
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstrate-controlled selectivity switch in a three-component reaction: sequential synthesis of spiro-oxazolidinedione-cyclopentenones and hydroxy enaminobarbiturates in water†
Mohammad Bagher Teimouri *,
Mahdi Heydari and
Kazem Mohammadi
*,
Mahdi Heydari and
Kazem Mohammadi
Faculty of Chemistry, Kharazmi University, Mofateh Ave., Tehran, Iran. E-mail: teimouri@khu.ac.ir
First published on 3rd April 2020
Abstract
An efficient eco-friendly catalyst-free three-component domino multicyclization for the synthesis of new spirobicyclic oxazolidinedione containing cyclopentenone moieties has been established by mixing amines, β-dicarbonyl compounds and N,N′-dimethylalloxan in water at room temperature. This domino process involves multiple reactions such as enamination/aldol-like reaction/Stork enamine annulation/intramolecular cyclization under mild conditions.
Introduction
The efficient assembly of complex heterocyclic compounds with chemical and biomedical importance is a challenging and hot topic in modern organic chemistry.1,2 Among these heterocycles, the structurally diverse oxazolidinedione is a versatile component of many biologically active compounds. A great number of oxazolidinedione-containing pharmaceuticals are being used for therapeutic and clinical utilization. As representative compounds, paramethadione (Paradione®), and trimethadione (Tridione®) are two anticonvulsant medications in the oxazolinedione family used commonly to treat seizures and epileptic conditions.3 It has also been reported recently for their bioactivities such as hypoglycaemic and hypolipidemic agents,4 muscarinic agonists,5 anticonvulsants6 and insulin sensitizers with antidiabetic activities7 elastase inhibitor,8 serine protease inhibitor,9 mineralocorticoid receptor (MR) antagonist,10 and anti-cancer.11 Furthermore, spiro-fused oxazolidinediones were frequently reported to possess aldose reductase inhibitory properties.12Spirocyclic five-membered carbocycles are not only the key structural motifs in a number of natural products but also serve as privileged building blocks to a variety of biologically significant class of compounds.13 For instance, amino cyclopentenones are particularly useful building blocks in the asymmetric synthesis of prostaglandins, prostanoid derivatives, jasmonates and other bioactive molecules.14 Although the synthesis of 2-cyclopenten-1-ones as a challenging area in methodological studies is accomplished in a variety synthetic routes, including more classical methods such as the aldol reaction,15 as well as newer methods such as the Nazarov cyclization reaction,16 Saegusa–Ito oxidation of cyclopentanones, ring-closing metathesis, oxidation of cyclic allylic alcohols, and the Pauson–Khand reaction,17 to the best of our knowledge, there are only few reports18 on the synthesis of 3-cyclopenten-1-ones especially their spiro-fused derivatives (Fig. 1).
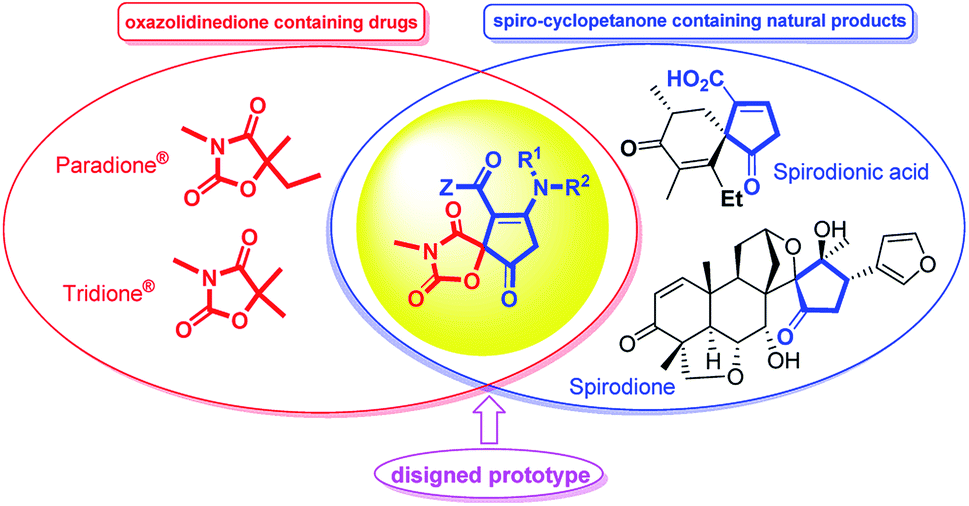 | ||
| Fig. 1 Some biologically important oxazolidinedione- and cyclopentane-containing drugs and designed cyclopentenone-fused spiro oxazolidinediones hybrid prototype. | ||
The “green chemistry” movement calls for redesigning new chemical processes and making reaction conditions safer that can potentially provide advantages for chemical syntheses in terms of waste treatment, energy and resource efficiency, product selectivity and environmental safety.19 Over the last decade multicomponent cascade reactions, particularly those performed in water as a cheap, non-flammable, nontoxic and environmentally friendly solvent, has witnessed considerable growth and development as a powerful tool in organic synthesis because of their expedient building up of molecular complexity and diversity, operational simplicity, convergence, atom economy, bond-forming efficiency and other suitable features from the point of view of green chemistry.20,21
In principle, alloxans (pyrimidine-2,4,5,6(1H,3H)-tetrones) should be regarded as suitable reaction partners for aldol-like reaction of enamines by virtue of the outstanding eletrophilicity of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond on the 5-position, and a series of biological important barbiturate derivatives can be assembled conveniently. Considering the efficiency as well as green chemistry aspects of aqueous-phase multicomponent reactions, recently our group have introduced a series of convenient methods with alloxans to provide biologically-interesting pyrimidine-containing heterocyclic compounds in water.22a–d It has been reported that when a mixture of alloxan derivatives and β-enaminoester derived in situ from the reaction of amines and acetylenic esters was stirred in water, the hydroxy enaminobarbiturates22c (Scheme 1, eqn (1)) and oxospirobicyclic butenolidobarbiturates22d (Scheme 1, eqn (2)) were produced. Inspired by the previous works as well as our continuous interests on the development of β-enaminocarbonyl-based synthesis of pharmaceutically important heterocycles, during this project, we found that when β-enaminoester employed in the above literatures were produced by reaction of amines and β-dicarbonyl compounds, the reaction occurred in another direction to form cyclopentenone-fused spiro oxazolidinediones in water that are important scaffolds for organic synthesis and drug design in pharmaceutical sciences (Scheme 1, eqn (3)). Surprisingly, no report has appeared for the synthesis of cyclopentenone-fused spiro oxazolidinediones. Encouraged by this initial result, we then made many efforts on optimizing reaction conditions.
O bond on the 5-position, and a series of biological important barbiturate derivatives can be assembled conveniently. Considering the efficiency as well as green chemistry aspects of aqueous-phase multicomponent reactions, recently our group have introduced a series of convenient methods with alloxans to provide biologically-interesting pyrimidine-containing heterocyclic compounds in water.22a–d It has been reported that when a mixture of alloxan derivatives and β-enaminoester derived in situ from the reaction of amines and acetylenic esters was stirred in water, the hydroxy enaminobarbiturates22c (Scheme 1, eqn (1)) and oxospirobicyclic butenolidobarbiturates22d (Scheme 1, eqn (2)) were produced. Inspired by the previous works as well as our continuous interests on the development of β-enaminocarbonyl-based synthesis of pharmaceutically important heterocycles, during this project, we found that when β-enaminoester employed in the above literatures were produced by reaction of amines and β-dicarbonyl compounds, the reaction occurred in another direction to form cyclopentenone-fused spiro oxazolidinediones in water that are important scaffolds for organic synthesis and drug design in pharmaceutical sciences (Scheme 1, eqn (3)). Surprisingly, no report has appeared for the synthesis of cyclopentenone-fused spiro oxazolidinediones. Encouraged by this initial result, we then made many efforts on optimizing reaction conditions.
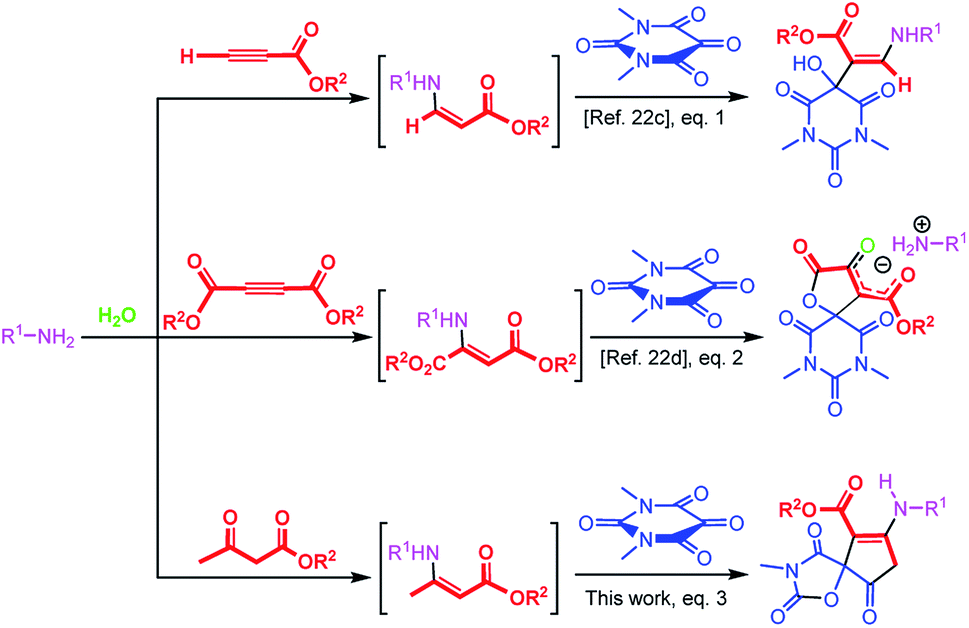 | ||
| Scheme 1 Recently reported β-enaminoester-based MCRs of N,N′-dimethylalloxan in water. | ||
Results and discussion
In optimization of the reaction conditions, first we focused on the efficient preparation of β-enaminoester or β-enaminones 5 from amines 1 and β-dicarbonyl compounds 2. Although a number of methods have been developed for the preparation of β-enaminocarbonyl compounds, considering the phrase, “the best solvent is no solvent and if a solvent (diluent) is needed then water is preferred” as clearly quoted by Sheldon in a review article,23 we chose solventless catalyst-free reaction conditions for the preparation of β-enaminoester and β-enaminones according to a previously reported procedure.24 At first the primary or secondary amine components reacted with β-dicarbonyl compounds without using any solvent or catalyst at room temperature for appropriate time to give the corresponding β-enaminocarbonyl compounds. The reaction time required for the enamination step are given in Table 1. Subsequently, an aqueous solution of N,N′-dimethylalloxan was added and the resulting mixture was stirred at room temperature for additional 1 h. The reactions were completed in all cases within only one hour and the products 4a–s were precipitated in the reaction mixture. The results are summarized in Table 1. 1H NMR and 13C NMR spectra of the crude products clearly indicated the formation of 4. It is also noteworthy to mention that this is the first report on the synthesis of 1-oxa-3-aza-spiro[4.4]non-6-enes that are important scaffolds for organic synthesis and drug design in pharmaceutical sciences.
|
|||||
|---|---|---|---|---|---|
| Entry | Amine | Z | Time (h) | Product | Yieldb (%) |
| a Reaction conditions: a solution of alloxan derivative 3 (1 mmol) in 3 mL water was added to the mixture of in situ-generated β-enaminocarbonyl compound from amine 1 (1 mmol) and β-dicarbonyl compound 2 (1 mmol) under solvent-free conditions at 25 °C (3–24 h) and the resulting mixture was stirred at room temperature for additional 1 h. The product 4 or 6 was separated by filtration.b Isolated yield. | |||||
| 1 |  |
 |
3 | 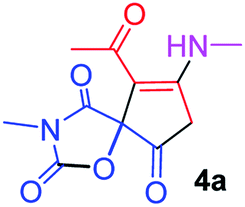 |
75 |
| 2 |  |
 |
3 | 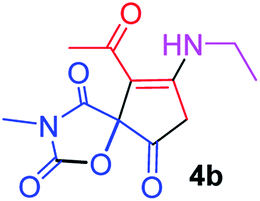 |
67 |
| 3 |  |
 |
3 | 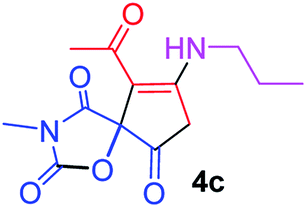 |
65 |
| 4 |  |
 |
3 | 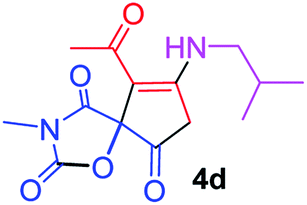 |
65 |
| 5 | 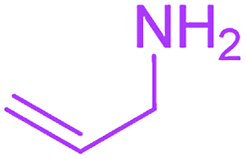 |
 |
3 | 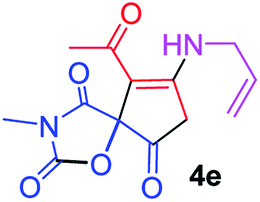 |
59 |
| 6 | 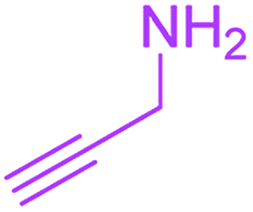 |
 |
3 | 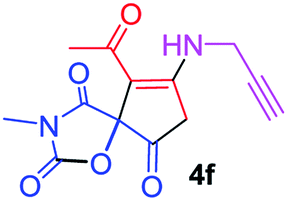 |
60 |
| 7 |  |
 |
3 | 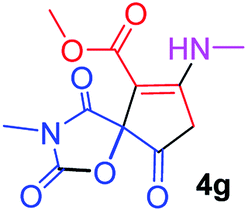 |
78 |
| 8 |  |
 |
3 | 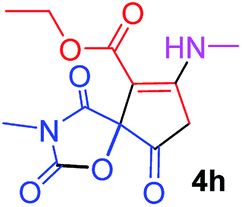 |
70 |
| 9 |  |
 |
3 | 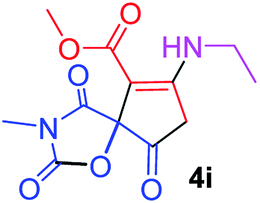 |
72 |
| 10 |  |
 |
3 | 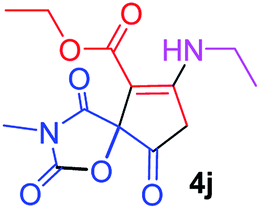 |
65 |
| 11 | 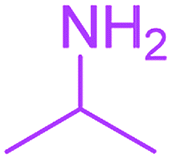 |
 |
3 | 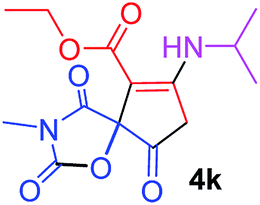 |
60 |
| 12 | 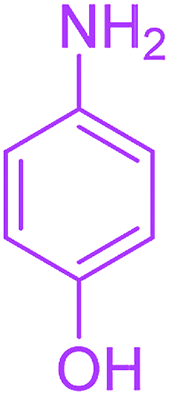 |
 |
3 | 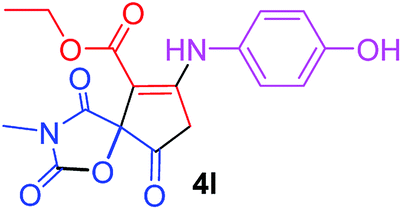 |
35 |
| 13 |  |
 |
3 | 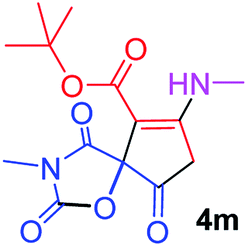 |
55 |
| 14 |  |
 |
24 | 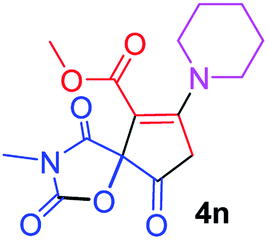 |
58 |
| 15 | 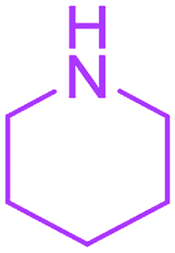 |
 |
24 | 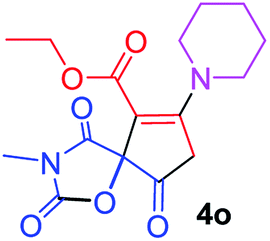 |
51 |
| 16 | 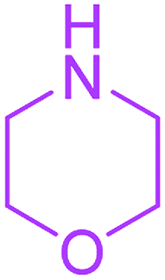 |
 |
16 | 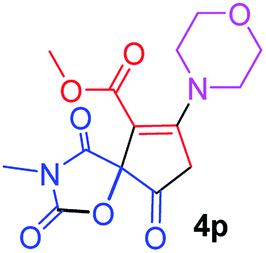 |
58 |
| 17 | 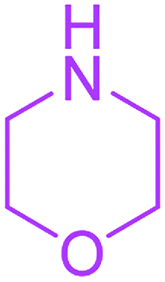 |
 |
16 | 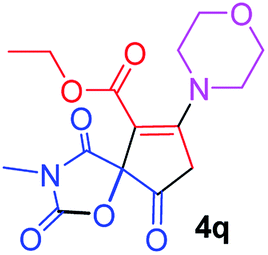 |
60 |
| 18 | 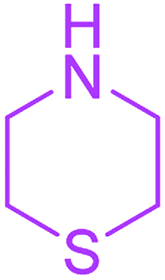 |
 |
24 | 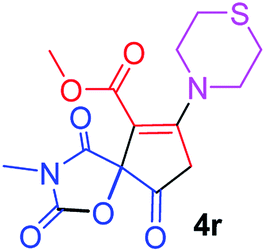 |
64 |
| 19 | 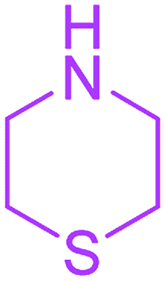 |
 |
24 | 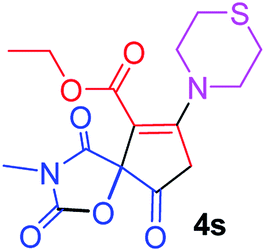 |
61 |
| 20 |  |
 |
3 | 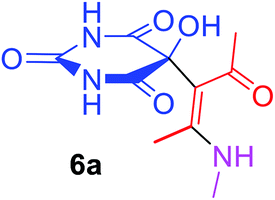 |
87 |
| 21 |  |
 |
3 | 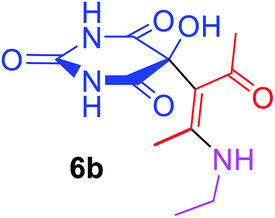 |
83 |
| 22 |  |
 |
3 | 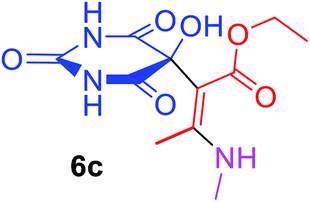 |
75 |
| 23 |  |
 |
3 | 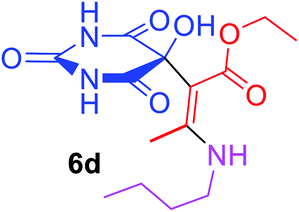 |
68 |
Compounds 4a–s are stable colorless solids whose structures were established by their analytical as well as spectral studies including FT-IR, 1H NMR, 13C NMR, and elemental analysis. For example, in the 1H NMR spectrum of 4a, the protons from OC–CH3 and two different NCH3 groups gave three singlets at δH 2.27, 3.14 and 3.16 ppm. An AB-quartet resonance was assigned (3.55 and 3.90 ppm, 2JHH 19.5 Hz) for the methylene of oxazolidinedione moiety. The NH group as a singlet (δH 5.84 ppm) was identified easily. The 13C{1H}-NMR spectrum of 4a showed 11 distinct resonances, which confirmed the proposed structure. The structural assignments made on the basis of the NMR spectra of compounds 4a was supported by the observed carbonyl frequency at 1853 cm−1 in FT-IR spectrum which is characteristic for oxazolidinedione ring.25
Under the optimized reaction conditions, a variety of primary and secondary aliphatic amines such as methylamine, ethylamine, n-propylamine, iso-butylamine, allylamine, propargyl amine, iso-propyl amine, piperidine, morpholine and thiomorpholine were employed and the reaction proceeded smoothly to give the corresponding spiro-oxazolidinedione-cyclopentenones 4a–s in moderate to good yields. Because the lower nucleophilicity and also lower hydrophilicity of aromatic amines (aniline derivatives) than that of aliphatic amines, unfortunately, our attempts to carry out this reaction under the same reaction conditions by using aromatic amines were unsuccessful and the reaction led to intractable mixture. Only with 4-hydroxyaniline (aniline bearing an strong electron-donating hydrophilic group) the corresponding 1-oxa-3-aza-spiro[4.4]non-6-enes 4l was obtained in 35% yield (Table 1, entry 12). Moreover, to extend the scope of this reaction with respect to β-dicarbonyl compounds, three alkyl acetoacetates and one 1,3-diketone were surveyed. The results show that the reaction is quite general with methyl acetoacetate, ethyl acetoacetate, tert-butyl acetoacetate, and acetylacetone affording 4, but failed to furnish the expected products with 1-phenylbutane-1,3-dione and ethyl 3-oxopentanoate. During further substrate screening, when the substrate combination was switched from N,N′-dimethylalloxan to alloxan using the optimized conditions, to our surprise the expected products 4 was not obtained and hydroxy enaminobarbiturates 6 was gained instead as the product (Table 1, entries 20–23).
As an economic and efficient protocol, the scale-up and operational simplicity of the three-component reaction have great practical significance for the preparation of spiro-oxazolidinedione-cyclopentenones in the academic laboratory, and even in industry. Thus, the different scale reactions of methyl amine, acetylacetone with N,N′-dimethylalloxan were carried out under the optimized reaction conditions. As shown in Scheme 2, the high-purity spiro-oxazolidinedione-cyclopentenone 4a can be simply obtained by filtration and washing with diethyl ether. This straightforward protocol followed environmentally-friendly GAP chemistry (group-assistant-purification chemistry), which avoids conventional purification procedures such as organic solvent extraction, silica gel column chromatography, and recrystallization.26 This is in conformity with the first of the principles of green chemistry, which is that it is better to prevent waste rather than to treat or clean up waste after it is formed.
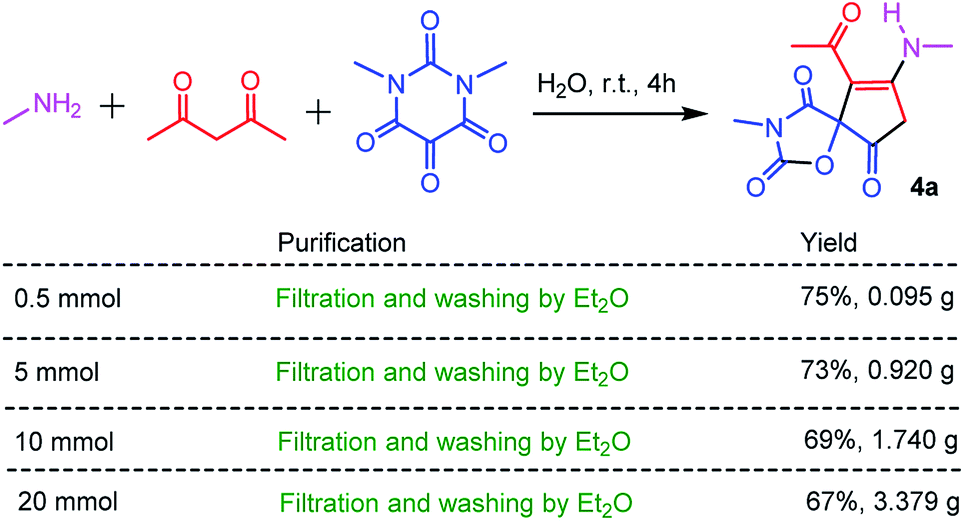 | ||
| Scheme 2 Gram-scale synthesis of 4a. | ||
Since the reusability of reaction medium is of great importance from the views of industry, economy and green chemistry, we examined the reusability of water after extraction of reaction residue with diethyl ether. We found that the reaction medium (water) after this extractive workup could be reused at least four times without significant loss of yield.
A possible mechanism was proposed as shown in Scheme 3. The initial step could be the dehydrative condensation of the amine 1 and 1,3-dicarbonyl compound 2 which affords the enaminone 5. Subsequently, a aldol-type addition of the enaminone 5 to the central carbonyl group of the alloxan derivative 3 occurs to generate the key iminium-oxyanion intermediate, which undergoes an iminium-enamine tautomerization and proton transfer to afford enamino hydroxybarbiturates 6 and 7 in an equilibrium. Now, the methyl group in 6 has been activated to enaminomethylene group in 7 by the isomerization. Then, intramolecular Stork enamine annulation between enaminomethylene moiety and imidic carbonyl group27 of 7 followed by ring opening of barbiturate moiety, iminium-enamine tautomerization and proton transfer afford the hydroxyl-cyclopentenone 8 and the latter undergoes intramolecular cyclization with concomitant methyl amine elimination to give the spiro-oxazolidinedione-cyclopentenone 4 (Scheme 3). The most exciting feature of this protocol is its mechanism that we have postulated here involving the unusual ring opening of a barbiturate moiety. This is probably because of the imidic carbonyl can be activated through the strong envelop-like five-membered hydrogen bonding with the vicinal hydroxyl group in intermediate 7. On the other hand, the leaving group which is similar to the conjugate base of N-methylurea can be a very good leaving group because it has two stable resonance structures.
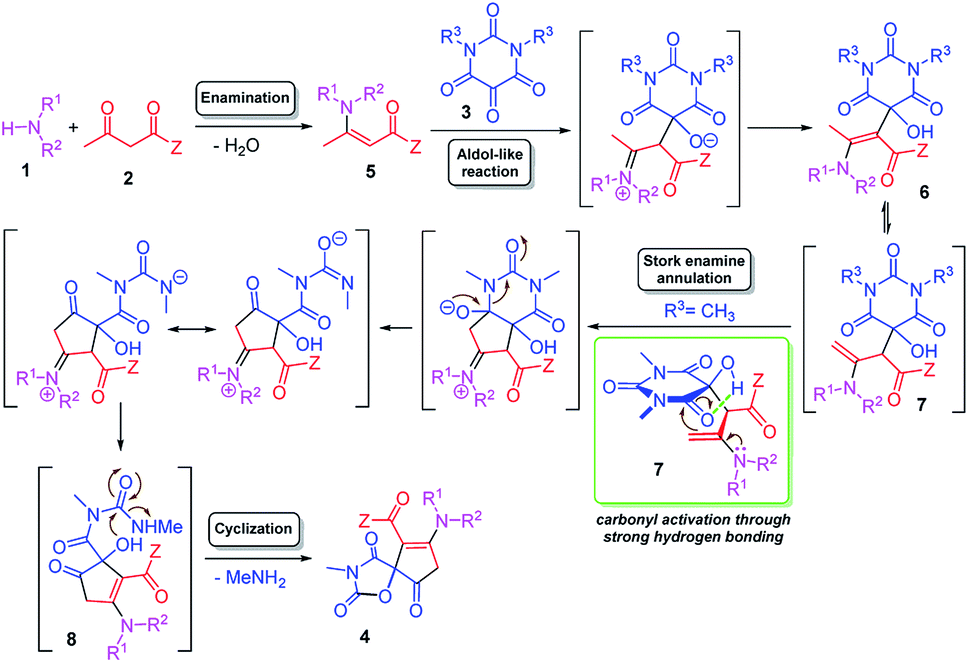 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In summary, we have developed an eco-friendly efficient strategy for the synthesis of spiro-oxazolidinedione-cyclopentenones and hydroxy enaminobarbiturates from readily available primary or secondary amines, β-enaminocarbonyl compounds and alloxan derivatives under catalyst-free conditions in water. At least four distinct reactions (enamination, aldol-type addition, Stork enamine annulation and cyclization) take place in this aqueous one-pot three-component process at room temperature to produce to produce the drug-like functionalized spiro-oxazolidinedione-cyclopentenones. All these reactions occur in an ordered manner to produce the final product with concomitant creation of four new bonds (two C–C, one C–N and one C–O) and two new rings.Experimental
General remarks
Melting points were measured on a Büchi 535 apparatus and are uncorrected. Elemental analyses were performed using an elementar vario EL III instrument. FT-IR Spectra were recorded on a Bruker Equinox-55 spectrometer. 1H and 13C NMR spectra were recorded on a Bruker DRX-300 Avance spectrometer at 300.13 and 75.47 MHz, respectively, with CDCl3 as solvent and calibrated using residual undeuterated solvent as an internal reference. Data for 1H NMR were presented as following: chemical shifts (δ, ppm), integration, multiplicity, coupling constant (Hz) and assignment of protons. The chemical shifts of resonances found were reported for 13C NMR spectra. Analytical TLC was carried out on precoated plates (Merck silica gel 60, F254) and visualized with UV light. All chemical reagents were obtained from Aldrich, Merck, Fluka or Acros and were used without further purification.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The resulting solid was removed by filtration, washed with water (5 mL) and diethylether (1 mL) and dried at 80 °C in air to give 4 or 6 as a white powder. The dried product thus obtained showed a single spot on TLC and was pure enough for all analytical purposes.
:1). The resulting solid was removed by filtration, washed with water (5 mL) and diethylether (1 mL) and dried at 80 °C in air to give 4 or 6 as a white powder. The dried product thus obtained showed a single spot on TLC and was pure enough for all analytical purposes.
9-Acetyl-3-methyl-8-(methylamino)-1-oxa-3-aza-spiro[4.4]non-8-ene-2,4,6-trione (4a). White powder (0.190 g, 75%); mp 131–133 °C (dec.); IR (KBr) (νmax, cm−1): 3470 (NH), 1853, 1735, 1703 and 1675 (C
O); 1H NMR (CDCl3, 300.1 MHz): δH 2.27 (3H, s, OC–CH3), 3.14 (3H, s, NCH3), 3.16 (3H, s, NCH3), 3.55 and 3.90 (2H, AB system, 2JHH 19.5 Hz, OC–CH2), 5.84 (1H, s, NH); 13C NMR (CDCl3, 75.5 MHz): δC 196.6, 169.4, 167.1, 153.4, 151.7, 102.7, 82.4, 34.3, 31.7, 28.3, 26.8; anal. calcd for C11H12N2O5 (252.23): C 52.38, H 4.80, N 11.11%; found: C 52.51, H 4.68, N 11.02%.
9-Acetyl-8-(ethylamino)-3-methyl-1-oxa-3-aza-spiro[4.4]non-8-ene-2,4,6-trione (4b). White powder (0.179 g, 67%); mp 137–139 °C (dec.); IR (KBr) (νmax, cm−1): 3457 (NH), 1834, 1747, 1734 and 1677 (C
O), 1591 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 1.27 (3H, t, 3JHH = 7.2 Hz, CH2CH3), 2.29 (3H, s, OC–CH3), 3.18 (3H, s, NCH3), 3.69 (2H, m, CH2CH3), 3.54 and 3.92 (2H, AB system, 2JHH 19.5 Hz, OC–CH2), 5.90 (1H, s, NH); 13C NMR (CDCl3, 75.5 MHz): δC 196.7, 169.5, 167.1, 153.9, 150.7, 102.3, 82.4, 36.9, 34.5, 31.8, 26.7, 11.7; anal. calcd for C12H14N2O5 (266.25): C 54.13, H 5.30, N 10.52%; found: C 53.85, H 5.25, N 10.54%.
9-Acetyl-3-methyl-8-(propylamino)-1-oxa-3-aza-spiro[4.4]non-8-ene-2,4,6-trione (4c). White powder (0.182 g, 65%); mp 138–140 °C (dec.); IR (KBr) (νmax, cm−1): 3479 (NH), 1832, 1754, 1739 and 1678 (C
O), 1592 (CC). 1H NMR (CDCl3, 300.1 MHz): δH 0.83 (3H, t, 3JHH = 7.30 Hz, CH2CH2CH3), 1.55 (2H, sex., CH2CH2CH3), 2.19 (3H, s, OC–CH3), 2.96 (3H, s, NCH3), 3.58 (2H, m, NH–CH2), 3.42 and 3.83 (2H, AB system, 2JHH 19.5 Hz, OC–CH2), 6.18 (1H, s, NH); 13C NMR (CDCl3, 75.5 MHz): δC 196.8, 170.1, 168.0, 154.6, 151.0, 102.9, 82.7, 42.5, 34.2, 31.5, 26.4, 19.4, 10.8; anal. calcd for C13H16N2O5 (280.28): C 55.71, H 5.75, N 10.00%; found: C 55.56, H 5.68, N 9.94%.
9-Acetyl-8-(isobutylamino)-3-methyl-1-oxa-3-azaspiro[4.4]non-8-ene-2,4,6-trione (4d). White powder (0.192 g, 65%); mp 178–180 °C (dec.); IR (KBr) (νmax, cm−1): 3476 (NH), 1829, 1754, 1737 and 1680 (C
O), 1603 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 0.93 and 0.96 (6H, 2 d, 3JHH 6.8 Hz, CHMe2), 2.12 (1H, m, CH), 2.27 (3H, s, OC–CH3), 3.16 (3H, s, NCH3), 3.44 (2H, m, NHCH2), 3.55 and 3.93 (2H, AB system, 2JHH 19.5 Hz, OC–CH2), 5.84 (1H, s, NH); 13C NMR (CDCl3, 75.5 MHz): δC 196.6, 169.6, 167.7, 154.0, 151.4, 102.7, 82.4, 49.0, 34.3, 31.8, 26.7, 26.1, 20.0, 19.8; anal. calcd for C14H18N2O5 (294.31): C 57.14, H 6.16, N 9.52%; found: C 56.98, H 6.13, N 9.45%.
9-Acetyl-8-(allylamino)-3-methyl-1-oxa-3-aza-spiro[4.4]non-8-ene-2,4,6-trione (4e). White powder (0.165 g, 59%); mp 140–143 °C (dec.); IR (KBr) (νmax, cm−1): 3467 (NH), 1834, 1753, 1737 and 1678 (C
O), 1650 and 1589 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 2.24 (3H, s, OC–CH3), 3.16 (3H, s, NCH3), 3.56 and 3.93 (2H, d of AB-system, 2JHH 19.5 Hz, 4JHH 1.5 Hz, OC–CH2), 4.22–4.27 (2H, m, NHCH2), 5.24 and 5.29 (2H, 2 d, 3JHH(trans) 17.2 Hz, 3JHH(cis) 10.4 Hz, CH2), 5.67–5.80 (1H, m, CH), 5.83 (1H, s, NH); 13C NMR (CDCl3, 75.5 MHz): δC 196.7, 169.6, 167.9, 153.8, 150.4, 128.4, 118.6, 103.3, 82.4, 43.9, 34.4, 31.8, 26.8; anal. calcd for C13H14N2O5 (278.26): C 56.11, H 5.07, N 10.07%; found: C 55.79, H 4.99, N 10.18%.
9-Acetyl-3-methyl-8-(prop-2-yn-1-ylamino)-1-oxa-3-aza-spiro[4.4]non-8-ene-2,4,6-trione (4f). White powder (0.166 g, 60%); mp 134–136 °C (dec.); IR (KBr) (νmax, cm−1): 3459 (NH), 3315 (
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H), 1837, 1748, 1684 (CO), 1597 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 2.28 (3H, s, OC–CH3), 2.35 (1H, t, 4JHH 2.5 Hz, CH), 3.15 (3H, s, NCH3), 3.58 and 3.92 (2H, AB-system, 2JHH 19.6 Hz, OC–CH2), 4.32 and 4.45 (2H, d of AB-system, 2JHH 17.6 Hz, 4JHH 2.5 Hz, HCC–CH2), 6.04 (1H, s, NH); 13C NMR (CDCl3, 75.5 MHz): δC 196.6, 169.2, 166.4, 153.7, 149.1, 103.9, 82.2, 74.3, 74.1, 34.3, 31.8, 31.1, 26.8; anal. calcd for C13H12N2O5 (276.25): C 56.52, H 4.38, N 10.14%; found: C 56.35, H 4.40, N 10.17%.
C–H), 1837, 1748, 1684 (CO), 1597 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 2.28 (3H, s, OC–CH3), 2.35 (1H, t, 4JHH 2.5 Hz, CH), 3.15 (3H, s, NCH3), 3.58 and 3.92 (2H, AB-system, 2JHH 19.6 Hz, OC–CH2), 4.32 and 4.45 (2H, d of AB-system, 2JHH 17.6 Hz, 4JHH 2.5 Hz, HCC–CH2), 6.04 (1H, s, NH); 13C NMR (CDCl3, 75.5 MHz): δC 196.6, 169.2, 166.4, 153.7, 149.1, 103.9, 82.2, 74.3, 74.1, 34.3, 31.8, 31.1, 26.8; anal. calcd for C13H12N2O5 (276.25): C 56.52, H 4.38, N 10.14%; found: C 56.35, H 4.40, N 10.17%.
Methyl 3-methy-7-(methylamino)-2,4,9-trioxo-1-oxa-3-aza-spiro[4.4]non-6-ene-6-carboxylate (4g). White powder (0.210 g, 78%); mp 199–202 °C (dec.); IR (KBr) (νmax, cm−1): 3390 (NH), 1836, 1745, 1743 and 1699, 1638 (C
O); 1H NMR (CDCl3, 300.1 MHz): δH 3.13 (3H, s, NCH3), 3.16 (3H, s, NCH3), 3.73 (3H, s, OCH3), 3.55 and 3.90 (2H, d of AB-system, 2JHH 19.3 Hz, 4JHH 1.8 Hz, OC–CH2), 5.43 (1H, t, 3JHH 1.8 Hz, NH); 13C NMR (CDCl3, 75.5 MHz): δC 169.5, 166.8, 166.5, 153.9, 152.4, 95.4, 82.4, 51.5, 33.5, 28.3, 26.8; anal. calcd for C11H12N2O6 (268.23): C 49.26, H 4.51, N 10.44%; found: C 49.47, H 4.57, N 10.37%.
Ethyl 3-methyl-7-(methylamino)-2,4,9-trioxo-1-oxa-3-aza-spiro[4.4]non-6-ene-6-carboxylate (4h). White powder (0.198 g, 70%); mp 139–141 °C (dec.), IR (KBr) (νmax, cm−1): 3389 (NH), 1828, 1748, 1747 and 1698, 1651 (C
O); 1H NMR (CDCl3, 300.1 MHz): δH 1.29 (3H, t, 3JHH 7.1 Hz, OCH2CH3), 3.13 (3H, s, NCH3), 3.16 (3H, s, NCH3), 3.55 and 3.90 (2H, d of AB-system, 2JHH 19.2 Hz, 4JHH 1.7 Hz, OC–CH2), 4.19 (2H, q, 3JHH 7.1 Hz, OCH2CH3), 5.43 (1H, t, 3JHH 1.7 Hz, NH); 13C NMR (CDCl3, 75.5 MHz): δC 169.5, 166.8, 166.1, 153.9, 152.1, 95.9, 82.5, 60.3, 33.5, 28.2, 26.8, 14.3; anal. calcd for C12H14N2O6 (282.25): C 51.07, H 5.00, N 9.93%; found: C 50.78, H 5.04, N 10.02%.
Methyl 7-(ethylamino)-3-methyl-2,4,9-trioxo-1-oxa-3-aza-spiro[4.4]non-6-ene-6-carboxylate (4i). White powder (0.204 g, 72%); mp 139–141 °C (dec.); IR (KBr) (νmax, cm−1): 3464 (NH), 1835, 1738, 1708 and 1629 (C
O), 1H NMR (CDCl3, 300.1 MHz): δH 1.23 (3H, t, 3JHH 7.2 Hz, OCH2CH3), 3.16 (3H, s, NCH3), 3.66 (2H, ABX3-system, NCH2CH3), 3.73 (3H, s, OCH3), 3.53 and 3.90 (2H, d of AB-system, 2JHH 19.3 Hz, 4JHH 1.8 Hz, OC–CH2), 5.46 (1H, t, 4JHH 1.8 Hz, NH); 13C NMR (CDCl3, 75.5 MHz): δC 169.5, 166.6, 153.9, 151.3, 95.0, 82.4, 51.4, 36.9, 33.7, 26.7, 11.6; anal. calcd for C12H14N2O6 (282.25): C 51.07, H 5.00, N 9.93%; found: C 50.79, H 4.94, N 9.88%.
Ethyl 7-(ethylamino)-3-methyl-2,4,9-trioxo-1-oxa-3-aza-spiro[4.4]non-6-ene-6-carboxylate (4j). White powder (0.193 g, 65%); mp 89–91 °C (dec.); IR (KBr) (νmax, cm−1): 3392 (NH), 1831, 1750, 1705, 1650 (C
O), 1H NMR (CDCl3, 300.1 MHz): δH 1.24 (3H, t, 3JHH 7.3 Hz, NCH2CH3), 1.29 (3H, t, 3JHH 7.3 Hz, OCH2CH3), 3.17 (3H, s, NCH3), 3.59 and 3.86 (2H, d of AB-system, 2JHH 19.2 Hz, 4JHH 1.8 Hz, OC–CH2), 3.62–3.71 (2H, ABX3-system, NCH2CH3), 4.18 (2H, q, 3JHH 7.3 Hz, OCH2CH3), 5.45 (1H, t, 4JHH 1.8 Hz, NH); 13C NMR (CDCl3, 75.5 MHz): δC 169.5, 166.6, 166.2, 153.9, 151.0, 95.4, 82.5, 60.3, 36.9, 33.6, 26.7, 14.3, 11.6; anal. calcd for C13H16N2O6 (296.28): C 52.70, H 5.44, N 9.46%; found: C 53.02, H 5.40, N 9.55%.
Ethyl 7-(isopropylamino)-3-methyl-2,4,9-trioxo-1-oxa-3-azaspiro[4.4]non-6-ene-6-carboxylate (4k). White powder (0.186 g, 60%); mp 175–177 °C (dec.); IR (KBr) (νmax, cm−1): 3366 (NH), 1821, 1755, 1705, 1660 (C
O), 1605 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 1.32–1.39 (9H, m, 3 CH3 overlapping), 3.12 (3H, s, NCH3), 3.02 and 3.33 (2H, AB-system, 2JHH 17.9 Hz, OC–CH2), 3.79–3.82 (1H, m, CH), 4.28 (2H, q, 3JHH 7.1 Hz, OCH2CH3), 9.36 (1H, d, 3JHH 7.7 Hz, NH); 13C NMR (CDCl3, 75.5 MHz): δC 184.1, 175.5, 170.9, 166.0, 154.7, 99.0, 85.4, 60.5, 47.5, 32.3, 26.5, 23.4, 23.3, 14.2; anal. calcd for C14H18N2O6 (310.31): C 54.19, H 5.85, N 9.03%; found: C 53.96, H 5.83, N 9.07%.
Ethyl 7-[(4-hydroxyphenyl)amino]-3-methyl-2,4,9-trioxo-1-oxa-3-aza-spiro[4.4]non-6-ene-6-carboxylate (4l). White powder (0.126 g, 35%); mp 196–200 °C (dec.); IR (KBr) (νmax, cm−1): 3398 (OH + NH), 1837, 1750, 1708 and 1641 (C
O), 1598 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 1.25 (3H, t, 3JHH 7.1 Hz, OCH2CH3), 3.19 (3H, s, NCH3), 3.68 and 4.05 (2H, d of AB-system, 2JHH 19.3 Hz, 4JHH 2.0 Hz, OC–CH2), 4.14 (2H, q, 3JHH 7.1 Hz, OCH2CH3), 5.21 (1H, t, 4JHH 1.8 Hz, NH), 6.60 (1H, brs, OH), 6.86 and 7.04 (4H, 2 d, 3JHH 7.1 Hz, –C6H4); 13C NMR (CDCl3, 75.5 MHz): 169.5, 167.0, 166.4, 157.3, 153.9, 153.2, 128.5, 124.5, 116.9, 97.7, 82.7, 60.4, 33.5, 26.8, 14.2; anal. calcd for C17H16N2O7 (360.31): C 56.67, H 4.48, N 7.77%; found: C 56.43, H 4.53, N 7.86%.
tert-Butyl 3-methyl-7-(methylamino)-2,4,9-trioxo-1-oxa-3-azaspiro[4.4]non-6-ene-6-carboxylate (4m). White powder (0.170 g, 55%); mp 183–186 °C (dec.), IR (KBr) (νmax, cm−1): 3436 (NH), 1834, 1759, 1746 and 1700 (C
O), 1644 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 1.49 (9H, s, C(CH3)3), 3.11 (3H, s, NCH3), 3.16 (3H, s, NCH3), 3.84 (2H, AB-system, 2JHH 19.2 Hz, OC–CH2), 5.35 (1H, s, NH). 13C NMR (CDCl3, 75.5 MHz): 169.6, 166.7, 165.4, 153.4, 150.3, 97.8, 82.6, 80.8, 33.4, 28.2, 26.8; anal. calcd for C14H18N2O6 (310.30): C 54.19, H 5.85, N 9.03%; found: C 53.98, H 5.90, N 8.95%.
Methyl 3-methyl-2,4,9-trioxo-7-piperidin-1-yl-1-oxa-3-azaspiro[4.4]non-6-ene-6-carboxylate (4n). White powder (0.187 g, 58%); mp 193–195 °C (dec.); IR (KBr) (νmax, cm−1): 1816, 1727, 1701, 1667 (C
O), 1592 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 1.79 (6H, m, CH2CH2CH2), 3.01 and 3.24 (2H, AB-system, 2JHH 16.6 Hz, OC–CH2), 3.12 (3H, s, NCH3), 3.54 (4H, m, CH2NCH2), 3.82 (3H, s, OCH3); 13C NMR (CDCl3, 75.5 MHz): δC 185.7, 170.9, 170.3, 164.3, 154.4, 103.7, 85.4, 53.2, 51.5, 50.7, 35.7, 26.0, 25.9, 25.2, 23.0; anal. calcd for C15H18N2O6 (322.32): C 55.90, H 5.63, N 8.69%; found: C 56.17, H 5.60, N 8.76%.
Ethyl 3-methyl-2,4,9-trioxo-7-piperidin-1-yl-1-oxa-3-azaspiro[4.4]non-6-ene-6-carboxylate (4o). White powder (0.172 g, 51%); mp 115–117 °C (dec.); IR (KBr) (νmax, cm−1): 1820, 1742, 1711, 1678 (C
O), 1568 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 1.32 (3H, t, 3JHH 7.1 Hz, OCH2CH3), 1.76 (6H, m, CH2CH2CH2), 3.00 and 3.22 (2H, AB-system, 2JHH 16.6 Hz, OC–CH2), 3.10 (3H, s, NCH3), 3.53 (4H, m, CH2NCH2), 4.26 (2H, q, 3JHH 7.1 Hz, OCH2CH3); 13C NMR (CDCl3, 75.5 MHz): δC 185.7, 170.7, 170.4, 164.2, 154.7, 103.2, 85.8, 60.9, 53.4, 51.0, 35.9, 26.4, 26.2, 25.6, 23.4, 14.2; anal. calcd for C16H20N2O6 (336.34): C 57.14, H 5.99, N 8.33%; found: C 56.88, H 6.08, N 8.28%.
Methyl 3-methyl-7-morpholin-4-yl-2,4,9-trioxo-1-oxa-3-azaspiro[4.4]non-6-ene-6-carboxylate (4p). White powder (0.188 g, 58%); mp 193–195 °C (dec.); IR (KBr) (νmax, cm−1): 1820, 1745, 1723, 1662 (C
O), 1576 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 3.04 and 3.24 (2H, AB-system, 2JHH 16.7 Hz, OC–CH2), 3.12 (3H, s, NCH3), 3.62–3.72 (4H, m, CH2NCH2), 3.81 (3H, s, OCH3), 3.88 (4H, m, CH2OCH2); 13C NMR (CDCl3, 75.5 MHz): δC 185.8, 171.4, 170.4, 163.8, 154.5, 103.1, 85.4, 66.4, 66.3, 52.4, 52.1, 50.0, 35.8, 26.5; anal. calcd for C14H16N2O7 (324.29): C 51.85, H 4.97, N 8.64%; found: C 51.67, H 5.05, N 8.53%.
Ethyl 3-methyl-7-morpholin-4-yl-2,4,9-trioxo-1-oxa-3-azaspiro[4.4]non-6-ene-6-carboxylate (4q). White powder (0.203 g, 60%); mp 177–180 °C (dec.); IR (KBr) (νmax, cm−1): 1820, 1724, 1722, 1663 (C
O), 1599 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 1.32 (3H, t, 3JHH 7.1 Hz, OCH2CH3), 3.03 and 3.22 (2H, AB-system, 2JHH 16.7 Hz, OC–CH2), 3.10 (3H, s, NCH3), 3.61–3.70 (4H, m, CH2NCH2), 3.86 (4H, s, CH2OCH2), 4.25 (2H, q, 3JHH 7.1 Hz, OCH2CH3); 13C NMR (CDCl3, 75.5 MHz): δC 186.1, 171.1, 170.5, 163.9, 154.6, 103.7, 85.5, 66.3, 61.1, 52.2, 49.9, 35.7, 26.5, 14.2; anal. calcd for C15H18N2O7 (338.32): C 53.25, H 5.36, N 8.28%; found: C 52.97, H 5.32, N 8.25%.
Methyl 3-methyl-2,4,9-trioxo-7-thiomorpholin-4-yl-1-oxa-3-azaspiro[4.4]non-6-ene-6-carboxylate (4r). White powder (0.218 g, 64%); mp 197–199 °C (dec.); IR (KBr) (νmax, cm−1): 1814, 1751, 1704, 1682 (C
O), 1578 (CC); 1H NMR (DMSO-d6, 300.1 MHz): δH 2.77 (4H, m, CH2SCH2), 2.87 (2H, m, OCCH2), 2.95 (3H, s, NCH3), 3.55–3.95 (4H, m, CH2NCH2), 3.67 (3H, s, OCH3); 13C NMR (DMSO-d6, 75.5 MHz): δC 186.1, 171.3, 170.8, 164.5, 155.0, 102.6, 85.8, 54.1, 52.2, 51.7, 35.5, 26.9, 26.2, 26.1; anal. calcd for C14H16N2O6S (340.35): C 49.41, H 4.74, N 8.23%; found: C 49.53, H 4.69, N 8.31%.
Ethyl 3-methyl-2,4,9-trioxo-7-thiomorpholin-4-yl-1-oxa-3-azaspiro[4.4]non-6-ene-6-carboxylate (4s). White powder (0.216 g, 61%); mp 195–197 °C (dec.); IR (KBr) (νmax, cm−1): 1820, 1743, 1725, 1667 (C
O), 1591 (CC); 1H NMR (CDCl3, 300.1 MHz): δH 1.31 (3H, t, 3JHH 7.1 Hz, OCH2CH3), 2.82–2.96 (4H, m, CH2SCH2), 3.02 and 3.20 (2H, AB-system, 2JHH 16.7 Hz, OC–CH2), 3.09 (3H, s, NCH3), 3.79–3.86 (4H, m, CH2NCH2), 4.25 (2H, q, 3JHH 7.1 Hz, OCH2CH3); 13C NMR (CDCl3, 75.5 MHz): δC 186.1, 170.7, 170.5, 164.2, 154.6, 104.2, 85.4, 61.2, 55.0, 52.6, 35.8, 27.8 and 27.1, 26.5, 14.2; anal. calcd for C15H18N2O6S (354.38): C 50.84, H 5.12, N 7.91%; found: C 51.05, H 5.18, N 8.01%.
5-[(1Z)-1-Acetyl-2-(methylamino)prop-1-en-1-yl]-5-hydroxypyrimidine-2,4,6(1H,3H,5H)-trione (6a). White powder (0.222 g, 87%); mp 149–151 °C (dec.); IR (KBr) (νmax, cm−1): 3439 and 3329 (NH), 3224 (OH), 1731, 1699 and 1622 (C
O), 1582 (CC); 1H NMR (DMSO-d6, 300.1 MHz): δH 2.10 (3H, s, OCCH3), 2.49 (3H, s, C–CH3 overlapping with H2O in DMSO-d6), 3.01 (3H, s, NCH3), 7.28 (1H, s, OH), 7.44 (1H, s, NH), 7.46 (1H, s, NH), 9.25 (1H, s, NH); 13C NMR (DMSO-d6, 75.5 MHz): δC 191.0, 174.0, 169.8, 159.4, 152.2, 117.5, 78.6, 29.2, 26.1, 13.0; anal. calcd for C10H13N3O5 (255.22): C 47.06, H 5.13, N 16.46%; found: C 46.88, H 5.21, N 16.59%.
5-[(1Z)-1-Acetyl-2-(ethylamino)prop-1-en-1-yl]-5-hydroxypyrimidine-2,4,6(1H,3H,5H)-trione (6b). White powder (0.223 g, 83%); mp 168–170 °C (dec.), IR (KBr) (νmax, cm−1): 3420 (NH), 3322 (OH), 1741, 1703, 1629 and 1607 (C
O), 1578 (CC); 1H NMR (DMSO-d6, 300.1 MHz): δH 1.07 (3H, t, 3JHH 7.0 Hz, NHCH2CH3), 2.10 (3H, s, OCCH3), 2.49 (3H, s, C–CH3 overlapping with H2O in DMSO-d6), 3.53 (2H, m, NHCH2CH3), 7.35 (1H, s, OH), 7.46 (2H, s, 2 NH), 9.21 (1H, s, NH). 13C NMR (DMSO-d6, 75.5 MHz): δC 191.1, 173.9, 169.8, 158.8, 152.1, 117.7, 78.5, 34.5, 29.3, 13.9, 12.7; anal. calcd for C11H15N3O5 (269.25): C 49.07, H 5.62, N 15.61%; found: C 48.80, H 5.65, N 15.53%.
Ethyl (2Z)-2-(5-hydroxy-2,4,6-trioxohexahydropyrimidin-5-yl)-3-(methylamino)but-2-enoate (6c). White powder (0.214 g, 75%); mp 175–177 °C (dec.), IR (KBr) (νmax, cm−1): 3414 and 3321 (NH), 3278 (OH), 1735, 1709, 1666 and 1622 (C
O), 1586 (CC); 1H NMR (DMSO-d6, 300.1 MHz): δH 1.13 (3H, t, 3JHH 7.1 Hz, OCH2CH3), 2.49 (3H, s, C–CH3 overlapping with H2O in DMSO-d6), 3.00 (3H, s, NCH3), 4.0 and 4.04 (2H, ABX3 system, 2JHH 17.9 Hz, 3JHH 7.1 Hz, OCH2CH3), 7.29 (1H, s, OH) 7.44 (2H, s, 2 NH), 8.97 (1H, s, NH); 13C NMR (DMSO-d6, 75.5 MHz): δC 174.6, 170.3, 162.6, 160.8, 152.6, 108.2, 78.5, 59.8, 26.6, 14.3, 12.7; anal. calcd for C11H15N3O6 (285.25): C 46.32, H 5.30, N 14.73%; found: C 46.50, H 5.37, N 14.65%.
Ethyl (2Z)-3-(butylamino)-2-(5-hydroxy-2,4,6-trioxohexahydropyrimidin-5-yl)but-2-enoate (6d). White powder (0.223 g, 68%); mp 191–193 °C (dec.), IR (KBr) (νmax, cm−1): 3369 and 3327 (NH), 3231 (OH), 1739, 1694, 1670 and 1631 (C
O); 1H NMR (DMSO-d6, 300.1 MHz): δH 0.87 (3H, t, 3JHH 7.2 Hz, NHCH2CH2CH2CH3), 1.13 (3H, t, 3JHH 7.1 Hz, OCH2CH3), 1.25 (2H, sex, 3JHH 7.1 Hz, NHCH2CH2CH2CH3), 1.45 (2H, qu, 3JHH 7.1 Hz, NHCH2CH2CH2CH3), 3.47 (2H, t, 3JHH 7.1, NHCH2CH2CH2CH3), 4.0 and 4.04 (2H, ABX3 system, 2JHH 17.9 Hz, 3JHH 7.1 Hz, OCH2CH3), 7.35 (1H, s, OH), 7.43 (2H, s, 2 NH), 8.93 (1H, s, NH); 13C NMR (DMSO-d6, 75.5 MHz): δC 174.4, 169.9, 162.2, 160.0, 152.0, 108.0, 77.9, 59.3, 30.6, 19.2, 13.8, 13.5, 12.0; anal. calcd for C14H21N3O6 (327.33): C 51.37, H 6.47, N 12.84%; found: C 51.53, H 6.55, N 12.81%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support of Kharazmi University (grant number D/2047) is gratefully acknowledged.Notes and references
- (a) A. D. Melhado, W. E. Brenzovitch, A. D. Lackner and F. D. Toste, J. Am. Chem. Soc., 2010, 132, 8885 CrossRef CAS PubMed; (b) G. L. Adams, P. J. Carroll and A. B. Smith, J. Am. Chem. Soc., 2013, 135, 519 CrossRef CAS PubMed; (c) S. A. Snyder, S. P. Breazzano, A. G. Ross, Y. Lin and A. L. Zografos, J. Am. Chem. Soc., 2009, 131, 1753 CrossRef CAS PubMed; (d) L. Yu, R. Qian, X. Deng, F. Wang and Q. Xu, Sci. Bull., 2018, 63, 1010 CrossRef CAS.
- (a) R. A. Yoder and J. N. Johnston, Chem. Rev., 2005, 105, 4730 CrossRef CAS PubMed; (b) J. K. Sutherland, in Comprehensive Organic Synthesis, ed. B. M. Trost, Pergamon Press, Elmsford, NY, 1991, vol. 1, pp. 341–377 Search PubMed.
- (a) A. Bergstrand, C. G. Bergstrand, N. Engström and K. M. Herrlin, Pediatrics, 1962, 30, 601 CAS; (b) J. W. C. Lewis, Chem. Rev., 1958, 58, 63 CrossRef; (c) P. C. Unangst, D. T. Connor, W. A. Cetenko, R. J. Sorenson, C. R. Kostlan, J. C. Sircar, C. D. Wright, D. J. Schrier and R. D. Dyer, J. Med. Chem., 1994, 37, 322 CrossRef CAS PubMed; (d) D. A. Heerding, L. T. Christmann, T. J. Clark, D. J. Holmes, S. F. Rittenhouse, D. T. Takata and J. W. Venslavsky, Bioorg. Med. Chem. Lett., 2003, 13, 3771 CrossRef CAS PubMed.
- T. Sohda, H. Ikeda and Y. Momose, Eur. Patent 643050, 1995.
- S. Tsukamoto, M. Ichihara, F. Wanibuchi, S. Usuda, K. Hidaka, M. Harada and T. Tamura, J. Med. Chem., 1993, 36, 2292 CrossRef CAS PubMed.
- J. W. Clark Lewis, Chem. Rev., 1958, 58, 63 CrossRef CAS.
- R. L. Dow, B. M. Bechle, T. Chou Thomas, D. A. Clark, B. Hulin and R. W. Stevenson, J. Med. Chem., 1991, 34, 1538 CrossRef CAS PubMed.
- R. Moreira, A. B. Santana, J. Iley, J. Neres, K. T. Douglas, P. N. Horton and M. B. Hursthouse, J. Med. Chem., 2005, 48, 4861 CrossRef CAS PubMed.
- A. B. Santana, S. D. Lucas, L. M. Gonçalves, H. F. Correia, A. F. Teresa, T. A. F. Cardote, R. C. Guedes, J. Iley and R. Moreira, Bioorg. Med. Chem. Lett., 2012, 22, 3993 CrossRef CAS PubMed.
- (a) C. Yang, H. C. Shen, Z. Wu, H. D. Chu, J. M. Cox, J. Balsells, A. Crespo, P. Brown, B. Zamlynny, J. Wiltsie, J. Clemas, J. Gibson, L. Contino, J. M. Lisnock, G. Zhou, M. Garcia-Calvo, T. Bateman, L. Xu, X. Tong, M. Crook and P. Sinclair, Bioorg. Med. Chem. Lett., 2013, 23, 4388 CrossRef CAS PubMed; (b) J. M. Cox, H. D. Chu, C. Yang, H. C. Shen, Z. Wu, J. Balsells, A. Crespo, P. Brown, B. Zamlynny, J. Wiltsie, J. Clemas, J. Gibson, L. Contino, J. M. Lisnock, G. Zhou, M. Garcia-Calvo, T. Bateman, L. Xu, X. Tong, M. Crook and P. Sinclair, Bioorg. Med. Chem. Lett., 2014, 24, 1681 CrossRef CAS PubMed.
- K. Harada, H. Kubo, A. Tanaka and K. Nishioka, Bioorg. Med. Chem. Lett., 2012, 22, 504 CrossRef CAS PubMed.
- (a) B. M. York, US Pat. 4,717,725, 1988; (b) B. M. York, US Pat. 5,070,100, 1991.
- (a) H. Cao, F. Chen, C. Su and L. Yu, Adv. Synth. Catal., 2019, 362, 438 CrossRef; (b) M. Zhang, J. Feng, X. Jia, J. Zhao, J. Liu, R. Chen, K. Xie, D. Chen, Y. Li, D. Zhang and J. Dai, Chin. Chem. Lett., 2019, 30, 435 Search PubMed; (c) S. P. Roche and D. J. Aitken, Eur. J. Org. Chem., 2010, 5339 CrossRef CAS; (d) B. M. Trost and A. B. Pinkerton, J. Org. Chem., 2001, 66, 7714 CrossRef CAS PubMed.
- (a) E. Baron, P. O'Brien and T. D. Towers, Tetrahedron Lett., 2002, 43, 723 CrossRef CAS; (b) M. M. Domostoj, E. Irving, F. Scheinmann and K. J. Hale, Org. Lett., 2004, 6, 2615 CrossRef CAS PubMed; (c) F. A. Davis and J. Deng, Org. Lett., 2005, 7, 621 CrossRef CAS PubMed; (d) D. P. Dickson and D. J. Wardrop, Org. Lett., 2009, 11, 1341 CrossRef CAS PubMed; (e) Y. Menjo, A. Hamajima, N. Sasaki and Y. Hamada, Org. Lett., 2011, 13, 5744 CrossRef CAS PubMed; (f) I. Takano, I. Yasuda, M. Nishijima, Y. Hitotsuyanagi, K. Takeya and H. Itokawa, J. Nat. Prod., 1996, 59, 965 CrossRef CAS; (g) F. Inagaki, M. Kinebuchi, N. Miyakoshi and C. Mukai, Org. Lett., 2010, 12, 1800 CrossRef CAS PubMed; (h) P. Magnus, M. R. Fielding, C. Wells and V. Lynch, Tetrahedron Lett., 2002, 43, 947 CrossRef CAS; (i) T. Luker, W. J. Koot, H. Hiemstra and W. N. Speckamp, J. Org. Chem., 1998, 63, 220 CrossRef CAS; (j) K. Namba, M. Inai, U. Sundermeier, T. J. Greshock and R. M. Williams, Tetrahedron Lett., 2010, 51, 6557 CrossRef CAS PubMed; (k) S. P. Simeonov, J. P. M. Nunes, K. Guerra, V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2016, 116, 5744 CrossRef CAS PubMed and references cited therein.
- (a) E. Wenkert, R. S. Greenberg and M. S. Raju, J. Org. Chem., 1985, 50, 4681 CrossRef CAS; (b) A. Pecunioso and R. Menicagli, J. Org. Chem., 1988, 53, 2614 CrossRef CAS.
- (a) W. He, X. Sun and A. J. Frontier, J. Am. Chem. Soc., 2003, 125, 14278 CrossRef CAS PubMed; (b) V. K. Aggarwal and A. J. Belfield, Org. Lett., 2003, 5, 5075 CrossRef CAS PubMed; (c) G. Liang, S. N. Gradl and D. Trauner, Org. Lett., 2003, 5, 4931 CrossRef CAS PubMed; (d) A. K. Basak, N. Shimada, W. F. Bow, D. A. Vicic and M. A. Tius, J. Am. Chem. Soc., 2010, 132, 8266 CrossRef CAS PubMed; (e) V. M. Marx and D. J. Burnell, Org. Lett., 2009, 11, 1229 CrossRef CAS PubMed; (f) M. Shindo, K. Yaji, T. Kita and K. Shishido, Synlett, 2007, 1096 CrossRef CAS; (g) T. N. Grant and F. G. West, J. Am. Chem. Soc., 2006, 128, 9348 CrossRef CAS PubMed.
- (a) P. Magnus, Tetrahedron Lett., 1985, 26, 4851 CrossRef CAS; (b) L.-J. Deng, J. Liu, J.-Q. Huang, Y. Hud, M. Chen, Y. Lan, J.-H. Chen, A. Lei and Z. Yang, Synthesis, 2007, 2565 CAS; (c) B.-U. Jaime, A. Loreto, P.-S. Leticia, D. Gema and P.-C. Javier, Chem. Soc. Rev., 2004, 33, 32 RSC; (d) H.-W. Lee and F.-Y. Kwong, Eur. J. Org. Chem., 2010, 789 CrossRef CAS.
- (a) E. J. Corey and S. W. Walinsky, J. Am. Chem. Soc., 1972, 94, 8932 CrossRef CAS; (b) G. R. Unruh and D. M. Birney, J. Am. Chem. Soc., 2003, 125, 8529 CrossRef CAS PubMed; (c) H. M. Hess and H. C. Brown, J. Org. Chem., 1967, 32, 4138 CrossRef CAS; (d) J. D. Lewis and J. Laane, Spectrochim. Acta, 1975, 31, 755 CrossRef.
- P. T. Anastas and J. C. Warner, in Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- P. Gunasekaran, J. Menéndez and S. Perumal, Synthesis of Heterocycles Through Multicomponent Reactions in Water, in Green Chemistry: Synthesis of Bioactive Heterocycles, ed. K. Ameta and A. Dandia, Springer, New Delhi, 2014, ch. 1, pp. 1–35 Search PubMed.
- (a) Z. Cao, Q. Zhu, Y.-W. Lin and W.-M. He, Chin. Chem. Lett., 2019, 30, 2132 CrossRef CAS; (b) C. Wu, X. Xin, Z.-M. Fu, L.-Y. Xie, K.-J. Liu, Z. Wang, W. Li, Z.-H. Yuan and W.-M. He, Green Chem., 2017, 19, 1983 RSC; (c) N. Gupta and D. Goyal, Chem. Heterocycl. Compd., 2015, 51, 4 CrossRef CAS; (d) S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS PubMed; (e) M. B. Gawande, V. D. B. Bonifacio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522 RSC and references therein.
- (a) M. B. Teimouri, P. Akbari-Moghaddam and M. Motaghinezhad, Tetrahedron, 2013, 69, 6804 CrossRef CAS; (b) M. B. Teimouri and M. Rezaei, J. Iran. Chem. Soc., 2017, 1, 1243 CrossRef; (c) M. B. Teimouri, T. Abbasi and H. Mivehchi, Tetrahedron, 2008, 64, 10425 CrossRef CAS; (d) M. B. Teimouri, T. Abbasi and H. R. Khavasi, J. Chem. Res., 2010, 310 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2005, 7, 267 RSC.
- R. K. Vohra, J.-L. Renaud and C. Bruneau, Synthesis, 2006, 731 Search PubMed.
- R. Maity, S. Naskar, K. Mal, S. Biswas and I. Das, Adv. Synth. Catal., 2017, 359, 4405 CrossRef CAS.
- (a) L.-Y. Xie, S. Peng, F. Liu, Y.-F. Liu, M. Sun, Z.-L. Tang, S. Jiang, Z. Cao and W.-M. He, ACS Sustainable Chem. Eng., 2019, 7, 7193 CrossRef CAS; (b) Z. Cao, Q. Zhu, Y.-W. Lin and W.-M. He, Chin. Chem. Lett., 2019, 30, 2132 CrossRef CAS; (c) L.-Y. Xie, S. Peng, J.-X. Tan, R.-X. Sun, X. Yu, N.-N. Dai, Z.-L. Tang, X. Xu and W.-M. He, ACS Sustainable Chem. Eng., 2018, 6, 16976 CrossRef CAS; (d) L.-Y. Xie, Y. Duan, L.-H. Lu, Y.-J. Li, S. Peng, C. Wu, K.-J. Liu, Z. Wang and W.-M. He, ACS Sustainable Chem. Eng., 2017, 5, 10407 CrossRef CAS.
- H. Zhang, M. Han, C. Yang, L. Yu and Q. Xu, Chin. Chem. Lett., 2019, 30, 263 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01699c |
| This journal is © The Royal Society of Chemistry 2020 |