
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic progress toward the marine natural product zamamiphidin A†
Hao Wanga,
Di Tianb,
Zhaoxiang Mengb,
Zhihao Chenb,
Fei Xue*b,
Xiao-Yu Liub,
Hao Songb and
Yong Qin *b
*b
aChongqing Key Laboratory of Natural Product Synthesis and Drug Research, School of Pharmaceutical Sciences, Chongqing University, Chongqing 401331, P. R. China
bKey Laboratory of Drug-Targeting and Drug Delivery System of the Education Ministry, Sichuan Engineering Laboratory for Plant-Sourced Drug, Sichuan Research Center for Drug Precision Industrial Technology, West China School of Pharmacy, Sichuan University, Chengdu 610041, P. R. China. E-mail: xuefei@scu.edu.cn; yongqin@scu.edu.cn
First published on 24th March 2020
Abstract
An asymmetric synthetic approach to the octahydrofuro[3,4-b]pyridine framework of marine natural product zamamiphidin A has been described. The key steps include an asymmetric Michael addition of (R)-N-tert-butanesulfinyl imidate with enamidomalonate to install the C10 stereocenter, an intramolecular alkoxide exchange/Michael addition/hydrogenation sequence to construct the bicyclic ring system.
Marine natural products have been recognized as an important source of potential lead compounds.1 Recently, the growing attention on innovative drug discovery has propelled the identification of numerous marine-derived bioactive compounds with superior chemical novelty.2 In 2013, Kobayashi and co-workers isolated a new macrocyclic diamine alkaloid zamamiphidin A (1, Fig. 1A) from an Okinawan marine sponge Amphimedon sp. (SS-1231),3 along with its biogenetically related manzamine alkaloids such as ircinic acid (2) and manzamine A (3). In the isolation work, the authors also demonstrated that compound 1 exhibited moderate antibacterial activity against Staphylococcus aureus (MIC, 32 μg mL−1).3 Architecturally, zamamiphidin A was characterized by an unprecedented heptacyclic framework, comprising a highly fused pentacyclic caged core, two 11-membered rings and seven contiguous stereocenters including one all-carbon quaternary center. More specifically, the fusion of 7-oxa-2-azabicyclo[2.2.1]heptane (4, Fig. 1B), azabicyclo[3.3.1]nonane (5) and octahydrofuro[3,4-b]pyridine (6) fragments as well as the presence of quaternary ammonium salt render zamamiphidin A unique among other macrocyclic diamine alkaloids.4 To the best of our knowledge, the chemical synthesis of 1 has yet to be disclosed. Herein, we report our recent synthetic efforts toward zamamiphidin A (1).
 | ||
| Fig. 1 Structures of (A) zamamiphidin A (1), its related manzamine alkaloids (2 and 3) and (B) fragments 4–6 of 1. | ||
From a retrosynthetic perspective (Scheme 1), we envisioned that zamamiphidin A would arise from the advanced intermediate 7 through late-stage formation of two 11-membered rings.4,5 Central to the synthesis of the target molecule (1) would be the construction of the pentacyclic and cage-like backbone 7. Structural analysis suggested the diazapentacyclic core of 7 could be simplified as a fully substituted tetrahydrofuran moiety containing two amine functionalities at C3 and C10 (Scheme 1, in blue), which thus could be traced back to the precursor 10. Specifically, disconnecting the C1–N2 bond in 7 revealed ketone 8; and the azabicyclo[3.3.1]nonane unit in 8 could be formed by a Mannich reaction and an intramolecular Dieckmann condensation from bicycle 9. Disassembly of the piperidine ring in 9 through an intramolecular Michael addition led back to the functionalized tetrahydrofuran 10. Based on our previous studies,6 10 could be easily traced back to the chiral malonate 12 bearing a pendant N-tert-butanesulfinyl imidate motif, relying on an intramolecular alkoxide exchange to forge the tetra-hydrofuran ring (11 to 10) and a Davis oxidation to install the C9 hydroxyl group (12 to 11). In turn, 12 could be accessed via a diastereoselective Michael addition of N-tert-butanesulfinyl imidate 13 to enamidomalonate 14.
 | ||
| Scheme 1 Retrosynthetic analysis of zamamiphidin A. | ||
Our synthetic approach started from the preparation of the chiral malonate 12 (Scheme 2). First, condensation of the known primary amine 157 with dimethyl methoxymethylenemalonate (16) in refluxing toluene delivered enamine 17 (92% yield), which was subsequently protected with Boc to give compound 14. The key asymmetric Michael addition of (R)-N-tert-butanesulfinyl imidate 13 to enamidomalonate 14 proceeded smoothly by employing LiHMDS as base,6,8–10 providing adduct 12 as the major diastereomer in 72% yield (d.r. = 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 at C10). We supposed that the addition would preferentially take place through Ts-18 to avoid the significant steric repulsion between the tert-butyl group of 13 and the N-Boc side chain of 14, thus favoring the generation of 12. The configuration of the newly generated C10 stereocenter in 12 could be confirmed later by further transformations (vide infra). As far as we knew, this example represents the first asymmetric conjugate addition of N-tert-butanesulfinyl metalloenamines by using enamidomalonate as the Michael acceptor, which would be useful for the preparation of β-amino acid.11
:1 at C10). We supposed that the addition would preferentially take place through Ts-18 to avoid the significant steric repulsion between the tert-butyl group of 13 and the N-Boc side chain of 14, thus favoring the generation of 12. The configuration of the newly generated C10 stereocenter in 12 could be confirmed later by further transformations (vide infra). As far as we knew, this example represents the first asymmetric conjugate addition of N-tert-butanesulfinyl metalloenamines by using enamidomalonate as the Michael acceptor, which would be useful for the preparation of β-amino acid.11
 | ||
| Scheme 2 Synthesis of the chiral malonate 12. | ||
With malonate 12 available, we next sought to construct the octahydrofuro[3,4-b]pyridine ring system of zamamiphidin A (Scheme 3). To this end, our first task was to introduce a hydroxyl group at C9 in 12. After exploring various oxidative conditions such as Mn(OAc)3/AcOH,12 IBX/DMSO/H2O,13 and O2/I2/NaOAc,14 we found that a Davis protocol15 (Davis' oxaziridine/NaH/THF, −50 °C) was successful to convert 12 to 11 with 82% yield. Formation of the tetrahydrofuran ring from 11 proceeded in the presence of Et3N/toluene at reflux temperature via an intramolecular alkoxide exchange, resulting in 19 (71% yield). Desilylation of 19 with HF afforded the primary alcohol 20 (85% yield). The latter then underwent a two-step synthetic sequence including oxidation of the hydroxyl group to aldehyde and Wittig olefination of the resulting aldehyde with methyl(triphenylphosphoranylidene)acetate (21), providing enoate 10 in 64% overall yield. Upon treating 10 with NaH in the presence of HMPA at −20 °C, an intramolecular Michael addition reaction occurred to enable the coupling of C4 and C4a, leading to the formation of the bicyclic intermediate 22 (72% yield). The structure of 22 was determined through extensive interpretation of the NMR data of its deprotected derivative 23. Unfortunately, we found that the configuration of the newly formed C4a stereocenter was opposite to that in the natural product zamamiphidin A. Inversion of the C4a stereochemistry through generation of the C4–C4a double bond (22 to 24) followed by hydrogenation (24 to 25) was investigated. When compound 22 was treated with LDA and PhSeCl followed by oxidation and spontaneous elimination under the conditions of H2O2/pyridine/CHCl3, the above reaction gave a complex mixture, with only 5–10% yield of the desired product 24 isolated. We attempted to improve this transformation by employing various bases (e.g., LiHMDS, NaHMDS, KHMDS, LiTMP) and different oxidants (e.g., m-CPBA, NaIO4, AcOOH), which all proved to be unsuccessful. These failures impeded further transformations.
 | ||
| Scheme 3 Synthesis of the tricyclic compound 28. | ||
On the other hand, we investigated the construction of the oxazolidine ring starting from the bicyclic intermediate 22 (Scheme 3). Thus, reduction of the imine double bond in 22 with borane dimethyl sulfide afforded sulfinamide 26 in 55% yield. Subjecting 26 to the conditions of HCl/MeOH16 followed by treatment of the resulting primary amine with 2-hydroxypyridine in refluxing toluene17 furnished an unexpected tricyclic compound 28. The structure of 28 was determined unambiguously through X-ray crystallographic analysis, which again confirmed the C4a stereochemistry generated in the aforementioned intramolecular Michael addition reaction (10 to 22). Formation of the undesired 28 was due to the lactamization occurred at C18 rather than at C1 that was initially envisaged to establish the oxazolidine ring. These results indicated that the correct configuration of C4a might be crucial for the subsequent synthesis.
In our revised synthetic approach, a propiolate group was employed to replace the originally used enoate for the Michael addition. We envisioned that hopefully reduction of the double bond generated in the Michael addition of the corresponding propiolate would secure the right configuration at the C4a position. As shown in Scheme 4, the known propargyl alcohol 29 was prepared on a decagram scale from 3-butyl-1-ol over two steps.18 After acylation of 29 and subsequent deprotection of the TBS group with TBAF, the resulting primary alcohol 30 was converted into phthalimide 31 via a Mitsunobu reaction in the presence of phthalimide/PPh3/DIAD (64% overall yield for three steps). Deacylation of 31 followed by silylation of the resulting primary alcohol with TBSCl/Et3N/DMAP provided 32 in 36% yield over two steps. Exposure of 32 to hydrazine hydrate in refluxing methanol resulted in removal of the phthalimide group and delivered a free amine, which was directly condensed with 16 to afford the secondary enamine 33 in 80% overall yield. Further protection of the amino group in 33 with Boc produced intermediate 34. Next, compound 34 underwent a similar three-step transformation to that performed on enamidomalonate 14, involving asymmetric 1,4-conjugate addition with imidate 13, Davis oxidation to install the C9 hydroxyl group (35 to 36), and cyclization with Et3N to assemble the tetrahydrofuran ring, yielding the intermediate 37 smoothly. To reach propiolate 39, the precursor of the key Michael addition for forming C4–C4a bond, further functional group manipulations of 37 were conducted. These included desilyation with HF (37 to 38), Dess–Martin oxidation, Pinnick oxidation, and esterification (38 to 39). Gratifyingly, upon treating 39 with LDA and HPMA in THF at −20 °C, compound 24 was formed with 42% yield through an intramolecular Michael addition and spontaneous migration of the double bond from C4a–C8a to C4–C4a.
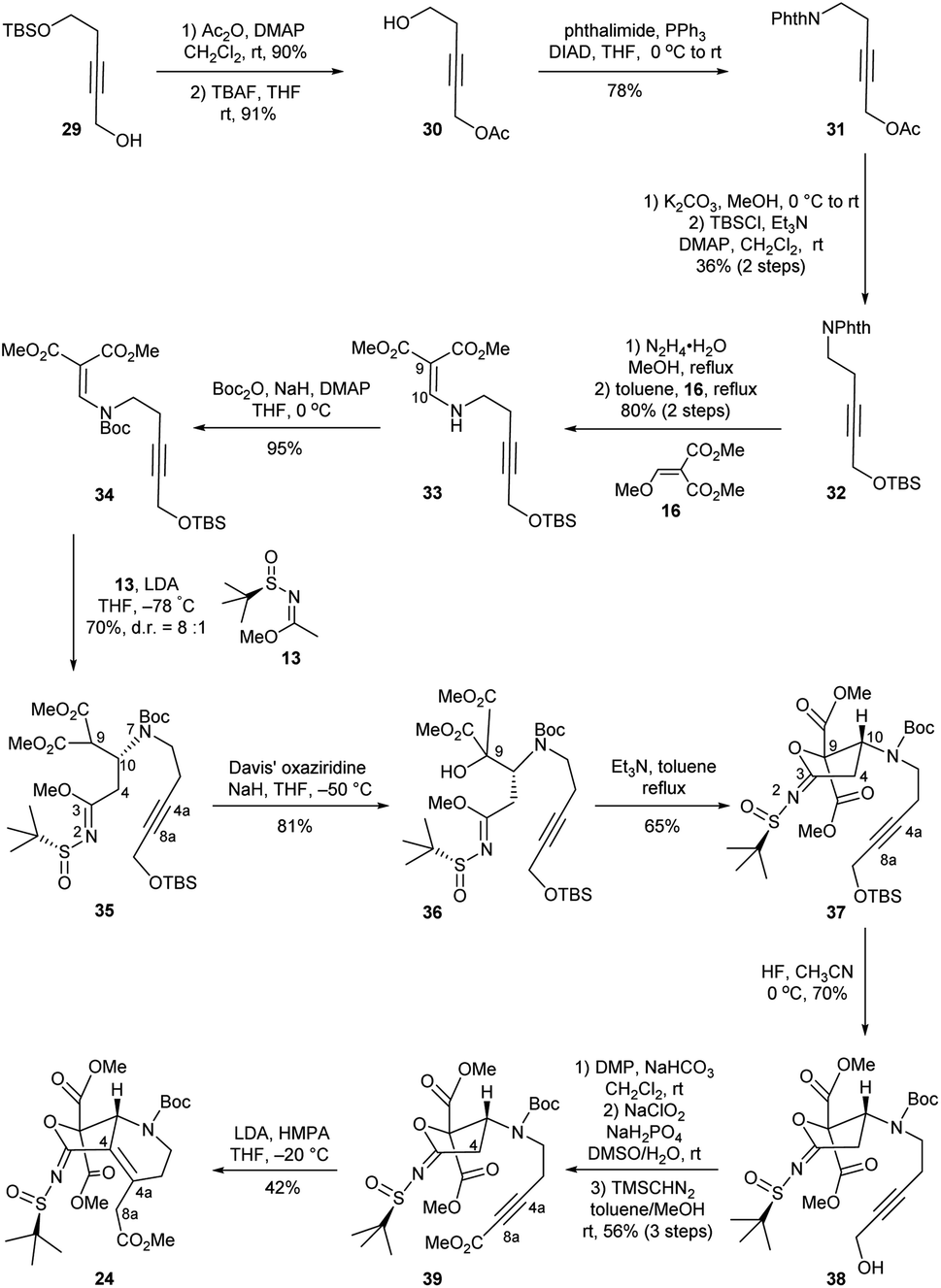 | ||
| Scheme 4 Synthesis of compound 24. | ||
With compound 24 in hand, selective reduction of the C4–C4a double bond was carried out under catalytic hydrogenation conditions (RANEY® Ni/H2) and furnished 25 (70% yield) as a single diastereomer with correct C4 and C4a configurations (Scheme 5). Ensuing reduction of the imine group in 25 using borane dimethyl sulfide delivered the tert-butyl sulfonamide 40 with excellent diastereoselectivity (single isomer, 72% yield). The structures of both 25 and 40 were extensively interpreted by NMR spectroscopy. As a consequence, the configurations at C3, C4, C4a and C10 were found to be consistent with the ones in zamamiphidin A.19 Furthermore, transformations of 25 or 40 into the core of the target zamamiphidin A were investigated. Subjecting 25 to various conditions (NaH, NaOMe, t-BuOK, LDA, LiHMDS, AlCl3, TiCl4, etc.) for a Dieckmann condensation failed to construct the C1–C8a bond and give the desired product 41. Instead, ring-opening of the tetrahydrofuran unit in 25 was observed under some circumstances, leading to the byproduct 42 via a retro-Michael reaction.
 | ||
| Scheme 5 Synthesis of 25 and attempted further transformations. | ||
Meanwhile, attempts to synthesize the tricyclic β-keto ester 43 or lactam 44 from compound 40 were also unsuccessful. Most of these experiments suffered from either no reaction or decomposition of the starting materials.
In conclusion, we have established a synthetic approach to the octahydrofuro[3,4-b]pyridine core of the complex natural product zamamiphidin A. The synthesis features an intermolecular asymmetric Michael addition (compounds 34 to 35) to form the first chiral center at C10 and a Michael addition/hydrogenation sequence (compounds 39 to 25) to secure the correct configurations at C4 and C4a. The highly functionalized bicyclic intermediate (i.e., 40) prepared in the present work contains four contiguous stereocenters and all the requisite heteroatoms at the right positions. While further transformations of related intermediates to zamamiphidin A were unfruitful, efforts to develop a feasible strategy for total synthesis of the target molecule are ongoing in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (21732005 and 21702140).Notes and references
- (a) T. F. Molinski, D. S. Dalisay, S. L. Lievens and J. P. Saludes, Nat. Rev. Drug Discovery, 2009, 8, 69–85 CrossRef CAS PubMed; (b) P. Kiuru, M. V. D'Auria, C. D. Muller, P. Tammela, H. Vuorela and J. Yli-Kauhaluoma, Planta Med., 2014, 80, 1234–1246 CrossRef CAS PubMed; (c) W. H. Gerwick and B. S. Moore, Chem. Biol., 2012, 19, 85–98 CrossRef CAS PubMed.
- (a) R. Montaser and H. Luesch, Future Med. Chem., 2011, 3, 1475–1489 CrossRef CAS PubMed; (b) J. W. Blunt, A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2018, 35, 8–53 RSC.
- T. Kubota, Y. Kamijyo, A. Takahashi-Nakaguchi, J. Fromont, T. Gonoi and J. Kobayashi, Org. Lett., 2013, 15, 610–612 CrossRef CAS PubMed.
- B. Cheng and J. Reyes, Nat. Prod. Rep., 2020 10.1039/C9NP00031C , advance article.
- Y. Wang, L. Y. Leng, G. Y. Dai, F. L. Xue, Z. H. Chen, J. Meng, G. H. Wen, Y. X. Xiao, X. Y. Liu and Y. Qin, Org. Lett., 2018, 20, 6701–6704 CrossRef CAS PubMed.
- H. J. Wang, P. Tang, Q. L. Zhou, D. Zhang, Z. T. Chen, H. X. Huang and Y. Qin, J. Org. Chem., 2015, 80, 2494–2502 CrossRef CAS PubMed.
- A. Padwa, P. Rashatasakhon, A. D. Ozdemir and J. Willis, J. Org. Chem., 2005, 70, 519–528 CrossRef CAS PubMed.
- H. X. Huang, H. J. Wang, L. Tan, S. Q. Wang, P. Tang, H. Song, X. Y. Liu, D. Zhang and Y. Qin, J. Org. Chem., 2016, 81, 10506–10516 CrossRef CAS PubMed.
- H. M. Peltier and J. A. Ellman, J. Org. Chem., 2005, 70, 7342–7345 CrossRef CAS PubMed.
- J. F. Wang, Y. Zhou, L. Zhang, Z. Li, X. J. Chen and H. Liu, Org. Lett., 2013, 15, 1508–1511 CrossRef CAS PubMed.
- M. P. Sibi and Y. Asano, J. Am. Chem. Soc., 2001, 123, 9708–9709 CrossRef CAS PubMed.
- L. Lamarque, A. Méou and P. Brun, Can. J. Chem., 2000, 78, 128–132 CAS.
- A. Duschek and S. F. Kirsch, Chem.–Eur. J., 2009, 15, 10713–10717 CrossRef CAS PubMed.
- C. B. Miao, M. Zhang, Z. Y. Tian, H. T. Xi, X. Q. Sun and H. T. Yang, J. Org. Chem., 2011, 76, 9809–9816 CrossRef CAS PubMed.
- (a) T. Tsubogo, S. Shimizu and S. Kobayashi, Chem.–Asian J., 2013, 8, 872–876 CrossRef CAS PubMed; (b) Z. Zhou, D. D. Dixon, A. Jolit and M. A. Tius, Chem.–Eur. J., 2016, 22, 15929–15936 CrossRef CAS PubMed; (c) S. W. B. Tan, C. L. L. Chai and M. G. Moloney, Org. Biomol. Chem., 2017, 15, 1889–1912 RSC.
- A. Watzke, R. M. Wilson, S. J. O'Malley, R. G. Bergman and J. A. Ellman, Synlett, 2007, 15, 2383–2389 Search PubMed.
- R. M. Williams, J. H. Cao, H. Tsujishima and R. J. Cox, J. Am. Chem. Soc., 2003, 125, 12172–12178 CrossRef CAS PubMed.
- (a) F. Glaus and K.-H. Altmann, Angew. Chem., Int. Ed., 2012, 51, 3405–3409 CrossRef CAS PubMed; (b) B. M. Trost and C. A. Kalnmals, Org. Lett., 2017, 19, 2346–2349 CrossRef CAS PubMed.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for new compounds. CCDC 1974253. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01580f |
| This journal is © The Royal Society of Chemistry 2020 |