
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA concise and sequential synthesis of the nitroimidazooxazole based drug, Delamanid and related compounds†
Sumit Sharmaab,
Radhika Ananda,
Pankaj Singh Chamab,
Sushil Rainaa,
Ram. A. Vishwakarmaab and
Parvinder Pal Singh *ab
*ab
aMedicinal Chemistry Division, CSIR-Indian Institute of Integrative Medicine, Canal Road, Jammu-180001, India. E-mail: ppsingh@iiim.ac.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad-201002, India
First published on 1st May 2020
Abstract
A concise, protection-group free and sequential route has been developed for the synthesis of the nitroimidazole based FDA-approved multi-drug resistant anti-tuberculosis drug, Delamanid and anti-leishmanial lead candidate VL-2098. The synthesis required chiral epoxides (11 and 17) as key intermediates. The chiral epoxide 11 was synthesised by sequential reaction cascades viz., allylation, selective N-arylation, Mitsunobu etherification, Sharpless asymmetric dihydroxylation and epoxidation, which do not require any special/dry reaction conditions. The steps involved towards the synthesis of epoxide also worked nicely in gram scales. After the synthesis of epoxide 11, the synthesis of Delamanid was achieved by reaction with 2-bromo-4-nitroimidazole 12 with an overall yield of 27%. Similarly, anti-leishmanial lead candidate VL-2098 was also synthesized in an overall yield of 36%.
Imidazoles are present in a wide variety of biologically relevant molecules exhibiting diverse pharmaceutical properties. Specifically, nitroimidazole containing compounds are active therapeutic agents against a wide variety of protozoan, bacterial (anaerobic) and leishmanial infections of humans and animals.1 The notable examples of drugs are fexindazole,2 metronidazole,3 benznidazole,4 tinidazole,5 etc. Nitroimidazole was also well explored in the area of tuberculosis (TB) drug discovery (the structure of nitroimidazole containing drugs and lead compounds shown in Fig. 1).6–9 CGI-17341 (I, developed by Ciba-Geigy, India) represents one of the very earliest outcomes but was not continued because of mutagenicity.6 Continued efforts to overcome the mutagenic liability led to the discovery of Delamanid (II, OPC-67683, a blockbuster against multi-drug resistant-TB, approved in 2014 by the EU)7 and Pretomanid8a (III, PA-824, against multi-drug-resistant-MTB, approved in 2019 by the USFDA).10 Our involvement in the area of the TB drug discovery program and nitroimidazole chemistry8b,c motivated us to develop a new concise and improved strategy towards the synthesis of Delamanid. The first synthesis of Delamanid was done by Tsubouchi et al. in 2004 (Otsuka Pharmaceutical Ltd. WO2004033463A1) which involved 16 steps (details shown in Fig. S1 of ESI†).11,12 The synthesis involves protection–deprotection strategy and made the process lengthy. Later on, in 2011, Otsuka had developed another concise route for Delamanid with improved yield (upper half of Fig. 2, details shown in Fig. S2 of ESI†). This method involved the Sharpless epoxidation of 2-methyl allylalcohol followed by ring opening with 4-bromophenol. Then coupling with 4(4-trifluoromethoxy phenyl)piperidine fragment under palladium catalyzed conditions, which in turn was synthesized in 2–5 steps.13 Considering the importance of Delamanid, It could be better if more concise route was developed. In this regard, here we devised a route which involves sequential addition of fragments which not only avoid protection and deprotection but also provides the advantages of avoidance of dry conditions and costly intermediates. The lower half of Fig. 2 represents the strategy of the present method used for the synthesis of Delamanid, which involve the sequential coupling with following cascade viz., allylation, selective N-arylation, Mitsunobu ether formation, Sharpless dihydroxylation, epoxidation, ring opening and cyclization using inexpensive and easily available starting materials.
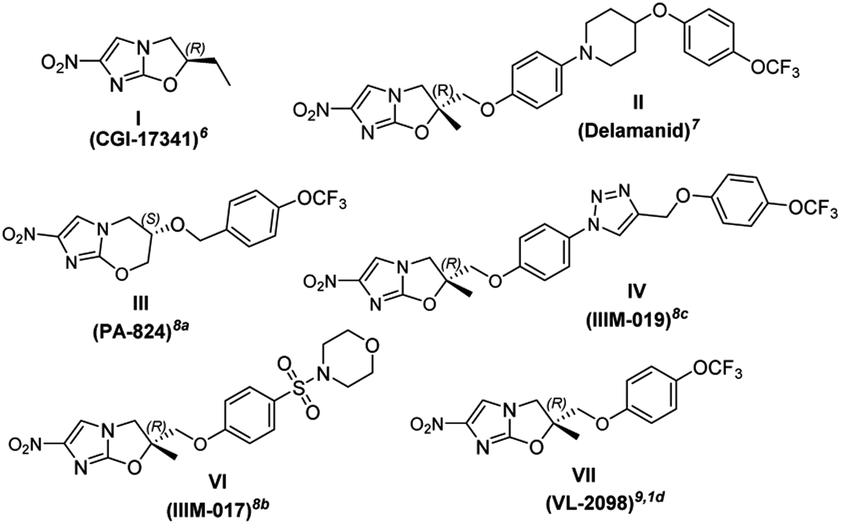 | ||
| Fig. 1 Nitroimidazole containing drugs and leads. | ||
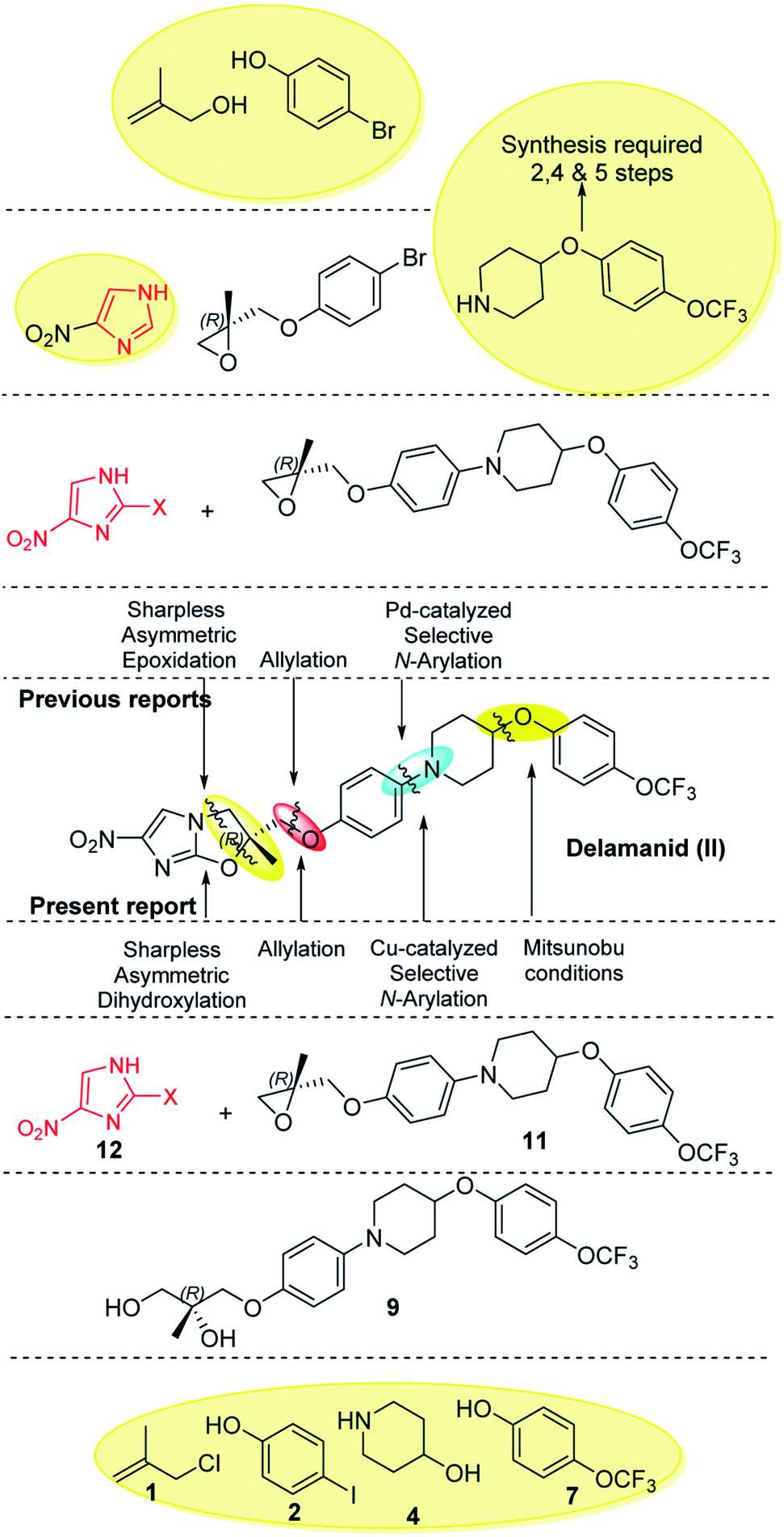 | ||
| Fig. 2 Previous and current approaches. | ||
The present synthesis started with 2-methylallyl chloride as first starting material and its selection is because of following reasons (i) as protecting group, (ii) inexpensive (33$ per 100 mL, Sigma) in comparison to 2-methylallyl alcohol (766$ per 100 mL, Sigma) which is used in earlier reported method and (iii) provide double bond functionality to generate epoxide via Sharpless dihydroxylation approach, which operates under open atmosphere conditions and avoids the anhydrous and dry environment as required for Sharpless epoxidation, used in the previous reported methods.7,11b,12
Result and discussion
The synthesis started with allylation of 4-iodophenol 2. The 2-methylallyl chloride 1 (or 3-chloro-2-methylprop-1-ene) reacted with 4-iodophenol 2 in the presence of potassium carbonate as base in DMF at 60 °C to furnish 1-iodo-4-((2-methylallyl)oxy)benzene 3 in 97% yield (Scheme 1). | ||
| Scheme 1 Allylation. Reagent and conditions: (i) compound 1 (100 mmol), compound 2 (200.0 mmol), K2CO3 (200.0 mmol), DMF, 60 °C, 97%. | ||
The second step involves the C–N bond formation between 1-iodo-4-((2-methylallyl)oxy)benzene 3 and 4-hydroxypiperidine 4. Here, our aim was to develop a condition for selective C–N bond formation in the presence of free hydroxyl group. In the literature, copper catalysis has been used for selective C–N bond formation,14 however, no such attempts were made in the synthesis of Delamanid, which on the other hand was achieved by Pd-catalysis.10,12–14 We started with copper catalysis and various attempts were made (details provided in Scheme 2, entries 1–10). The optimization started by using CuI as catalyst in the presence of different ligands as well as solvents. The reaction with CuI (10 mol%), phenanthroline (10 mol%) and Cs2CO3 (2 equiv.) gave the desired N-arylated product 5 in a yield of 55% without any O-arylated product 5′ (entry 1). When the same reaction was performed without any base, it gives 53% yield of 5 (entry 2). In the next attempt, reaction was performed by using bipyridine as ligand which gave 5 in a yield of 60% (entry 3). After that, pyrrolidine was attempted which gave poor yield of 35% of 5 (entry 4). Then, combination of CuI (10 mol%) and L-proline (10 mol%) was tried at room temperature which gave the yield of desired N-arylated product 5 in a yield of 80% (entry 6). The reaction with CuI (10 mol%) and L-proline (10 mol%) at 80 °C furnished the N-arylated product 5 and O-arylated product 5′ in a yield of 40% and 20% respectively (entry 7). Then varying the molar ratio of ligand (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20%) at 80 °C did not show any improvement and desired product 5 was obtained in a yield of 48% along with 25% of O-arylated product 5′ (entry 8). In further refinement experiments (entries 9–11), different molar ratio of ligand and catalyst was tried, where CuI (20 mol%) and L-proline (40 mL%) gave highest yield of 5 (85%). After optimization, the synthesis of desired N-aryalted product 5 was performed at 100 mmol scale which worked nicely and delivered the required product 5 in a yield of 81%.
:20%) at 80 °C did not show any improvement and desired product 5 was obtained in a yield of 48% along with 25% of O-arylated product 5′ (entry 8). In further refinement experiments (entries 9–11), different molar ratio of ligand and catalyst was tried, where CuI (20 mol%) and L-proline (40 mL%) gave highest yield of 5 (85%). After optimization, the synthesis of desired N-aryalted product 5 was performed at 100 mmol scale which worked nicely and delivered the required product 5 in a yield of 81%.
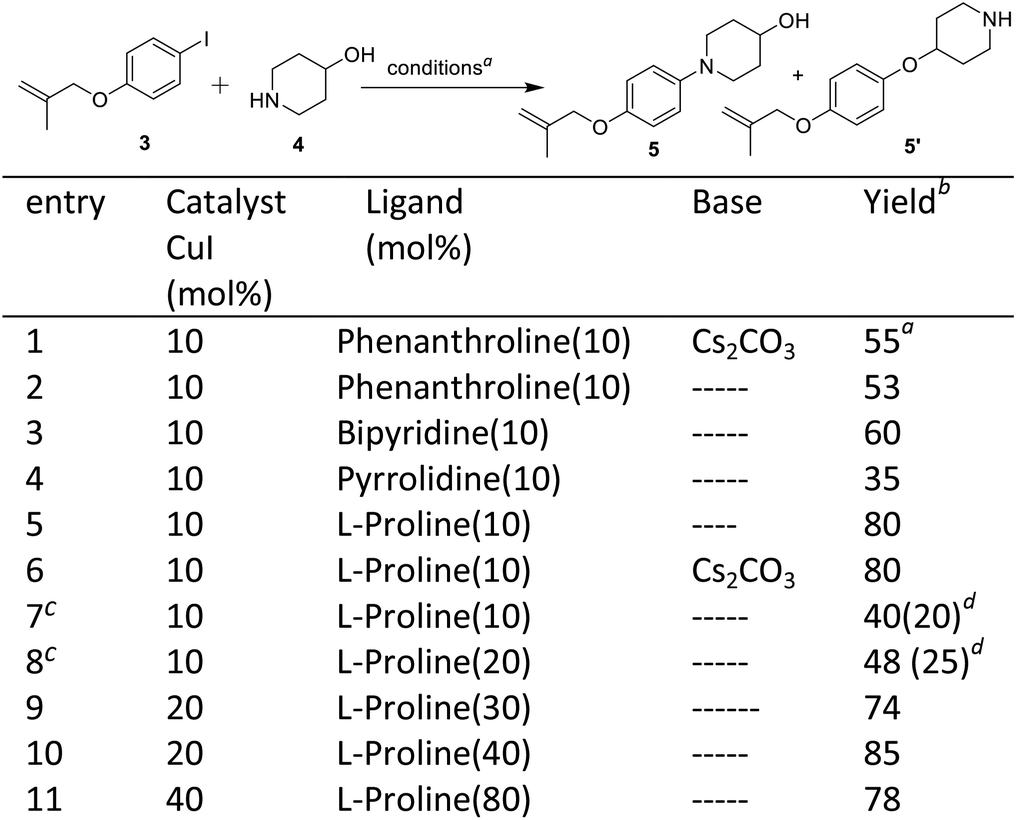 | ||
| Scheme 2 C–N bond formation. aReagents and conditions: compound 3 (0.5 mmol), compound 4 (1 mmol), DMF, 10–24 h; bisolated yields; creaction performed at 80 °C; dyield of O-arylated product 5′. | ||
The third step involves the ether bond formation between 1-(4-((2-methylallyl)oxy)phenyl)piperidin-4-ol 5 and 4-trifluoromethoxyphenol 7. In the first instance, the ether formation was tried via mesylation of hydroxyl group followed by nucleophilic substitution with 4-trifluoromethoxyphenol 7. The mesylation of 1-(4-((2-methylallyl)oxy)phenyl)piperidin-4-ol 5 was carried out by treating with mesyl chloride in presence of triethylamine which afforded the O-mesylated product 6 in the yield of 93%. The coupling of O-mestlated product 6 with 4-trifluoromethoxyphenol 7 have been tried by using different solvents (N,N-diemthylformamide, toluene) and bases (Cs2CO3, K2CO3, NaH) combination, but none gave satisfactorily yields of coupled product 8 (Scheme 3, entries 1–5). Then, we switch over to Mitsunobu reaction conditions using triphenyl phosphine (PPh3) and diethyl azodicarboxylate (DEAD) for ether formation. Again various attempts have been made under different solvents (Scheme 4).14 By screening different solvents, THF as solvent and 2.0 equivalents of PPh3 and DEAD gave best result towards the formation of required key intermediate 8 in a yield of 70%. Taking the best conditions, the ether formation was tried in 20 mmol scale which afforded the key product 8 in a yield of 72%. Here, we have provided a new approach for the ether formation. The use of environmental friendly version of Mitsunobu (polymeric or catalytic variants) will be tried during upscaling.15
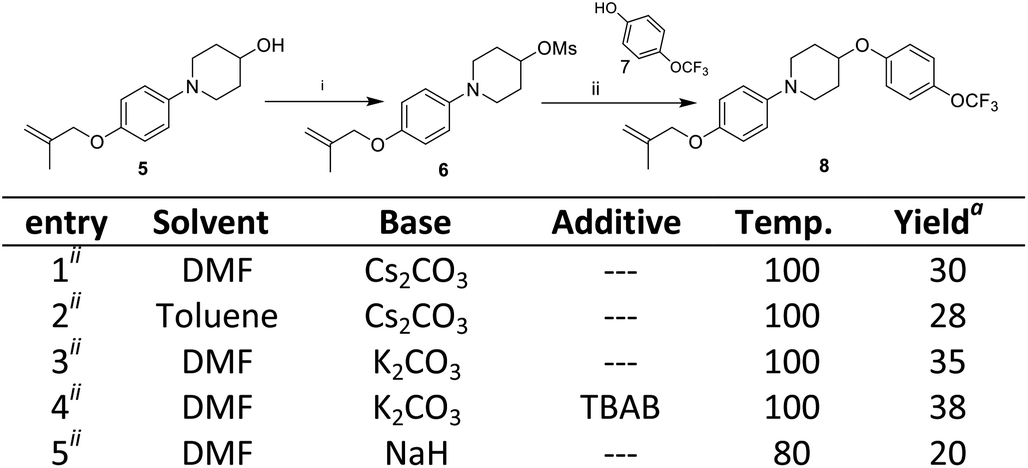 | ||
| Scheme 3 Nucleophilic substitution for ether formation. Reagent and conditions: (i) compound 5 (4 mmol), mesyl chloride (4.4 mmol), Et3N (6 mmol), DCM, 0 °C to rt; (ii) compound 6 (0.31 mmol), phenol 7 (0.31 mmol), base (0.62), 2–12 h; aisolated yield. | ||
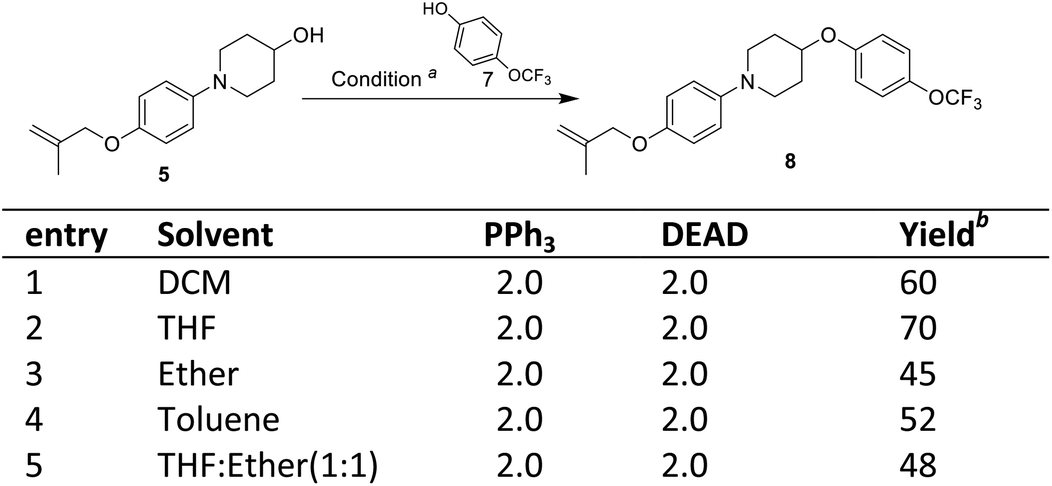 | ||
| Scheme 4 Mitsunobu reaction for ether formation. aReagents and conditions: compound 5 (0.8 mmol), compound 7 (0.8 mmol), PPh3 (1.14 mmol), DEAD (0.97 mmol), rt, 24 h; bisolated yield. | ||
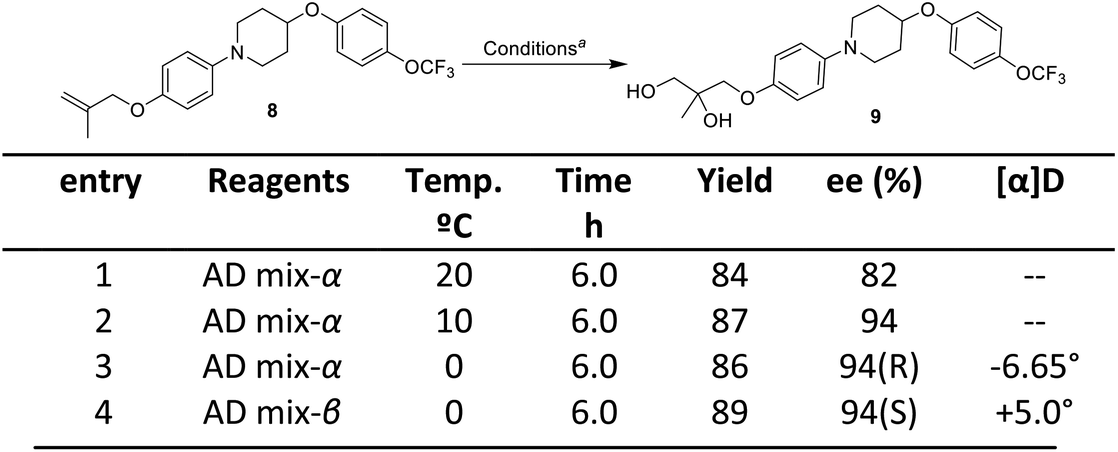 | ||
| Scheme 5 Sharpless asymmetric dihydroxylation. aReagent and conditions: compound 8 (2.0 mmol), AD mix-α or β (2.8 g, 1.4 g for 1 mmol of alkene), tert-butanol:water (1:1). | ||
The next step involved the generation of enantiopure epoxide which in present case was achieved via Sharpless asymmetric dihydroxylation using AD mix-α and AD mix-β.16 The optimization started with the reaction of 1-(4-((2-methylallyl)oxy)phenyl)-4-(4-trifluoromethoxy) phenoxy)piperidine 8 with AD mix-α at 20 °C which gave diol product 9 in a yield of 84% with an ee (enantiomeric excess) of 82%. Decrease in the temperature conditions from 20 °C to 0 °C improved the enantioselctivity where the reaction at 0 °C gave diol product 9 in a yield of 86% with ee of 94% (HPLC detail provided in ESI†). The synthesized diol 9 has shown an optical rotation of [α]25D −6.65°. In order to confirm the R-configuration of diol 9, the same has also been synthesized by reported method (detail provided in Fig. S3 of ESI†), which is in well agreement with our synthesized diol 9.9,17 In the further exploration, when the reaction was attempted with AD mix-β, the diol product was obtained with yield of 89% having ee of 94% (and shown opposite optical rotation [α]25D +5° and in agreement with literature reported S-configured diol. The present approach provides the opportunity to synthesize both the enantiomers which can be exploited in hit to lead optimization strategy of medicinal chemistry program. As the present strategy required R-isomer, our next steps are focused with R-isomer. The temperature played an important role in this reaction to get the better enantioselctivity. The Sharpless asymmetric dihydroxylation conditions operated under open atmospheric conditions, which provided another advantage of present method over the existing method.
The diol R-(−)-9 was converted into a key intermediate epoxide 11 via mesylation and ring formation as per the conditions provided in Scheme 6.
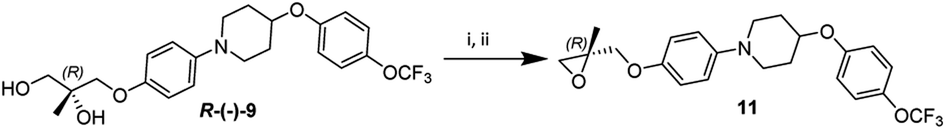 | ||
| Scheme 6 Epoxide formation. Reagent and conditions: (i) compound R-(−)-9 (3 mmol), mesyl chloride (3.6 mmol), Et3N (6 mmol), DCM, 0 °C to rt; (ii) compound 10 (0.31 mmol), DBU (0.62 mmol), rt, 2 h, yield 82%. | ||
The 2-bromo-4-nitroimidazole 12, another key fragment and coupling partner was synthesized from 4-nitroimidazole (detail shown in Fig. S4 of ESI†). After the successful synthesis of both the key intermediates viz., epoxide 11 and 2-bromo-4-nitroimidazole 12, the final attempt towards the suitable coupling conditions were made (details given Table S1 of ESI†) where DIPEA (act as base as well as solvent) gave the coupled product 13 which subsequent treated with Cs2CO3 as base successfully furnished the final construct Delamanid II with an overall yield of 63% (Scheme 7).
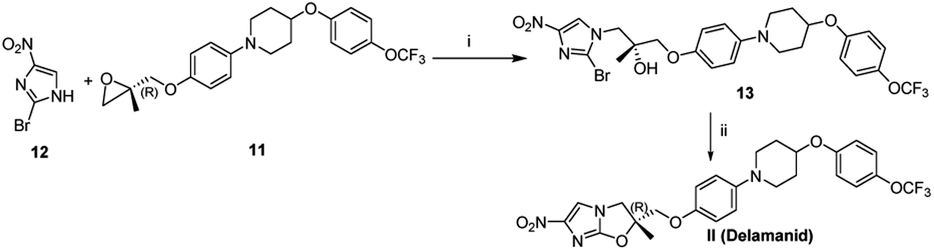 | ||
| Scheme 7 Final coupling. Reagent and conditions: (i) compound 12 (3 mmol), compound 11 (3 mmol), DIPEA, 115 °C, yield 90%; (ii) compound 13 (1 mmol), Cs2CO3 (2 mmol), DMF, 50 °C, 2 h, yield 70%. | ||
After finishing this, we further extended the present approach to VL-2098 (VII) which is another nitroimidazole based lead reported against visceral leishmaniasis.1d As per the literature, two routes are reported for its synthesis. The first synthesis was reported by Tsubouchi et al. (2004) which involved 7 steps (details shown in Fig. S5 of ESI†).11,18 Later on, in 2017, Pati et. al. developed another alternative route with an overall yield of 19% (details shown in Fig. S6 of ESI†) (Fig. 3).9
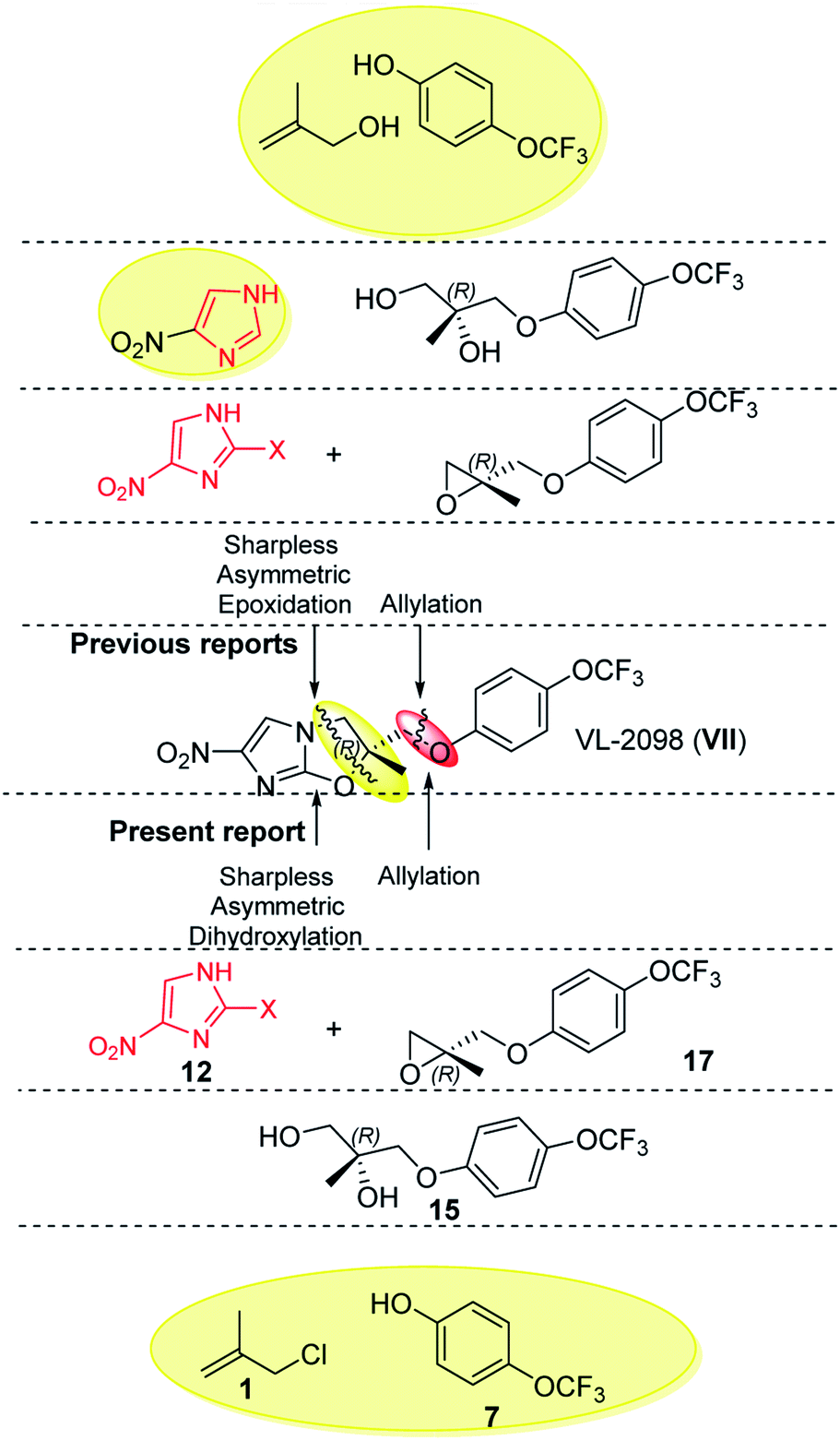 | ||
| Fig. 3 Previous and current approaches for VL-2098. | ||
To synthesized VL-2098, 2-methylallyl chloride 1 (or 3-chloro-2-methylprop-1-ene) reacted with 4-trifluoromethoxy phenol 7 under basic conditions to furnished 1-((2-methylallyl)oxy)-4-(trifluoromethoxy)benzene 14 in 97% yield. The next step involved the Sharpless asymmetric dihydroxylation using AD mix-α.16 The reaction started with the 1-((2-methylallyl)oxy)-4-(trifluoromethoxy)benzene 14 with AD mix-α at 20 °C which gave diol product R-(+)-15 with an yield of 88%. The synthesized diol has shown an optical rotation of [α]25D +8.83°. The diol R-(+)-15 was converted into a key intermediate epoxide 17 via mesylation as per the conditions provided in Scheme 8. The final step involved the coupling of key fragment epoxide 17 with 2-bromo-4-nitroimidazole 12 to get the desired product VL-2098 (VII) with an optical rotation of [α]25D +7° ([α]25D +8.445°).9 This new approach has an overall yield of 36%.
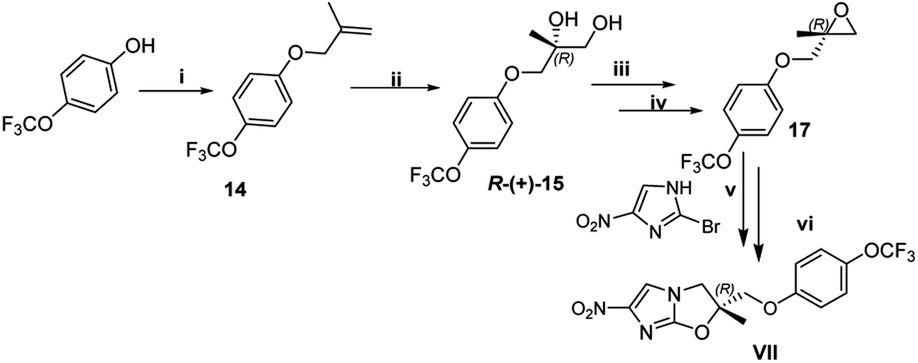 | ||
| Scheme 8 Synthesis of VL-2098. Reagent and conditions: (i) Compound 7 (28 mmol), K2CO3 (56 mmol), DMF, 60–65 °C, 2 h, 97%; (ii); compound 14 (4.3 mmol), AD-mix α (3 mmol), t-BuOH–H2O (1:1), rt, 2 h, 88%; (iii) compound R-(+)-15 (3.7 mmol), MsCl (2.2 mmol), Et3N (4 mmol), EtOAc, <15 °C – rt, 2 h; (iv) DBU (4 mmol), EtOAc, rt, 2 h, 89%; (v) compound 17 (1 mmol), DIPEA,110 °C, 2 h; (vi) Cs2CO3 (2 mmol), DMF, 50 °C, 2 h, 48%. | ||
In summary, we have developed an alternative concise route for the synthesis of nitroimidazole based drug molecule Delamanid and lead candidate VL-2098 with an overall yield of 27% and 36% respectively. The present strategy avoids the use of protecting groups and expensive reagents. The present strategy also provides the opportunity towards the generation of opposite enantiomers and manipulation of functionalities/groups, which is presently being pursued by our group to generate best-in-class nitroimidazole based TB drug free of hERG liability and better drug-like properties.
Experimental section
General information
All product mixtures were analysed by thin layer chromatography silica TLC plates. UV-active compounds were detected with a UV lamp (λ = 254 nm). For column chromatography, silica gel (60–120 mesh) was used as stationary phase. 1H NMR, 13C and DEPT NMR spectra were recorded on 400 MHz spectrometer using CDCl3, DMSO solvent. ESI-MS and HRMS spectra were recorded on LC-MS/MS and HRMS-6540-UHD machines.Synthesis of Delamanid
Procedure for the synthesis of the 1-iodo-4-((2-methylallyl)oxy)benzene (3). The 4-iodophenol 2 (21.9 g, 100 mmol, 1 equiv.) and potassium carbonate (K2CO3) (200 mmol, 2 equiv.) were taken in N,N-dimethylformamide (DMF) and then methallyl chloride 1* (200 mmol, 1.3 equiv.) was added to the reaction mixture and heated to 60 °C for a period of 2 h. Upon cooling the reaction mixture, it was diluted with EtOAc and washed twice with H2O and brine. Then organic layer was evaporated under vacuum and purified by column chromatography on a silica gel (60–120) with hexane/ethyl acetate (9
:1) as the eluent to get the desired product 3 as oily liquid. (26.5 g, yield 97%). TLC Rf = 0.5 (20% EtOAc:hexane) 1H NMR (400 MHz, CDCl3) δ 7.56–7.54 (d, J = 8.0 Hz, 2H), 6.72–6.70 (d, J = 8.0 Hz, 2H), 5.08 (s, 1H), 5.00 (s, 1H), 4.41 (s, 2H), 1.83 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 158.74, 140.53, 138.27, 117.35, 113.07, 83.03, 71.87, 19.48; GC-MS(EI) m/z (relative intensity) 274 (M+, 100%).#The care has to be taken while handling methallyl chloride as it is a reported to have mutagenic properties.
Synthesis of the 1-(4-((2-methylallyl)oxy)phenyl)piperidin-4-ol (5) (Scheme 2, entry 10). An oven-dried round bottom flask was charged with copper iodide (19 mg, 0.1 mmol) and L-proline (23 mg, 0.4 mmol). The flask was evacuated with N2 atmosphere & then fitted with a rubber septum. DMF (2 mL) was next added, as well as the aryl iodide 3 (136 mg, 0.5 mmol), which was added at this stage. The resulting blue solution was stirred for 10 min before adding 4-hydroxypiperidine 4 (202 mg, 2 mmol). Upon completion, sodium hydroxide (1 M aqueous solution) was added to the reaction mixture and extracted twice with ethyl acetate. Anhydrous sodium sulphate was added to the organic layer, filtered and concentrated to get the desired pure product as solid 5 (104 mg, yield 85%).
Scale up reaction
An oven-dried flask was charged with copper iodide (3.8 g, 20 mmol) and L-proline (4.6 g, 40 mmol). The flask was evacuated with N2 atmosphere & then fitted with a rubber septum. DMF (120 mL) was next added, as well as the aryl iodide 3 (27 g, 100 mmol), which was added at this stage. The resulting blue solution was stirred for 10 min before adding 4-hydroxypiperidine 4 (40 g, 400 mmol). Upon completion, sodium hydroxide (1 M aqueous solution) was added to the reaction mixture and extracted twice with ethyl acetate. Anhydrous sodium sulphate was added to the organic layer, filtered and concentrated to get the desired pure product as solid 5 (19.8 g, yield 81%). TLC Rf = 0.5 (40% EtOAc:hexane).
1H NMR (400 MHz, CDCl3) δ 6.84–6.76 (m, 4H), 5.00 (s, 1H), 4.89 (s, 1H), 4.31 (s, 2H), 3.76–3.72 (m, 1H), 3.34–3.31 (m, 2H), 2.78–2.71 (m, 2H), 1.96–1.92 (m, 2H), 1.75 (s, 3H), 1.68–1.60 (m, 2H); 13C NMR (101 MHz, CDCl3): δ (ppm) = 153.01, 145.92, 141.32, 118.69, 115.49, 112.43, 72.19, 48.94, 34.47, 29.70, 19.28; HRMS (ESI+) calcd for C15H21NO2 248.1651 ([M + H]+), found 248.1621.
Synthesis of 1-(4-((2-methylallyl)oxy)phenyl)-4-(4-(trifluoromethoxy)phenoxy)piperidine (8) (Scheme 4, entry 2). To a solution of 4-hydroxypiperidine derivative 5 (200 mg, 0.8 mmol) in THF (3 mL) was added PPh3 (420 mg, 1.6 mmol) and further a solution of 4-trifluoromethoxyphenol 7 (144 mg, 0.8 mmol) and DEAD (324 mg, 1.6 mmol) was added slowly dropwise below 5 °C, and stirred at room temperature for 24 hours. The reaction mixture was allowed to concentrate and aqueous sodium hydroxide solution was added, followed by extraction with EtOAc. Then anhydrous sodium sulfate was added to the organic layer and concentrated under reduced pressure and purified by silica gel column chromatography (eluent; hexane
:EtOAc = 4:1 (v/v)) to obtain 1-(4-((2-methylallyl)oxy)phenyl)-4-(4-(trifluoromethoxy)phenoxy)piperidine as light brown solid material 8 (231 mg, yield 70%).
Scale up reaction
To a solution of 4-hydroxypiperidine derivative 5 (5 g, 20 mmol) in THF (30 mL) was added PPh3 (10.5 g, 40 mmol) and further a solution of 4-trifluoromethoxyphenol 7 (3.56 g, 20 mmol) and DEAD (8.1 g, 40 mmol) was added slowly dropwise below 5 °C, and stirred at room temperature for 24 hours. The reaction mixture was allowed to concentrate and aqueous sodium hydroxide solution was added, followed by extraction with EtOAc. Then anhydrous sodium sulfate was added to the organic layer and concentrated under reduced pressure and purified by silica gel column chromatography (eluent; hexane:EtOAc = 4:1 (v/v)) to obtain 1-(4-((2-methylallyl)oxy)phenyl)-4-(4-(trifluoromethoxy)phenoxy)piperidine as light brown solid material 8 (5.93 g, yield 72%). TLC Rf = 0.5 (5% EtOAc:hexane).
1H NMR (400 MHz, CDCl3) δ 7.17–7.14 (d, J = 12.0 Hz, 2H), 6.96–6.87 (m, 6H), 5.10 (s, 1H), 4.99 (s, 1H), 4.40 (s, 3H), 3.41–3.36 (m, 2H), 3.03–2.97 (m, 2H), 2.16–2.11 (m, 2H), 2.01–1.93 (m, 2H), 1.84 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 155.97, 153.16, 145.86, 142.84, 141.32, 122.48, 119.33 (q, JC–F = 256.54 Hz), 118.71, 116.93, 115.53, 112.52, 72.96, 72.30, 48.27, 30.77, 19.41; HRMS (ESI+) calcd for C22H24F3NO3 408.1787 ([M + H]+), found 408.1796.
Synthesis of (R)-2-methyl-3-(4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)phenoxy)propane-1,2-diol (R-(−)-9) (Scheme 5, entry 3). In a round bottom flask, AD-mix-α (12 g) was taken in tert-butyl alcohol
:water (1:1, 30 mL) and stir at room temperature to dissolve the AD-mix-α. The mixture was cooled to 0 °C and then added 1-(4-((2-methylallyl) oxy)phenyl)-4-(4-(trifluoromethoxy)phenoxy) piperidine 8 (5 g, 12.28 mmol) and again stir vigorously at 0 °C for 6 h and then 1.93 g (15.3 mmol) of sodium sulphite was added. The reaction mixture was allowed to stir at room temperature for 1 h and diluted with 20 mL of DCM and 40 mL of water and extracted with portions of DCM. The anhydrous MgSO4 was added to the organic phase, filtered and concentrated to afford the crude oil. The residue was purified by chromatography on a silica gel column with gradient elution with 4:1 (hexanes/ethyl acetate) as brownish gummy material R-(−)-9. Yield (4.7 g, 86%). Enantiomeric excess was determined by chiral HPLC (chiral pack ADH normal phase column, 4.6 × 250 mm, 5 μm; enantiomeric excess (ee): 94%). TLC Rf = 0.5 (70% EtOAc:hexane). [α]25D −6.65° (c 1, chloroform).1H NMR (400 MHz, CDCl3) δ 7.08–7.06 (d, J = 8.0 Hz, 2H), 6.87–6.82 (m, 4H), 6.80–6.76 (m, 2H), 4.37–4.32 (m, 1H), 3.84–3.78 (dd, J = 12.0 Hz, 1H), 3.66–3.63 (d, J = 12.0 Hz, 1H), 3.51–3.48 (d, J = 12.0 Hz, 1H), 3.33–3.27 (m, 2H), 2.95–2.89 (m, 2H), 2.06–2.01 (m, 2H), 1.90–1.84 (m, 2H), 1.21 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 155.89, 152.65, 146.28, 142.86, 122.46, 119.33(q, JC–F = 260.58 Hz), 118.72, 116.93, 115.39, 73.53, 72.91, 72.01, 67.83, 48.14, 30.60, 21.49; HRMS (ESI+) calcd for C22H27F3NO5 442.1841 ([M + H]+), found 442.1843.
Synthesis of (S)-2-methyl-3-(4-(4-(4-(trifluoromethoxy)phenoxy) piperidin-1-yl)phenoxy)propane-1,2-diol (S-(+)-9) (Scheme 5, entry 4). In a round-bottomed flask equipped with magnetic stirring bar, AD-mix-β (12 g) was taken in tert-butyl alcohol
:water (1:1, 30 mL) and stir at room temperature to dissolve the AD-mix-β. The mixture was cooled to 0 °C and then added the 1-(4-((2-methylallyl)oxy)phenyl)-4-(4-(trifluoromethoxy)phenoxy)piperidine 8 (5 g, 12.28 mmol) and again stir vigorously at 0 °C for 6 h and then 1.93 g (15.3 mmol) of sodium sulphite was added. The reaction mixture was allowed to stir at room temperature for 1 h and diluted with 20 mL of DCM and 40 mL of water and the reaction mixture was extracted with portions of DCM. The anhydrous MgSO4 was added to the organic phase, filtered and concentrated to afford the crude oil. The residue was purified by chromatography on a silica gel column with gradient elution with 4:1 hexanes/ethyl acetate as brownish gummy material S-(+)-9. Yield (4.8 g, 89%). Enantiomeric excess was determined by using chiral HPLC (chiral pack ADH, normal phase column, 4.6 × 250 mm, 5 μm; enantiomeric excess (ee): 95%). [α]25D +5° (c 1, chloroform).1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 8.0 Hz 2H), 6.93–6.97 (m, 4H), 6.87–6.90 (d, J = 12.0 Hz, 2H), 4.41 (m, 1H), 3.87 (d, J = 12.0 Hz, 1H), 3.95 (d, J = 12.0 Hz, 1H), 3.74 (d, J = 12.0 Hz, 1H), 3.60 (d, J = 12.0 Hz, 1H), 3.38 (m, 2H), 3.0 (m, 2H), 2.1 (m, 2H), 1.99 (m, 2H), 1.30 (s, 3H).
Synthesis of (R)-2-hydroxy-2-methyl-3-(4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)phenoxy)propyl methanesulfonate 10 and (R)-1-(4-((2-methyloxiran-2-yl)methoxy)phenyl)-4-(4-trifluoromethoxy)phenoxy) (11) (Scheme 6). In a 100 mL round-bottomed flask, (R)-2-methyl-3-(4-(4-(4-(trifluoromethoxy)phenoxy) piperidin-1-yl)phenoxy)propane-1,2-diol R-(−)-9 (0.9 g, 2 mmol) was taken into DCM solvent and add triethylamine (0.570 mL, 4 mmol) followed by the addition of methanesulfonyl chloride (0.170 mL, 2.2 mmol) dropwise at 0 °C and stirred at room temperature for a period of 2 h. Then solvent was evaporated and extract with ethyl acetate and aqueous layer. The organic solvent was evaporated to afford the mesylated product 10 as gummy material and used as such for the next reaction. In the next step, mesylated compounds 10 was dissolved in ethyl acetate and DBU (0.596 mL, 4 mmol) was added and stirred the reaction mixture for a period of 2 h and then water was added to the reaction mixture and extract with ethyl acetate and evaporate the organic layer under rotary evaporator followed by purification over 100–200 silica gel and ethyl acetate
:hexane (40:60) as eluent to afford the compound 11 as brown gummy liquid. (0.7 g, yield 82%).1H NMR (400 MHz, CDCl3) δ 7.15–7.13 (d, J = 8 Hz, 2H), 6.94–6.91 (dd, J = 4 Hz, 4H), 6.88–6.86 (d, J = 8 Hz, 2H), 4.44–4.38 (m, 1H), 4.00–3.90 (dd, J = 16 Hz, 2H), 3.40–3.35 (m, 2H), 3.02–2.96 (m, 2H), 2.86–2.85 (d, J = 4.0 Hz, 1H), 2.72–2.71 (d, J = 4 Hz, 1H), 2.14–2.09 (m, 2H), 1.99–1.92 (m, 2H), 1.48 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 156.02, 152.94, 146.28, 142.86, 122.46, 119.40 (q, JC–F = 256.54 Hz), 118.66, 116.97, 115.53, 73.01, 72.20, 55.63, 51.97, 48.14, 30.77, 18.49; HRMS (ESI+) calcd for C22H24F3NO4 424.1730 ([M + H]+, found 424.1733.
Synthesis of 1-(2-bromo-4-nitro-1H-imidazol-1-yl)-2-methyl-3-(4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)phenoxy)propan-2-ol (13) (Scheme 7). A mixture of epoxide 11 (424 mg, 1 mmol), 2-bromo-4-nitro-1H-imidazole 12 (190 mg, 1 mmol), and DIPEA (2.0 mL) were taken into a sealed tube and heated to 115 °C for a period of 12 h. Upon cooling the reaction mixture, dichloromethane was added and evaporated to dryness under reduced pressure and then residue was purified on silica gel 100–200 and then eluted with 0.5% EtOAc/CH2Cl2 gave the desired compound 13 (552 mg) in a yield of 90%.
1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 7.16–7.14 (d, J = 8.0 Hz, 2H), 6.95–6.91 (m, 4H), 6.83–6.81 (d, J = 8.0 Hz, 2H), 4.43 (m, 1H), 4.31–4.28 (d, J = 16 Hz, 1H), 4.20–4.16 (d, J = 16 Hz, 1H), 3.87–3.79 (m, 2H), 3.42–3.35 (m, 2H), 3.02 (m, 2H), 2.17–2.06 (m, 2H), 2.00–1.91 (m, 2H), 1.33 (s, 3H). Mass (m/z): 615 (M + H)+.
Synthesis of 2-methyl-6-nitro-2-((4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)phenoxy)methyl)-2,3-dihydroimidazo[2,1-b]oxazoles (II, Delamanid, Scheme 7). To the stirred solution of compound 13 (307 mg, 0.5 mol) in anhydrous DMF, caesium carbonate was taken and stirred the reaction mixture for 2 h at 50 °C and then upon completion of reaction, add water and extract with ethyl acetate. Then evaporate the solvent and purified the reaction mixture with silica gel 100–200 eluting with 40% ethyl acetate: dichloromethane gave compound (Delamanid II) as white solid (185 mg, 70%). Mp 195–196 °C. 1H NMR (CDCl3) δ 7.58 (s, 1H), 7.17–7.15 (d, J = 8.0 Hz, 2H), 6.84–6.91 (m, 4H), 6.81–6.78 (d, J = 12.0 Hz, 2H), 4.53–4.50 (d, J = 12.0 Hz, 1H), 4.46–4.41 (1H, m), 4.21–4.18 (d, J = 10.2 Hz, 1H), 4.07–4.04 (dd, J = 12.0 Hz, 2H), 3.41–3.35 (m, 2H), 3.04–2.98 (m, 2H), 2.14–2.09 (m, 2H), 1.99–1.93 (m, 2H), 1.78 (s, 3H); 19F NMR (CDCl3, 376 MHz): δ −58.32; 13C NMR (101 MHz, CDCl3) δ 155.92, 151.71, 146.89, 122.50, 119.29 (q, JC–F = 256.54 Hz) 118.56, 116.84, 115.57, 112.54, 93.24, 72.70, 72.23, 51.43, 47.86, 30.57, 23.16; [α]25D −12° (c 1, chloroform) [[α]25D −9.9°];7 HRMS (ESI+) calcd for C25H25F3N4O6 535.180 ([M + H]+, found 535.179.
Synthesis of 2,5-dibromo-4-nitro-1H-imidazole S2 (Fig. S4 of ESI†). In a three necked round bottom flask (3 L), 4-nitroimidazole S1 was taken into water (400 mL) and stir the mixture to temperature below 5 °C. Then sodium bicarbonate (86 g, 1 mol) was added followed by the slow addition of bromine (160 g, 1.0 mmol) to reaction mixture over a period of 4.0 h by keeping the temperature below 0 °C and then stir it at 65 °C for 6.0 h. After completion of reaction, pH of the reaction mixture was adjusted with aqueous hydrochloric acid (6.0 N, 50 mL) to pH 2–3 and stirred it for 1.0 h below 5 °C and filtered. The compound S2 obtained as a pale-yellow solid (104 g, 88%). Mp 229–231 °C.
1H NMR: (400 MHz, DMSO-d6) δ 7.98 (s, 1H); mass (m/z): 271.4 (M + H)+.
Synthesis of 2-bromo-4-nitro-1H-imidazole (12) (Fig. S4 of ESI†). In a 3 L capacity round bottom flask, 2,5-dibromo-4-nitro-1H-imidazole, S2 (100 g, 378 mmol) was taken into glacial acetic acid (1.2 L) and stirred it at room temperature. Then potassium iodide (94 g, 567 mmol), and sodium sulfite (71 g, 567 mmol) was added to the reaction mixture and heated to 120–125 °C for 16 h. Upon completion, glacial acetic acid was distill off and water (500 mL) was added to the reaction mixture and stirred for 10 min, followed by the addition of aqueous sodium bisulfite solution (5%) to quench excess iodide. The reaction mixture was extracted with ethyl acetate (3 × 250 mL) and washed with water (2 × 250 mL), dried over magnesium sulphate, and concentrated to get 2-bromo-4-nitroimidazole, 12 as a yellow solid (72 g, 65%). Mp 240 °C.
1H NMR: (400 MHz, DMSO-d6) δ 14.14 (bs, 1H), 8.41 (s, 1H); HRMS (ESI+) calcd for C3H3BrN3O2 191.9409 ([M + H]+, found 191.9402.
Synthesis of VL-2098
Procedure for the synthesis of the 1-((2-methylallyl)oxy)-4-(trifluoromethoxy)benzene (14) (Scheme 8). The starting material 4-trifluoromethoxyphenol 7 (5 g, 28 mmol, 1 equiv.) and K2CO3 (7.8 g, 56 mmol, 2 equiv.) were taken in DMF and then methallyl chloride 1 (3 mL, 36.4 mmol, 1.3 equiv.) was added and heated the reaction mixture to 60 °C for a period of 2 h. Upon cooling the reaction mixture, it was diluted with EtOAc and washed twice with H2O and brine. Then organic layer was evaporated under vacuum and purified by column chromatography on a silica gel (60–120) with hexane/ethyl acetate (9
:1) as the eluent to get the desired product (6.32 g, yield 97%). 1H NMR (400 MHz, CDCl3) δ 7.12 (d, J = 8.5 Hz, 2H), 6.89 (d, J = 8.5 Hz, 2H), 5.09 (s, 1H), 5.00 (s, 1H), 4.40 (s, 2H), 1.82 (s, 3H). 19F NMR (CDCl3, 376 MHz): δ −58.32. 13C NMR (101 MHz, CDCl3) δ 157.40, 142.87, 140.55, 124.52, 122.32, 121.98, 119.44 (q, JC–F = 256.54 Hz), 115.54, 112.88, 72.06, 19.14. GC-MS(EI) m/z (relative intensity) 232 (M+, 100%).
Synthesis of (R)-2-methyl-3-(4-(trifluoromethoxy)phenoxy)propane-1,2-diol R-(+)-15 (Scheme 8). In a 100 mL round-bottomed flask AD-mix-α (4 g) was taken in tert-butyl alcohol
:water (1:1, 30 mL) and stir at room temperature to dissolve the AD-mix-α. Add 1-((2-methylallyl)oxy)-4-(trifluoromethoxy)benzene 14 (1 g, 4.31 mmol) at 0 °C and stir it for a period of 6–12 h and then sodium sulphite (1.93 g, 15.3 mmol) was added. The reaction mixture was allowed to stir at room temperature for 1 h and diluted with DCM (20 mL) and water (40 mL) and extracted with DCM. The combined organic phase was dried over anhydrous MgSO4, filtered and concentrated to get crude oil. The residue was purified through column chromatography on a silica gel (100–200 mesh size) with gradient elution with 4:1 hexane/ethyl acetate as white solid material R-(+)-15. Yield 1.0 g (88%). [α]25D +8.83°.1H NMR (400 MHz, CDCl3) δ 7.14 (d, J = 8.8 Hz, 2H), 6.90 (d, J = 9.0 Hz, 2H), 3.90 (q, J = 8.9 Hz, 2H), 3.71 (dd, J = 11.0 Hz, 1H), 3.57 (dd, J = 11.0 Hz, 1H), 2.82 (s, 1H), 2.44 (s, 1H), 1.29 (s, 3H). LC-MS (ESI+): m/z calcd for (M–OH)+ 249.20.
Synthesis of (R)-2-hydroxy-2-methyl-3-(4-(trifluoromethoxy)phenoxy)propyl methanesulfonate 16 and (R)-1-(4-((2-methyloxiran-2-yl)methoxy)phenyl)-4-(4-trifluoromethoxy)phenoxy) (17) (Scheme 8). In a 100 mL round-bottomed flask, (R)-2-methyl-3-(4-(trifluoromethoxy)phenoxy)propane-1,2-diol R-(+)-15 (0.9 g, 3.7 mmol) in DCM solvent was taken triethylamine (0.570 mL, 4 mmol) and then methanesulfonyl chloride (0.170 mL, 2.2 mmol) was added dropwise at 0 °C and stir the reaction mixture at room temperature for a period of 2 h. Then solvent was evaporated and extract with ethyl acetate and aqueous layer. Then evaporation of organic phase gave the mesylated product 16 as gummy material and used as such for the next reaction. In the next step, mesylated compounds 16 was dissolved in ethyl acetate and DBU (0.596 mL, 4 mmol) was added and stirred the reaction mixture for a period of 2 h and then water was added to the reaction mixture and extract with ethyl acetate and evaporate the organic layer under rotary evaporator followed by purification over 100–200 silica gel and ethyl acetate
:hexane (40:60) as eluent to afford the compound 17 as yellow clear liquid (0.75 g) with the yield of 89%.1H NMR (400 MHz, CDCl3) δ 7.10 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.8 Hz, 2H), 4.00 (d, J = 10.5 Hz, 1H), 3.87 (d, J = 10.5 Hz, 1H), 2.82 (d, J = 4.6 Hz, 1H), 2.69 (d, J = 4.6 Hz, 1H), 1.44 (s, 3H). LC-MS (ESI+): m/z calcd for (M + H)+ 249.20.
Synthesis of (R)-2-methyl-6-nitro-2-((4-(trifluoromethoxy)phenoxy)methyl)-2,3-dihydroimidazo[2,1-b]oxazole (VL-2098, VII) (Scheme 8). A mixture of epoxide 17 (248 mg, 1 mmol), 2-bromo-4-nitro-1H-imidazole (12, 190 mg, 1 mmol), and DIPEA (2.0 mL) were taken in a sealed tube and heated at 115 °C for a period of 2 h. Upon cooling the reaction mixture, dichloromethane was added and evaporated to get crude product and used for the next reaction. To the stirred solution of crude product in anhydrous DMF was added caesium carbonate and stirred the reaction mixture for 2 h at 50 °C and then upon completion of reaction, add water and extract the reaction mixture with ethyl acetate. Then evaporate the solvent and purified the reaction mixture with silica gel 100–200 eluting with 40% ethyl acetate
:dichloromethane gave compound VII as white solid (172 mg, 48%). [α]25D +7° (c 1, chloroform) [[α]25D +8.445°].91H NMR (400 MHz, CDCl3) δ 7.53 (s, 1H), 7.11 (d, J = 8.8 Hz, 2H), 6.81 (d, J = 9.1 Hz, 2H), 4.46 (d, J = 10.3 Hz, 1H), 4.19 (d, J = 10.1 Hz, 1H), 4.05 (dd, J = 10.0, 8.0 Hz, 2H), 1.75 (s, 3H). LC-MS (ESI+): m/z calcd for (M + H)+ 360.20.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S. thanks to CSIR and ICMR, RA and PS thanks to CSIR for their research fellowship, respectively. This research work was financially supported by ICMR (GAP-2165) and CSIR, New Delhi (HCP-0008, BSC0205), IIIM Publication No. CSIR-IIIM/IPR/00167.Notes and references
- (a) K. Nepali, H.-Y. Lee and J.-P. Liou, J. Med. Chem., 2019, 62, 2851–2893 CrossRef CAS PubMed; (b) C. W. Ang, A. M. Jarrad, M. A. Cooper and M. A. T. Blaskovich, J. Med. Chem., 2017, 60, 7636–7657 CrossRef CAS PubMed; (c) L. G. Dover and G. D. Coxon, J. Med. Chem., 2011, 54, 6157–6165 CrossRef CAS PubMed; (d) S. Gupta, V. Yardley, P. Vishwakarma, R. Shivahare, B. Sharma, D. Launay, D. Martin and S. K. Puri, J. Antimicrob. Chemother., 2014, 70, 518–527 CrossRef PubMed.
- E. D. Deeks, Drugs, 2019, 79, 215–220 CrossRef CAS PubMed.
- P. L. Lin, V. Dartois, P. J. Johnston, C. Janssen, L. Via, M. B. Goodwin, E. Klein, C. E. Barry and J. L. Flynn, Proc. Natl. Acad. Sci., 2012, 109, 14188–14193 CrossRef CAS PubMed.
- M.-J. Pinazo, J. Muñoz, E. Posada, P. López-Chejade, M. Gállego, E. Ayala, E. del Cacho, D. Soy and J. Gascon, Antimicrob. Agents Chemother., 2010, 54, 4896–4899 CrossRef CAS PubMed.
- M. L. Nigro and M. Carballo, Toxicol. Lett., 2008, 180, 46–52 CrossRef PubMed.
- R. G. S. Kuppuswamy Nagrajan, S. Rajappa, S. J. Shenoy and R. Costa-Pereira, Eur. J. Med. Chem., 1989, 24, 631–633 CrossRef.
- H. Sasaki, Y. Haraguchi, M. Itotani, H. Kuroda, H. Hashizume, T. Tomishige, M. Kawasaki, M. Matsumoto, M. Komatsu and H. Tsubouchi, J. Med. Chem., 2006, 49, 7854–7860 CrossRef CAS PubMed.
- (a) A. M. Thompson, A. Blaser, R. F. Anderson, S. S. Shinde, S. G. Franzblau, Z. Ma, W. A. Denny and B. D. Palmer, J. Med. Chem., 2009, 52, 637–645 CrossRef CAS PubMed; (b) K. R. Yempalla, G. Munagala, S. Singh, G. Kour, S. Sharma, R. Chib, S. Kumar, P. Wazir, G. D. Singh, S. Raina, S. S. Bharate, I. A. Khan, R. A. Vishwakarma and P. P. Singh, ACS Med. Chem. Lett., 2015, 6, 1059–1064 CrossRef CAS PubMed; (c) G. Munagala, K. R. Yempalla, S. Singh, S. Sharma, N. P. Kalia, V. S. Rajput, S. Kumar, S. D. Sawant, I. A. Khan, R. A. Vishwakarma and P. P. Singh, Org. Biomol. Chem., 2015, 13, 3610–3624 RSC.
- V. S. Satam, S. R. Pedada, P. Kamaraj, N. Antao, A. Singh, R. M. Hindupur, H. N. Pati, A. M. Thompson, D. Launay and D. Martin, Org. Process Res. Dev., 2017, 21, 52–59 CrossRef CAS PubMed.
- https://www.tballiance.org/news.
- (a) S. H. Tsubouchi Hidetsugu, H. Kuroda, I. Motohiro, T. Hasegawa, Y. Haraguchi, T. Kuroda, M. Takayuki, K. Tai, K. Makoto, M. Matsumoto, H. Hiroyuki, T. Tatsuo, Seike, M. Kawasaki, S. Takumi and M. Shin, 2,3-Dihydro-6-nitroimidazo[2,1-b]oxazoles, WO/2004/033463A1, 2004; (b) M. Matsumoto, H. Hashizume, T. Tomishige, M. Kawasaki and Y. Shimokawa, Antituberculous composition comprising oxazole compounds, JP2006/320239, 2007.
- (a) A. M. Thompson, W. A. Denny, A. Blaser and Z. Ma, Nitroimidazooxazine and nitroimidazooxazole analogues and their uses, US20110028466 A1, 2011; (b) H. Hashizume, M. Kawasaki, M. Matsumoto, Y. Shimokawa and T. Tomishige, Antituberculous composition comprising oxazole compounds, WO/2007/043542A1, Otsuka Pharmaceutical Co Ltd, 2006.
- (a) M. Kreis, Producing (N-heterocyclic) aryl ethers, EP2179988A1, 2010; (b) A. A. Masahiro Miyake and T. Okado, Method for producing the 1-4-(Hydroxyphenyl)-4-(4-(trifluoromethoxy)phenoxy)piperidine or salt thereof, US Pat., 2018/0065931 A1, 2018; (c) D. F. Bruhn, M. S. Scherman, L. K. Woolhiser, D. B. Madhura, M. M. Maddox, A. P. Singh, R. B. Lee, J. G. Hurdle, M. R. McNeil, A. J. Lenaerts and B. Meibohm, PloS One, 2014, 9, e87909 CrossRef PubMed.
- (a) L. Z. Xing, C.W. Haoxiong, L. Yaoyu and Y. Xiaona, Method for preparing highly pure Delamanid intermediate, CN106699635A, 2015; (b) X. Lu, J. Tang, S. Cui, B. Wan, S. G. Franzblauc, T. Zhang, X. Zhang and K. Ding, Eur. J. Med. Chem., 2017, 125, 41–48 CrossRef CAS PubMed.
- R. H. Beddoe, K. G. Andrews, V. Magné, J. D. Cuthbertson, J. Saska, A. L. Shannon-Little, S. E. Shanahan, H. F. Sneddon and R. M. Denton, Science, 2019, 365, 910–914 CrossRef CAS PubMed.
- E. J. Corey, A. Guzman-Perez and M. C. Noe, J. Am. Chem. Soc., 1995, 117, 10805–10816 CrossRef CAS.
- S. Koichi, Y. Akihiro, F. Nobuhisa, A. Shinji, O. Shin and U. Naoto, Synthetic intermediate of oxazole compound and method for producing the same, WO 2011/093529 Al, 2011.
- F. Goto, N. Takemura, T. Otani, T. Hasegawa, H. Tsubouchi, N. Utsumi, S. Fujita, H. Kuroda, T. Shitsuta and H. Sasaki, 1-Substituted 4-nitroimidazole compound and process for producing the same, US Pat., USOO7368579B2, 2005.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR, 13C NMR and mass spectra of synthesized compounds. See DOI: 10.1039/d0ra01662d |
| This journal is © The Royal Society of Chemistry 2020 |