
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed highly regioselective mono and double Sonogashira cross-coupling reactions of 5-substituted-1,2,3-triiodobenzene under ambient conditions†
Raed M. Al-Zoubi *a,
Mothana K. Al-Omaria,
Walid K. Al-Jammala and
Michael J. Fergusonb
*a,
Mothana K. Al-Omaria,
Walid K. Al-Jammala and
Michael J. Fergusonb
aDepartment of Chemistry, Jordan University of Science and Technology, P. O. Box 3030, Irbid, 22110, Jordan. E-mail: rmzoubi@just.edu.jo; Fax: +962-2-7201071; Tel: +962-2-7201000-ext. 23651
bDepartment of Chemistry, Gunning-Lemieux Chemistry Centre, University of Alberta, Edmonton, Alberta T6G2G2, Canada
First published on 24th April 2020
Abstract
An efficient synthesis of 2,3-diiodinated diphenylacetylene and iodinated meta-terphenylacetylene derivatives through highly regioselective mono and double Sonogashira cross-coupling reactions of 5-substituted-1,2,3-triiodobenzene is reported. Significantly, the regioselectivity of coupling reactions is exclusively performed at the terminal C–I bonds, the less sterically hindered and the most regioactive positions. The highest isolated yields were achieved from reactions of electron-poor/neutral 1,2,3-triiodoarene and electron-rich arylacetylene derivatives. The use of 2.0 equiv. of arylacetylenes in one-pot fashion afforded the iodinated meta-terphenylacetylenes in excellent site selectivity and in good isolated yields. Different functional groups were found to be suitable under optimized conditions. This report discloses the first method to synthesize hitherto unknown 2,3-diiodinated diphenylacetylenes and iodinated meta-terphenylacetylenes that is facile, highly regioselective, general in scope and produces remarkable building blocks for other chemical transformations.
Introduction
Harnessing the regioselectivity to quickly access highly functionalized molecules by means of site-selective functional group transformation from adaptable building blocks is essential in synthetic chemistry and biology. Inspired by the broad biological activities of 2,3-dihalogenated phenylacetylene derivatives,1–5 only limited synthetic protocols have been reported and none for diiodinated motifs thus far.6–14 For instance, a 2,3-diiodinated phenylacetylene derivative (1, Fig. 1) is reported to have cathepsin K inhibitory action.15 Additionally, 2,3-difluorinated diphenylacetylene derivative 2, is reported as allosteric modulator for metabotropic glutamate receptor subtype 5 (mGluR5) for treating neural and psychiatric disorders associated with glutamate dysfunction.3 Furthermore, another 2,3-difluorinated diphenylacetylene derivative (3, Fig. 1) are reported to inhibit the proliferation of LS174T colon cancer by inhibition of c-myc and induction of the cyclin-dependent kinase inhibitor.2,16 Lastly, 2,3-dichlorinated phenylacetylene derivative (4, Fig. 1), is found to be useful as selectively active antagonists of N-methyl-D-aspartate (NMDA) receptor subtypes for treating conditions such as central nervous system trauma, hypoglycemia, anxiety, stroke, convulsions, cerebral ischemia, chronic pain or neurodegenerative disorders as Alzheimer's Disease, Parkinsonism and Huntington's disease.5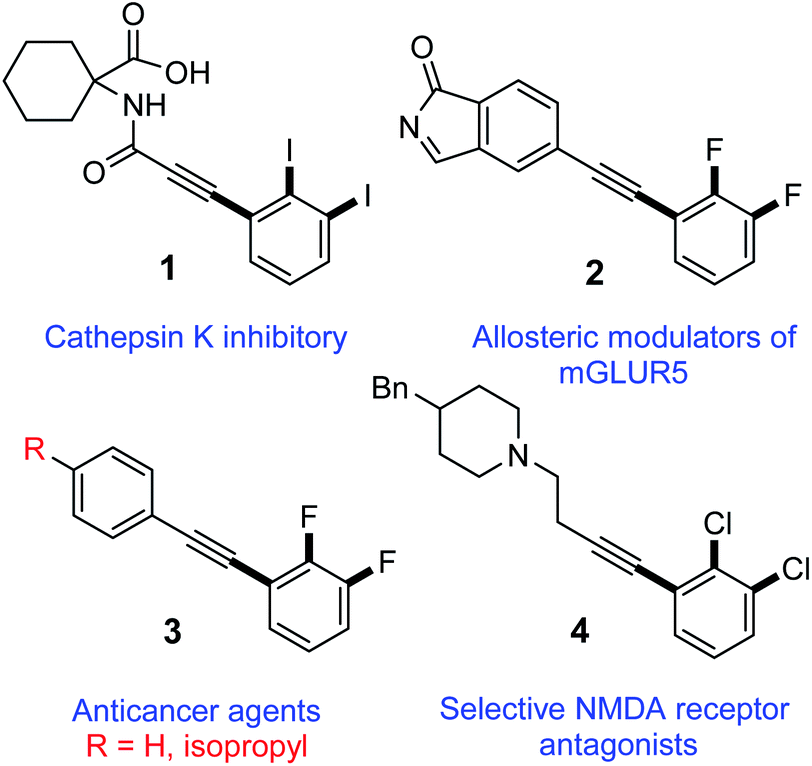 | ||
| Fig. 1 Some biologically active 2,3-dihalogenated phenylacetylene compounds in medicine. | ||
The status quo of these 2,3-dihalogenated phenylacetylenes and other derivatives17,18 with their remarkable applications in medicine encouraged us for developing a new method to access 2,3-diiodinated phenylacetylene molecules. Although few reports for the synthesis of 2,3-dihalogenated phenylacetylenes were published, no protocol for the synthesis of 2,3-diiodinated diphenylacetylene motifs is reported to date. Herein, we report the first synthetic method to access hitherto unknown 2,3-diiodinated diphenylacetylene derivatives by regioselective Sonogashira cross-coupling of 5-substituted-1,2,3-triiodobenzene that is efficient, scalable and affords moderate to good yields.
A broad functional groups were examined under the optimized reaction conditions and found tolerant providing the desired terminal coupling products in highly regioselective manner that are indeed difficult to make by alternative means.
Results and discussion
Considering the remarkable bioactivities of many 2,3-dihalogenated phenylacetylene derivatives in literature and also to our finding in regioselective Suzuki–Miyaura cross-coupling of 5-substituted-1,2,3-triiodobenzene,19 we felt impelled to examine the Sonogashira cross-couplings on 5-substituted-1,2,3-triiodobenzenes aiming forward to diiodinated phenylacetylene derivatives. 5-Substituted-1,2,3-triiodobenzene starting materials were prepared according to our previous procedures from anilines or benzoic acids.20–22Several regioselective Sonogashira of polyhalogenated arene systems are found in literature.23–32 For instance, Langer and co-workers published the synthesis of quinolino[3′,4′:4,5]pyrrolo[1,2-f]phenanthridines via regioselective Sonogashira of 4-chloro-3-iodo-2-methylquinoline with different aryl acetylenes in excellent regioselectivity towards the iodo substituent in moderate to good yields.23 Usuki and co-workers reported the total synthesis of desmosine, a biomarker and elastin cross-linker, in 13 steps and 11% overall yield through sequential regioselective Sonogashira cross-coupling reactions of 3,4,5-trihalopyridine.25,26 The Sonogashira cross-coupling reaction of 5-substituted-1,2,3-triiodobenzene bearing two regiochemically unsymmetrical C–I positions provides at most two possible regioisomeric coupling products, the internal and terminal diiodinated diphenylacetylenes (Scheme 1). Consequently, the satisfactory use of 1.0 mol equivalent of aryl acetylene is adequate to couple one of the three iodo substituents providing the desired diiodinated diphenylacetylene products.
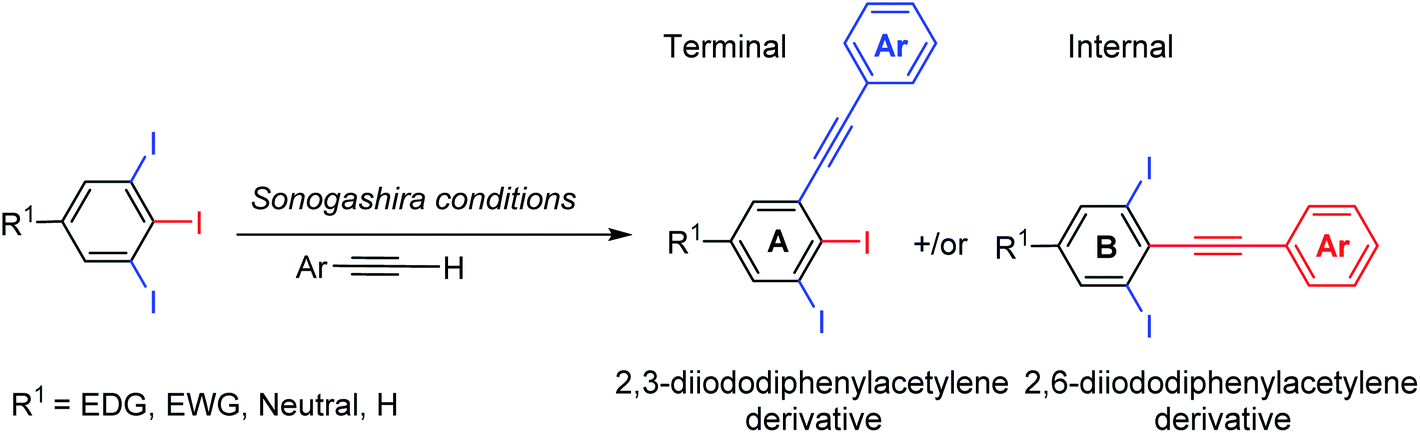 | ||
| Scheme 1 Possible diiodinated diphenylacetylene regioisomers from Sonogashira cross-coupling of 5-substituted-1,2,3-triiodoarenes. | ||
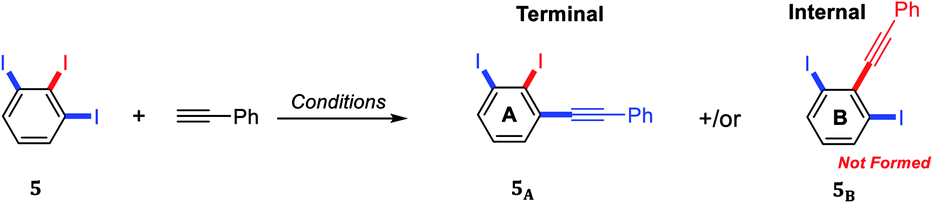
A thorough optimization using 1,2,3-triiodobenzene 5 and phenylacetylene as model substrates to explore this hypothesis as shown in Table 1. Happily and as expected, the reaction was found to be highly regioselective to the terminal C–I bond, the more accessible and less hindered position, providing only the 2,3-diiododiphenylacetylene 5A. The order of addition proved to be essential to improve the yield and to minimize the Glaser Homo-coupling side reaction. Temperature and solvent were also found to be essential parameters in our optimization. The reaction of 1,2,3-triiodobenzene 5 with 1.2 equiv. of phenylacetylene in DMF at 100 °C provided a non clean reaction forming other side reactions and Glaser homo-coupling product (Table 1, entries 1 and 2). It is worth mentioning that due to the low polarity of the desired product, a difficult purification was observed in the presence of Glaser homo-coupling product. At elevated temperature such as 100 °C, it is not surprising that significant amount of multiple coupling reactions with other C–I bonds and other side reactions were possibly formed. Lowering the reaction temperature to 50 °C and 25 °C were found to be unsuccessful (Table 1, entry 3 and 4). Changing the base to 4-(dimethylamino)pyridine (DMAP) shutdown the reaction (Table 1, entry 5). Changing the solvent to anhydrous toluene provided the wanted product 5A in 15% yield (Table 1, entry 6). Elongate reaction time to 24 hours at same temperature provided 29% (Table 1, entry 7). Increasing the Pd(PPh3)4 loading to 20 mol% gave 21% yield of the desired product with ∼5% of the meta bis-coupled product (Table 1, entry 8). While increasing the CuI loading to 20 mol% was found to be beneficial provided 37% yield of the desired product 5A (Table 1, entry 9). The highest isolated yield for this transformation was achieved by the use of 10 mol% of Pd(PPh3)4, 20 mol% of CuI as a co-catalysis, 7.0 equiv. of Cs2CO3 as a base in anhydrous toluene at 25 °C for 24 h (Table 1, entry 10). Other conditions were found to be unsuccessful to further enhance the reaction (Table 1, entries 11–13). The coupling reaction over and above performed nicely on large scale (Table 1, entry 14). The scope of the regioselective Sonogashira reaction was then studied under the optimized conditions. Therefore, a variety of 5-substituted-1,2,3-triiodobenzene starting materials were examined under the optimized conditions providing the terminal coupling products in excellent site-selectivity (Scheme 2).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst, co-catalyst (mol%) | Alkyne (equiv.) | Base (equiv.) | Solvent | T (°C) | Time (h) | % yield of 5Ab (%) |
| a Conditions: all recations were carried out using 0.65 mmol (1.0 equiv., 0.08 M) of 1,2,3-triiodobenzene (5) in 8.0 mL anhyd. solvent.b Isolated yield.c NS: not separable mixture of products.d 5% of bis-terminal coupling product was isolated.e 1.0 gram scale (2.19 mmol). | |||||||
| 1 | Pd(PPh3)4 (10%), CuI (10%) | 1.2 | Cs2CO3 (4) | DMF | 100 | 12 | NSc |
| 2 | Pd(PPh3)4 (10%), CuI (10%) | 1.0 | Cs2CO3 (4) | DMF | 100 | 12 | NSc |
| 3 | Pd(PPh3)4 (10%), CuI (10%) | 1.0 | Cs2CO3 (4) | DMF | 50 | 12 | NSc |
| 4 | Pd(PPh3)4 (10%), CuI (10%) | 1.0 | Cs2CO3 (4) | DMF | 25 | 12 | NSc |
| 5 | Pd(PPh3)4 (10%), CuI (10%) | 1.0 | DMAP (4) | DMF | 25 | 12 | 0% |
| 6 | Pd(PPh3)4 (10%), CuI (10%) | 1.0 | Cs2CO3 (4) | Toluene | 25 | 12 | 15% |
| 7 | Pd(PPh3)4 (10%), CuI (10%) | 1.0 | Cs2CO3 (4) | Toluene | 25 | 24 | 29% |
| 8 | Pd(PPh3)4 (20%), CuI (10%) | 1.0 | Cs2CO3 (4) | Toluene | 25 | 24 | 21%d |
| 9 | Pd(PPh3)4 (10%), CuI (20%) | 1.0 | Cs2CO3 (4) | Toluene | 25 | 24 | 37% |
| 10 | Pd(PPh3)4 (10%), CuI (20%) | 1.0 | Cs2CO3 (7) | Toluene | 25 | 24 | 60% |
| 11 | Pd(PPh3)4 (10%), CuI (20%) | 1.0 | Cs2CO3 (10) | Toluene | 25 | 24 | 58% |
| 12 | Pd(PPh3)4 (5%), CuI (20%) | 1.0 | Cs2CO3 (7) | Toluene | 25 | 24 | 42% |
| 13 | Pd(PPh3)4 (10%), CuI (20%) | 1.0 | K2CO3 (7) | Toluene | 25 | 24 | 49% |
| 14e | Pd(PPh3)4 (10%), CuI (20%) | 1.0 | Cs2CO3 (7) | Toluene | 25 | 24 | 53% |
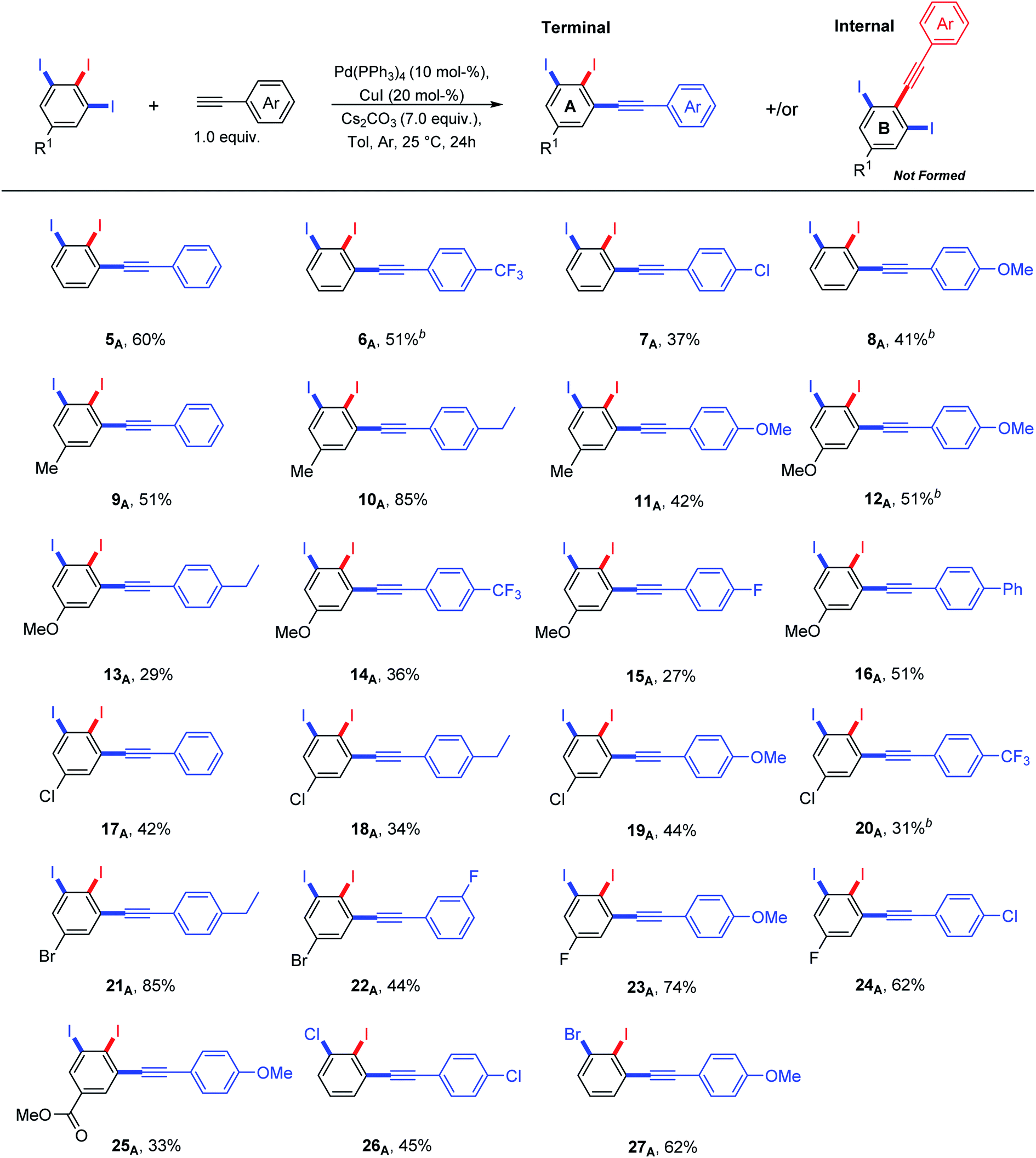 | ||
| Scheme 2 Terminal vs. internal diiodinated diphenylacetylene derivatives via regioselective Sonogashira cross-coupling of 5-substituted-1,2,3-triiodobenzenes and arylcetylenes. aYields are given for isolated compounds (reaction scale: 0.65 mmol). b5–10% of meta bis-terminal coupling product was isolated. | ||
It was found that the nature of R1 substituent has a big impact on the reactivity of Sonogashira cross-coupling reactions but not on regioselectivity. A combination between electron-poor/neutral 1,2,3-triiodoarenes and electron-rich arylacetylenes provided the terminal coupling products in high isolated yields (Scheme 2: 10A, 21A, 23A and 24A). In contrast, electron-rich 1,2,3-triiodoarenes afforded moderate yields (Scheme 2: 12A–16A). The formation of the internal coupling regioisomer was not detected in all examples (Scheme 2: B). Gratefully, the coupling reaction with bromo or chloro substituents was not observed (Scheme 2: 17A–22A and 26A–27A) and it can tolerate a wide range of functional groups. 5–10% of the meta bis-terminal coupling products were isolated in some cases (Scheme 2: 6A, 8A, 12A and 20A).
The structure of 2,3-diiodinated diarylacetylene compounds are further confirmed by X-ray diffraction methods for three coupling products, 1,2-diiodo-3-((4-(trifluoromethyl)phenyl)ethynyl)benzene 6A, 5-chloro-1,2-diiodo-3-((4-(trifluoromethyl)phenyl)ethynyl) benzene 20A and 5-fluoro-1,2-diiodo-3-((4-methoxyphenyl)ethynyl) benzene 23A as shown in Fig. 2.33
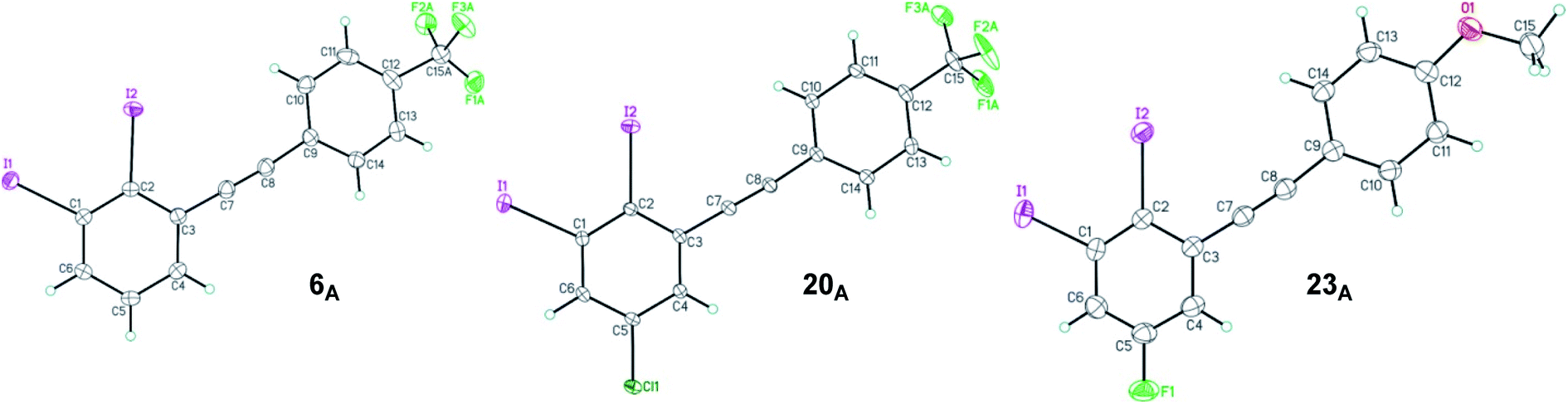 | ||
| Fig. 2 ORTEP view of 1,2-diiodo-3-((4-(trifluoromethyl)phenyl)ethynyl)benzene 6A, 5-chloro-1,2-diiodo-3-((4-(trifluoromethyl)phenyl)ethynyl)benzene 20A and 5-fluoro-1,2-diiodo-3-((4-mehthoxyphenyl)ethynyl)benzene 23A. Thermal Gaussian ellipsoids at 30% probability level. | ||
Although various approaches for regioselective Sonogashira cross-coupling have been previously reported,34–46 a plausible catalytic cycle for regioselective Sonogashira cross-coupling of 5-substituted-1,2,3-triiodobenzene is proposed in Scheme 3. Oxidative addition by Pd0 at the terminal C–I position, the less sterically hindered and more accessible position forming PdII intermediate A. The transmetallation of PdII intermediate A with copper(I) acetylide B, which was in situ generated with base and CuII, to afford PdII intermediate C. Reductive elimination afforded the terminal coupled product D and regenerated the catalyst for another catalytic cycle. We then turn our attentions to examine the reactivity order of the other iodo groups under the optimized conditions. Therefore, one-pot double Sonogashira cross-coupling reactions of 5-substituted-1,2,3-triiodobenzene with 2.0 equiv. of arylacetylenes were performed. The excellent regioselectivity of the first coupling, vide supra, promoted the second coupling reaction to occur at the other terminal position providing exclusively the iodinated meta-terphenyl products in moderate to good yields with excellent regioselectivity (Scheme 4: 28A–35A). We did not observe the ortho bis-coupled products in all reactions. It is believed that the reaction may proceed via a reversible oxidative addition step.
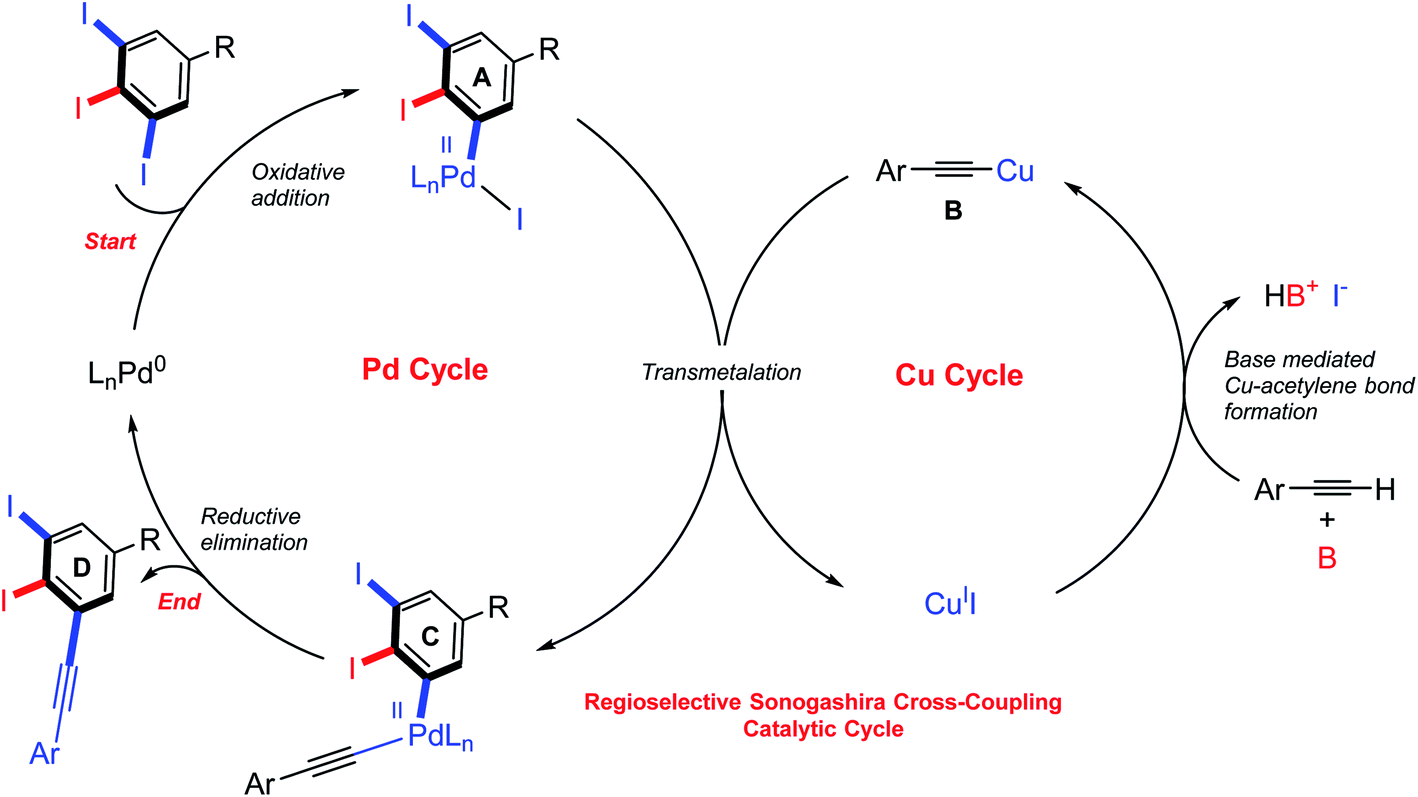 | ||
| Scheme 3 Proposed catalytic cycle for regioselective Sonogashira cross-coupling reaction of 5-substituted-1,2,3-triiodobenzene. | ||
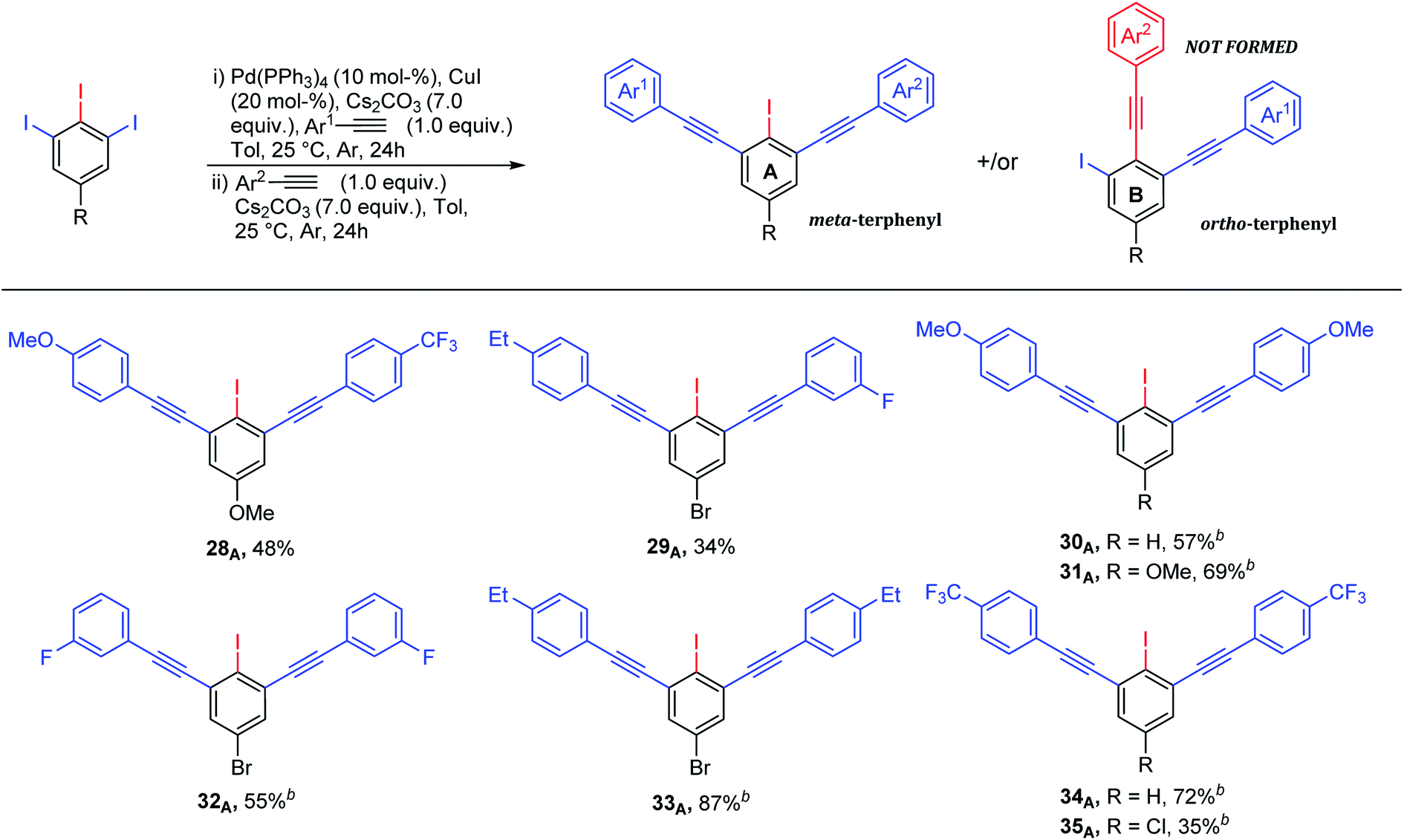 | ||
| Scheme 4 Iodinated terphenyls via one-pot double Sonogashira cross-coupling reaction of 1,2,3-triiodoarenes and arylacetylenes. aYields are given for isolated yields (reaction scale 0.66 mmol). b2.0 equiv. of arylacetylene was used. | ||
The structure of bis-coupling products was further supported by X-ray crystallography of one of the derivative, 4,4′-((5-chloro-2-iodo-1,3-phenylene)bis(ethyne-2,1-diyl))bis((trifluoromethyl)benzene) 35A as shown in Fig. 3.33
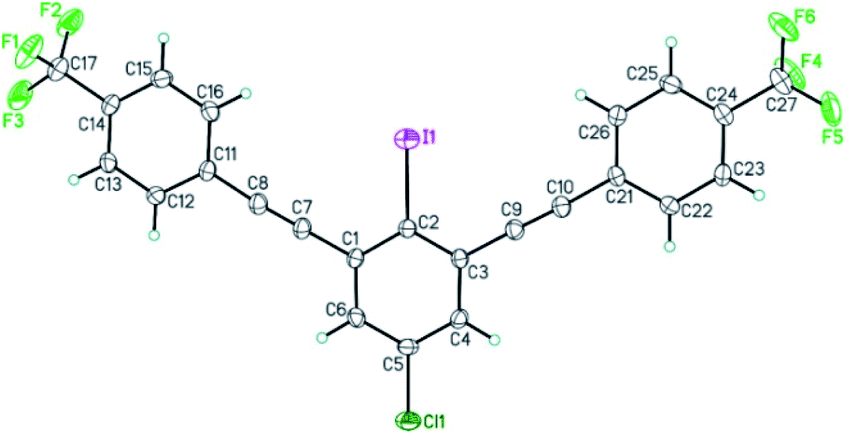 | ||
| Fig. 3 ORTEP view of 4,4′-((5-chloro-2-iodo-1,3-phenylene)bis(ethyne-2,1-diyl))bis((trifluoromethyl)benzene) 35A. Thermal Gaussian ellipsoids at 30% probability level. | ||
The quickly access of highly functionalized compounds from available starting materials is crucial in academia and industry. The 1,2,3-trisubstituted benzenes are demanded and challenging derivatives in literature. Therefore, 2,3-diiododiphenylacetylene 5A was used to quickly access 1,2,3-trisubstitutedbenzene derivatives using Suzuki–Miyaura cross-coupling reactions (Scheme 5).
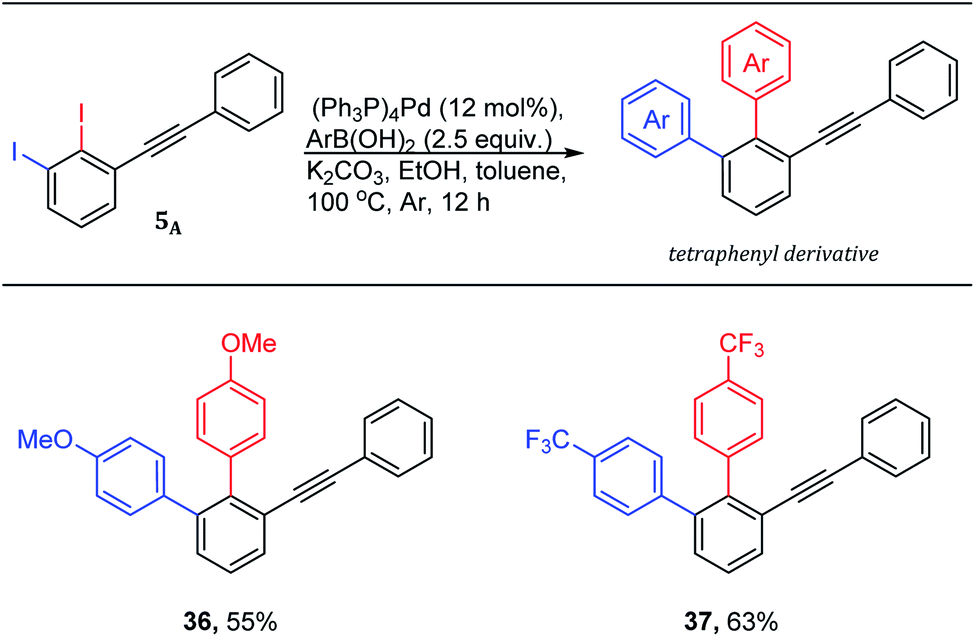 | ||
| Scheme 5 Tetraphenyls via one-pot double Suzuki–Miyaura cross-coupling reaction of 1,2-diiodo-3-(phenylethynyl)benzene 5A. aYields are given for isolated yields (reaction scale 0.66 mmol). | ||
The use of 2.0 mol equiv. of arylboronic acid under the optimized conditions,19 provided the trisubstituted derivatives (Scheme 5: 36–37) through one-pot double Suzuki–Miyaura cross-coupling in good isolated yields. It is worth noting that the use of 1.0 mol equiv. of arylboronic acid under the same conditions is found to be inefficient providing a non separable mixture of ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of both coupling products.
:1 of both coupling products.
Conclusion
In summary, we reported the first synthesis of hitherto unknown 2,3-diiodinated diphenylacetylene and iodinated meta terphenylacetylene derivatives via highly regioselective mono and double Sonogashira cross-coupling reaction from 5-substituted-1,2,3-triiodobenzene. The desired products were isolated in moderate to good yields (17–85%) and the reaction tolerated a broad range of functional groups. No coupling reaction with bromo or chloro groups was observed. The regioselectivity of coupling reactions exclusively performed at the terminal C–I bonds, the less sterically hindered and the most regioactive positions. The highest isolated yields were achieved from reactions of electron-poor/neutral 1,2,3-triiodoarene and electron-rich arylacetylene derivatives. The structure of the products was further supported by X-ray diffraction methods. Having other iodo substituents on these products, further chemical elaborations could easily be explored.Experimental
General
All commercial reagents and chromatography solvents were used as obtained unless otherwise stated. Ethanol, toluene, ethyl acetate, hexanes, anhydrous sodium sulfate (Na2SO4, BDH), CuI (Sigma-Aldrich), Pd(PPh3)4 (Sigma-Aldrich) were used as received. Anhydrous solvents were distilled over appropriate drying agents prior to use. Analytical thin layer chromatography (TLC) was performed on Merck silica gel 60 F254. Merck silica gel 60 (0.063–0.2 mm) was used for column chromatography. Visualization of TLC was accomplished with UV light (254 nm). NMR spectra were recorded on a Bruker-Avance 400 MHz spectrometer. The residual solvent protons (1H) or the solvent carbon (13C) were used as internal standards. 1H-NMR data are presented as follows: chemical shift in ppm (δ) downfield from trimethylsilane (multiplicity, integration, coupling constant). The following abbreviations are used in reporting NMR data: s, singlet; bs, broad singlet; d, doublet; t, triplet; q, quartet; dq, doublet of quartets; dd, doublet of doublets; m, mutiplet. High resolution mass spectra were recorded using Chemical Ionization (CI) and electrospray ionization (ESI) techniques.General procedure for Sonogashira cross-coupling reactions of 5-substituted-1,2,3-triiodobenzenes
A flame-dried Schlenk flask was charged with 5-substituted-1,2,3-triiodobenzene (0.65 mmol, 0.08 M, 1.0 equiv.), aryl acetylene (1.0 equiv.), Cs2CO3 (7.0 equiv.) in 8.0 mL dry toluene. The mixture was stirred under argon at room temperature for 20 min. Tetrakis(triphenylphosphine)palladium (0) (10 mol%) and copper iodide (20 mol%) were added, capped with septum, carefully degassed with argon, and the reaction flask was wrapped with aluminum foil and stirred at room temperature for 24 h. The reaction mixture was then diluted with EtOAc and filtered over Celite 545®. Distilled water (100 mL) was added and extracted with EtOAc (2 × 50 mL). The organic layers were combined, washed with brine, dried with anhyd. Na2SO4, filtered and evaporated under reduced pressure. The crude product was purified by flash chromatography (100% hexane) to yield the pure desired product.Synthesis of 1,2-diiodo-3-(phenylethynyl)benzene (5A)
The title compound was synthesized using the general procedure and isolated in 60% yield as colorless oil after flash chromatography. IR (cast film, cm−1) 3087, 3046, 2201, 1599, 1523, 912, 837, 749, 624. δH (400 MHz, CDCl3) δ: 7.81 (d, 1H, J = 7.9 Hz), 7.50–7.65 (m, 2H), 7.47 (d, 1H, J = 7.6 Hz), 7.30–7.40 (m, 3H), 7.03 (dd, 1H, J = 7.8 Hz). δC (100 MHz, CDCl3) δ: 138.9, 131.9, 131.8, 131.3, 129.2, 129.0, 128.6, 122.8, 114.7, 108.9, 93.5, 93.1. HRMS (EI) m/z for C14H8I2 [M]+: calcd 429.8715; found, 429.8709.Synthesis of 1,2-diiodo-3-((4-(trifluoromethyl)phenyl)ethynyl) benzene (6A)
The title compound was synthesized using the general procedure and isolated in 51% yield as white solid after flash chromatography. IR (cast film, cm−1) 3109, 3046, 2204, 1588, 1541, 1042, 943, 812, 738, 661. δH (400 MHz, CDCl3) δ: 7.85 (d, 1H, J = 7.9 Hz), 7.68 (d, 2H, J = 8.0 Hz), 7.62 (d, 2H, J = 8.2 Hz), 7.48 (d, 1H, J = 7.6), 7.06 (dd, 1H, J1 = 7.8 Hz, J2 = 7.7 Hz). δC (100 MHz, CDCl3) δ: 139.5, 132.0, 131.6, 131.2, 130.7 (q, JC–F = 33 Hz), 129.3, 126.6, 125.5 (q, JC–F = 4 Hz), 124.0 (q, JC–F = 271 Hz), 114.8, 109.1, 95.6, 91.4. Mp: 92–94 °C. HRMS (EI) m/z for C15H7F3I2 [M]+: calcd 497.8589; found, 497.8576.Synthesis of 1-((4-chlorophenyl)ethynyl)-2,3-diiodobenzene (7A)
The title compound was synthesized using the general procedure and isolated in 37% yield as white solid after flash chromatography. IR (cast film, cm−1) 3086, 3012, 2203, 914, 824, 748, 638. δH (400 MHz, CDCl3) δ: 7.81 (d, 1H, J = 7.8 Hz), 7.40–7.60 (m, 3H), 7.32 (d, 2H, J = 8.4 Hz), 7.03 (dd, 1H, J = 7.8 Hz, J = 7.7 Hz). δC (100 MHz, CDCl3) δ: 139.2, 135.2, 133.0, 131.6, 131.4, 129.2, 129.0, 121.2, 114.7, 109.0, 94.4, 91.9. Mp: 101–103 °C. HRMS (EI) m/z for C14H7ClI2 [M]+: calcd 463.8326; found, 463.8317.Synthesis of 1,2-diiodo-3-((4-methoxyphenyl)ethynyl)benzene (8A)
The title compound was synthesized using the general procedure and isolated in 41% yield as white solid after flash chromatography. IR (cast film, cm−1) 3087, 3024, 2215, 1606, 1546, 1324, 1157, 973, 843, 642. δH (400 MHz, CDCl3) δ: 7.78 (d, 1H, J = 7.8 Hz), 7.51 (d, 2H, J = 8.7 Hz), 7.44 (d, 1H, J = 7.6 Hz), 7.02 (dd, 1H, J1 = 7.7 Hz,, J2 = 7.8 Hz), 6.89 (d, 2H, J = 8.7 Hz). δC (100 MHz, CDCl3) δ: 160.3, 138.6, 133.3, 132.3, 131.1, 129.2, 114.9, 114.6, 114.3, 108.9, 93.4, 92.5, 55.5. Mp: 82–84 °C. HRMS (EI) m/z for C15H10I2O [M]+: calcd 459.8821; found, 459.8814.Synthesis of 1,2-diiodo-5-methyl-3-(phenylethynyl)benzene (9A)
The title compound was synthesized using the general procedure and isolated in 51% yield as colorless oil after flash chromatography. IR (cast film, cm−1) 3086, 3015, 2209, 1604, 1573, 784, 652, 507. δH (400 MHz, CDCl3) δ: 7.67 (d, 1H, J = 1.1 Hz), 7.56–7.59 (m, 2H), 7.36–7.37 (m, 3H), 7.30 (d, 1H, J = 1.1 Hz), 2.24 (s, 3H). δC (100 MHz, CDCl3) δ: 139.9, 139.5, 132.4, 131.8, 131.3, 129.0, 128.6, 122.8, 110.5, 108.8, 93.5, 92.7, 20.4. HRMS (EI) m/z for C15H10I2 [M]+: calcd 443.8872; found, 443.8867.Synthesis of 1-((4-ethylphenyl)ethynyl)-2,3-diiodo-5-methyl benzene (10A)
The title compound was synthesized using the general procedure and isolated in 85% yield as colorless oil after flash chromatography. IR (cast film, cm−1) 3104, 3086, 3042, 2198, 1594, 1546, 964, 862, 779, 634. δH (400 MHz, CDCl3) δ: 7.65 (s, 1H), 7.49 (d, 2H, J = 8.0 Hz), 7.29 (d, 1H, J = 1.0 Hz), 7.19 (d, 2H, J = 8.0 Hz), 2.64–2.70 (m, 2H), 2.23 (s, 3H), 1.23–1.27 (m, 3H). δC (100 MHz, CDCl3) δ: 145.5, 139.7, 139.5, 132.7, 132.3, 131.8, 131.5, 128.1, 120.0, 110.5, 108.7, 93.0, 29.1, 20.4, 15.5. HRMS (EI) m/z for C17H14I2 [M]+: calcd 471.9185; found, 471.9182.Synthesis of 1,2-diiodo-3-((4-methoxyphenyl)ethynyl)-5-methyl benzene (11A)
The title compound was synthesized using the general procedure and isolated in 42% yield as white solid after flash chromatography. IR (cast film, cm−1) 3107, 3056, 2205, 1614, 1573, 1168, 1021, 983, 673. δH (400 MHz, CDCl3) δ: 7.64 (s, 1H), 7.51 (d, 2H, J = 8.7 Hz), 7.27 (s, 1H), 6.89 (d, 2H, J = 8.7 Hz), 3.83 (s, 3H), 2.23 (s, 3H). δC (100 MHz, CDCl3) δ: 160.2, 139.5, 139.4, 133.3, 132.1, 131.6, 114.9, 114.2, 110.4, 108.7, 92.9, 92.5, 55.5, 20.4. Mp: 91–93 °C HRMS (EI) m/z for C16H12I2O [M]+: calcd 473.8978; found, 473.8974.Synthesis of 1,2-diiodo-5-methoxy-3-((4-methoxyphenyl)ethynyl)benzene (12A)
The title compound was synthesized using the general procedure and isolated in 51% yield as white solid after flash chromatography (5% EtOAc/hexane) using the general procedure. IR (cast film, cm−1) 3042, 3012, 2195, 1594, 1548, 1209, 1168, 1023, 918, 652. δH (400 MHz, CDCl3) δ: 7.52 (d, 2H, J = 8.6 Hz), 7.39 (d, 1H, J = 2.8 Hz), 7.02 (d, 1H, J = 2.7 Hz), 6.89 (d, 2H, J = 8.6 Hz), 3.83 (s, 3H), 3.78 (s, 3H). δC (100 MHz, CDCl3) δ: 160.3, 159.4, 133.3, 131.9, 125.6, 117.1, 114.7, 114.3, 108.8, 103.6, 93.1, 92.5, 55.8, 55.5. Mp: 83–85 °C. HRMS (EI) m/z for C16H12I2O2 [M]+: calcd exact 489.8927; found 489.8921.Synthesis of 1-((4-ethylphenyl)ethynyl)-2,3-diiodo-5-methoxy benzene (13A)
The title compound was synthesized using the general procedure and isolated in 29% yield as white solid after flash chromatography. IR (cast film, cm−1) 3059, 3024, 2212, 1584, 1542, 1145, 1048, 956, 768. δH (400 MHz, CDCl3) δ: 7.50 (d, 2H, J = 8.0 Hz), 7.40 (d, 1H, J = 2.8), 7.20 (d, 2H, J = 7.8), 7.04 (d, 1H, J = 2.8), 3.78 (s, 3H), 2.6–2.7 (m, 2H), 1.24 (t, 3H, J = 7.6). δC (100 MHz, CDCl3) δ: 159.4, 145.7, 131.8, 128.2, 125.8, 119.8, 117.3, 108.8, 103.7, 93.2, 93.0, 55.8, 29.1, 15.5, (missing one peak due to overlapping). Mp: 96–98 °C. HRMS (EI) m/z for C17H14I2O [M]+: calcd 487.9134; found, 487.9121.Synthesis of 1,2-diiodo-5-methoxy-3-((4-(trifluoromethyl)phenyl)ethynyl)benzene (14A)
The title compound was synthesized using the general procedure and isolated in 36% yield as white solid after flash chromatography. IR (cast film, cm−1) 3102, 3075, 3005, 2209, 1597, 1578, 1310, 1125, 993, 867, 631, 523. δH (400 MHz, CDCl3) δ: 7.68 (d, 2H, J = 8.2 Hz), 7.62 (d, 2H, J = 8.4 Hz), 7.44 (d, 1H, J = 2.9 Hz), 7.06 (d, 1H, J = 2.8 Hz), 3.8 (s, 3H). δC (100 MHz, CDCl3) δ: 159.5, 132.1, 130.9, 130.8 (q, JC–F = 30 Hz), 126.5, 126.4, 125.5 (q, JC–F = 4 Hz), 124.0 (q, JC–F = 270 Hz), 117.7, 109.1, 103.7, 95.5, 91.1, 55.9. Mp: 92–94 °C. HRMS (EI) m/z for C16H9F3I2O [M]+: calcd 527.8695; found, 527.8682.Synthesis of 1-((4-fluorophenyl)ethynyl)-2,3-diiodo-5-methoxybenzene (15A)
The title compound was synthesized using the general procedure and isolated in 27% yield as white solid after flash chromatography. IR (cast film, cm−1) 3104, 3078, 2204, 1608, 1599, 1359, 1146, 984, 873, 749, 653. δH (400 MHz, CDCl3) δ: 7.54–7.58 (m, 2H), 7.41 (d, 1H, J = 2.9 Hz), 7.07 (t, 2H, J = 8.7 Hz), 7.03 (d, 1H, J = 2.8 Hz), 3.78 (s, 3H). δC (100 MHz, CDCl3) δ: 163.0 (d, JC–F = 149 Hz), 159.4, 133.8, 133.8 (d, JC–F = 9 Hz), 125.9, 118.8 (d, JC–F = 4 Hz), 117.4, 115.9 (d, JC–F = 21 Hz), 108.9, 103.6, 93.1, 91.7, 55.8. Mp: 70–72 °C. HRMS (EI) m/z for C15H9FI2O [M]+: calcd 477.8727; found, 477.8713.Synthesis of 4-((2,3-diiodo-5-methoxyphenyl)ethynyl)-1,1′-biphenyl (16A)
The title compound was synthesized using the general procedure and isolated in 51% yield white solid after flash chromatography. IR (cast film, cm−1) 3145, 3048, 2213, 1609, 1588, 1189, 1077, 961, 632. δH (400 MHz, CDCl3) δ: 7.55–7.70 (m, 5H), 7.47 (dd, 2H, J1 = 7.3 Hz, J2 = 7.8 Hz), 7.42 (d, 1H, J = 2.8 Hz), 7.37 (dd, 2H, J1 = 7.4 Hz, J2 = 7.2 Hz), 7.07 (d, 1H, J = 2.8),3.80 (s, 3H). δC (100 MHz, CDCl3) δ: 159.5, 141.8, 140.4, 132.3, 131.7, 129.1, 127.9, 127.3, 127.2, 125.9, 121.5, 117.4, 108.9, 103.8, 94.1, 92.8, 55.9. Mp: 85–87 °C. HRMS (EI) m/z for C21H14I2O [M]+: calcd 535.9134; found, 535.9119.Synthesis of 5-chloro-1,2-diiodo-3-(phenylethynyl)benzene (17A)
The title compound was synthesized using the general procedure and isolated in 42% yield as white solid after flash chromatography. IR (cast film, cm−1) 3104, 3086, 2207, 1602, 1071, 943, 827, 634. δH (400 MHz, CDCl3) δ: 7.79 (d, 1H, J = 2.4 Hz), 7.57–7.59 (m, 2H), 7.46 (d, 1H, J = 2.3 Hz), 7.38–7.39 (m, 3H). δC (100 MHz, CDCl3) δ: 138.3, 134.6, 132.6, 131.9, 131.2, 129.4, 128.6, 122.3, 112.5, 109.2, 94.3, 92.5. Mp: 90–92 °C. HRMS (EI) m/z for C14H7ClI2 [M]+: calcd 463.8326; found, 463.8323.Synthesis of 5-chloro-1-((4-ethylphenyl)ethynyl)-2,3-diiodo benzene (18A)
The title compound was synthesized using the general procedure and isolated in 34% yield as white solid after flash chromatography. IR (cast film, cm−1): 3104, 3089, 3018, 2212, 1579, 1542, 928, 813, 742, 642. δH (400 MHz, CDCl3) δ: 7.78 (d, 1H, J = 2.2 Hz), 7.49 (d, 2H, J = 8.0 Hz), 7.45 (d, 1H, J = 2.2 Hz), 7.21 (d, 2H, J = 7.9 Hz), 2.67 (q, 2H, J = 7.5 Hz), 1.25 (t, 3H, J = 7.6 Hz). δC (100 MHz, CDCl3) δ: 146.0, 138.0, 134.5, 132.8, 131.9, 131.0, 128.2, 119.4, 112.5, 109.2, 94.6, 92.0, 29.1, 15.5. Mp: 87–89 °C. HRMS (EI) m/z for C16H11ClI2 [M]+: calcd 491.8639; found, 491.8632.Synthesis of 5-chloro-1,2-diiodo-3-((4-methoxyphenyl)ethynyl)benzene (19A)
The title compound was synthesized using the general procedure and isolated in 44% yield as white solid after flash chromatography. IR (cast film, cm1): 3121, 3049, 2209, 1613, 1597, 1310, 1149, 976, 842, 742, 619. δH (400 MHz, CDCl3) δ: 7.76 (d, 1H, J = 2.2 Hz), 7.51 (d, 2H, J = 8.7 Hz), 7.43 (d, 1H, J = 2.2 Hz), 6.89 (d, 2H, J = 8.8 Hz), 3.48 (s, 3H). δC (100 MHz, CDCl3) δ: 160.6, 137.9, 134.5, 133.6, 133.5, 132.9, 130.9, 114.3, 112.3, 109.1, 94.6, 91.6, 55.5. Mp: 112–114 °C. HRMS (EI) m/z for C15H9ClI2O [M]+: calcd 493.8431; found, 493.8425.Synthesis of 5-chloro-1,2-diiodo-3-((4-(trifluoromethyl)phenyl)ethynyl)benzene (20A)
The title compound was synthesized using the general procedure and isolated in 31% yield as white solid after flash chromatography. IR (cast film, cm−1) 3107, 3088, 3012, 2196, 1612, 1579, 1012, 983, 867, 764, 642, 521. δH (400 MHz, CDCl3) δ: 7.81 (d, 1H, J = 2.4 Hz), 7.60–7.70 (m, 4H), 7.46 (d, 1H, J = 2.4 Hz). δC (100 MHz, CDCl3) δ: 138.8, 134.7, 133.0, 132.1, 131.8, 131.4, 131.0 (q, J = 32 Hz), 125.6 (q, J = 4 Hz), 123.9 (q, JC–F = 271 Hz), 112.6, 109.5, 94.4, 92.4. Mp: 116–118 °C. HRMS (EI) m/z for C15H6ClF3I2 [M]+: calcd 531.8199; found, 531.8188.Synthesis of 5-bromo-1-((4-ethylphenyl)ethynyl)-2,3-diiodobenzene (21A)
The title compound was synthesized using the general procedure and isolated in 85% yield as colorless oil after flash chromatography. IR (cast film, cm1): 3142, 3048, 3013, 2201, 1592, 943, 841, 746, 529. δH (400 MHz, CDCl3) δ: 7.93 (d, 1H, J = 2.2 Hz), 7.67 (d, 1H, J = 2.2 Hz), 7.47 (d, 2H, J = 8.1 Hz), 7.20 (d, 2H, J = 8.0 Hz), 2.68 (q, 2H, J = 7.6 Hz), 1.25 (t, 3H, J = 7.6 Hz). δC (100 MHz, CDCl3) δ: 146.0, 141.6, 135.4, 132.0, 128.2, 125.5, 120.1, 119.4, 99.3, 96.7, 85.0, 29.1, 15.5, (missing one peak due to overlapping). HRMS (EI) m/z for C16H11BrI2 [M]+: calcd 535.8133; found, 535.8128.Synthesis of 5-bromo-1-((3-fluorophenyl)ethynyl)-2,3-diiodo benzene (22A)
The title compound was synthesized using the general procedure and isolated in 44% yield as white solid after flash chromatography. IR (cast film, cm−1) 3097, 3048, 2214, 1598, 948, 812, 794, 653. δH (400 MHz, CDCl3) δ: 7.95 (d, 1H, J = 2.2 Hz), 7.66 (d, 1H, J = 2.2 Hz), 7.32–7.34 (m, 2H), 7.23 (d, 1H, J = 9.2 Hz), 7.10–7.11 (m, 1H). δC (100 MHz, CDCl3) δ: 161.5 (d, JC–F = 246 Hz), 142.2, 139.3, 135.6, 130.2 (d, JC–F = 8 Hz), 129.9 (d, JC–F = 3 Hz), 124.8, 124.0 (d, JC–F = 10 Hz), 120.2, 118.8 (d, JC–F = 22 Hz), 116.8 (d, JC–F = 22 Hz), 99.4, 94.8 (d, JC–F = 3 Hz), 86.2. Mp: 68–70 °C HRMS (EI) m/z for C14H6BrFI2 [M]+: calcd 525.7726; found, 525.7719.Synthesis of 5-fluoro-1,2-diiodo-3-((4-methoxyphenyl)ethynyl)benzene (23A)
The title compound was synthesized using the general procedure and isolated in 74% yield as white solid after flash chromatography. IR (cast film, cm−1) 3102, 3042, 2191, 1608, 1597, 1023, 894, 742, 691. δH (400 MHz, CDCl3) δ: 7.51–7.57 (m, 3H), 7.20 (dd, 1H, J1 = 2.7 Hz, J2 = 8.6 Hz), 6.90 (d, 2H, J = 8.6 Hz), 3.84 (s, 3H). δC (100 MHz, CDCl3) δ: 161.6 (d, JC–F = 251 Hz), 160.6, 133.5, 132.9 (d, JC–F = 10 Hz), 130.9, 126.3 (d, JC–F = 24 Hz), 118.4 (d, JC–F = 23 Hz), 108.7 (d, JC–F = 4 Hz), 108.6, 108.5, 94.5, 91.8 (d, JC–F = 3 Hz), 55.5. Mp: 87–90 °C. HRMS (EI) m/z for C15H9FI2O [M]+: calcd 477.8727; found, 477.8718.Synthesis of 1-((4-chlorophenyl)ethynyl)-5-fluoro-2,3-diiodo benzene (24A)
The title compound was synthesized using the general procedure and isolated in 42% yield as white solid after flash chromatography. IR (cast film, cm−1) 3089, 3041, 2201, 1597, 1524, 1016, 927, 813, 691. δH (400 MHz, CDCl3) δ: 7.57 (dd, 1H, J1 = 7.7 Hz, J2 = 2.4 Hz), 7.48 (d, 2H, J = 8.3 Hz), 7.34 (d, 2H, J = 8.2 Hz), 7.20 (dd, 1H, J1 = 8.6 Hz, J2 = 2.4 Hz). δC (100 MHz, CDCl3) δ: 161.5 (d, JC–F = 252 Hz), 135.5, 133.1, 132.1 (d, JC–F = 10 Hz), 129.0, 126.8 (d, JC–F = 24 Hz), 120.8, 118.8 (d, JC–F = 23 Hz), 108.9 (d, JC–F = 4 Hz), 108.8 (d, JC–F = 8 Hz), 93.5 (d, JC–F = 3 Hz), 92.9. Mp: 113–114 °C. HRMS (EI) m/z for C14H6ClFI2 [M]+: calcd 481.8231; found, 481.8228.Synthesis of methyl 3,4-diiodo-5-((4-methoxyphenyl)ethynyl)benzoate (25A)
The title compound was synthesized using the general procedure and isolated in 33% yield as white solid after flash chromatography. IR (cast film, cm−1) 3104, 3085, 2195, 1765, 1604, 1586, 1415, 1204, 1112, 876, 743. δH (400 MHz, CDCl3) δ: 8.37 (d, 1H, J = 1.8 Hz), 8.05 (d, 1H, J = 1.8 Hz), 7.52 (d, 2H, J = 8.8 Hz), 6.90 (d, 2H, J = 8.7 Hz), 3.92 (s, 3H), 3.84 (s, 3H). δC (100 MHz, CDCl3) δ: 165.1, 160.5, 138.7, 133.4, 132.5, 131.5, 131.1, 120.6, 114.5, 114.3, 108.9, 94.4, 91.9, 55.5, 52.8. Mp: 173–175 °C. HRMS (EI) m/z for C17H12I2O3 [M]+: calcd 517.8876; found, 517.8874.Synthesis of 1-chloro-3-((4-chlorophenyl)ethynyl)-2-iodobenzene (26A)
The title compound was synthesized using the general procedure and isolated in 45% yield as white solid after flash chromatography. IR (cast film, cm−1) 3078, 3024, 2208, 1601, 1598, 1107, 867, 523. δH (400 MHz, CDCl3) δ: 7.52 (d, 2H, J = 8.4 Hz), 7.34–7.40 (m, 4H), 7.26 (dd, 1H, J = 7.7 Hz, J = 7.9 Hz). δC (100 MHz, CDCl3) δ: 139.8, 135.2, 133.9, 133.0, 132.5, 130.4, 129.0, 128.9, 121.3, 105.5, 93.0, 92.5. Mp: 90–92 °C. HRMS (EI) m/z for C14H7Cl2I [M]+: calcd 371.8969; found, 371.8960.Synthesis of 1-bromo-2-iodo-3-((4-methoxyphenyl)ethynyl) benzene (27A)
The title compound was synthesized using the general procedure and isolated in 62% yield as white solid after flash chromatography. IR (cast film, cm−1) 3097, 3021, 2207, 1578, 1543, 1204, 1079, 894, 742. δH (400 MHz, CDCl3) δ: 7.53 (d, 3H, J = 8.2 Hz), 7.41 (d, 1H, J = 7.6 Hz), 7.17 (dd, 1H, J1 = 7.9, J2 = 7.8 Hz), 6.90 (d, 2H, J = 8.3 Hz), 3.84 (s, 3H). δC (100 MHz, CDCl3) δ: 160.3, 133.3, 133.2, 131.8, 130.7, 130.5, 129.1, 114.8, 114.3, 108.3, 93.9, 91.6, 55.5. Mp: 53–55 °C. HRMS (EI) m/z for C15H10BrIO [M]+: calcd 411.8960; found, 411.8954.General procedure for one-pot double Sonogashira cross-coupling reactions of 5-substituted-1,2,3-triiodobenzenes
A flame-dried Schlenk flask was charged with 5-substituted-1,2,3-triiodobenzene (0.65 mmol, 0.08 M, 1.0 equiv.), aryl acetylene (1.0 equiv.), Cs2CO3 (7.0 equiv.) in 8.0 mL dry toluene under argon. The mixture was stirred at room temperature for 20 min. Tetrakis(triphenylphosphine)palladium (0) (10 mol%) and copper iodide (20 mol%) were added, capped with septum, carefully degassed with argon, and the reaction flask was wrapped with aluminum foil and stirred at room temperature for 24 h. Aryl acetylene (1.0 equiv.) and Cs2CO3 (7.0 equiv.) were added to the mixture, carefully degassed with argon, wrapped with aluminum foil and stirred at room temperature for another 24 h. The reaction mixture was diluted with EtOAc and filtered over Celite 545®. Distilled water (100 mL) was added and extracted with EtOAc (2 × 50 mL). The organic layers were combined washed with brine, dried with anhyd. Na2SO4, filtered and evaporated under reduced pressure. The crude product was purified by flash chromatography (100% hexane) to yield the pure desired product.Synthesis of 2-iodo-5-methoxy-1-((4-methoxyphenyl)ethynyl)-3-((4-(trifluoromethyl)phenyl)ethynyl)benzene (28A)
The title compound was synthesized using the one-pot general procedure and isolated in 48% yield as pale yellow oil after flash chromatography. IR (cast film, cm−1) 3124, 3086, 3006, 2203, 1601, 1583, 1204, 1139, 976, 867, 647. δH (400 MHz, CDCl3) δ: 7.71 (d, 2H, J = 8.1 Hz), 7.62 (d, 2H, J = 8.1 Hz), 7.56 (d, 2H, J = 8.5 Hz), 7.06 (d, 1H, J = 2.7 Hz), 7.04 (d, 1H, J = 2.8 Hz), 6.90 (d, 2H, J = 8.5 Hz), 3.84 (s, 3H), 3.82 (s, 3H). δC (100 MHz, CDCl3) δ: 160.3, 159.2, 133.4, 131.5 (q, JC–F = 134 Hz), 132.0, 130.5 (q, JC–F = 33 Hz), 126.8, 125.5, 125.4 (q, JC–F = 4 Hz), 122.7, 118.2, 118.0, 114.9, 114.3, 97.0, 94.3, 93.7, 91.3, 90.8, 55.8, 55.5. HRMS (EI) m/z for C25H16F3IO2 [M]+: calcd 532.0147; found, 532.0133.Synthesis of 5-bromo-1-((4-ethylphenyl)ethynyl)-3-((3-fluoro phenyl)ethynyl)-2-iodobenzene (29A)
The title compound was synthesized using the one-pot general procedure and isolated in 34% yield as white solid after flash chromatography. IR (cast film, cm−1) 3125, 3073, 3041, 2201, 1623, 1579, 1042, 941, 837, 622. δH (400 MHz, CDCl3) δ: 7.64 (dd, 2H, J1 = 10.9 Hz, J2 = 2.1 Hz), 7.50 (d, 2H, J = 7.9 Hz), 7.28–7.36 (m, 3H), 7.21 (d, 2H, J = 7.8 Hz), 7.00–7.15 (m, 1H), 2.67 (q, 2H, J = 7.6 Hz), 1.26 (m, 3H). δC (100 MHz, CDCl3) δ: 162.5 (d, JC–F = 245 Hz), 136.6, 135.4, 135.0, 132.0, 130.2 (d, JC–F = 8 Hz), 128.2, 127.9 (d, JC–F = 2 Hz), 126.0, 125.1, 124.3 (d, JC–F = 9 Hz), 119.5, 119.4, 118.7 (d, JC–F = 23 Hz), 116.6 (d, JC–F = 21 Hz), 96.8, 94.6, 85.8, 84.3, 29.1, 15.5, (missing one peak due to overlapping). Mp: 83–85 °C. HRMS (EI) m/z for C24H15BrFI [M]+: calcd 527.9386; found, 527.9382.Synthesis of 4,4′-((2-iodo-1,3-phenylene)bis(ethyne-2,1-diyl))bis(methoxybenzene) (30A)
The title compound was synthesized using the one-pot general procedure and isolated in 57% yield as pale yellow oil after flash chromatography. IR (cast film, cm−1) 3124, 3082, 3012, 2214, 1624, 1602, 967, 962, 841, 723. δH (400 MHz, CDCl3) δ: 7.55 (d, 4H, J = 8.7 Hz), 7.40 (d, 2H, J = 7.5 Hz), 7.28 (d, 1H, J = 8.0 Hz), 6.90 (d, 4H, J = 8.7 Hz), 3.8 (s, 6H). δC (100 MHz, CDCl3) δ: 160.2, 133.3, 131.5, 131.2, 127.8, 115.2, 114.3, 107.7, 93.5, 91.2, 55.5. HRMS (EI) m/z for C24H17IO2 [M]+: calcd 464.0273; found, 464.0269.Synthesis of 4,4′-((2-iodo-5-methoxy-1,3-phenylene)bis(ethyne-2,1-diyl))bis(methoxybenzene) (31A)
The title compound was synthesized using the one-pot general procedure and isolated in 69% yield as colorless oil after flash chromatography. IR (cast film, cm−1) 3124, 3075, 3042, 2198, 1614, 1587, 1345, 1207, 1184, 986, 748, 625. δH (400 MHz, CDCl3) δ: 7.55 (d, 4H, J = 8.6 Hz), 7.02 (s, 2H), 6.90 (d, 4H, J = 8.6 Hz), 3.84 (s, 6H), 3.82 (s, 3H). δC (100 MHz, CDCl3) δ: 160.2, 159.2, 133.4, 131.9, 117.5, 115.1, 114.3, 96.9, 93.3, 91.1, 55.8, 55.5. HRMS (EI) m/z for C25H19IO3 [M]+: calcd 494.0379; found, 494.0371.Synthesis of 3,3′-((5-bromo-2-iodo-1,3-phenylene)bis(ethyne-2,1-diyl))bis(fluorobenzene) (32A)
The title compound was synthesized using the one-pot general procedure and isolated in 55% yield as white solid after flash chromatography. IR (cast film, cm−1) 3124, 3057, 2195, 1604, 1578, 684, 742, 628. δH (400 MHz, CDCl3) δ: 7.66 (s, 2H), 7.33–7.36 (m, 4H), 7.22–7.30 (m, 2H), 7.07–7.12 (m, 2H). δC (100 MHz, CDCl3) δ: 162.6 (d, JC–F = 245 Hz), 136.8, 135.5, 130.3 (d, JC–F = 8 Hz), 127.9 (d, JC–F = 2 Hz), 125.4, 124.2 (d, JC–F = 9 Hz), 119.5, 118.8 (d, JC–F = 22 Hz), 116.7 (d, JC–F = 21 Hz), 94.9 (d, JC–F = 3 Hz), 85.6. Mp: 122–124 °C. HRMS (EI) m/z for C22H10BrF2I [M]+: calcd 517.8979; found, 517.8975.Synthesis of 4,4′-((5-bromo-2-iodo-1,3-phenylene)bis(ethyne-2,1-diyl))bis(ethylbenzene) (33A)
The title compound was synthesized using the one-pot general procedure and isolated in 87% yield as colorless oil after flash chromatography. IR (cast film, cm−1) 3106, 3086, 3015, 2186, 1612, 1592, 837, 467. δH (400 MHz, CDCl3) δ: 7.61 (s, 2H), 7.44 (d, 4H, J = 7.8 Hz), 7.16–7.21 (m, 4H), 2.60–2.75 (m, 4H), 1.20–1.30 (m, 6H). δC (100 MHz, CDCl3) δ: 145.5, 133.8, 132.7, 131.9, 128.2, 125.7, 119.9, 91.6, 86.7, 81.7, 29.0, 15.4. HRMS (EI) m/z for C26H20BrI [M]+: calcd 537.9793; found, 537.9784.Synthesis of 4,4′-((2-iodo-1,3-phenylene)bis(ethyne-2,1-diyl))bis ((trifluoromethyl)benzene) (34A)
The title compound was synthesized using the one-pot general procedure and isolated in 72% yield as white solid after flash chromatography. IR (cast film, cm−1) 3108, 3086, 3046, 2216, 1587, 1537, 976, 891, 642. δH (400 MHz, CDCl3) δ: 7.72 (d, 4H, J = 8.1 Hz), 7.64 (d, 4H, J = 8.2 Hz), 7.50 (d, 2H, J = 7.6 Hz), 7.36 (dd, 1H, J1 = 7.9, J2 = 7.5 Hz). δC (100 MHz, CDCl3) δ: 132.5, 132.1, 130.9, 130.8, 128.1, 126.7, 125.6 (q, JC–F = 4 Hz), 122.7, 108.1, 94.1, 91.9. Mp: 92–94 °C. HRMS (EI) m/z for C24H11F6I [M]+: calcd 539.9810; found, 539.9798.Synthesis of 4,4′-((5-chloro-2-iodo-1,3-phenylene)bis(ethyne-2,1-diyl))bis((trifluoromethyl)benzene) (35A)
The title compound was synthesized using the one-pot general procedure and isolated in 55% yield as white solid after flash chromatography. IR (cast film, cm−1) 3097, 3064, 2207, 1599, 1548, 948, 816, 724, 634. δH (400 MHz, CDCl3) δ: 7.70 (d, 4H, J = 8.3 Hz), 7.64 (d, 4H, J = 8.9 Hz), 7.47 (s, 2H). δC (100 MHz, CDCl3) δ: 138.8, 134.3, 133.0, 132.2 (q, JC–F = 34 Hz), 132.1 (q, JC–F = 3 Hz), 131.0 (q, JC–F = 33 Hz), 125.6 (q, JC–F = 3 Hz), 124.0 (q, JC–F = 171 Hz), 105.5, 93.0, 92.9. Mp: 116–118 °C. HRMS (EI) m/z for C24H10ClF6I [M]+: calcd 573.9420; found, 573.9412.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was generously funded by Jordan University of Science and Technology (JUST) – Deanship of Research – Jordan (Grant No. 336/2018 for R.M.A.). We thank the department of chemistry at University of Alberta for X-ray crystallographic analysis.Notes and references
- T. P. Selby, N. R. Deprez, A. X. Ding, S. F. McCann, A. D. Satterfield, K. A. Hughes and D. A. Travis, World Patent WO2015191377A1, E. I. du Pont de Nemours and Company, USA, 2015.
- V. M. Sviripa, W. Zhang, L. M. Kril, A. X. Liu, Y. Yuan, C.-G. Zhan, C. Liu and D. S. Watt, Bioorg. Med. Chem. Lett., 2014, 24, 3638–3640 CrossRef CAS PubMed.
- P. J. Conn, C. W. Lindsley, S. R. Stauffer, C. K. Jones, S. Conde-Ceide, H. M. Tong, J. M. Bartolome-Nebreda and G. J. Macdonald, World Patent WO2012078817A1, Vanderbilt University, USA, 2012.
- R. Apodaca, J. G. Breitenbucher, N. A. Hawryluk, W. M. Jones, J. M. Keith, J. E. Merit, M. S. Tichenor and A. K. Timmons, World Patent WO2008153752A2, Janssen Pharmaceutica N.V., Belg. Netherland, 2008.
- C. F. Bigge, S. X. Cai, E. Weber, R. Woodward, N. C. Lan, J. F. W. Keana, Z.-L. Zhou and J. Wright, World Patent WO9723214A1, Warner-Lambert Company, CoCensys, Inc., USA, 1997.
- G. Csjernyik, S. Karlstrom, A. Kers, K. Kolmodin, M. Nylof, L. Ohberg, L. Rakos, L. Sandberg, F. Sehgelmeble, P. Soderman, B. M. Swahn and S. Von Berg, World Patent WO 2012/087237A1, A. B. Astrazeneca, 2012.
- A. Daina, C. Giuliano, C. Pietra, J. Wang, Y. Chi, Z. Zou, F. Li, Z. Yan, Y. Zhou, A. Guainazzi, S. Garcia Rubio and V. Zoete, J. Med. Chem., 2018, 61, 11039–11060 CrossRef CAS PubMed.
- C. Giuliano, S. G. Rubio, A. Daina, A. Guainazzi and C. Pietra, World Patent WO2015/134839 A1, Helsinn Healthcare SA, Switzerland, 2015.
- A. Fonari, J. C. Roeder, H. Shen, T. V. Timofeeva and K. P. C. Vollhardt, Synlett, 2014, 25, 2429–2433 CrossRef CAS.
- S. Reimann, P. Ehlers, M. Sharif, K. Wittler, A. Spannenberg, R. Ludwig and P. Langer, Catal. Commun., 2012, 25, 142–147 CrossRef CAS.
- J. Panteleev, L. Zhang and M. Lautens, Angew. Chem., Int. Ed., 2011, 50, 9089–9092 CrossRef CAS PubMed.
- K. Miki, H. Kuge, R. Umeda, M. Sonoda and Y. Tobe, Synth. Commun., 2011, 41, 1077–1087 CrossRef CAS.
- W. A. Carroll, D. M. Kalvin, A. Perez Medrano, A. S. Florjancic, Y. Wang, D. L. Donnelly-Roberts, M. T. Namovic, G. Grayson, P. Honore and M. F. Jarvis, Bioorg. Med. Chem. Lett., 2007, 17, 4044–4048 CrossRef CAS PubMed.
- D. L. Musso, M. J. Clarke, J. L. Kelley, G. E. Boswell and G. Chen, Org. Biomol. Chem., 2003, 1, 498–506 RSC.
- K. Tsuneo, K. Hisatomo and K. Nobuo, World Patent WO2007JP50152, Seikagaku Kogyo Co Ltd, Japan, 2008.
- V. M. Sviripa, W. Zhang, C. Liu and D. Watt, US Pat. 2015/272908, University of Kentucky Research Foundation, Pharmaceutical Sciences Faculty Patents USA, Lexington, KY, 2018.
- J. Frackenpohl, T. Mueller, J. Dittgen, D. Schmutzler, J. P. Ruiz-Santaella Moreno and M. J. Hills, World Patent WO2014086751A1, Bayer CropScience AG, Germany, 2014.
- G. P. Lahm and B. K. Smith, World Patent WO2012177668A1, E. I. du Pont de Nemours and Company, USA, 2012.
- R. M. Al-Zoubi, W. K. Al-Jammal, M. Y. El-Khateeb and R. McDonald, Eur. J. Org. Chem., 2015, 3374–3384 CrossRef CAS.
- R. M. Al-Zoubi, H. Al-Mughaid, M. A. Al-Zoubi, K. T. Jaradat and R. McDonald, Eur. J. Org. Chem., 2015, 5501–5508 CrossRef CAS.
- R. M. Al-Zoubi, H. Al-Mughaid and R. McDonald, Aust. J. Chem., 2015, 68, 912–918 CrossRef CAS.
- R. M. Al-Zoubi, H. A. Futouh and R. McDonald, Aust. J. Chem., 2013, 66, 1570–1575 CrossRef CAS.
- N. N. Pham, G. A. Salman, M. B. Ponce, T. T. Dang, A. Spannenberg, P. Ehlers and P. Langer, Eur. J. Org. Chem., 2017, 3865–3873 CrossRef CAS.
- R. Balderrama-Martinez-Sotomayor, M. Flores-Jarillo and A. Alvarez-Hernandez, ARKIVOC, 2016, 36–47 Search PubMed.
- T. Usuki, H. Yamada, T. Hayashi, H. Yanuma, Y. Koseki, N. Suzuki, Y. Masuyama and Y. Y. Lin, Chem. Commun., 2012, 48, 3233–3235 RSC.
- H. Yamada, T. Hayashi and T. Usuki, Bull. Chem. Soc. Jpn., 2015, 88, 673–683 CrossRef CAS.
- P. Ehlers, T. T. Dang, T. Patonay, A. Villinger and P. Langer, Eur. J. Org. Chem., 2013, 2000–2007 CrossRef CAS.
- I. Malik, Z. Ahmad, S. Reimann, M. Nawaz, T. Patonay and P. Langer, Synlett, 2012, 23, 1463–1466 CrossRef CAS.
- K. C. Majumdar and S. Nath, Synthesis, 2011, 1413–1418 CrossRef CAS.
- N. Ibrahim, F. Chevot and M. Legraverend, Tetrahedron Lett., 2011, 52, 305–307 CrossRef CAS.
- A. El Akkaoui, I. Bassoude, J. Koubachi, S. Berteina-Raboin, A. Mouaddib and G. Guillaumet, Tetrahedron, 2011, 67, 7128–7138 CrossRef CAS.
- D. Thibeault, M. Auger and J.-F. Morin, Eur. J. Org. Chem., 2010, 3049–3067 CrossRef CAS.
- CCDC-1982244–1982247 contain the supplementary crystallographic data for compounds 35A, 6A, 23A and 20A respectively.
- I. J. S. Fairlamb, C. T. O'Brien, Z. Lin and K. C. Lam, Org. Biomol. Chem., 2006, 4, 1213–1216 RSC.
- R. Chinchilla and C. Najera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- L. Dokladalova, F. Bures, W. Kuznik, I. V. Kityk, A. Wojciechowski, T. Mikysek, N. Almonasy, M. Ramaiyan, Z. Padelkova, J. Kulhanek and M. Ludwig, Org. Biomol. Chem., 2014, 12, 5517–5527 RSC.
- P. Ehlers, A. Petrosyan, A. Neubauer, T. Broese, S. Lochbrunner, T. V. Ghochikyan, A. S. Saghyan and P. Langer, Org. Biomol. Chem., 2014, 12, 8627–8640 RSC.
- A. R. Kapdi and D. Prajapati, RSC Adv., 2014, 4, 41245–41259 RSC.
- A. K. Danodia, R. K. Saunthwal, M. Patel, R. K. Tiwari and A. K. Verma, Org. Biomol. Chem., 2016, 14, 6487–6496 RSC.
- N. Park, K. C. Ko, H.-W. Shin, S. M. Lee, H. J. Kim, J. Y. Lee and S. U. Son, J. Mater. Chem. A, 2016, 4, 8010–8014 RSC.
- P. O. Delaye, M. Penichon, H. Allouchi, C. Enguehard-Gueiffier and A. Gueiffier, Org. Biomol. Chem., 2017, 15, 4199–4204 RSC.
- M. Yamaguchi and K. Manabe, Org. Biomol. Chem., 2017, 15, 6645–6655 RSC.
- K. M. Saini, R. K. Saunthwal and A. K. Verma, Org. Biomol. Chem., 2017, 15, 10289–10298 RSC.
- Y. Sasai, H. Tsuchida, T. Kakuta, T. Ogoshi and Y. Morisaki, Mater. Chem. Front., 2018, 2, 791–795 RSC.
- O. S. Miljanic, K. P. C. Vollhardt and G. D. Whitener, Synlett, 2003, 29–34 CAS.
- G. Mao, A. Orita, D. Matsuo, T. Hirate, T. Iwanaga, S. Toyota and J. Otera, Tetrahedron Lett., 2009, 50, 2860–2864 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1982244–1982247. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra01569e |
| This journal is © The Royal Society of Chemistry 2020 |