
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAttachment of hybridizable oligonucleotides to a silica support and its application for selective extraction of unmodified and antisense oligonucleotides from serum samples†
Sylwia Studzińska *a,
Magdalena Skoczylasa,
Szymon Bociana,
Anna Dembskab and
Bogusław Buszewskia
*a,
Magdalena Skoczylasa,
Szymon Bociana,
Anna Dembskab and
Bogusław Buszewskia
aChair of Environmental Chemistry and Bioanalytics, Faculty of Chemistry, Nicolaus Copernicus University in Toruń, 7 Gagarin St., 87-100 Toruń, Poland
bLaboratory of Bioanalytical Chemistry, Faculty of Chemistry, Adam Mickiewicz University in Poznań, 8 Uniwersytetu Poznanskiego St., 61-614 Poznań, Poland. E-mail: kowalska@chem.umk.pl
First published on 23rd April 2020
Abstract
The main aim of the present study was the synthesis of an oligonucleotide-based material with high chemical stability, repeatability and specificity to complementary oligonucleotides. The oligonucleotides were attached to a silica gel surface modified with amino acids during one-step synthesis. The amount of the oligonucleotides immobilized on the support surface had an impact on adsorption effectiveness, due to steric interference. The adsorption capacity corresponds to 4.7 μg of complementary oligonucleotide per 1 mg of material, which reflects 50% of immobilized oligonucleotides. The presented results contain comprehensive studies on hybridization and release of fully complementary, partially complementary, non-complementary and antisense oligonucleotides from the newly synthesized adsorbent. The salt concentration and time period were the most influential parameters in the case of adsorption, while high temperature and low salt content were indispensable for effective desorption. Selectivity studies revealed that the adsorption percentage increases with the decreasing number of base mismatches. Consequently, the desorption of low complementarity oligonucleotides was always greater in comparison with the fully complementary sequence. Furthermore, it was shown that oligonucleotide-based materials may be successfully used for the extraction of antisense oligonucleotides and their metabolites from serum samples with recoveries ranging between 65 and 73%.
1. Introduction
Synthetic oligonucleotides (OGNs) are single-stranded short nucleic acid fragments.1 Nowadays they are applied in many different scientific areas, e.g. as modifiers of solid supports.2–5 Hybridization of complementary strands to OGNs immobilized on solid supports is increasingly studied. The earliest supports were DNA-modified celluloses used for purification of polynucleotides.3 Later, DNA oligonucleotides were attached to agarose, Teflon fibers, latex beads, glass, ceramics, nylon, polymers, membranes and other media for DNA affinity chromatography.4,5 Lately, magnetic or mesoporous silica nanoparticles have also been applied as a supports for oligonucleotide attachment.6,7 It has opened the door to a range of applications especially biomedical, such as: cell uptake studies (e.g. cancer cells) due to their traceability by microscopy; stimuli-responsive release systems with nucleic acids as caps; the detection of specific genes in a DNA sensor format; adsorption of specific oligonucleotides; siRNA detection and quantitation via hybridization assays.4,6–10 These achievements result from the specific properties of oligonucleotides, such as synthetic availability, conformational polymorphism, good biocompatibility, high physicochemical robustness and remarkable capability of molecular recognition.6,8,10Different immobilization strategies, including adsorption and chemical binding are used to bound oligonucleotides onto surfaces. Noncovalent immobilization methods cause high background resulting from nonspecific adsorption.10 For this reason well-developed covalent attachment methods are commonly used to link oligonucleotides: 5′-thiol-terminated oligonucleotides react with supports bearing activated thiol groups; amino terminal oligonucleotides react with isothiocyanate or carboxylated supports, 5′-terminal carboxylic acid-derivatized OGNs react with aliphatic amino groups at support surface.10–13 These covalent bonding is based on the formation of amide, disulphide and phosphodiester bond.10 It was shown that synthesis of materials with immobilized OGNs by the creation of amide bond has many advantages, such as: quantitative reaction under mild conditions with stable products, feasibility, the possibility of incorporation of terminal carboxylic acid functional group.
Despite the fact that attachment of OGNs to a solid support is of interest for many biotechnological applications, these materials are not widespread used or the selective extraction of OGNs based on hybridization. Lately, Bartlett et al.14 have used streptavidin magnetic beads with biotinylated capture strand (5′-AAAGAAAATATCATCT-3′) to selectively capture eluforsen and its metabolites. This drug is antisense oligonucleotide (ASO) so it is a modified (at phosphate group or sugar molecules) derivative of OGNs. ASOs are used in antisense therapy, which is based on their binding to complementary fragments of nucleic acids, similarly to their hybridization with OGNs immobilized at solid supports. However, Bartlett et al.14 have concluded that their application is limited due to difficulties associated with the loss of affinity of a variety of metabolite from a capture strand despite their high specificity and good recovery (80–95%). On the other hand in another paper it was shown that OGNs probe provided very effective cross-hybridization with 3′ n-1 and n-2 metabolites.5 Consequently, it seems that the quantitation of the full-length antisense strand may be overestimated. Regardless, hybridization assay was able to bind full-length antisense, 3′ n-1 and n-2 sequences.5,6 It was also shown that the sensitivity and specificity of the assay depend on the choice of the methods/reagents and the ONGs sequences.14–16 On the other hand hybridization based techniques provides the best sensitivity and throughput compared with other methods.5,6,13–16
The limited application of hybridization based techniques for selective extraction is due to problems associated with the reproducibility, long-term stability, non-specific binding of DNA, special care during designing capture probes to ensure appropriate affinity for the OGN analyte. To solve these problems, we have made an attempt to synthesize DNA-based material with high chemical stability, high repeatability and fewer nonspecific surface adsorption. We developed a method for attaching oligonucleotides to silica gel modified with amino acid and applied them for the selective extraction of unmodified antisense oligonucleotides, as well as their metabolites from solutions as well as serum. The presented work describes a comparative study upon complex optimization of OGN attachment to silica gel as well as extraction conditions. The hybridization and release of a complementary, non-complementary and antisense OGNs to the solid-phase was done to demonstrate the integrity of the immobilized oligonucleotide to silica gel.
2. Experimental section
2.1. Materials and reagents
The solid support of laboratory-prepared stationary phases was Kromasil 100, with particle diameter 5 μm, pore diameter 100 Å, pore volume 0.9 mL g−1, and surface area 310 m2 g−1 (Akzo Nobel, Bohus, Sweden).Following reagents: γ-aminopropyltrimethoxysilane, blocked aspartic acid Fmoc-Asp(OtBu)-OH, N,N'-dicyclohexylcarbodiimide (DCC), piperidine, trifluoroacetic acid (TFA), anhydrous dichloromethane (DCM), anhydrous N,N-dimethylformamide (DMF), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), morpholineethanesulfonic acid (MES) and ammonium acetate were obtained from Sigma Aldrich (Sigma-Aldrich, St. Louis, MO, USA). Sodium chloride, sodium citrate, and hydrochloric acid (for MES pH correction) were purchased from POCH S.A. (Gliwice, Poland). OGNs in lyophilized form were purchased from Sigma Aldrich (unmodified and phosphorothioates) and Eurogentec (Seraing, Belgium) (2′-O-methyl and 2′-O-(2-methoxyethyl) modifications). Their sequences, modification types, and masses were presented in Table 1. Each of riboses was modified with 2′-O-methyl and 2′-O-(2-methoxyethyl) groups in the case of ME and MOE OGNs. Phosphorothioate modification was included at each phosphate group in the structure of studied PS OGNs. HPLC gradient grade methanol (MeOH) (Merck, Darmstadt, Germany) and deionized water (Milli-Q system, Millipore, El Passo, TX, USA) were also used during the chromatographic studies.
| Abbreviation | Type of modification | Molecular mass [g mol−1] | Sequence 5′–3′ |
|---|---|---|---|
| DNA1 | Unmodified DNA | 6063 | GCCCAAGCTGGCATCCGTCA |
| DNA | DNA modified with aminoalkyl group | 6383 | TGA CGG ATG CCA GCT TGG GC–(CH2)7NH2 |
| OL1 | Unmodified DNA | 6092 | ATCGATCGATCGATCGATCT |
| OL2 | Unmodified DNA | 3696 | AAAAAAAAAAAA |
| OL3 | Unmodified DNA | 6157 | ATCGATCGATGGATCGATCG |
| OL4 | Unmodified DNA | 6110 | ATTGGAACCTTGGAACCCAA |
| OL5 | Unmodified DNA | 6031 | GCCCAAGCTAACATCCGTCA |
| OL6 | Unmodified DNA | 6045 | GCCCTTGCTGGCATCCGTCA |
| OL7 | Unmodified DNA | 6183 | GGGGAAGCTGGCATCCGTCA |
| OL8 | Unmodified DNA | 6152 | GGGGAAGCTGGCAGCCGTCA |
| DNA18 | Unmodified DNA | 5461 | CGGGTTCGACCGTAGGCA |
| DNA16 | Unmodified DNA | 4827 | CGGGTTCGACCGTAGG |
| MOE | 2′-O-(2-Methoxyethyl) | 7657 | CGGGTTCGACCGTAGGCAGT |
| ME | 2′-O-Methyl | 6621 | CGGGTTCGACCGTAGGCAGT |
| ME18 | 2′-O-Methyl | 6278 | CGGGTTCGACCGTAGGCA |
| ME16 | 2′-O-Methyl | 5959 | CGGGTTCGACCGTAGGC |
| PS | Phosphorothioate | 6368 | CGGGTTCGACCGTAGGCAGT |
2.2. Apparatus and experimental conditions for chromatography and circular dichroism
The amount of OGN immobilized, adsorbed and desorbed from modified silica surface was determined by measuring the concentration of OGN in the supernatant with the application of ultra high performance liquid chromatography (UHPLC). The Thermo Fisher Scientific™ Vanquish™ Horizon UHPLC system equipped with a diode array detector (Thermo Fisher Scientific, CA, USA) was used for this purpose. The data were collected with the use of Chromeleon 7 program. The ACE Excel C18Ar (1.7 μm, 100 × 2.1 mm) column purchased from Advanced Chromatography Technologies Ltd. (Aberdeen, UK) was utilized during the research. Mobile phase contained methanol (MeOH) and 5 mM ammonium acetate. The gradient elution was used during the study with three different gradient programs. In case of single OGNs solutions the following gradient elution program was applied: 5–25% v/v of MeOH in 6 min. For the mixtures of DNA1 with other OGNs (OL1–OL10): 10–20% v/v of MeOH in 6 min was used, while for modified ME, MOE and PS the gradient elution was changed and 10–40% v/v of MeOH in 6 min was applied. In the case of DNA1, DNA18, DNA16 mixture the 10–20% v/v of MeOH in 6 min was used, while for ME, ME18, ME16 mixture 15–20% v/v of MeOH in 6 min. The detection wavelength was λ = 260 nm. The injection volume equaled 1 μL. Autosampler and column temperature was 30 °C, while mobile phase flow rate 0.3 mL min−1.FTIR spectra were registered using Bruker Alpha Platinum ATR spectrophotometer equipped with a single-reflection platinum ATR module (Bruker Corporation, Billerica, MA, USA). Spectrum was recorded in the range of 400–4000 cm−1. Due to low carbon content in investigated material, spectrum were averaged over 4000 scans. The data was analyzed using OPUS software (Bruker Corporation).
Circular dichroism spectra were recorded on a Jasco J-715 spectropolarimeter equipped with a Peltier Jasco accessory (Jasco, Tokyo, Japan). Each measurement was the average of three repeated scans recorded: (1) from 200 nm to 320 nm at a scan speed of 200 nm min−1; at 25 °C and (2) from 200 nm to 360 nm at a scan speed of 200 nm min−1; at 95 °C. All experiments were performed in quartz cuvettes with a path length of 1 cm. The scan of the corresponding buffer solution was subtracted from the average scan for each sample.
Samples centrifugation was performed with the use of 5424 microliter Eppendorf AG (Hamburg, Germany) and CentriVap vacuum concentrator (Labconco, Kansas City, MO, USA). Heating–cooling dry block (Grant Instruments Ltd., Royston, Great Britain) and laboratory pH meter (Elmetron CP-505, Zabrze, Poland) were also applied.
2.3. Synthesis of Si-ASP adsorbent
Before the chemical modification of bare silica gel, a sample of adsorbent was placed in a specially designed glass reactor protecting against the contact of the reagents with the external environment. Silica gel was treated at 180 °C under vacuum (10−2 Pa) for 10 h in order to remove physically adsorbed water. Then, the temperature was decreased to 120 °C and γ-aminopropyltrimethoxysilane was added. Silica support surface modification with aminopropyl ligands has been carried out in non-solvent conditions described in detail in ref. 17 and 18.After 12 h, the reaction products were successively washed out with toluene, methanol and hexane, and dried. The aminopropyl silica was placed in a glass reactor and dried at 100 °C. Bonding of aspartic acid to prepared support was done using aspartic acid with free C-terminal group and with a protected amino group and second carboxylic group (Fmoc-Asp(OtBu)-OH) in the presence of DCC (activator of carboxyl group) in anhydrous DMF and DCM at 40 °C during 24 h.
Product was washed with toluene and methanol on a Schott funnel, in order to remove the excess reactants. The next stage of the synthesis was the deprotection of the bonded aspartic acid. The removal of the Fmoc groups was carried out using 50 mL of a 20% piperidine solution in anhydrous DMF. The resulting suspension was stirred for 15 min, and then the product was filtered and washed with toluene and methanol consecutively. The removal of the t-butyl (tBu) group was done by the treatment with 40% solution of trifluoroacetic acid (TFA) in anhydrous DCM. The product was filtered and washed with toluene and methanol and finally dried.18 The scheme of Si-ASP adsorbent synthesis was presented in Fig. S1 (ESI†). Its characterization was presented in ref. 18.
2.4. DNA OGN immobilization at the Si-ASP surface
The one-step immobilization procedure was applied for the DNA immobilization. This OGN was modified with short alkyl chain (seven methyl groups) with terminal amino group at the 3′ end. 200 μL of DNA and EDC in 0.1 M MES (pH 4.5) buffer solution was added to 2 mg of Si-ASP. The suspension was mixed and left at 30 °C for 2 h. The scheme of Si-DNA adsorbent synthesis was presented in Fig. 1. The impact of various parameters on the synthesis efficiency was tested, namely: EDC concentration (5–40 mg), DNA concentration (5.0–22.5 μM), synthesis time. After the synthesis the mixture was centrifuged for 15 minutes at 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g and supernatant was analyzed using RP UHPLC. Moreover, the remaining, newly synthesized adsorbent was washed with the use of 200 μL 0.05 M sodium citrate with 0.5 M sodium chloride in order to remove all non-specifically bonded DNA. It has to be underline that each synthesis was performed at least twice.
000g and supernatant was analyzed using RP UHPLC. Moreover, the remaining, newly synthesized adsorbent was washed with the use of 200 μL 0.05 M sodium citrate with 0.5 M sodium chloride in order to remove all non-specifically bonded DNA. It has to be underline that each synthesis was performed at least twice.
 | ||
| Fig. 1 Schematic presentation of Si-DNA synthesis from Si-ASP. | ||
Optimized Si-DNA synthesis conditions were as follows: 2 mg of Si-ASP, 15 mg of EDC and 40 μL of 100 μM DNA1 in 200 μL of 0.1 M MES (pH 4.5). Synthesis was performed during 1 hour at 30 °C.
2.5. OGN adsorption and desorption from Si-DNA
Adsorption of different OGNs onto the Si-DNA was done in solution of sodium chloride containing sodium citrate at pH 7.0 (saline sodium citrate buffer – SSC). OGNs dissolved in SSC at various concentrations were added to 2 mg Si-DNA and left for 2 h at 30 °C. Next, the sample was centrifuged for 15 minutes at 11000g and the supernatant was removed. After adsorption, Si-DNA was washed several times with 200 μL of 0.03 M sodium citrate in 0.3 M sodium chloride (2× SSC) in order to remove non-specifically bounded OGN. Every time the suspension was centrifuged at 11000g for 15 minutes and supernatant was removed and analyzed by RP UHPLC.
The adsorption process conditions were optimized for DNA1 (Table 1), which was complementary to DNA immobilized at the silica surface. The influence of several parameters was investigated, namely: SSC concentration (0–0.5 M sodium chloride and 0–0.05 M sodium citrate), number of washing cycles (1–4). Each adsorption experiment was done at least twice.
OGNs desorption from Si-DNA was done using SSC at high temperature. The impact of SSC concentration (0–0.5 M sodium chloride and 0.05 M sodium citrate), temperature (70–100 °C) and desorption time (1–24 h) was studied with regard to OGN recovery. The amount of OGN released from the adsorbent was determined using RP UHPLC. Each experiment was performed in two repeats.
Finally optimized adsorption conditions were as follows: 2 mg Si-DNA and 10 μM DNA1 in 3.3× SSC (total volume 200 μL) during 40 minutes at 30 °C. Next, adsorbent was washed with the use of two portions of 200 μL 3.3× SSC. The desorption was performed with 200 μL of water at 80 °C for 1 hour (Table S1†).
2.6. Sorption kinetics and adsorption capacity
Sorption kinetics and adsorption capacity tests were carried out under the optimized experimental conditions. The 2 mg of Si-DNA was weighted and 10 μM DNA1 was adsorbed from 3.3× SSC solution. The amount of adsorbed OGN was measured at intervals during two hours (10, 15, 30, 50, 75, 120 minutes) using RP UHPLC (as a difference in peak area for the 10 μM DNA1 standard solution and peak area for DNA1 obtained in the supernatant at different time intervals). Each experiment was performed in three repeats.The amount of DNA1 adsorbed on the Si-DNA during the experiments was calculated using the mass-balance equation:
 | (1) |
The sorption effectiveness was calculated by the following formula:
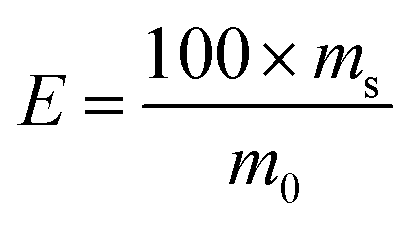 | (2) |
The adsorption capacity of Si-DNA for DNA1 was determined for three different amounts of adsorbent. 1, 2 or 3 mg of Si-DNA were added to 200 μL of different concentrations of DNA1 (from 5 to 15 μM). The tests were performed for two (in case of 1 and 3 mg) or four weighted portions (in case of 2 mg). The value of adsorption capacity Qe, [μmol mg−1] in equilibrium was calculated in accordance with the following equation:
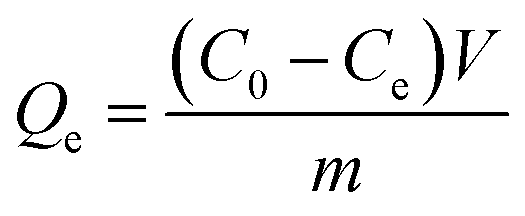 | (3) |
2.7. Selectivity tests
The selectivity (in terms of hybridization specificity) of Si-DNA to OGNs with various sequences (possessing different degree of complementarity) was tested in the present work. Non-complementary OL1–OL4 as well as OGNs with single-base, two-base, three-base, five-base mismatches (OL5–OL8) were examined (Table 1). Moreover, shorter (18 mer and 16 mer) OGNs (DNA18, DNA16) with complementary sequences were studied (Table 1). The tests were done by mixing of 10 μM of DNA with the same concentration of one of chosen OGNs. Next, the adsorption and desorption of OGNs was done under optimized, equilibrium conditions. Each experiment was performed in two repeats.2.8. ASO adsorption and desorption
The 10 μM solutions of PS, ME or MOE (Table 1) OGNs were used in order to study their adsorption and desorption from Si-DNA. All tests were performed under optimized, equilibrium conditions summarized in paragraphs 2.4 and 2.5. Each experiment was performed in four repeats.2.9. Extraction of unmodified and modified OGNs from spiked serum samples
Human serum was diluted at the ratio 1:5 with the use of 3.3× SSC. All experiments with human serum were performed in accordance with relevant guidelines of Ludwik Rydgier Collegium Medicum of Nicolaus Copernicus University in Toruń and approved by the Bioethical Commission of the Nicolaus Copernicus University (permission no. 707/2019). Informed consents were obtained from human participants of this study. The sample was spiked with the mixture of DNA, DNA18, DNA16 or the mixture of ME, ME18, ME16. The final concentration of each OGN in 600 μL of tested sample was 3.7 μM. These samples were firstly purified with the use of liquid\liquid extraction (LLE) with the use of phenol/chloroform/isoamyl alcohol (25:24:1, v/v/v) (1:1). Next, they were centrifuged at 11000g for 40 minutes. The upper, aqueous layer was washed three times with chloroform at the ratio of 1:4. Subsequently, the OGNs were selectively extracted with the use of method developed during present study. The blank samples were also prepared and analyzed with the use of similar procedure. The blank samples were of the same composition as the test samples, however they did not contained OGN, instead of which water was added (to maintain a constant total sample volume).
3. Results and discussion
3.1. DNA immobilization on the Si-ASP
Our purpose was to immobilize linear OGN at the silica surface, thus further hybridization of complementary strand would be more effective. Before DNA immobilization and complementary DNA1 hybridization, the structure of both OGNs was studied. The circular dichroism (CD) technique was used for this purpose, since it is applied for the study of the secondary structures. CD spectroscopy is sensitive to OGN conformation, especially within the 180 to 320 nm wavelength range. Therefore, CD was used in order to confirm DNA (in MES and EDC solution, as well as in 3× SSC solution) and DNA1 (in 3× SSC solution) linearity and lack of the secondary structures. CD spectra from 320 to 200 nm of these OGNs are shown in Fig. S2 (ESI†). As expected, CD spectra of DNA and DNA1 showed a band with positive ellipticity at λmax around = 280 nm and a negative peak of lower intensity at approx. 250 nm, and are in good agreement with that of other single-stranded OGNs, e.g. d(GT)12.19The synthesis method used for OGN immobilization at the Si-ASP surface was very simple. The amino modified DNA was used in order to perform a carbodiimide mediated acylation. Additionally, the acidic pH of MES buffer was used in order to obtain the most efficient crosslinking.10
Obtained material was investigated using FTIR spectroscopy to confirm the bonding of DNA molecules. Due to the low masses of synthesized sorbent it was analyzed in attenuated total reflectance (ATR) mode that is unfortunately less sensitive. Nevertheless, the spectra were obtained with the most abundant bands. The comparison of spectra of silica gel modified with aspartic acid (Si-ASP) and final product with bonded DNA molecule (Si-DNA) are presented in Fig. 2.
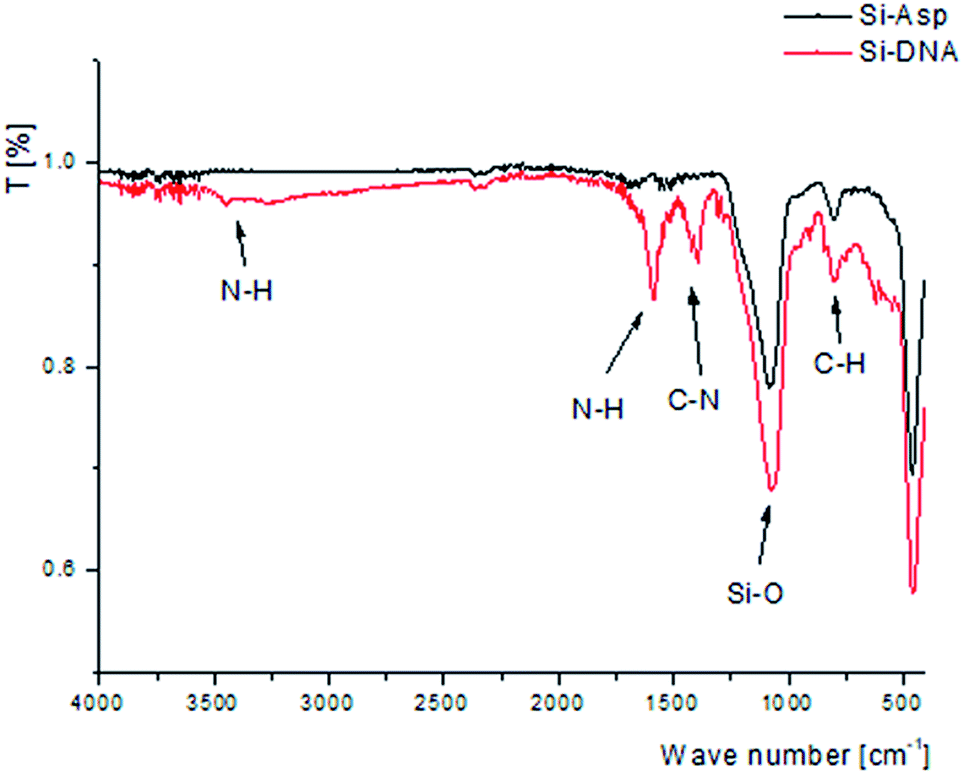 | ||
| Fig. 2 FTIR spectra of Si-ASP and Si-DNA. | ||
After the modification of Si-ASP material by bonding DNA molecules additional signals were obtained that confirm the presence of nitrogen–hydrogen and nitrogen–carbon bonds presented in nucleotides structure. Bands of C–N stretch vibration may be observed at around 1400 cm−1. Signals of N–H bend and N–H stretch vibration are observed at 1600 and around 3400 cm−1, respectively. The presence of N–H and C–N bonds confirms the successful bonding of DNA molecules to the support.
The efficiency of the immobilization was determined by measuring the amount of DNA remaining in the solution after the synthesis. The effect of EDC concentration on the DNA immobilization on Si-ASP was studied using 20 μM DNA in 200 μL of 0.1 M MES buffer (pH 4.5), 2 mg of adsorbent and 5–40 mg of EDC (Table 2). It has to be pointed out that when EDC was not used for the synthesis the concentration of DNA in the solution after synthesis was 100% as initial. With increasing EDC amount, the concentration of studied OGN in the supernatant decreased. Approximately 90% of DNA was bonded to the silica support when EDC amount was in the range of 30–40 mg (Table 2). However, at this step of investigation the most optimal EDC concentration was not selected yet, since also the impact of EDC concentration on the further hybridization step should be considered.
| EDC [mg] | Immobilized DNA [μM] | Adsorbed DNA1 [μM] |
|---|---|---|
| 5 | 10.8 ± 1.2 | 5.1 ± 1.8 |
| 15 | 14.2 ± 1.5 | 7.4 ± 1.2 |
| 20 | 14.8 ± 1.3 | 7.5 ± 1.3 |
| 30 | 16.8 ± 0.9 | 5.9 ± 1.1 |
| 40 | 18.6 ± 1.4 | 4.4 ± 1.9 |
To investigate the influence of DNA concentration on its immobilization, four different concentration of OGN (5.0, 10.0, 20.0, 22.5 μM) were tested using 15 mg of EDC. As expected, an increase in the concentration of DNA available for immobilization, there was an increase in the total amount bonded to the Si-ASP. In case of 5 and 10 μM of DNA all of OGN in the solution was immobilized, but the immobilization efficiency decreased to 76% at the highest tested concentration (22.5 μM). The immobilization efficiency of 20 μM DNA is 74%, which is in agreement with data presented in Table 2.
The synthesis time (1, 2, 4, 12 hours) was also optimized with regard to the immobilization efficiency. The amount of unbounded DNA was similar after one and twelve hours of synthesis. Consequently, all further syntheses were carried out just one hour.
The synthesis in optimized conditions (20 μM DNA in 200 μL of 0.1 M MES buffer pH 4.5, 2 mg of adsorbent and 15 mg of EDC) was repeated 10 times and immobilization efficiency of 20 μM DNA was in the range of 70–75% indicating repeatability of the process and material.
3.2. Adsorption of complementary DNA1 at Si-DNA
The 10 μM of DNA1 was used for the optimization of adsorption conditions. Our results showed that a complementary DNA1 was hybridized to immobilized Si-DNA. Generally, the surface density of Si-DNA at the modified silica surface influences the steric hindrance impeding the adsorption of a complementary OGN based of hybridization. The greater the number of immobilized DNA molecules, the higher the probability of steric interference. Therefore, we have investigated the impact of increasing the DNA concentration at the silica surface on the DNA1 adsorption efficiency. The obtained data are presented in Table 2. These results demonstrate that initially with an increase in the amount of DNA bounded to the surface, there was an increase in the amount of adsorbed DNA1. For optimized conditions as mentioned before, the 14.2 μM of DNA was immobilized and 7.4 μM of DNA1 was adsorbed at Si-DNA surface. However, at surface concentrations greater than 16 μM, there was a significant decrease in hybridization efficiency (Table 2), which reflects the steric hindrance. It has to be reminded, that the pore size of the silica used was 100 Å and bonded DNA has occupied significant space inside the pores that result in worse penetration of complementary DNA1 into the pores and thus the lower adsorption. During the optimization step we have used 20 μM of DNA, however later we have changed this value and DNA concentration for immobilization was always selected in such a way that the amount of DNA bounded to the Si-ASP surface did not exceed 14.2 μM.The impact of saline sodium citrate concentration on the DNA1 adsorption was also tested. Three attempts were done: hybridization from water solution, from 2× SSC and 3.3× SSC. SSC was selected as the most often used buffer in hybridization experiments, e.g. Southern blotting. The sodium ions stabilize the hybrid-formation by decreasing the electrostatic repulsion of the two single strands. Its concentration has a significant effect on OGN adsorption based on complementary strands hybridization. The 70.2 ± 1.1% adsorption of 10 μM DNA1 was observed when 3.3× SSC was implemented. This value was very similar (71.0 ± 1.4%) for 2× SSC, while different results were obtained when DNA1 was adsorbed from pure water solution (only 42.4 ± 1.9%). Consequently, 3.3× SSC buffer was selected for further studies (Table S1†).
After DNA1 hybridization, the adsorbent was washed 4 times with 200 μL of 3.3× SSC before desorption step. The aim of washing was to remove non hybridized DNA1. The number of washing cycles was optimized and it appeared that are satisfactory.
Time-dependent sorption of DNA1 by Si-DNA is shown in Fig. 3. The equilibrium was reached after 40 minutes (Fig. 3). The amount of adsorption increases rapidly in 10 min. The most of the OGN (about 50%) is adsorbed within ten minutes, making the process relatively quick. When the DNA1 is transported into the Si-DNA surface the further diffusion becomes more difficult due to the steric hindrance, thus the amount of adsorbed DNA1 tends to increase very slowly and finally reaches an equilibrium state (Fig. 3). The time needed for hybridization was equal 40 minutes.
 | ||
| Fig. 3 Kinetics of DNA1 sorption by Si-DNA under static conditions (for experimental conditions see in 2.6 Section). | ||
The adsorption capacity was also determined based on the eqn (3). The tests were performed for three different Si-DNA masses and different DNA1 concentrations. The adsorption capacity of Si-DNA for DNA1 was determined to be 0.000205 μmol mg−1 (Table 3). It corresponds to 4.7 μg DNA1 per 1 mg of Si-DNA, while 1 mg of Si-ASP surface was modified with 9.1 μg of DNA. The hybridization of OGN was found to be approximately 50% lower than that of immobilized one. This reflects the steric interference of the Si-DNA surface density to DNA1, as well as the blocking of a certain volume of pores.
| Mass of Si-DNA [mg] | DNA1 concentration used for experiment [μM] | DNA1 adsorbed at the surface [μmol L−1] | Adsorption capacity Qe [μmol mg−1] |
|---|---|---|---|
| 1 | 5 | 3.95 ± 0.22 | 0.000210 |
| 2 | 10 | 7.92 ± 0.41 | 0.000208 |
| 2 | 10 | 7.81 ± 0.39 | 0.000209 |
| 3 | 15 | 12.12 ± 0.63 | 0.000193 |
Since Si-DNA was synthesized by the modification of Si-ASP, an attempt to verify if DNA1 is adsorbed just by the hybridization with DNA was performed. The polar amino and carboxyl groups are present at the surface of Si-ASP and they can interact with OGNs by hydrogen bonding, because probably not all of carboxyl groups were blocked with DNA. In result, the non-specific adsorption may occur during the extraction of DNA1, contrary to very specific hybridization. We have studied the adsorption of DNA1 at the surface of unmodified Si-ASP under conditions similar to hybridization at Si-DNA. The adsorption attempt was done for 10 μM DNA1 during 1 hour at 30 °C from 3.3× SSC solution (for 2 mg Si-ASP). It appeared that DNA1 was not adsorbed at Si-ASP surface. These results indicated that DNA1 adsorption at Si-DNA occurs due to hybridization between DNA1 and complementary OGN attached to silica and it is not a consequence of non-specific interactions with residual polar groups remaining at the Si-DNA surface (after last step of synthesis).
After hybridization of OGN, the Si-DNA was washed 4 times with 200 μL of 2× SSC before DNA1 release (Table S1†). The unadsorbed DNA1 was effectively removed during the second wash cycle, therefore two cycles of washing were selected for the optimized procedure applied during further study.
3.3. DNA1 desorption from Si-DNA surface
For the release of DNA1 from Si-DNA, the temperature was raised up to 70 °C and maintained for two hours. The 52 ± 3% of adsorbed DNA1 was desorbed. In order to improve this value several parameters were changed, namely SSC concentration, temperature and time. Results obtained during this part of study were collected in Table 4.| Desorption conditions | DNA1 recovery [%] | ||
|---|---|---|---|
| Time [hours] | SSC (sodium chloride/sodium citrate) concentration [M] | Temperature [°C] | |
| 2 | 0.3/0.03 | 70 | 52.2 ± 3.0 |
| 2 | 0.3/0.03 | 80 | 66.2 ± 1.1 |
| 2 | 0.3/0.03 | 100 | 66.5 ± 2.0 |
| 2 | 0.5/0.05 | 80 | 62.4 ± 3.1 |
| 2 | 0.1/0.01 | 80 | 70.1 ± 1.2 |
| 2 | H2O | 80 | 72.2 ± 1.4 |
| 0.5 | H2O | 80 | 72.3 ± 2.1 |
| 0.5 | 0.3/0.03 | 80 | 59.4 ± 2.0 |
| 1 | 0.3/0.03 | 80 | 65.3 ± 3.9 |
| 1.5 | 0.3/0.03 | 80 | 66.5 ± 1.5 |
| 3 | 0.3/0.03 | 80 | 66.2 ± 2.8 |
| 4.5 | 0.3/0.03 | 80 | 67.3 ± 1.7 |
| 5.5 | 0.3/0.03 | 80 | 66.4 ± 1.6 |
The increase of desorption temperature about 10 °C resulted in the recovery increase to 66%, however further temperature increasing already had negligible effect (Table 4). In contrast, increasing SSC concentration provided a decrease in the amount of desorbed DNA1. Extending the desorption time from 0.5 to 1 hour increased the value of DNA1 recovery from the surface of Si-DNA from 59 to 65% (Table 4). However, for more than one hour, the recovery value did not change and equaled about 66%. The greatest recovery (73%) was determined for the application of pure water as the desorption solvent or for very low SSC concentration (Table 4). These effects are probably results of salt absence (salt ions screen electrostatic repulsion between negatively charged OGNs), causing electrostatic repulsion between negatively charged backbones of DNA1 and DNA leading to base pairs destabilization and release of DNA1 from Si-DNA surface. Based on results collected in Table 4 release of OGN was performed in water at 80 °C for 0.5 hour (Table S1†).
3.4. Si-DNA selectivity
:1). Fig. S3 (ESI†) presents exemplary chromatograms obtained during the analysis of DNA1 and OL1 mixture at various stages of experiment: standards, adsorption (hybridization) and desorption (release). Similar results were noticed for mixture of DNA1 with OL2, OL3 or OL4.OGNs with low degree of complementarity to immobilized DNA were not adsorbed at the modified silica surface, contrary to complementary DNA1 (70%). These results suggest that Si-DNA can be apparently and successfully used for the separation of OGN with sequence complementary to the one immobilized at adsorbent surface from other, non-complementary OGNs.
 | ||
| Fig. 4 (A) The quantity of adsorbed and desorbed DNA1 and other OGNs with different-base mismatches; (B) the impact of OGN modification on their hybridization with complementary OGN immobilized at adsorbent surface (Si-DNA). Notation: PS – phosphorothioate, ME – 2′-O-methyl, MOE – 2′-O-(2-methoxyethyl). | ||
In each case, both OGNs (complementary DNA1 and other OGN with a lower degree of complementarity) were adsorbed at Si-DNA surface. However, DNA1 adsorption was more efficient than OL5, OL6, OL7 or OL8. It is also characteristic that as the degree of complementarity decreases (from double-base mismatch to several-base mismatch), the amount of adsorbed not completely complementary OGN also decrease (Fig. 4A), while the amount of adsorbed DNA1 increase slightly. What is more important, the amount of adsorbed DNA1 was lower (50–55%) compared to hybridization results obtained from solution containing only DNA1 (Fig. 3). This is probably the result of competition for active sites between DNA1 and OL5–OL8.
The sorption effectiveness of OGNs with double-base mismatch to DNA-Si (OL5, OL6) was only 23–25% (Fig. 4A), while in the case of OGN containing five mismatch nucleotides in the sequence this value dropped to 13%.
These results indicate the relatively high selectivity of Si-DNA, because only OGNs with small differences in sequence will adsorb on its surface. At the same time, their adsorption will be two to five times lower compared to the adsorption of the fully complementary OGN (Fig. 4A). Consequently, the Si-DNA application allows to obtain much greater selectivity of OGN extraction compared to methods commonly used in the isolation of this group of compounds (e.g. liquid–liquid extraction, solid phase extraction), since they are used to extract all OGNs present in the sample.
Regarding the desorption of hybridized OGNs, as expected OL5, OL6, OL7 and OL8 were released with almost 100% of recovery (Fig. 4A). Reducing the degree of complementarity of these OGNs to Si-DNA1, results in a very effective desorption. Contrary to these findings, the recovery of DNA1 was lower and equaled 60–65% (Fig. 4A).
3.5. Application of Si-DNA for the adsorption and desorption of ASO
Si-DNA can be successfully used to extract unmodified OGNs. However, ASO are of greater interest among researchers investigating such kind of compounds. The mechanism of ASO action is based on the complementarity of base pairs, similar as is the case of Si-DNA adsorbent. Therefore, one of our research goals was the attempt to use synthesized material (DNA-Si) for the extraction of ASO.OGNs with different types of modification were used for this purpose: phosphorothioate (PS), 2′-O-methyl (ME) and 2′-O-(2-methoxyethyl) (MOE). Their sequence was complementary to the DNA sequence immobilized at the silica surface (Table 1). The amount of adsorbed compound was similar regardless of the ASO type (about 70%) with very low standard deviations (Fig. 4B). Apparently modification type has not influenced adsorption based on complementarity. The modification of ASO strand also has not affected the desorption, since recovery for each of three ASO (PS, ME, MOE) equaled about 60% (Fig. 4B). Moreover standard deviation was very low, therefore Si-DNA may be applied in the extraction of ASO.
3.6. Application of Si-DNA for the OGN extraction from serum samples
The developed methods were used for the determination of DNA, ME and their 3′ shortened metabolites (n-2 and n-4) in extracts obtained from enriched human serum samples (they were enriched to 3.7 μM of each OGN). The basic validation parameters were collected in Table 5. The linearity was determined by the calculation of the correlation coefficient (R2) for the calibration curves in the range of 0.1–5.0 μM (6 different concentrations) of each OGN. The LOD and LOQ of the method were determined by the signal-to-noise ratio using the eqn (3) S/N and 10 S/N respectively. The LOQ values were in the range of 0.1–0.2 μM. The repeatability and precision was expressed in terms of relative standard deviation (RSD). Method repeatability was obtained from RSD value by repeating the 7 times analysis on the same day for intraday precision for three different concentrations (0.4, 1.5, 5.0 μM). It was lower than 0.9% for DNA and ME mixtures. Intermediate precision was determined 5 injections per three different concentrations (0.4, 1.5, 5.0 μM) on the first, fourth and seventh day. This parameter did not exceeded 2.2%.| Oligonucleotide | Concentration range [μM] | Calibration curve equation | R2 | LOQ [μM] | RSD [%] inter-day 0.4 μM | RSD [%] inter-day 1.5 μM | RSD [%] intra-day 3.5 μM | RSD [%] intra-day 0.4 μM | RSD [%] intra-day 1.5 μM | RSD [%] inter-day 3.5 μM | Recovery [%] |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DNA | 0.1–5.0 | y = 0.9815x − 0.0595 | 0.9997 | 0.10 | 0.7 | 0.5 | 0.9 | 1.1 | 1.1 | 1.3 | 62 ± 1 |
| DNA18 | y = 1.1472x − 0.0603 | 1.0000 | 0.11 | 0.7 | 0.7 | 0.8 | 1.4 | 1.2 | 1.5 | 59 ± 2 | |
| DNA16 | y = 0.8798x − 0.0496 | 0.9999 | 0.10 | 0.9 | 1.0 | 0.9 | 1.2 | 1.5 | 1.0 | 62 ± 1 | |
| ME | 0.2–5.0 | y = 0.6281x − 0.0301 | 0.9999 | 0.15 | 0.6 | 0.6 | 0.8 | 1.9 | 1.9 | 1.9 | 55 ± 0.3 |
| ME18 | y = 0.8398x − 0.0139 | 0.9998 | 0.20 | 0.9 | 0.8 | 0.9 | 1.6 | 1.0 | 1.8 | 63 ± 0.3 | |
| ME16 | y = 0.5494x − 0.0161 | 0.9990 | 0.20 | 0.5 | 0.8 | 0.5 | 1.1 | 1.6 | 2.2 | 65 ± 2.7 |
The matrix effect was determined based on the ratio of peak areas obtained for 3.7 μM standard OGN mixture (first mixture was composed of DNA, DNA18, DNA16 and the second of ME, ME18, ME16) and for the blank matrix sample enriched post-extraction with the same concentration of OGN standards. Matrix effect values were in the range of 89–102% for DNA, DNA18, DNA16 mixture and 89–94% for ME, ME18, ME16. These effects are very low, since 100% indicate no matrix effect. However these effects were taken into account when OGN recoveries form serum samples were calculated (Table 5).
The fortified and diluted plasma samples were pre-treated by LLE extraction with phenol/chloroform/isoamyl alcohol mixture in order to remove proteins. Next, extracts were washed with chloroform to remove residual phenol. Then, the method developed during presented study was applied for selective OGN extraction. The extraction procedure was monitored at various stages: after first LLE, after washing with chloroform, after hybridization and desorption. The LLE allowed for effective protein and phenol removement. Moreover, all of studied OGN (each at 3.7 μM) were completely adsorbed at the Si-DNA surface. Fig. 5 presents chromatograms obtained for DNAs and MEs extracts from serum samples after extraction with the use of Si-DNA. The recoveries from enriched serum samples were 62.5 ± 0.9%, 59.9 ± 1.2%, 62.7 ± 1.1% for DNA, DNA18 and DNA16 respectively. For ME, ME18 and ME16 these values were as follows 55 ± 2%, 63 ± 1%, 65 ± 2%. The developed method can be successfully applied to serum samples containing both unmodified and modified ASO, but external calibration has limitations over standard addition method. It should be applied when industry or regulatory agencies will utilize method developed during present investigation.
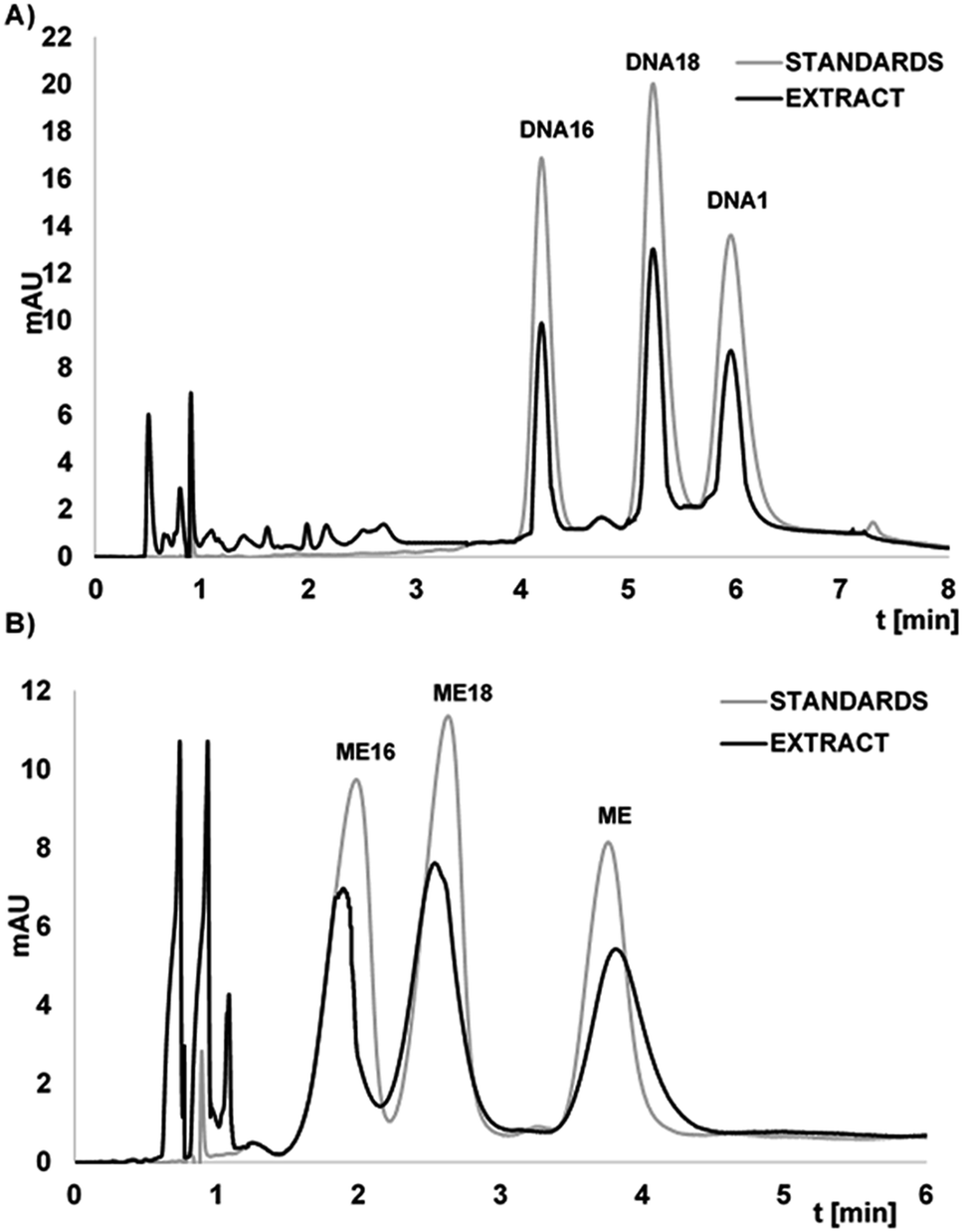 | ||
| Fig. 5 Chromatograms for the separation of: (A) DNA1, DNA18, DNA16; (B) ME, ME18, ME16 for standards mixture (3.7 μM each), as well as for the extracts from serum samples (enriched to the concentration of 3.7 μM). Experimental conditions: ACE Excel C18Ar (1.7 μm, 100 × 2.1 mm) column; mobile phase: MeOH and 5 mM ammonium acetate; gradient elution: for DNA's mixture: 10–20% v/v of MeOH in 6 min; for ME's mixture: 15–20% v/v of MeOH in 6 min; UV detection at λ = 260 nm; column temperature 30 °C, autosampler temperature 30 °C, flow rate 0.3 mL min−1, injection volume 2 μL. | ||
4. Conclusions
The attachment of OGN to silica gel modified with amino acid was developed and shown as an effective interface for selective oligonucleotide hybridization and extraction. The amount of the OGN immobilized on the silica gel surface had an impact on adsorption effectiveness: high DNA content lead to low hybridization of a complementary OGN due to steric interference. The latter proved the importance of controlling synthesis process with regard to adsorption of complementary OGN. The adsorption capacity of adsorbent corresponds to 4.7 μg of complementary OGN per 1 mg of Si-DNA. This result reflects to 50% of immobilized OGN and it is a consequence of steric hindrance.The optimization of extraction process revealed that SSC concentration and time were the most influential parameters in case of hybridization, while high temperature and low salt content are indispensable for effective release of OGN. The desorption of complementary OGN from adsorbent was as high as 70%.
The results obtained during selectivity study of synthesized adsorbent showed that no non-complementary OGNs are retained at the surface indicating its usefulness in the e.g. OGN fractionation. However, the adsorption increase with the decreasing number of the sequence base mismatches. Consequently, it was the greatest for OGN differing by two nucleotides, and lower in case of OGN containing three or five base mismatch. On the other hand, the desorption of these oligonucleotides was always with higher efficiency in comparison with OGN with fully complementary sequence. The selectivity of adsorbent was also tested towards shorter complementary strands. The positive results indicated that Si-DNA adsorbent can be successfully used for the extraction of shorter OGNs, typically metabolites. Most importantly, it has been shown that the synthesized materials can be used for extraction of ASO with sequences complementary to one immobilized at the silica gel surface. Moreover, the adsorption of ASO is independent of the type of oligonucleotide strand modification. This result is very promising due to therapeutic application of these compounds and the growing need for new methods of selective sample preparation for ASO.
Silica based adsorbents with immobilized OGNs may be successfully used for the selective extraction of studied OGN and its metabolites from serum samples. Such extraction proved to be not only selective but also effective, since recoveries were in range of 65–73% for both unmodified OGNs and ASOs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Science Centre (Cracow, Poland) within the Sonata Bis project (2016/22/E/ST4/00478). Special acknowledgements to Mrs Anna Kaczmarkiewicz and Mr Łukasz Nuckowski for their support.References
- C. I. E. Smith and R. Zain, Annu. Rev. Pharmacol. Toxicol., 2019, 59, 605–630 CrossRef CAS PubMed.
- N. Rublack, H. Nguyen, B. Appel, D. Springstubbe, D. Strohbach and S. Müller, J. Nucleic Acids, 2011, 805253 Search PubMed.
- P. Gilham, J. Am. Chem. Soc., 1964, 86, 4982 CrossRef CAS.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS PubMed.
- M. B. Thayer, J. M. Lade, D. Doherty, F. Xie, B. Basiri, O. Barnaby, N. Bala and B. M. Rock, Sci. Rep., 2019, 9, 3566 CrossRef PubMed.
- S. C. Humphreys, M. B. Thayer, J. M. Lade, B. Wu, K. Sham, B. Basiri, Y. Hao, X. Huang, R. Smith and B. M. Rock, Drug Metab. Dispos., 2019, 47, 1174 CrossRef CAS PubMed.
- M. Chen, S. Yang, X. He, K. Wang, P. Qiu and D. He, J. Mater. Chem. B, 2014, 2, 6064 RSC.
- A. Leidner, S. Weigel, J. Bauer, J. Reiber, A. Angelin, M. Grösche, T. Scharnweber and C. M. Niemeyer, Adv. Funct. Mater., 2018, 28, 1707572 CrossRef.
- M. Song, Y. Ding, M. A. Snyder and J. Mittal, Langmuir, 2016, 32, 10017 CrossRef CAS PubMed.
- M. K. Walsh, X. Wang and B. C. Weimer, J. Biochem. Biophys. Methods, 2001, 47, 221 CrossRef CAS PubMed.
- J. B. Wheatley, M. H. Lyttle, M. D. Hocker and D. E. Schmidt, J. Chromatogr., 1996, 726, 77 CrossRef CAS PubMed.
- Z. Guo, R. Guilfoyle, A. J. Thiel, R. Wang and L. M. Smith, Nucleic Acids Res., 1994, 22, 5456 CrossRef CAS PubMed.
- C. P. Vary, Clin. Chem., 1992, 38, 687 CrossRef CAS.
- J. Kim, B. Basiri, C. Hassan, C. Punt, E. van der Hage, C. den Besten and M. G. Bartlett, Mol. Ther.–Nucleic Acids, 2019, 17, 714 CrossRef CAS PubMed.
- L. Wang, Bioanalysis, 2011, 3, 1299 CrossRef CAS PubMed.
- L. Sips, E. N. Ediage, B. Ingelse, T. Verhaeghe and L. Dillen, Bioanalysis, 2019, 11, 1941 CrossRef CAS PubMed.
- M. Skoczylas, S. Bocian and B. Buszewski, RSC Adv., 2016, 6, 96389 RSC.
- B. Buszewski, M. Jezierska, M. Wełniak and R. Kaliszan, J. Chromatogr. A, 1999, 845, 433 CrossRef CAS.
- C. L. Clark, P. K. Cecil, D. Singh and D. M. Gray, Nucleic Acids Res., 1997, 25, 4098 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01620a |
| This journal is © The Royal Society of Chemistry 2020 |