
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical proton-coupled electron transfer of an anthracene-based azo dye†
Amanda N. Oldacre ab and
Elizabeth R. Young*b
ab and
Elizabeth R. Young*b
aDepartment of Chemistry, St. Lawrence University, Canton, New York, 13617, USA. E-mail: ery317@lehigh.edu; Fax: +1-610-758-6536
bDepartment of Chemistry, Lehigh University, Bethlehem, Pennsylvania 18015, USA
First published on 14th April 2020
Abstract
Herein, we report the thermodynamics, kinetics, and mechanism for electrochemical proton-coupled electron transfer involving the anthracene-based azo dye azo-OMe. The peak reduction potential of azo-OMe with organic acids spanning the pKa range of 2.6–23.51 shows a dependence upon the pKa of the acid when the acid pKa falls between 8 and 20 (in acetonitrile). A potential-pKa diagram is constructed and used to estimate the pKa of the azo-OMe species. Heterogeneous electron-transfer rate constants are obtained using rotating disk electrode voltammetry in combination with Koutecký–Levich and Tafel analysis. Electrochemical analysis shows that the reactions are diffusion limited and are kinetically controlled by the electron-transfer step. Kinetic isotope studies indicate a concerted proton, electron transfer event occurs in the pKa-dependent range when using trifluoroacetic acid.
Introduction
Nature has evolved a mechanistic advantage in carrying out electron-transfer steps, as well as associated chemical reactions, by coupling the movement of electrons along reaction pathways to proton motions. The coupling of proton and electron movements play an important role in energy conversion reactions, energy storage, and small molecule activation reactions.1–5 Detailed mechanistic understanding of charge transfer and proton-coupled electron transfer (PCET) can enable a sustainable carbon-neutral energy economy. Fundamental mechanistic studies, wherein the PCET mechanism can be controlled, are of vast importance for energy conversion reactions.Over the past decade, PCET at electrochemical interfaces has gained attention due to the significant role electrochemical PCET plays in energy conversion reactions and catalysis.6–15 Experimental, theoretical, and computational work has shown non-innocence of protons at electrochemical interfaces.14–25 Indeed, many chemical reactions, including reactions involving homogeneous catalysis and heterogeneous catalysis at electrode surfaces show a clear dependence on the pH of the solution.15
Protons influence the Fermi level at the electrochemical interface thereby controlling the thermodynamics and, as a result, the kinetics of electrochemical PCET reactions. Transition state theory makes this connection between activation energy and reaction kinetics. The activation energy of stepwise PCET compared to the activation energy of concerted PCET dictates the observed mechanism. For example, if the activation energy of the concerted PCET reaction is less than the activation energy of the sum of the individual proton- and electron-transfer steps, then the concerted reaction will be kinetically favoured. It follows, then that the dependence of the reactions on pH results from a decoupling of the electron and proton transfer steps in PCET reactions caused by one step of the reaction (ET or PT) becoming more thermodynamically favoured. With this in mind, catalysts and reactions can be designed such that changes in pH result in changing the mechanism of PCET between stepwise or concerted PCET.5,26–29
The nature of the proton (either solvated or at chemically accessible sites from an acidic bond or organic acids) can be the driver for tuning the thermodynamics of the proton-transfer step in the chemical reactivity. By tuning the pKa of organic acids, the rate and mechanism of PCET in small-molecule inorganic complexes can be systematically dialled in.22,23,30,31 Further, strong electronic coupling between molecular sites on electrodes has been shown to cause reactivity that fundamentally diverges from that of solution-phase or surface-tethered analogues and can be correlated to the acid/base behaviour of the molecular analogue.17 Many recent mechanistic studies involved photo-induced PCET of bio-mimic model systems.32 Photo-induced PCET model systems, both molecular and biological, have been used to quantify the influence of proton-transfer driving force and transfer distance on the kinetics of PCET.33–38 Because of the complex nature of PCET, it remains important to understand mechanisms and factors that govern electron and proton transfer at electrochemical interfaces of a wide variety of molecular and materials systems.
Synthetically facile molecular model systems are important to form the basis for the study for PCET. Azo dyes possess such facile synthetic procedures that offer rich synthetic versatility and allow for the development of a wide range of model systems in which thermodynamic properties can be systematically tuned. Applications of azo dyes include organic solar cells, molecular photoswitches, protein probes, molecular machines, and non-linear optical components, which encompasses a wide range of applications-driven systems.39–42
In previous work from our group, we described the design and study of (anthracen-2-yl)-2-(4-methoxyphenyl)diazene (azo-OMe, Fig. 1.) as a model system for the study of photo-induced PCET. Although the photochemistry was not perturbed in the presence of 1 equivalents (eq.) of trifluoroacetic acid (TFA), we found a large positive potential shift in the presence of 1 eq. TFA versus 0 eq. TFA.43 This work builds upon these initial electrochemical measurements to fully understand the electrochemical PCET reactivity of this anthracene-based azo dye. Towards this end, we have probed the electrochemical reaction of azo-OMe using several different organic acids with acidity constants spanning the pKa range of 2.6–23.51 in acetonitrile using the combination of cyclic voltammetry, differential pulse voltammetry, and rotating-disk electrode voltammetry along with a kinetic isotope dependence study. Each of the electrochemical reduction reactions are show to be diffusion controlled at the electrode surface, and are kinetically limited by the electron-transfer step. We show that the PCET mechanism is tuned by the variation of the organic acid pKa, which serves a proxy (or stand in) for pH in non-aqueous conditions. Using a combination of rotating disk voltammetry and kinetic isotope effect studies, we show that in solutions with a less acidic acid (i.e. acids with pKa values greater than the pKa of azo-OMe), azo-OMe undergoes a concerted 1H+/1e− transfer PCET redox reaction. The activation energy of the concerted reaction is less than the sum of the individual proton- and electron-transfer steps. However, in the presence of a highly acidic organic acid (i.e. acids with pKa values less than the pKa of azo-OMe), such as HDMF OTf, a proton transfer occurs prior to the electrochemical electron transfer, indicating that the activation energy of the proton-transfer step is sufficiently decreased to decouple the concerted PCET reaction.
 | ||
| Fig. 1 (Anthracen-2-yl)-2-(4-methoxyphenyl)diazene (azo-OMe). | ||
Results and discussion
Electrochemical reduction potentials depend upon the pKa of added acids
The redox properties of azo-OMe were measured with cyclic voltammetry (CV) and differential pulse voltammetry (DPV). To probe the electrochemical PCET reaction, nine organic acids were used, with a pKa range in acetonitrile of 2.6–23.51 (Table S1, ESI†) in order to tune the proton-dependent thermodynamics of the reaction while measuring the electron-transfer reactivity. These data may be used to construct a potential-pKa diagram (vide infra) that yields a complete ET/PT thermodynamic picture of azo-OMe. A 10 mM stock solution of acid with 0.5 mM azo-OMe in acetonitrile (100 mM TBAPF6) was titrated into a solution of 0.5 mM azo-OMe (100 mM TBAPF6) at 100 and 500 μL increments (0.19–2.33 eq. of acid). CV was acquired upon each addition of acid and DPV was acquired at 1 and 2 eq. of acid.Consistent with previous results, the reversible reduction peak of azo-OMe in acetonitrile under inert and aprotic conditions was E1/2 = −1.7 V vs. Fc+/0 before addition of any acid.43 Upon the addition of small amounts (<∼0.5 equivalents) of each acid, the reversible reduction peak disappeared concomitant with the growth of an irreversible reduction peak at more positive potentials. As additional acid was added (>0.5 equivalents), the initial reversible peak continued to disappear and the first irreversible reduction peak also disappeared to yield another, even more positively shifted irreversible reduction peak The irreversible peaks were shifted to positive potentials, meaning that reduction became more facile upon the addition of acid (Fig. 2 and S1–S8, ESI†). As the acid titrations reached the 2 equivalent acid added point (and up to 2.33 equivalents) the reduction peak ceased to significantly shift.
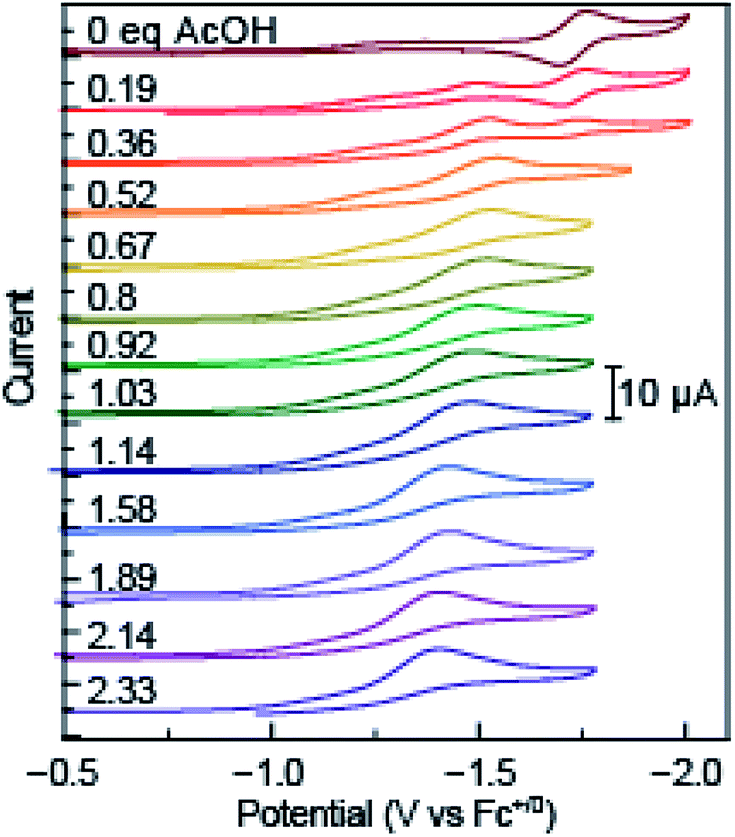 | ||
| Fig. 2 Overlaid cyclic voltammetry scans of azo-OMe acid titration with 0 equivalent to 2.33 equivalent acetic acid (pKa (MeCN) = 23.51). Inset: scale of current response in μA. | ||
As expected, the ΔE of the peak shift depended intimately on the pKa of the acid added, a hallmark of proton-coupled electron-transfer reactions. Acids with higher pKa values (in acetonitrile) resulted in a smaller positive shift of the reduction potential. For example, addition of acetic acid (pKa (MeCN) = 23.51) resulted in only a 300 mV shift from E1/2 = −1.7 V vs. Fc+/0 (no added acid) to E1/2 = −1.38 V vs. Fc+/0 upon addition of 2 equivalents of acid. Whereas, acids with lower pKa values (in acetonitrile) resulted in significantly larger changes in the reduction potential. Specifically, dimethylformamidium trifluoromethanesulfonic acid (HDMF) (pKa (MeCN) = 2.6) resulted in a 950 mV shift to E1/2 = −0.45 V vs. Fc+/0 upon addition of 2 equivalents of acid. By utilizing organic acids with pKa values spanning this range, the thermodynamics of azo-OMe and any reactions that occur using it can be experimentally determined.
Potential-pKa diagram
Potential-pKa diagrams have been developed to understand PCET reactions in non-aqueous solutions for which Pourbaix diagrams that are used for aqueous systems cannot be employed. Because solvated protons do not exist in organic solvents the way they do in aqueous media, the pH scale of a Pourbaix diagram must be modified. Instead of pH, the pKa of an organic acid can be used as the proxy for the thermodynamic availability of the proton in a potential-pKa diagram.44 The potential-pKa diagram plots the electrochemical reduction or oxidation potential of a process of interest versus the pKa of a range of added organic acids. These plots show regions in which various electrochemical and protonation states of the analyte, in this case the azo-OMe, exist as a function of proton availability (from an added organic acid).To construct the potential-pKa diagram for azo-OMe, the reduction potential of azo-OMe was measured using cyclic voltammograms that were acquired with 0.5 mM azo-OMe, 2 equivalents of acid, and 100 mM TBAPF6 in acetonitrile. Fig. 3 shows the peak potential of azo-OMe (vs. Fc+/0) plotted versus the corresponding pKa of each added acid. The potential-pKa diagram for azo-OMe reveals one pKa-dependent and two pKa-independent regions. One pKa-independent region occurred when strongly acidic acids (pKa 2.6 to 8.6) are used. In this region, the results suggests that azo-OMe is protonated prior to reduction and that in this case, the reduction potential of the protonated azo-OMe dye is measured directly. The other pKa-independent region occurred when weaker acids (pKa 20.35 to 23.51) were used. In this region azo-OMe is likely not significantly protonated prior to or during the reduction event. The acids with pKa > 20.35 are not acidic enough to protonate either the reduced or neutral azo-OMe species. A pKa-dependent region is observed between (pKa 2.6 to 8.6). The slope of the pKa-dependent region (pKa 8.6 to 20.35) is 75 mV per decade, which indicates that this process involves 1H+/1e− transfer event. The difference from the idealized Nernstian slope (59 mV per decade) is not substantially large and due to homoconjugation of organic acids in acetonitrile.44 While the process could be a 2H+/2e− process, the current response associated with the azo-OMe PCET reaction does not in fact double when the acid concentration is increased from 1 to 2 equivalents of acid, thereby ruling out that possibility.
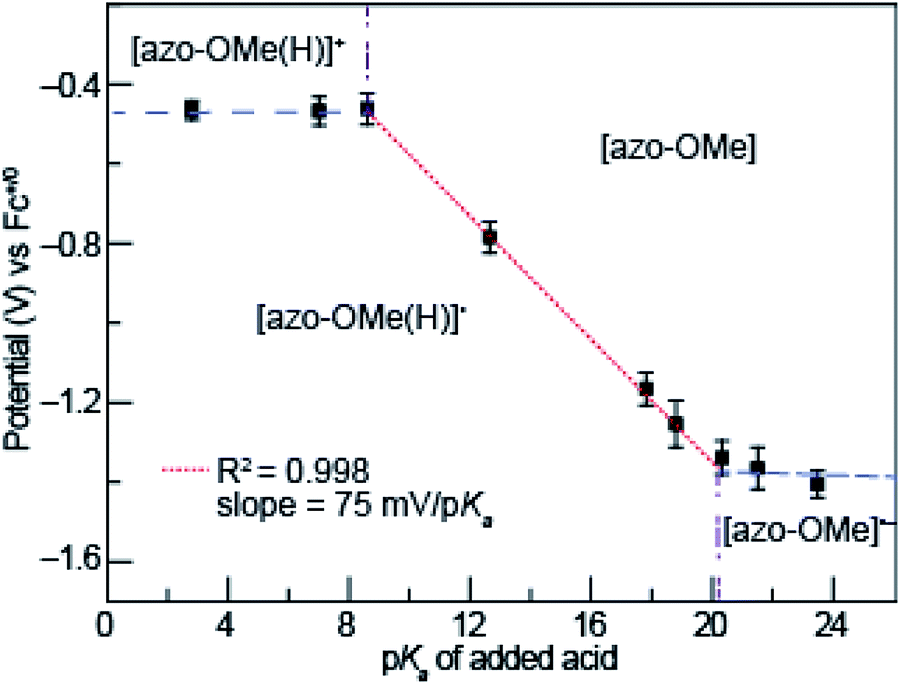 | ||
| Fig. 3 Potential-pKa diagram of azo-OMe with 2 eq. of acid. | ||
The intersection of the pKa dependent and independent regions can be used to predict the pKa of [azo-OMe(H)]+ and [azo-OMe(H)], which are approximately 8.6 and 20.1 respectively. Taken together, lines drawn through each pKa dependent (Fig. 3, blue dashed) and independent regions (Fig. 3, red dotted), as well as vertical lines at their intersection (Fig. 3, dot-dashed purple) can be used create the organic-solvent version of the Pourbaix diagram, the potential-pKa diagram.
Rotating disk electrode voltammetry
To garner mechanistic understanding of electrochemical PCET processes, rotating disk electrode (RDE) voltammograms were acquired with solutions of 0.5 mM azo-OMe and 2 eq. of acid (HDMF OTf, or TFA) at various rotation rates (ω = 100, 200, 500, 1000, 1500, and 2000 rpm). HDMF OTf and TFA were chosen because they fall into the two relevant pKa ranges for PCET reactions with azo-OMe, according to the potential-pKa plot. HDMF OTf is highly acidic and protonates azo-OMe even when not reduced, and TFA falls into the pKa-dependent region and is acidic enough to protonate the reduced azo-OMe species. Fig. 4 shows RDE voltammograms of HDMF OTf (top) and TFA (bottom). For 2 eq. HDMF OTf voltammograms, the current increases as more reducing potentials are applied at a given rpm until a plateau region is reached at approximately −0.4 V vs. Fc+/0. For the 2 eq. TFA voltammograms, the onset of current is shifted to more reducing potentials (∼−0.6 V vs. Fc+/0 rather than −0.2 V vs. Fc+/0 for the HDMF voltammograms). The plateau current was not observed in the RDE plot with 2 eq. of TFA because proton reduction occurs via the glassy carbon electrode at −1.2 V vs. Fc+/0, which begins to dominate the current response. The disk current of azo-OMe with 2 eq. of HDMF OTf and 2 eq. of TFA exhibited Levich behaviour, which is the increase in current response as the rotation rate is increased. The Levich behaviour observed when the rotation rate was increased (Fig. 4), indicates that the reaction is diffusion controlled. RDE voltammograms were used to garner information about the kinetics and mechanism of the PCET reaction of azo-OMe and 2 equivalents of acid through a Koutecký–Levich analysis (vide infra).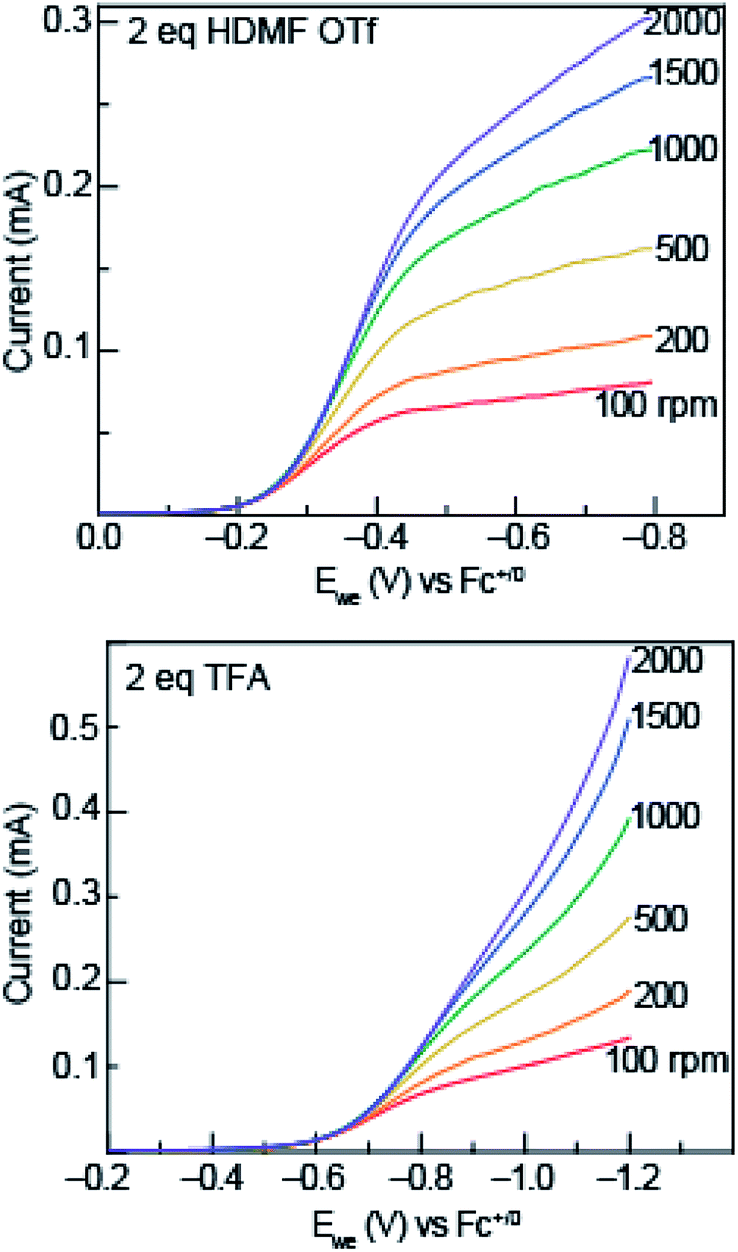 | ||
| Fig. 4 Representative rotating ring disk voltammograms of 0.5 mM azo-OMe with 2 eq. HDMF OTf (top) and TFA (bottom) in 100 mM TBAPF6 (MeCN) at rotations rates 100, 200, 500, 1000, 1500, and 2000 rpm and are labelled directly on the plot. Ewe is the potential at the working electrode. Inset: scale of current response in mA. | ||
Koutecký–Levich and Tafel analysis
The data obtained from RDE experiments were plotted using the Koutecký–Levich (KL) eqn (1)
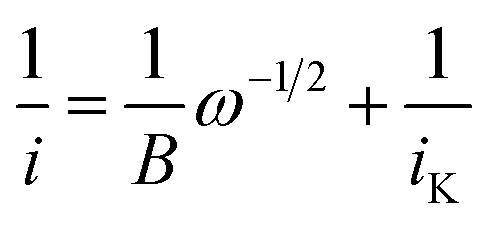 | (1) |
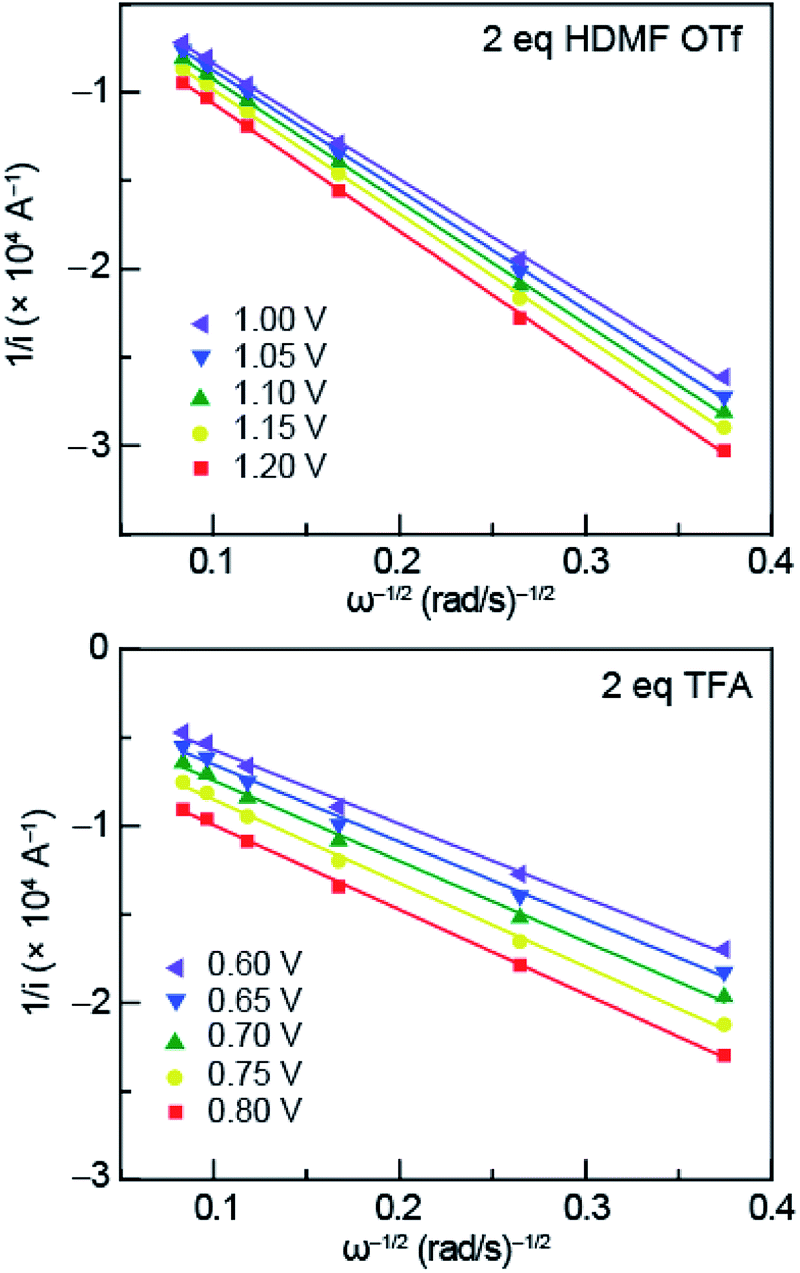 | ||
| Fig. 5 Representative Koutecký–Levich plots of 0.5 mM azo-OMe with 2 eq. HDMF OTf (top) and TFA (bottom) in 100 mM TBAPF6 (MeCN) at various overpotentials. The data was plotted using the linear relationship described by the Koutecký–Levich equation (eqn (1)). | ||
The reciprocal of measured currents, obtained from the rotating-disk voltammograms over a range of overpotentials that fall within the plateau region, were plotted versus the reciprocal of the square root of rotation rate. The reaction overpotential (η) is defined as the difference between the peak potential of azo-OMe0/− (E) in the presence of acid and the half potential of azo-OMe0/− (−1.7 V = Eeq.) eqn (2).
| η = E − Eeq. | (2) |
The KL plots at various overpotentials do not intersect the origin of the y-axis, which indicates that the reaction is also kinetically limited by the electron-transfer step. The linear KL plots were extrapolated to yield the y-intercept, 1/iK, where current would be kinetically limited, but not limited under diffusion control. The separation of the kinetically-limited and diffusion-limited processes using KL treatment allows for the determination of heterogeneous electron-transfer rate constants.
To obtain the exchange current, i0, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) iK (iK = inverse of the KL y-intercept) was plotted versus overpotential, shown in Fig. 6, using the Tafel equation, eqn (3),
iK (iK = inverse of the KL y-intercept) was plotted versus overpotential, shown in Fig. 6, using the Tafel equation, eqn (3),
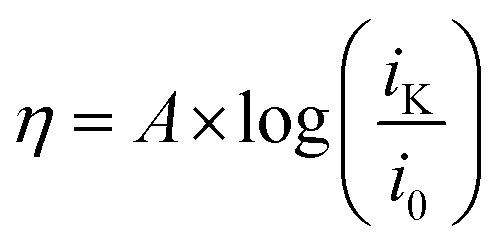 | (3) |
| i0 = ksFAC | (4) |
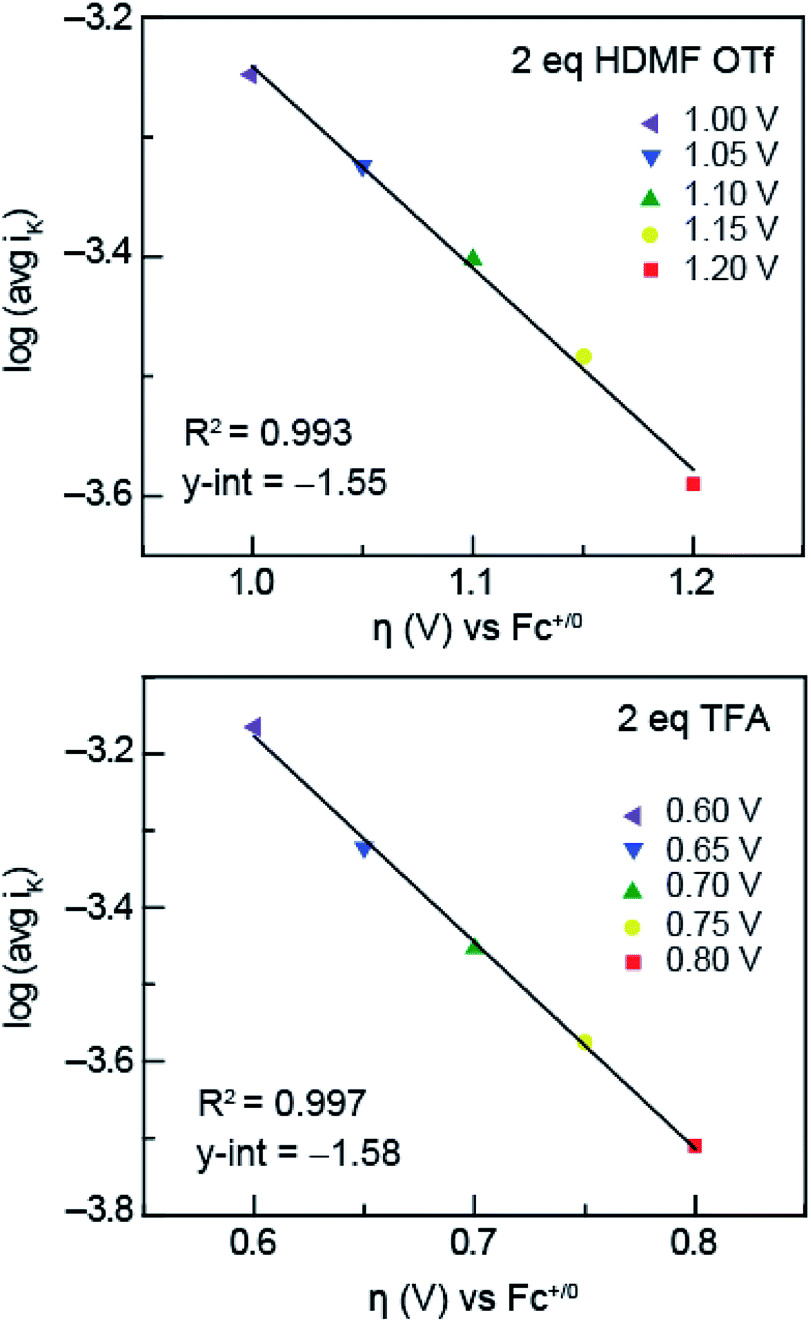 | ||
| Fig. 6 Tafel plots of HDMF OTf (top) and TFA (bottom) used to obtain ks. The inverse of the KL y-intercept and overpotential was plotted using the relationship furnished by the Tafel equation (eqn (3)). | ||
| Acid | ks (cm s−1) |
|---|---|
| HDMF OTf | 0.614 ± 0.008 |
| TFA | 0.587 ± 0.005 |
| d1-TFA | 0.245 ± 0.007 |
In solutions of 2 equivalents HDMF OTf with azo-OMe, the electron transfer is found to be faster (ks = 0.614 ± 0.008 cm s−1) than the electron transfer than when 2 equivalents of TFA with azo-OMe is used (ks = 0.587 ± 0.005 cm s−1). This observation can be understood by considering the thermodynamics of proton transfer before the electron transfer is initiated. The pKa (MeCN) of HDMF OTf is 2.6, four orders of magnitude more acidic than the azo-OMe, which is estimated to be 8.6 from the potential-pKa analysis (Fig. 3). Therefore, azo-OMe is overwhelmingly protonated in solution of HDMF OTf prior to an ET event, which allows for a faster electron transfer. In contrast, the pKa of TFA (pKa = 12.6 in MeCN) is less than that of azo-OMe, meaning that the azo-OMe is not appreciably protonated before the electron transfer event. However, upon reduction, the azo-OMe will be certainly be protonated (pKa [azo-OMe]− = ∼20 in MeCN) because the reduced species is far less acidic than the TFA. Because both the electron and proton transfer must occur in this event, the electron transfer is ks = 0.587 ± 0.005 cm s−1, slower than the more acidic solutions. With 2 equivalents of TFA, azo-OMe is not protonated prior to electron transfer, but from this analysis, it is not clear whether there is concerted electron–proton transfer (CPET) or stepwise electron, then proton transfer (ETPT). To distinguish between these processes, kinetic isotope experiments were performed.
Kinetic isotope effect with deuterated TFA
To garner information about the proton-coupled electron-transfer mechanism of less acidic acids (acids that fall into the pKa-dependent range of the potential-pKa plot of Fig. 3), RDE experiments were performed with solutions of 0.5 mM azo-OMe and 2 equivalent of deuterated TFA at various rotation rates (Fig. 7, left). In similar fashion to those of the protonated acids, the RDE voltammograms with deuterated TFA also show Levich behavior, which indicates that the deuterated reaction is also under diffusion control.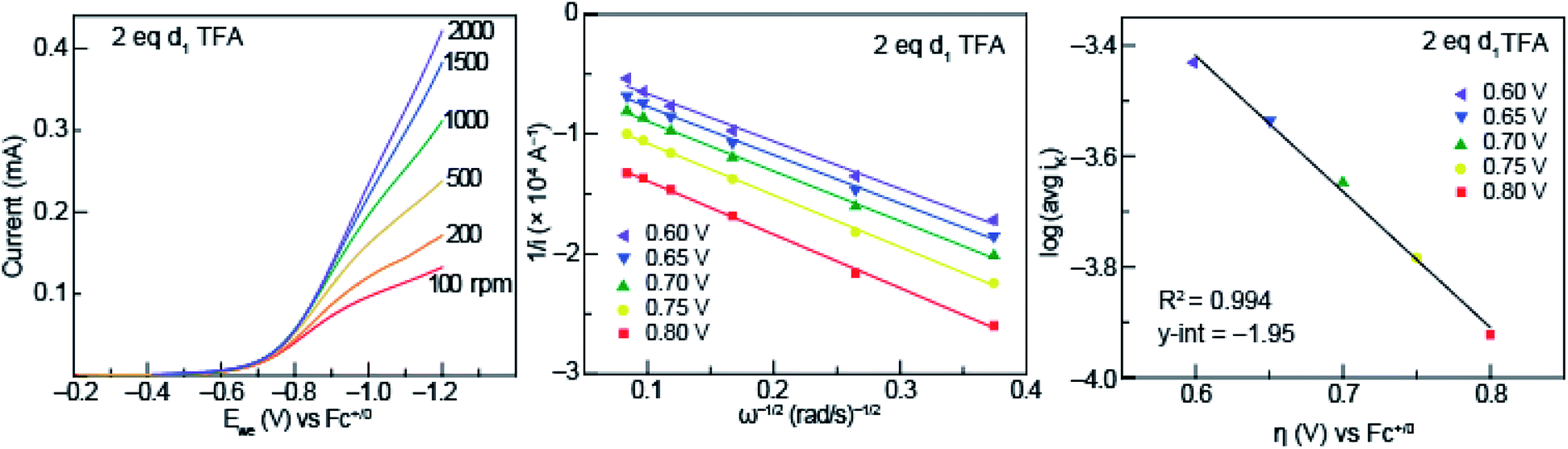 | ||
| Fig. 7 (Left) Representative rotating ring disk voltammograms of 0.5 mM azo-OMe with 2 eq. d1 TFA in 100 mM TBAPF6 (MeCN) at rotations rates 100, 200, 500, 1000, 1500, and 2000 rpm. Ewe is the potential at the working electrode. Inset: scale of current response in mA. (Center) Representative Koutecký–Levich plots of 0.5 mM azo-OMe with 2 eq. d1 TFA in 100 mM TBAPF6 (MeCN) at various overpotentials (per eqn (1)). (Right) Tafel plot of d1 TFA used to obtain ks (per eqn (3)). | ||
The data obtained from rotating disk experiments was plotted with Koutecký–Levich and Tafel analysis, shown in Fig. 7 (center and right, respectively). The y-intercept of the Koutecký–Levich plots with d1-TFA did not intersect the origin, which indicates the electron-transfer step is also kinetically limited for the deuterated reaction. The heterogeneous rate constant obtained from Tafel analysis is shown in Table 1.
A KIE (ks,H/ks,D) of 1.8 was found, which is consistent with a concerted proton- and electron-transfer mechanism.24,32 Previous reports by Costentin et al. indicated an intramolecular electrochemical CPET of a substituted-phenol with a nearby amine group (KIE of 1.8).45
Along the pKa-dependent region of the potential-pKa diagram, a PTET mechanism can be excluded due to the difference in pKa of [azo-OMe(H)]+ (≈8.6 in MeCN) and weak organic acids, such as TFA (ΔpKa = pKa(TFA) − pKa([azo-OMe(H)]+) ≈ 4). A protonation prior to an electron-transfer in this regime would be too energetically uphill and a concerted mechanism would bypass that high energy intermediate. An ETPT mechanism can be excluded based on the magnitude of KIE (although small), which is consistent with the involvement of PT in the rate-determining electron transfer step.46,47
Conclusions
To understand the electrochemical PCET reactivity, this work builds upon initial electrochemical measurements of azo-OMe previously reported by our laboratory.43 In this work, we have probed the proton-dependence of the electrochemical reaction of azo-OMe using nine organic acids, with acidity constants spanning the pKa range of 2.6–23.51 in acetonitrile, using a combination of electrochemical techniques. Our goal was to control the kinetics and mechanism of the PCET reaction at an electrode surface by tuning the thermodynamic (and by extension of transition state theory) the kinetics of the PCET reaction.The data obtain from cyclic voltammetry experiments was used to construct a potential-pKa diagram, which indicated a 1H+/1e− PCET process occurrs in the presence of acids with pKa values spanning pKa 8 to 20. The potential-pKa diagram was also used to predict the pKa of [azo-OMe(H)]+ and [azo-OMe(H)], which are approximately 8.6 and 20.1 respectively.
Rotating disk voltammetry with Koutecký–Levich and Tafel analysis was used to obtain a mechanistic understanding of the PCET process azo-OMe undergoes in the presence of added organic acids. These analyses shows that the PCET reactions are diffusion limited and are kinetically controlled by the electron-transfer step. Due to the pKa difference between azo-OMe and HDMF OTf (pKa (MeCN) = 2.6), azo-OMe is overwhelmingly protonated in solution of 2 equivalents HDMF OTf prior to an electron transfer event, which allows for a faster electron transfer (ks = 0.614 ± 0.008 cm s−1). In solution of 2 equivalents TFA (pKa (MeCN) = 12.6), ks = 0.587 ± 0.005 cm s−1, slower than the more acidic solutions. To understand this result, kinetic isotope effect studies were conducted with deuterated TFA to distinguish if the PCET event in the less acidic TFA was concerted (CPET) or stepwise (ETPT). A KIE of 1.8 was obtained, which indicates that the electron and proton are transferred in a concerted step.
Our results demonstrate that azo-based dyes exhibit rich electrochemical PCET chemistry – the mechanism of which can be tuned using the proton-transfer thermodynamics. These results represent some of the first examples using azo-based dyes that examine the PCET mechanism at electrode surfaces in which the proton originates from organic acids introduced into the solution, rather than via an intramolecular processes, which have been studied in a limited fashion prior to this.20 Ongoing efforts to vary the identity of arene on the anthracene side of the azo bond as well as the R group on the phenyl side are currently underway. Further, work is underway to probe the influence of azo bond protonation on the ultra-fast excited state dynamics of the azo dyes for which the thermodynamic properties reported in this work are essential.
Experimental
Materials and synthetic procedures
All reagents were obtained from commercial sources and used as received without further purification, unless otherwise specified. The anthracene-based azo dye, (anthracen-2-yl)-2-(4-methoxyphenyl)diazene (azo-OMe) was synthesized according to literature procedure.43Cyclic voltammetry and differential pulse voltammetry
All cyclic voltammetry (CV) and differential pulse voltammetry (DPV) measurements were recorded on a CH Instruments potentiostat/galvanostat. CV and DPV were performed using a glassy carbon disk (3 mm diameter) working electrode, a platinum wire counter electrode, and a silver wire quasi reference electrode. The glassy carbon working electrode was polished with 0.05 μM alumina powder/water slurry, rinsed with water and acetone, and allowed to air dry prior to each experiment. Tetrabutylammonium hexafluorophospate (TBAPF6) was recrystallized with hot ethanol three times, dried under vacuum, and used as the supporting electrolyte. Acetonitrile was pre-dried with 4 Å molecular sieves, distilled over calcium hydride, and stored over 4 Å molecular sieves prior to use. Ferrocene was used as an internal standard and all voltammograms were reported vs. Fc+/0 couple. The scan rate used for CV was 100 mV s−1 unless otherwise specified. All samples were sparged with N2 for 10 minutes before CV and DPV and kept under a N2 blanket during acquisition.Rotating disk voltammetry
All rotating disk experiments were acquired with a Bio-Logic SP-300 bipotentiostat/galvanostat and a Pine MSR rotator. A glassy carbon fixed-disk PEEK shroud working electrode and a platinum mesh counter electrode were used for RDE experiments. The glassy carbon disk was polished with 0.05 μM alumina powder/water slurry, rinsed with water and acetone, and allowed to air dry prior to each experiment. Acetonitrile was purified from a Pure Process Technology free standing solvent purification system. The scan rate used for RDE was 20 mV s−1 with rotation rates (ω) of 100, 200, 500, 1000, 1500, and 2000 rpm. All solutions were sparged with N2 for 10 minutes before RDE and were blanketed with N2 during acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
ANO and ERY thank Prof. Timothy R. Cook at the University of Buffalo for use of bio-logic potentiostat and PINE rotator.Notes and references
- A. Pannwitz and O. S. Wenger, Photoinduced Electron Transfer Coupled to Donor Deprotonation and Acceptor Protonation in a Molecular Triad Mimicking Photosystem II, J. Am. Chem. Soc., 2017, 139, 13308–13311 CrossRef CAS PubMed.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Proton-Coupled Electron Transfer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- D. Motoyama, K. Yoshikawa, H. Ozawa, M. Tadokoro and M. Haga, Energy-Storage Applications for a pH Gradient between Two Benzimidazole-Ligated Ruthenium Complexes That Engage in Proton-Coupled Electron-Transfer Reactions in Solution, Inorg. Chem., 2017, 56, 6419–6428 CrossRef CAS PubMed.
- C. N. Brodsky, D. Kwabena Bediako, C. Shi, T. P. Keane, C. Costentin, J. L. Billinge and S. G. Nocera, Proton–Electron Conductivity in Thin Films of a Cobalt–Oxygen Evolving Catalyst, ACS Appl. Energy Mater., 2018, 2, 3–12 CrossRef.
- K. J. Lee, C. T. Gruninger, K. M. Lodaya, S. Qadeer, B. E. Griffith and J. L. Dempsey, Analysis of Multi-Electron, Multi-Step Homogeneous Catalysis by Rotating Disc Electrode Voltammetry: Theory, Application, and Obstacles, Analyst, 2020, 145, 1258–1278 RSC.
- S. Ki Kim, Y.-J. Zhang, H. Bergstrom, R. Michalsky and A. Peterson, Understanding the Low-Overpotential Production of CH4 from CO2 on Mo2C Catalysts, ACS Catal., 2016, 6, 2003–2013 CrossRef.
- K. Yan, T. A. Maark, A. Khorshidi, V. A. Sethuraman, A. A. Peterson and P. R. Guduru, The Influence of Elastic Strain on Catalytic Activity in the Hydrogen Evolution Reaction, Angew. Chem., Int. Ed., 2016, 55, 6175–6181 CrossRef CAS PubMed.
- S. K. Kim, Y. Qiu, Y.-J. Zhang, R. Hurt and A. Peterson, Nanocomposites of Transition-Metal Carbides on Reduced Graphite Oxide as Catalysts for the Hydrogen Evolution Reaction, Appl. Catal., B, 2018, 235, 36–44 CrossRef CAS.
- R. Michalsky, Y.-J. Zhang and A. A. Peterson, Trends in the Hydrogen Evolution Activity of Metal Carbide Catalysts, ACS Catal., 2014, 4, 1274–1278 CrossRef CAS.
- G. Kastlunger, P. Lindgren and A. A. Peterson, Controlled-Potential Simulation of Elementary Electrochemical Reactions: Proton Discharge on Metal Surfaces, J. Phys. Chem. C, 2018, 122, 12771–12781 CrossRef CAS.
- K. Yan, S. K. Kim, A. Khorshidi, P. R. Guduru and A. A. Peterson, High Elastic Strain Directly Tunes the Hydrogen Evolution Reaction on Tungsten Carbide, J. Phys. Chem. C, 2017, 121, 6177–6183 CrossRef CAS.
- S. Ghosh, A. V. Soudackov and S. Hammes-Schiffer, Electrochemical Electron Transfer and Proton-Coupled Electron Transfer: Effects of Double Layer and Ionic Environment on Solvent Reorganization Energies, J. Chem. Theory Comput., 2016, 12, 2917–2925 CrossRef CAS PubMed.
- M. T. Huynh, S. Jimena Mora, M. Villalba, M. E. Tejeda-Ferrari, P. A. Liddell, B. R. Cherry, A.-L. Teillout, W. Machan, C. P. Kubiak and D. Gust, et al., Concerted One-Electron Two-Proton Transfer Processes in Models Inspired by the Tyr-His Couple of Photosystem II, ACS Cent. Sci., 2017, 3, 372–380 CrossRef CAS PubMed.
- C. Venkataraman, A. V. Soudackov and S. Hammes-Schiffer, Theoretical Formulation of Nonadiabatic Electrochemical Proton-Coupled Electron Transfer at Metal–Solution Interfaces, J. Phys. Chem. C, 2008, 112, 12386–12397 CrossRef CAS.
- M. T. M. Koper, Theory of Multiple Proton–electron Transfer Reactions and Its Implications for Electrocatalysis, Chemcial Science, 2013, 4, 2710–2723 RSC.
- M. N. Jackson, S. Oh, C. J. Kaminsky, S. B. Chu, G. Zhang, T. Miller and Y. Surendranath, Strong Electronic Coupling of Molecular Sites to Graphitic Electrodes via Pyrazine Conjugation, 2018 Search PubMed.
- M. N. Jackson, M. L. Pegis and Y. Surendranath, Graphite-Conjugated Acids Reveal a Molecular Framework for Proton-Coupled Electron Transfer at Electrode Surfaces, ACS Cent. Sci., 2019, 5, 831–841 CAS.
- W. Schmickler, A Unified Model for Electrochemical Electron and Ion Transfer Reactions, Chem. Phys. Lett., 1995, 237, 152–160 CrossRef CAS.
- J. Grimminger, S. Bartenschlager and W. Schmickler, A Model for Combined Electron and Proton Transfer in Electrochemical Systems, Chem. Phys. Lett., 2005, 416, 316–320 CrossRef CAS.
- J.-M. Savéant and C. Tard, Proton-Coupled Electron Transfer in Azobenzene/Hydrazobenzene Couples with Pendant Acid–Base Functions. Hydrogen-Bonding and Structural Effects, J. Am. Chem. Soc., 2014, 136, 8907–8910 CrossRef PubMed.
- M. T. M. Koper, Theory of the Transition from Sequential to Concerted Electrochemical Proton-Electron Transfer, Phys. Chem. Chem. Phys., 2013, 15, 1399–1407 RSC.
- T. Huang, E. S. Rountree, A. P. Traywick, M. Bayoumi and J. L. Dempsey, Switching between Stepwise and Concerted Proton-Coupled Electron Transfer Pathways in Tungsten Hydride Activation, J. Am. Chem. Soc., 2018, 140, 14655–14669 CrossRef CAS PubMed.
- S. Ott, T. Liu, R. Tyburski, S. Wang, R. Ferna and L. Hammarstrom, Elucidating Proton-Coupled Electron Transfer Mechanisms of Metal Hydrides with Free Energy- and Pressure-Dependent Kinetics, J. Am. Chem. Soc., 2019, 141, 17245–17259 CrossRef PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Concerted Proton–Electron Transfers: Electrochemical and Related Approaches, Acc. Chem. Res., 2010, 43, 1019–1029 CrossRef CAS PubMed.
- S. Hammes-Schiffer and A. a. Stuchebrukhov, Theory of Coupled Electron and Proton Transfer Reactions, Chem. Rev., 2010, 110, 6939–6960 CrossRef CAS PubMed.
- J. C. Lennox and J. L. Dempsey, Influence of Proton Acceptors on the Proton-Coupled Electron Transfer Reaction Kinetics of a Ruthenium–Tyrosine Complex, J. Phys. Chem. B, 2017, 121, 10530–10542 CrossRef CAS PubMed.
- N. Elgrishi, B. D. Mccarthy, E. S. Rountree and J. L. Dempsey, Reaction Pathways of Hydrogen-Evolving Electrocatalysts: Electrochemical and Spectroscopic Studies of Proton-Coupled Electron Transfer Processes, ACS Catal., 2016, 6, 3644–3659 CrossRef CAS.
- E. S. Rountree and J. L. Dempsey, Reactivity of Proton Sources with a Nickel Hydride Complex in Acetonitrile: Implications for the Study of Fuel-Forming Catalysts, Inorg. Chem., 2016, 55, 5079–5087 CrossRef CAS PubMed.
- B. D. Mccarthy, D. J. Martin, E. S. Rountree, A. C. Ullman and J. L. Dempsey, Electrochemical Reduction of Brønsted Acids by Glassy Carbon in Acetonitrile – Implications for Electrocatalytic Hydrogen Evolution, Inorg. Chem., 2014, 53, 8350–8361 CrossRef CAS PubMed.
- J. C. Lennox, D. A. Kurtz, T. Huang and J. L. Dempsey, Excited-State Proton-Coupled Electron Transfer: Different Avenues for Promoting Proton/Electron Movement with Solar Photons, ACS Energy Lett., 2017, 2, 1246–1256 CrossRef CAS.
- D. A. Kurtz and J. L. Dempsey, Proton-Coupled Electron Transfer Kinetics for the Photoinduced Generation of a Cobalt(III)-Hydride Complex, Inorg. Chem., 2019, 58, 16510–16517 CrossRef CAS PubMed.
- A. Pannwitz and O. S. Wenger, Recent Advances in Bioinspired Proton-Coupled Electron Transfer, Dalton Trans., 2019, 48, 5861–5868 RSC.
- L. Hammarström and S. Styring, Proton-Coupled Electron Transfer of Tyrosines in Photosystem II and Model Systems for Artificial Photosynthesis: The Role of a Redox-Active Link between Catalyst and Photosensitizer, Energy Environ. Sci., 2011, 4, 2379 RSC.
- M. Zhang, T. Irebo, O. Johansson and L. Hammarstr, Proton-Coupled Electron Transfer from Tyrosine: A Strong Rate, J. Am. Chem. Soc., 2011, 133, 13224–13227 CrossRef CAS PubMed.
- S. D. Glover, G. A. Parada, T. F. Markle, S. Ott and L. Hammarstrom, Isolating the Effects of the Proton Tunneling Distance on Proton-Coupled Electron Transfer in a Series of Homologous Tyrosine-Base Model Compounds, J. Am. Chem. Soc., 2017, 139, 2090–2101 CrossRef CAS PubMed.
- S. D. Glover, R. Tyburski, L. Liang, C. Tommos and L. Hammarstrom, Pourbaix Diagram, Proton-Coupled Electron Transfer, and Decay Kinetics of a Protein Tryptophan Radical: Comparing the Redox Properties of W32˙ and Y32˙ Generated Inside the Structurally Characterized α3W and α3Y, J. Am. Chem. Soc., 2018, 140, 185–192 CrossRef CAS PubMed.
- P. Dongare, S. Maji and L. Hammarström, Direct Evidence of a Tryptophan Analogue Radical Formed in a Concerted Electron–Proton Transfer Reaction in Water, J. Am. Chem. Soc., 2016, 138, 2194–2199 CrossRef CAS PubMed.
- J. Soetbeer, D. Prateek and L. Hammarström, Marcus-Type Driving Force Correlations Reveal the Mechanism of Proton-Coupled Electron Transfer for Phenols and [Ru(bpy)3]3+ in Water at Low pH, Chem. Sci., 2016, 7, 4607–4612 RSC.
- D. Bandara and S. Burdette, Photoisomerization in Different Classes of Azobenzene, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- Y. Ali, S. A. Hamid and U. Rashid, Biomedical Applications of Aromatic Azo Compounds, Mini-Rev. Med. Chem., 2018, 18, 1548–1558 CrossRef CAS PubMed.
- L. Zhang and J. M. Cole, TiO2-Assisted Photoisomerization of Azo Dyes Using Self-Assembled Monolayers: Case Study on Para-Methyl Red Towards Solar-Cell Applications, ACS Appl. Mater. Interfaces, 2014, 6, 3742–3749 CrossRef CAS PubMed.
- P. S. Addy, Y. Zheng, J. S. Italia and A. Chatterjee, A “Quenchergenic” Chemoselective Protein Labeling Strategy, ChemBioChem, 2019, 20, 1659–1663 CrossRef CAS PubMed.
- A. N. Oldacre, C. A. Pointer, S. M. Martin, A. Kemmerer and E. R. Young, Anthracene-Based Azo Dyes for Photo-Induced Proton-Coupled Electron Transfer, Chem. Commun., 2019, 55, 5875–5877 RSC.
- B. D. McCarthy and J. L. Dempsey, Decoding Proton-Coupled Electron Transfer with Potential–pKa Diagrams, Inorg. Chem., 2017, 56, 1225–1231 CrossRef CAS PubMed.
- C. Costentin, M. Robert and J.-M. Savéant, Electrochemical and Homogeneous Proton-Coupled Electron Transfers: Concerted Pathways in the One-Electron Oxidation of a Phenol Coupled with an Intramolecular Amine-Driven Proton Transfer, J. Am. Chem. Soc., 2006, 128, 4552–4553 CrossRef CAS PubMed.
- J. Ravensbergen, C. L. Brown, G. F. Moore, R. N. Frese, R. Van Grondelle, D. Gust, T. A. Moore, A. L. Moore and J. T. M. Kennis, Kinetic Isotope Effect of Proton-Coupled Electron Transfer in a Hydrogen Bonded Phenol-pyrrolidino[60]fullerene, Photochem. Photobiol. Sci., 2015, 14, 2147–2150 RSC.
- T. Irebo, O. Johansson and L. Hammarström, The Rate Ladder of Proton-Coupled Tyrosine Oxidation in Water: A Systematic Dependence on Hydrogen Bonds and Protonation State, J. Am. Chem. Soc., 2008, 130, 9194–9195 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01643h |
| This journal is © The Royal Society of Chemistry 2020 |