
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of temperature-controlled chiral hybrid structures constructed from copper(II)-monosubstituted Keggin polyoxoanions and copper(II)-organoamine complexes in enantioselective sensing of tartaric acid†
Mu-Xiu Yang,
Meng-Jie Zhou,
Jia-Peng Cao,
Ye-Min Han,
Ya-Lin Hong and
Yan Xu *
*
College of Chemical Engineering, State Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, Nanjing 210009, P. R. China. E-mail: yanxu@njtech.edu.cn
First published on 4th April 2020
Abstract
Temperature usually occupies a crucial position in the construction of chiral compounds. By controlling the temperature of the reaction system, chiral and non-chiral compounds can be designed and synthesized. Given the above, three new chiral and non-chiral compounds based on copper(II) monosubstituted polyoxoanions and Cu(en) complexes (en = ethylenediamine), D/L-[Cu(H2O)(en)2]2{[Cu(H2O)2(en)][SiCuW11O39]}·5H2O (1, D-1 and L-1) and [Cu(H2O)(en)2]{[Cu(en)2]2[SiCuW11O39]}·2.5H2O (2), were successfully synthesized under hydrothermal conditions. The main synthesis conditions of compound 1 (D-1 and L-1) and compound 2 are the same, however, the only difference is that the reaction temperatures are 80 °C and 140 °C, respectively. What's more, compounds 1 and 2 can form a 1D chiral chain by Cu–O and W/Cu–O–W/Cu bonds, respectively, and further obtain a 3D-supramolecular framework through hydrogen bonding interaction. Meanwhile, due to the asymmetry of chiral compound 1, optical second-harmonic generation (SHG) was used to investigate the second-order nonlinear optical effect and it was found that the observed SHG efficiency of compound 1 is 0.3 times that of urea. To further investigate the chiral properties, D-1 and L-1 were used in the electrochemical enantioselective sensing of D-/L-tartaric acid (D-/L-tart) molecules, respectively, which demonstrates that D-1 and L-1 have a good application prospect in sensing chiral substances.
Introduction
Polyoxometalates (POMs), one of the most significant metal–oxygen cluster materials, have raised many concerns because of their structural variability and potential applications in many fields of nonlinear optics,1 catalysis,2 magnetism,3 materials science,4 luminescence,5 etc.Chiral polyoxometalates (CPOMs), as a special subclass of POMs, contain intriguing architectures, and are a good research prospect.6 As the previous reported literature,7 chiral POM-based frameworks can be approached via two synthetic strategies. The first method can be realized by the use of enantiomerically pure chiral inducement, including chiral metal–organic fragments and organic ligands.8 The second tactic is using achiral precursors via spontaneous resolution to yield a polymer consisting of a pair of enantiomers, while the entire polymer is racemic.9 Notably, most efforts so far have been made to explore the first method, while the second method has rarely been developed. Although many groups have committed to synthesize chiral POMs through spontaneous resolution, and the studies of achiral structures to chiral structures in metal–organic frameworks (MOFs) have developed rapidly in the last few decades,10 limited examples of POMs in such aspects have been reported.11 The information associated with the key factors which dominates the construction of chiral and achiral compounds has also been uncommonly reported until now. Moreover, spontaneous resolution cannot be predicted in advance since the laws of physics which determines the process outputs have not been completely understood. Although there are many examples of chiral MOFs12 as well as their spontaneous resolution,13 it is more difficult to design and build chiral POM-based compounds. The data show that merely 5–10% racemates can become the conglomerate, indicating that spontaneous resolution of achiral components in chiral POM chemistry is a great challenge.14
To the best of our knowledge, temperature usually occupies a crucial position in the construction of POMs. Furthermore, by adjusting the reaction temperature, chiral or non-chiral POMs compounds can be synthesized.10a,11 Zheng's group have reported a chiral vanadium phosphonate compound, synthesising from non-chiral materials at 120 °C. Upon heating, chirality of the compound releases and leads to an achiral compound.15 An and co-workers documented the [Co2Mo10H4O38]6− as a model and constructed chiral and non-chiral compounds at 85 °C and 25 °C, respectively, which is an intriguing example of chiral POMs to achiral POMs transformation.16 In addition, the chiral POMs had good applications in catalysis, enantioselective identification, non-linear optics, etc.,6,17 but the investigation of chiral POMs is still relatively limited. Thus, it is urgent to explore the property of chiral POMs.
Given the above mentioned, we designed and synthesized chiral conglomerates D- and L-[Cu(H2O)(en)2]2{[Cu(H2O)2(en)][SiCuW11O39]}·5H2O (D-1, L-1), synthesising from achiral precursors via spontaneous resolution, obtaining at 80 °C. While compound [Cu(H2O)(en)2]{[Cu(en)2]2[SiCuW11O39]}·2.5H2O (2) was obtained at 140 °C, and the chirality disappeared, becoming an achiral compound. They represent the example of chiral compounds to achiral compounds in the area of POMs and it also manifests that the reaction temperature is a crucial factor to mediate the formation of achiral compounds and chiral compounds. The chiral POMs were obtained from achiral precursors via the spontaneous resolution method by controlling the temperature, which may have a certain guiding effect on the subsequent chiral POMs synthesis. In addition, electrochemical measurements demonstrate that carbon paste electrodes prepared by D- and L-1 (D- and L-cpe) exhibited remarkable electrochemical enantioselective sensing to chiral substances (D/L-tart molecule).
Experimental section
Materials and physical measurements
K8[α-SiW11O39]·13H2O was prepared according to the literature,18 while other chemicals were purchased and put to use without further purification. Elemental analyses (C, H, and N) were performed with an elemental analyzer (PerkinElmer 2400). IR spectra of compounds 1–2 were recorded on a Nicolet Impact 410 FTIR spectrometer in the range of 400–4000 cm−1 with pressed KBr pellets. In flowing N2 atmosphere, thermogravimetric measurement (TG) was performed on a diamond thermogravimetric analyzer, and the test temperature increased from 25 to 700 °C at a rate of 10 °C min−1. Bruker D8X diffractometer equipped with monochromatized Cu-Kα (λ = 1.5418 Å) radiation was used to record Powder X-ray diffraction (PXRD) at room temperature, and data were collected in the range of 5° to 50°. The circular dichroism spectra were obtained with the use of JASCOJ-810 spectropolarimeter.X-ray crystallography
Bruker Apex II CCD using Mo-Kα radiation (λ = 0.71073 Å) was applied to collect the crystallographic analyses data of compounds 1 and 2 under room temperature. In addition, the SHELX-2014 program package was taken to resolve the crystal structures with direct methods, and F2-based full-matrix least-squares methods were used to further refine the crystal structure. Refine the H atoms of organic molecules with specified isotropic thermal parameters, but hydrogen atoms for water were not situated. Compared with W atoms, the N, C and O atoms are very light, and not stable during refinement. The thermal parameters of some N, C and O atoms are restrained. The structural determination and crystal data for three compounds are disposed and presented in Table 1. Selected bond lengths (Å) and angles (°) and hydrogen bonds are shown in Tables S2–S4 and Tables S5–S7,† respectively.| Compound | D-1 | L-1 | 2 |
|---|---|---|---|
| a R1 = ∑‖Fo| − |Fc‖/∑|Fo|.b wR2 = ∑[w(F2o − F2c)2]/∑[w(F2o)2]1/2. | |||
| Formula | C10H58N10O48SiW11Cu4 | C10H58N10O48SiW11Cu4 | C12H55N12O42.5SiW11Cu4 |
| Formula weight | 3391.26 | 3391.26 | 3352.28 |
| Crystal system | Triclinic | Triclinic | Monoclinic |
| Space group | P1 | P1 | P2(1)/c |
| a (Å) | 10.970(4) | 10.9871(11) | 18.900(5) |
| b (Å) | 12.408(4) | 12.3354(12) | 23.208(6) |
| c (Å) | 12.818(4) | 12.7954(13) | 26.175(7) |
| α (deg.) | 101.383(4) | 101.8070(10) | 90 |
| β (deg.) | 111.028(4) | 110.3310(10) | 96.397(3) |
| γ (deg.) | 108.089(4) | 107.7180(10) | 90 |
| V (Å3) | 1451.3(8) | 1452.5(3) | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 410(5) 410(5) |
| Z | 1 | 1 | 8 |
| Dc (mg m−3) | 3.880 | 3.877 | 3.903 |
| F (000) | 1516 | 1516 | 11960 |
| Crystal size (mm3) | 0.120 × 0.100 × 0.100 | 0.130 × 0.110 × 0.100 | 0.150 × 0.130 × 0.120 |
| Limiting indices | −13 ≤ h ≤ 12, −14 ≤ k ≤ 14, −12 ≤ l ≤ 15 | −13 ≤ h ≤ 12, −14 ≤ k ≤ 14, −15 ≤ l ≤ 15 | −22 ≤ h ≤ 22, −27 ≤ k ≤ 27, −31 ≤ l ≤ 31 |
| Reflections collected/unique | 10382/8081 |
10722/8269 |
79061/20131 |
| R(int) | 0.0478 | 0.0329 | 0.2186 |
| Data/restraints/parameters | 8081/456/748 | 8269/445/757 | 20131/589/1486 |
| GOF | 1.029 | 1.100 | 1.052 |
| Absolute structure parameter | 0.06(2) | 0.02(2) | |
| R1a, wR2b [I > 2σ(I)] | R1 = 0.0517, wR2 = 0.1249 | R1 = 0.0483, wR2 = 0.1304 | R1 = 0.0718, wR2 = 0.1717 |
| R1, wR2 (all data) | R1 = 0.0565, wR2 = 0.1356 | R1 = 0.0591, wR2 = 0.1697 | R1 = 0.1031, wR2 = 0.1841 |
Synthesis of compounds
(D-1 and L-1). Cu(OAc)2·H2O (0.1002 g, 0.5 mmol) and K8[α-SiW11O39]·13H2O (0.3012 g, 0.094 mmol) were placed into 3 ml ethanol and 6 ml CH3COOH–CH3COONa buffer solution and stirred for 0.5 h. Then ethylenediamine (0.0502 g 0.83 mmol) was added drop by drop to the previous mixture and continued to be stirred for about 2 hours. The mixed solution was poured into a 25 ml Teflon-lined autoclave and then put in the 80 °C oven for 7 days. After cooling to room temperature, purple plate crystals were collected by washing with distilled water (yield 47.2% based on W). Elemental anal. found (%): C, 3.55; H, 1.42; N, 4.14. Calcd (%): C, 3.54; H, 1.71; N, 4.13.
Results and discussion
Synthesis
There are many methods for synthesizing POM-based compounds, among which hydrothermal synthesis is a commonly used and effective method. The reaction process of hydrothermal synthesis is complicated and unpredictable, and the final products will be certainly influenced by plenty of factors. Typical factors include starting substances (the type of reagents, concentrations of solvents, etc.) and the reaction conditions (temperature, pH, time, etc.). In our experiment, the reaction temperature is a crucial factor of the reaction system, contributing to the synthesis of three compounds. The main synthesis conditions of compound 1 (D-1 and L-1) and compound 2 are the same, apart from the reaction temperature. D-1 and L-1 were obtained at lower temperature 80 °C, with an increase of the temperature to 140 °C, compound 2 was formed.The IR spectra of compounds 1–2 were measured in the range of ν = 4000–400 cm−1 (Fig. S5 and S6†). The characteristic peaks at 790, 895 cm−1 (1) and 792, 897 cm−1 (2) for ν(W–O–W); 946 cm−1 (1) and 940 cm−1 (2) for ν(W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1102 cm−1 (1) and 1033 cm−1 (2) for ν(Si–O). Additionally, in the range of 1383–1593 cm−1 (1) and 1383–1590 cm−1 (2) could be ascribed to the characteristic peaks of the ethylenediamine molecules. Whereas the broad peak at about 3449 cm−1 (1) and 3464 cm−1 (2) demonstrates the presence of water molecules. The peaks discussed above are corresponding to the result of single crystal structural analyses.
O); 1102 cm−1 (1) and 1033 cm−1 (2) for ν(Si–O). Additionally, in the range of 1383–1593 cm−1 (1) and 1383–1590 cm−1 (2) could be ascribed to the characteristic peaks of the ethylenediamine molecules. Whereas the broad peak at about 3449 cm−1 (1) and 3464 cm−1 (2) demonstrates the presence of water molecules. The peaks discussed above are corresponding to the result of single crystal structural analyses.
As shown in Fig. S9 and S10,† the experimental PXRD patterns of samples 1–2 had relatively good agreement with the simulated PXRD patterns, while only slight differences in intensity were observed, revealing that compounds 1 and 2 have better phase purity.
Structure of D- and L-[Cu(H2O)(en)2]2{[Cu(H2O)2(en)][SiCuW11O39]}·5H2O (D-1 and L-1) (1)
Due to crystallographic data of compounds D-1 and L-1 collected from single-crystal X-ray diffractometer are almost same, and the structures of the D-1 and L-1 are mirror symmetrical. Hence, we just take D-1 as an example to analyze the structure detailedly. Compound D-1 crystallizes in the chiral triclinic crystal system with a space group P1. The basic unit of D-1 contains a {[Cu(H2O)2(en)][SiCuW11O39]}4− anion (Fig. 1), two independent [Cu(H2O)(en)2]2+ cations, five lattice water molecules. Four separate copper cations adopt two different types of coordination geometry patterns. Cu1 coordination cation incorporated into the defect site of the polyoxoanion [α-SiW11O39]8− is six-coordinated, possessing a distorted octahedron geometry, in which five coordination oxygen atoms are derived from one O-donor [α-SiW11O39]8− and the last one coordination oxygen atom from the adjacent polyoxoanion. Both Cu3 and Cu4 in the independent [Cu(H2O)(en)2]2+ cation display pentacoordinate distorted square pyramid geometry, which are coordinated with four nitrogen atoms from two ethylenediamine molecules and one coordination oxygen atom from lattice water molecules. The coordination mode around Cu2 in the [Cu(H2O)2(en)]2+ cation is the same as that of Cu3 and Cu4, the distinction is wherein the source of the coordination oxygen atoms, two of them are supplied by lattice water molecules and the last one is provided by the O-donor polyoxoanion (Fig. S1†). Many hybrid materials based on monosubstituted Keggin anions are achiral as well as show subtle type of disorder in the solid state. It is worth mentioning that the hybrid materials constructed from [SiCuW11O39]6− anions in this article are chiral, which is different from the previous reports. The Flack parameters (0.06(2) for D-1, 0.02(2) for L-1) and circular dichroism spectroscopy also fully demonstrate that the chiral absolute configurations of D-1 and L-1 are correct. | ||
| Fig. 1 The {[Cu(H2O)2(en)][SiCuW11O39]}4− anion of L-1 (a) and D-1 (b) (all H atoms omitted for clarity). | ||
In D-1, chiral subunits {[Cu(H2O)2(en)][SiCuW11O39]}4− are bridged together to come into being a 1D chiral chain through Cu1–O19 (Fig. 2). The bond length of Cu1–O19 interaction in compound 1 is 2.40(2) Å. Due to the axial extension of the Jahn–Teller effect of copper(II) ions, which is considered that a usual coordination bond is formed between Cu1–O19. And the adjacent right-handed chains are joined up together by strong hydrogen bonds (N–H⋯O), generating a 2D supramolecular chiral layer (Fig. 3). Moreover, 2D chiral sheets are linked together through hydrogen-bonding interactions (N–H⋯O) to come into being a 3D supramolecular chiral network (Fig. 4). The bond lengths of Cu–O and Cu–N are in the scope of 1.950(2)–2.440(3) Å and 1.948(4)–2.055(3) Å, respectively. The valence of the Cu cations is +2, which is obtained from the bond valence sum (BVS) calculations (Table S1†). Compound 1 can be manually separated under a polarizing microscope.
 | ||
| Fig. 2 Polyhedral and ball-and-stick representation of the 1D right-handed chain in D-1. | ||
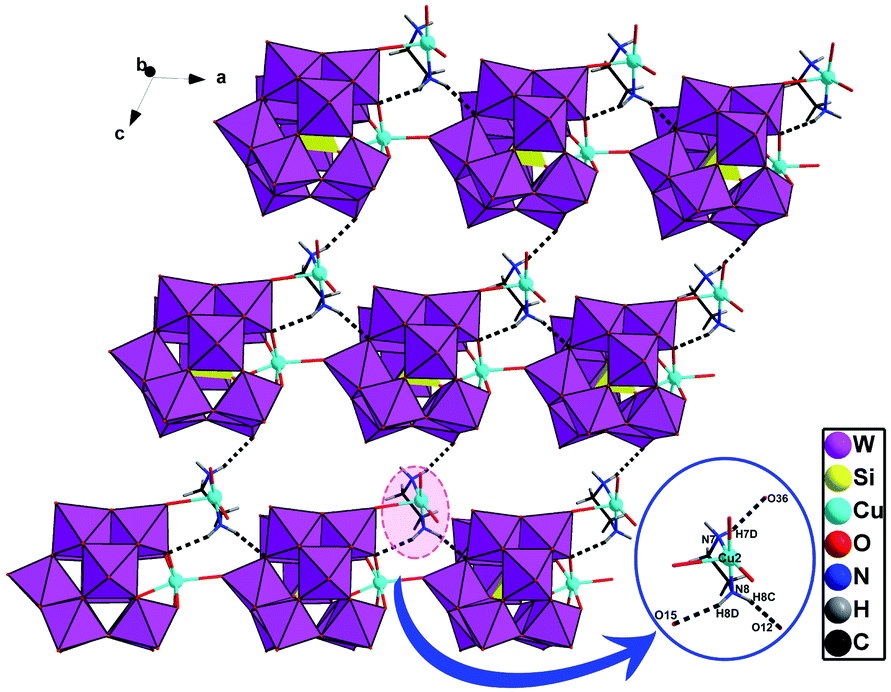 | ||
| Fig. 3 The 2D homochiral sheet in D-1 (the hydrogen-bonding between the polyoxoanions and organic ligands are demonstrated). | ||
 | ||
| Fig. 4 The 3D supramolecular homochiral network in D-1 (the hydrogen-bonding between the polyoxoanions and organic ligands are demonstrated). | ||
Structure of [Cu(H2O)(en)2]{[Cu(en)2]2[SiCuW11O39]}·2.5H2O (2)
When adjusting the reaction temperature from 80 °C to 140 °C under the similar conditions, achiral compound 2 was obtained. Compound 2 crystallizes in the monoclinic system and space group P21/c. The asymmetric structural unit of compound 2 is occupied by a {[Cu(en)2]2[(SiW11CuO39)]}2− anion, one free [Cu(H2O)(en)2]2+ cation, and 2.5 lattice water molecules (Fig. 5). The coordination environments of four Cu2+ cations in compound 2 are similar to that of compound 1. In addition, Cu and W atoms in [(SiW11CuO39)]6− anion are statistically disordered, occupying half of the Cu/W positions in the cluster, respectively. | ||
| Fig. 5 Polyhedral and ball-and-stick representation of asymmetric unit in compound 2. | ||
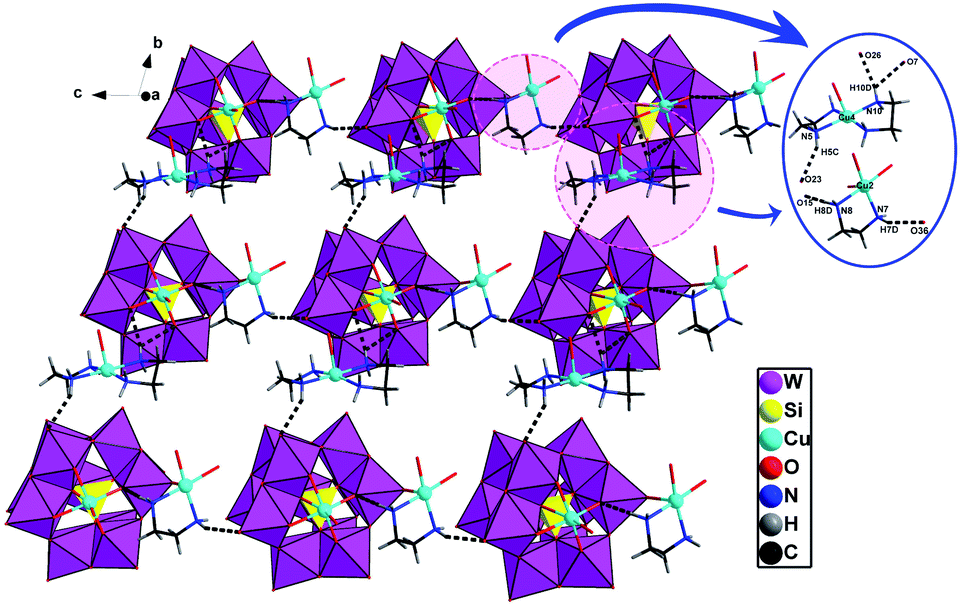
As shown in Fig. 6, turning one [(SiW11CuO39)]6− ion ∼45° around the anionic equatorial plane and then connected the adjacent planar [(SiW11CuO39)]6− ion by terminal oxygen atoms of [(SiW11CuO39)]6− ion, generating a one-dimensional chain construction. The bond length and the bond angle of W/Cu–O–W/Cu were 3.7779(26) Å and 158.253(13)°, respectively. Adjacent chiral chains are interconnected by hydrogen-bonding (N–H⋯O) to produce a 2D supramolecular achiral layer (Fig. 7). Furthermore, these 2D sheets are further connected into 3D open framework by hydrogen bonds (Fig. 8).
 | ||
| Fig. 6 Polyhedral representation of the 1D chain in 2 (the dimethylamine molecules and H atoms are omitted for clarity). | ||
 | ||
| Fig. 7 2D achiral sheet in compound 2 (the hydrogen-bonding between the polyoxoanions and organic ligands are demonstrated). | ||
 | ||
| Fig. 8 View of 3D supramolecular achiral framework in 2 (all dimethylamine molecules and H atoms omitted for clarity), the hydrogen-bonding interactions between the adjacent 2D achiral sheets. | ||
CD spectrum
The crystal samples of compound 1 show dark and bright forms under polarizing microscope with polarized light (Fig. 9), while their crystal colors, shapes and sizes are almost identical under normal microscopes. Therefore, D-1 and L-1 can be manually separated under a polarizing microscope to further measure their solid state circular dichroism (CD) spectra,19 which is employed to further discuss the chiral optical activity of them. The CD spectra of D-1 and L-1 samples are symmetrical to each other (Fig. 10). Within the range of 150–700 nm, D-1 first presented a negative Cotton effects at λ = 281 nm, and then presented a positive Cotton effects λ = 475 nm. As expected, at the same wavelength, L-1 exhibits the opposite cotton effects to D-1, demonstrating that they are enantiomers.20 | ||
| Fig. 9 The display of light D-1 and dark L-1 under polarizing microscope. | ||
 | ||
| Fig. 10 CD spectra for D-1 and L-1. | ||
Nonlinear optical measurement
Nonlinear optical properties belong to a research direction of chiral polyoxometalates.17d–g Considering the asymmetry of chiral compound 1, optical second-harmonic generation (SHG) was used to investigate second-order nonlinear optical effect of mixed crystal samples (D-1 and L-1). The SHG intensity data of compound 1 was measured on the Q-switched Nd:YAG laser (λ = 1064 nm), and microcrystalline urea was selected as standard nonlinear optical material. The result showed that the observed SHG efficiency of compound 1 is 0.3 times that of urea.Meanwhile, third-order NLO properties of D-1 and L-1 were investigated by the open-aperture Z-scan technique. Fig. S11 and S12† showed the Z-scan data of V measurements of D-1 and L-1, respectively. The TPA absorption coefficient β of D-1 and L-1 were calculated as 0.0337 and 0.0279 cm/GW, respectively. Furthermore, the TPA cross section σ was computed as 805 GM for D-1 and 817 GM for L-1 (1 GM = 10−50 cm4 s per photon). Ju also reported similar work,17c and the 2 PA cross section σ of D-1 and L-1 was 392 GM and 389 GM, respectively, which lower than that of this work. Based on this, chiral compound 1 is expected to be applied in the field of nonlinear optics.
Electrochemical measurements
The electrochemical impedance spectroscopy (EIS) measurements and linear sweep voltammogram (LSV) tests of D-1 and L-1 were carried out, in order to evaluate their electrochemical enantioselective sensing abilities for chiral substances (D/L-tart molecule). According to the previous literature,21 a carbon paste working electrode D/L-CPE (D/L-1 (20 mg) and graphite powder (0.2 g)) was established, and the electrochemical measurement of D/L-cpe was carried out by the conventional three-electrode method. As shown in Fig. 11, the EIS Nyquist plots and LSV curves at different stages were studied on the electrode surface of D-CPE, L-CPE and bare CPE. Obviously, in the mixture of Na2SO4 (9 ml, 1 M) and D/L-tart (1 ml, 0.1 M) solutions, the EIS Nyquist plot of D-CPE for D-tart appeared a semicircle at the beginning, indicating that an interfacial charge transfer was taking place between D-CPE and Na2SO4 solution (Fig. 11a, red line).22 However, the EIS Nyquist plot of D-CPE for L-tart only showed a straight line in the whole region, demonstrating that only mass diffusion occurred on the D-CPE surface. Beyond that, the same process takes place in the L-CPE system (Fig. 11a, black line). When l-1 was used as the electrode material, the EIS Nyquist plot of L-CPE for L-tart appeared a semicircle at the beginning, showing that typical interfacial charge transfer was taking place between L-CPE and the mixture of Na2SO4 (9 ml, 1 M) and L-tart (1 ml, 0.1 M) solutions (Fig. 11b, black line). While the interfacial electron transfer of L-CPE for D-tart was not observed in the mixture of Na2SO4 (9 ml, 1 M) and D-tart (1 ml, 0.1 M) solutions (Fig. 11b, red line). | ||
| Fig. 11 EIS and LSV of D-CPE (a and d), L-CPE (b and e) and bare CPE (c and f) in the mixture of Na2SO4 (9 ml, 1 M) and D/L-tart (1 ml, 0.1 M) solutions at the sweep rate of 100 mV s−1 (D-tart and L-tart for red and black line, respectively). | ||
In order to further prove that chiral compounds have electrochemical enantioselective sensing ability, a blank experiment was carried out. After replacing D-CPE and L-CPE with bare CPE, the EIS Nyquist plots of bare CPE for D- and L-tart only showed straight lines in the whole process (Fig. 11c), which was a typical feature of mass diffusion process, indicating that bare CPE had no enantioselective sensing for D- and L-tart. At the same time, the LSV of D-1 and L-1 were also analyzed to fully confirm their enantioselective sensing ability for D/L-tart molecules. In the mixed solution of Na2SO4 and D/L-tart, the LSV curve of bare CPE showed that almost no current was generated (Fig. 11f), while the LSV curve of D/L-CPE exhibited peak current in the range of 0–0.6 V (Fig. 11d and e). In addition, the peak current of D-CPE for D-tart is higher than that of L-tart, and the peak current of L-CPE for L-tart is higher than that of D-tart. The result indicates that D-CPE has better enantioselective sensing ability to D-tart, L-CPE has better enantioselective sensing ability to L-tart. However, bare CPE has no selectivity for D- and L-tart molecules.
Taking above discussion into consideration, it can be concluded that the D-CPE possesses enantioselective sensing ability toward the D-tart. Meanwhile, the L-CPE exhibits good enantioselective sensing performance toward the L-tart. LSV tests of D-CPE and L-CPE were consistent with that of impedance experiments, which further corroborate the electrochemical enantioselective sensing abilities of D-1 and L-1 for D/L-tart. In addition, By introducing D-1 or L-1 into the electrode can also facilitate the electron transfer and promotes enantioselective ability for D/L-tart comparing with the bare CPE.
Conclusions
In summary, in the reaction system of polyoxoanion [α-SiW11O39]8−, copper cations and ethylenediamine ligands by changing the temperatures, compounds 1 and 2 have been designed and synthesized. Samples 1 and 2 are the rare examples of temperature-controlled compounds in the field of POMs, which exhibit as well as good nonlinear optical properties. Moreover, the EIS and LSV measurements have revealed that the D-CPE and L-CPE possess a remarkable enantioselective sensing abilities and electrocatalytic abilities toward D-tart and L-tart, respectively. In light of the success in constructing three compounds, we would not only further synthesize new chiral POMs with spontaneous resolution, but also apply deeper researches to these compounds to develop multifunctional materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant 21571103) and Jiangsu province (BK20191359) the Major Natural Science Projects of the Jiangsu Higher Education Institution (Grant 16KJA150005).Notes and references
- (a) A. Dolbecq, E. Dumas, C. R. Mayer and P. Mialane, Chem. Rev., 2010, 110, 6009–6048 CrossRef CAS PubMed; (b) Y. Dong, G. Hu, H. Miao, X. He, M. Fang and Y. Xu, Inorg. Chem., 2016, 55, 11621–11625 CrossRef CAS PubMed; (c) X.-M. Luo, N.-F. Li, Z.-B. Hu, J.-P. Cao, C.-H. Cui, Q.-F. Lin and Y. Xu, Inorg. Chem., 2019, 58, 2463–2470 CrossRef CAS PubMed.
- (a) S.-S. Wang and G.-Y. Yang, Chem. Rev., 2015, 115, 4893–4962 CrossRef CAS PubMed; (b) D.-L. Long, R. Tsunashima and L. Cronin, Angew. Chem., Int. Ed., 2010, 49, 1736–1758 CrossRef CAS PubMed; (c) L.-N. Zhang, S.-H. Li, H.-Q. Tan, S. U. Khan, Y.-Y. Ma, H.-Y. Zang, Y.-H. Wang and Y.-G. Li, ACS Appl. Mater. Interfaces, 2017, 9, 16270–16279 CrossRef CAS PubMed; (d) X. Wei, J. Wei, L. Huang, T. Yan and F. Luo, Inorg. Chem. Commun., 2017, 81, 10–14 CrossRef CAS; (e) M.-X. Jiang and C.-G. Liu, J. Phys. Chem. C, 2017, 121, 12735–12744 CrossRef CAS.
- (a) M. A. AlDamen, J. M. Clemente-Juan, E. Coronado, C. Martí-Gastaldo and A. Gaita-Arino, J. Am. Chem. Soc., 2008, 130, 8874–8875 CrossRef CAS PubMed; (b) U. Kortz, A. Müller, J. van Slageren, J. Schnack, N. S. Dalal and M. Dressel, Coord. Chem. Rev., 2009, 253, 2315–2327 CrossRef CAS; (c) D. Zhang, F. Cao, P. Ma, C. Zhang, Y. Song, Z. Liang, X. Hu, J. Wang and J. Niu, Chem.–Eur. J., 2015, 21, 17683–17690 CrossRef CAS PubMed; (d) J. Wang, Y. Niu, M. Zhang, P. Ma, C. Zhang, J. Niu and J. Wang, Inorg. Chem., 2018, 57, 1796–1805 CrossRef CAS PubMed.
- (a) E. Rafiee and S. Shahebrahimi, J. Mol. Struct., 2017, 1139, 255–263 CrossRef CAS; (b) H. Wu, Y.-Q. Zhang, M.-B. Hu, L.-J. Ren, Y. Lin and W. Wang, Langmuir, 2017, 33, 5283–5290 CrossRef CAS PubMed.
- (a) A. M. Kaczmarek, K. Van Hecke and R. Van Deun, Inorg. Chem., 2017, 56, 3190–3200 CrossRef CAS PubMed; (b) N. Shi, J. Tan, X. Wan, Y. Guan and J. Zhang, Chem. Commun., 2017, 53, 4390–4393 RSC; (c) A. Maalaoui, O. Pérez, M. Rzaigui and S. T. Akriche, J. Alloys Compd., 2017, 695, 1061–1072 CrossRef CAS.
- D.-Y. Du, L.-K. Yan, Z.-M. Su, S.-L. Li, Y.-Q. Lan and E.-B. Wang, Coord. Chem. Rev., 2013, 257, 702–717 CrossRef CAS.
- (a) H. Tan, Y. Li, Z. Zhang, C. Qin, X. Wang, E. Wang and Z. Su, J. Am. Chem. Soc., 2007, 129, 10066–10067 CrossRef CAS PubMed; (b) F. Xiao, J. Hao, J. Zhang, C. Lv, P. Yin, L. Wang and Y. Wei, J. Am. Chem. Soc., 2010, 132, 5956–5957 CrossRef CAS PubMed; (c) J. Zhang, J. Luo, P. Wang, B. Ding, Y. Huang, Z. Zhao, J. Zhang and Y. Wei, Inorg. Chem., 2015, 54, 2551–2559 CrossRef CAS PubMed.
- (a) Y. Wang, L. Shi, Y. Yang, B. Li and L. Wu, Dalton Trans., 2014, 43, 13178–13186 RSC; (b) L. Shi, B. Li and L. Wu, Chem. Commun., 2015, 51, 172–175 RSC; (c) R. Ishimoto, K. Kamata, K. Suzuki, K. Yamaguchi and N. Mizuno, Dalton Trans., 2015, 44, 10947–10951 RSC; (d) Y.-L. Wang, J.-W. Zhao, Z. Zhong, J.-J. Sun, X.-Y. Li, B.-F. Yang and G.-Y. Yang, Inorg. Chem., 2019, 58, 4657–4664 CrossRef CAS PubMed.
- (a) Y. Hou, H. An, T. Xu, S. Zhao and J. Luo, New J. Chem., 2016, 40, 10316–10324 RSC; (b) X. Wang, M.-M. Zhang, X.-L. Hao, Y.-H. Wang, Y. Wei, F.-S. Liang, L.-J. Xu and Y.-G. Li, Cryst. Growth Des., 2013, 13, 3454–3462 CrossRef CAS; (c) Z.-M. Zhang, X. Duan, S. Yao, Z. Wang, Z. Lin, Y.-G. Li, L.-S. Long, E.-B. Wang and W. Lin, Chem. Sci., 2016, 7, 4220–4229 RSC.
- (a) P. Cui, L. Ren, Z. Chen, H. Hu, B. Zhao, W. Shi and P. Cheng, Inorg. Chem., 2012, 51, 2303–2310 CrossRef CAS PubMed; (b) M. C. Bernini, F. Gandara, M. Iglesias, N. Snejko, E. Gutierrez-Puebla, E. V. Brusau, G. E. Narda and M. A. Monge, Chem.–Eur. J., 2009, 15, 4896–4905 CrossRef CAS PubMed.
- (a) H. An, L. Wang, Y. Hu and F. Fei, CrystEngComm, 2015, 17, 1531–1540 RSC; (b) H. An, L. Wang, Y. Hu, T. Xu and Y. Hou, Inorg. Chem., 2016, 55, 144–153 CrossRef CAS PubMed.
- (a) Y. Ma, Z. Han, Y. He and L. Yang, Chem. Commun., 2007, 4107–4109 RSC; (b) C. Zhuo, Y. Wen, S. Hu, T. Sheng, R. Fu, Z. Xue, H. Zhang, H. Li, J. Yuan, X. Chen and X. Wu, Inorg. Chem., 2017, 56, 6275–6280 CrossRef CAS PubMed; (c) X.-H. Bu, W. Chen, M. Du, K. Biradha, W.-Z. Wang and R.-H. Zhang, Inorg. Chem., 2002, 41, 437–439 CrossRef CAS PubMed; (d) J.-R. Li, Y. Tao, Q. Yu, X.-H. Bu, H. Sakamoto and S. Kitagawa, Chem.–Eur. J., 2008, 14, 2771–2776 CrossRef CAS PubMed.
- (a) I. Katsuki, Y. Motoda, Y. Sunatsuki, N. Matsumoto, T. Nakashima and M. Kojima, J. Am. Chem. Soc., 2002, 124, 629–640 CrossRef CAS PubMed; (b) B.-Q. Song, D.-Q. Chen, Z. Ji, J. Tang, X.-L. Wang, H.-Y. Zang and Z.-M. Su, Chem. Commun., 2017, 53, 1892–1895 RSC; (c) X.-D. Zheng, M. Zhang, L. Jiang and T.-B. Lu, Dalton Trans., 2012, 41, 1786–1791 RSC; (d) X.-L. Tong, T.-L. Hu, J.-P. Zhao, Y.-K. Wang, H. Zhang and X.-H. Bu, Chem. Commun., 2010, 46, 8543–8545 RSC.
- (a) J. Zhang, J. Hao, Y. Wei, F. Xiao, P. Yin and L. Wang, J. Am. Chem. Soc., 2010, 132, 14–15 CrossRef CAS PubMed; (b) Y. Hou, X. Fang and C. L. Hill, Chem.–Eur. J., 2007, 13, 9442–9447 CrossRef CAS PubMed; (c) S. Liu and X. Qu, Appl. Surf. Sci., 2017, 412, 189–195 CrossRef CAS.
- X.-J. Yang, S.-S. Bao, T. Zheng and L.-M. Zheng, Chem. Commun., 2012, 48, 6565–6567 RSC.
- H. An, Y. Hu, L. Wang, E. Zhou, F. Fei and Z. Su, Cryst. Growth Des., 2015, 15, 164–175 CrossRef CAS.
- (a) Q. Han, B. Qi, W. Ren, C. He, J. Niu and C. Duan, Nat. Commun., 2015, 6, 10007 CrossRef PubMed; (b) Q. Han, C. He, M. Zhao, B. Qi, J. Niu and C. Duan, J. Am. Chem. Soc., 2013, 135, 10186–10189 CrossRef CAS PubMed; (c) W.-W. Ju, H.-T. Zhang, X. Xu, Y. Zhang and Y. Xu, Inorg. Chem., 2014, 53, 3269–3271 CrossRef CAS PubMed; (d) J. D. Compain, P. Mialane, A. Dolbecq, J. Marrot, A. Proust, K. Nakatani, P. Yu and F. Sécheresse, Inorg. Chem., 2009, 48, 6222–6228 CrossRef CAS PubMed; (e) W. Cheng, F.-C. Shen, Y.-S. Xue, X. Luo, M. Fang, Y.-Q. Lan and Y. Xu, ACS Appl. Energy Mater., 2018, 1, 4931–4938 CrossRef CAS; (f) J. Cao, Y. Xiong, X. Luo, L. Chen, J. Shi, M. Zhou and Y. Xu, Dalton Trans., 2018, 47, 6054–6058 RSC; (g) J.-P. Cao, Y.-S. Xue, Z.-B. Hu, X.-M. Luo, C.-H. Cui, Y. Song and Y. Xu, Inorg. Chem., 2019, 58, 2645–2651 CrossRef CAS PubMed.
- A. Téazéa, G. Hervéa, R. G. Finke and D. K. Lyon, Inorg. Synth., 1990, 27, 85–96 Search PubMed.
- X. He, Y. Liu, Y. Lv, Y. Dong, G. Hu, S. Zhou and Y. Xu, Inorg. Chem., 2016, 55, 2048–2054 CrossRef CAS PubMed.
- G. Zhang, B. Wang, Y. Wei, Q. Zhang, K. Cai, X. Zhang, Z. Sun and Y. Shi, Chem. Commun., 2016, 69, 70–74 CAS.
- G. Zhang, H. Hu, H. Li, F. Zhao, Y. Liu, X. He, H. Huang, Y. Xu, Y. Wei and Z. Kang, CrystEngComm, 2013, 15, 3288–3291 RSC.
- (a) L. Hu, Y. Sun, Y. Zhou, L. Bai, Y. Zhang, M. Han, H. Huang, Y. Liu and Z. Kang, Inorg. Chem. Front., 2017, 4, 946–953 RSC; (b) Y. Zhang, L. Hu, Y. Sun, C. Zhu, R. Li, N. Liu, H. Huang, Y. Liu, C. Huang and Z. Kang, RSC Adv., 2016, 6, 59956–59960 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: TG curves, IR spectra, PXRD patterns, nonlinear optical measurement. CCDC 1837268–1837270 for compounds 1–2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01904f |
| This journal is © The Royal Society of Chemistry 2020 |