L. Simison, C. D. Stokes, J. J. Harrison and R. F. Storey, Macromolecules, 2006, 39, 2481 CrossRef CAS.
- D. Held, B. Ivan, A. H. E. Müller, F. de Jong and T. Graafland, ACS Symp. Ser., 1997, 665, 63 CrossRef CAS.
- Y. C. Bae and R. Faust, Macromolecules, 1997, 30, 7341 CrossRef CAS.
- M. Vierle, Y. Zhang, E. Herdtweck, M. Bohnenpoll, O. Nuyken and F. E. Kühn, Angew. Chem., Int. Ed., 2003, 42, 1307 CrossRef CAS PubMed.
- A. Guerrero, K. Kulbaba and M. Bochmann, Macromolecules, 2007, 40, 4124 CrossRef CAS.
- A. K. Hijazi, N. Radhakrishnan, K. R. Jain, E. Herdtweck, O. Nuyken, H.-M. Walter, P. Hanefeld, B. Voit and F. E. Kühn, Angew. Chem., Int. Ed., 2007, 46, 7290 CrossRef CAS PubMed.
- M. R. Lichtenthaler, A. Higelin, A. Kraft, S. Hughes, A. Steffani, D. A. Plattner, J. M. Slattery and I. Krossing, Organometallics, 2013, 32, 6725 CrossRef CAS.
- H. Y. Yeong, Y. Li, F. E. Kühn and B. Voit, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 158–167 CrossRef CAS.
- S. V. Kostjuk, F. N. Kaputsky, V. P. Mardykin, L. V. Gaponik and L. M. Antipin, Polym. Bull., 2002, 49, 251 CrossRef CAS.
- S. V. Kostjuk, Polym. Bull., 2004, 51, 277 CrossRef CAS.
- S. V. Kostjuk, A. Yu. Dubovik, I. V. Vasilenko, A. N. Frolov and F. N. Kaputsky, Eur. Polym. J., 2007, 43, 968 CrossRef CAS.
- A. N. Frolov, S. V. Kostjuk, I. V. Vasilenko and F. N. Kaputsky, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3736 CrossRef CAS.
- P. W. Ambler, P. K. G. Hodgson and N. J. Stewart, EP 0558187, BP Chem. Ltd, 1993.
- A. Abdul-Sada, P. W. Ambler, P. K. G. Hodgson, K. R. Seddon and N. J. Stewart, WO 9521871, BP Chem. Ltd, 1995.
- V. Murphy, WO 00/32685, Symyx Tech. Inc., 2000.
- A. Abdul-Sada, K. P. Seddon and N. J. Stewart, WO 9521872, BP Chem. Ltd, 1995.
- X. Li, Y. Wu, J. Zhang, S. Li, M. Zhang, D. Yang, H. Wang, Y. Shang, W. Guo and P. Yan, Polym. Chem., 2019, 10, 201 RSC.
- D. I. Shiman, I. V. Vasilenko and S. V. Kostjuk, Polym. Chem., 2019, 10, 5998 RSC.
| This journal is © The Royal Society of Chemistry 2020 |