
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative radical coupling of hydroquinones and thiols using chromic acid: one-pot synthesis of quinonyl alkyl/aryl thioethers†
T. P. Adarsh Krishna ,
Sakthivel Pandaram,
Suresh Chinnasamy and
Andivelu Ilangovan*
,
Sakthivel Pandaram,
Suresh Chinnasamy and
Andivelu Ilangovan*
School of Chemistry, Bharathidasan University, Tiruchirappalli, Tamilnadu-620024, India. E-mail: ilangovanbdu@yahoo.com
First published on 21st May 2020
Abstract
An efficient, simple and practical protocol for one-pot sequential oxidative radical C–H/S–H cross-coupling of thiols with hydroquinones (HQs) and oxidation leading to the formation of quinonyl alkyl/aryl thioethers using H2CrO4 was developed. This cross-coupling of thiyl and aryl radicals offers mono thioethers in good to moderate yield and works well with a wide variety of thiols. Similarly, this method works well for coupling of 2-amino thiophenol and HQs to form phenothiazine-3-ones 5a–c. C–S bond formation via thioether synthesis was observed using a chromium reagent for the first time. Theoretical studies on the pharmacokinetic properties of compounds 5a–c revealed that due to drug-like properties, compound 5b strongly binds with Alzheimer's disease (AD) associated AChE target sites.
Introduction
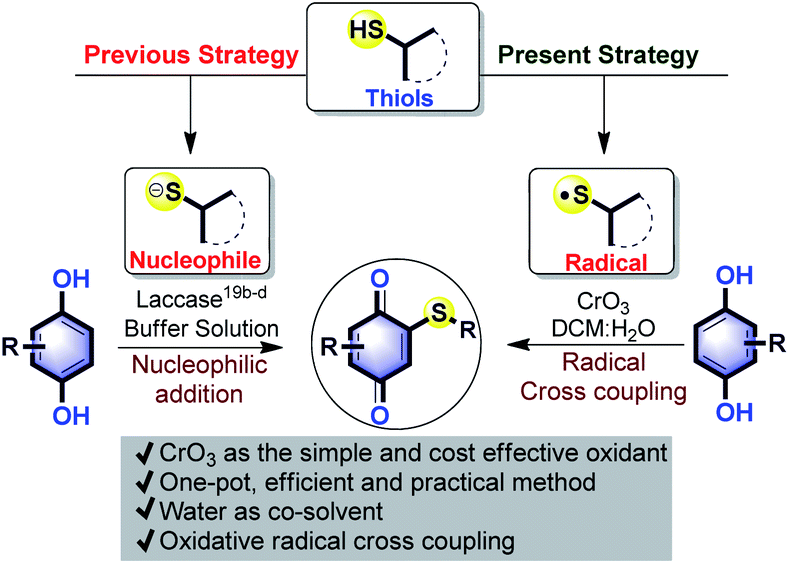
The quinonoid class of compounds finds applications in several fields such as synthetic chemistry,1 medicinal chemistry,2 natural products,3 and functional materials.4 The chemical reactivity and catalytic performance of quinones depends upon their electron-accepting character, redox behavior,5 and the nature of functional group(s) present.6 Hetero-functionalized quinones, especially quinonyl thioethers, display good structural stability,7 and useful pharmaceutical8 and other biological activities.9 Around thirty-one currently marketed drugs are thioether derivatives (Fig. 1),10 used for the treatment of cancer, HIV, diabetes, Alzheimer's, inflammatory diseases, etc. In the case of materials, the thioether group contributes to physical, electronic and surface properties.11 | ||
| Fig. 1 Bioactive molecules containing a thioether linkage. | ||
In general, number of methods known for C–S12 bond formation via C–H functionalization only less compared to C–N or C–O bond formation. One of the potential reasons could be lack of transition metal catalysts and reagents that can tolerate sulfur poisoning. Classical approaches for the construction of C–S bond are the direct coupling of organic halides or acids with thiols/disulfides/arylsulfonyl halides and addition of thiols to unsaturated C–C bonds under free radical or metal (Mn, Fe, Ni, Cu, Ru, Rh, Pd, Ag and Cs) catalyzed conditions.12
A good number of methods are known for the introduction of the thioether group on quinones (Q–S–R). Aryl quinonyl thioethers (Q–S–R) were prepared by reaction of arylsulfonyl chlorides with quinone–CuI–PPh3,13a aryl disulfides (R–S–S–R) with quinone–AgOAc–dpp13b and CuI catalyzed oxidative addition of aryl boronic acids,14a and aryl iodides with aryl thiols.14b We recently demonstrated I2-DMSO15 mediated thiomethylation (–SMe) of quinone. Moreover, the conjugate addition of thiol on quinone was demonstrated using CuI–O2,16 and Co(OAc)2–O2 (ref. 7) system.
Conjugate addition of thiols or its derivatives to quinones,17 usually leads to a mixture of thiolated hydroquinones (HQs).18 This followed by re-oxidation provides quinonyl thioethers. A popular method for synthesis of quinone is the oxidation of HQ. In direct conversion of HQs into quinonyl thioethers, two such transformations should take place in one pot. To the best of our knowledge, there are only two such reports known. In one case, carcinogenic S-alkylisothiouronium salt19a was used for thioalkylation and in another study, an enzyme laccase was used to catalyse thiol addition on naphthohydroquinone19b and HQ19c,d (Scheme 1). Formation of poly thioalkylated products and poor yield are the main drawbacks of the enzymatic reaction. C–S bond formation on HQ via oxidative radical cross-coupling (ORCC), to form quinonyl thioethers was seldom explored.
 | ||
| Scheme 1 Quinonyl thioethers from thiol and hydroquinone. | ||
Chromium reagents are powerful oxidants which finds application in polymerization, aldol and Diels–Alder reactions.20a However, compared to other transition metals, its application in organic synthesis is undeveloped. To avoid poisonous effects due to contamination, chromium reagents are immobilized on zeolite.20b It is well known that chromic acid formed from CrO3 dimerizes thiol oxidatively to form disulfides through thio radical,20c and also oxidizes hydroquinone to quinone.20d Based on this and in continuation of our research interest on C–H functionalization of quinones,15,21 we hypothesized that HQ may undergo cross dehydrogenative oxidative radical cross-coupling (CDORCC) with thiol in the presence of CrO3. CDORCC is highly advantageous because it is atom economic,22 no need for pre-functionalization, and could be used for C–S bond forming reaction.23 Under the present study, we observed the formation of quiononyl thioether in the presence of in situ generated chromic acid from CrO3. To the best of our knowledge, this is the first example of C–S bond-forming reaction observed using a chromium reagent. Herein we present the results.
Result and discussion
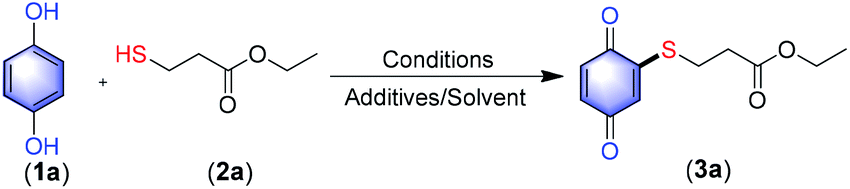
To check our hypothesis, a solution of HQ (1a, 1.0 equiv.) and ethyl 3-mercaptopropanoate (2a, 1.5 equiv.) in DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1 v/v, 4 mL) was treated with different oxidizing agents (1.0 equiv.) and the results are summarized in Table 1. In the case of oxidants such as KMnO4, NBS, Na2O2 (entry 1) and iodine reagents like PIDA, PIFA, IBX or NaIO4 (entry 2) the reaction failed to deliver the expected product 3a. Interestingly, with K2S2O8 (1.0 equiv., entry 3) both oxidation of HQ to quinone as well as C–S bond-forming reaction took place to afford the product 3a in 10% isolated yield. Based on this encouraging result, we tried the same reaction using (NH4)2S2O8 (1.0 equiv., entry 4) and CrO3 (1.0 equiv., entry 5), to get product 3a in 20% and 30% respectively. Further, when 3.0 equiv. of CrO3 (entry 5) was used the yield improved to 75%. However, with higher equivalents (3.0 equiv.) of K2S2O8 (35%) and (NH4)2S2O8 (48%) the product 3a was obtained in less isolated yield as compared with CrO3 (entries 3–5). By decreasing (2.0 equiv., entry 6) or increasing (4.0 equiv., entry 6) the quantity of CrO3 the yield did not improve. Additionally, when the reaction was carried out with only DCM (no reaction) or H2O (12%, entry 7), the result was not encouraging. This proves that solvent combinations are more favorable and water is essential for this reaction. Further, when the reaction was carried out in a combination of H2O with various solvents such as DCE, DMF and ACN (Table 1, entries 8–10) the reaction yield diminished significantly. Finally, the effects of different acids as additive was evaluated.
:H2O (1:1 v/v, 4 mL) was treated with different oxidizing agents (1.0 equiv.) and the results are summarized in Table 1. In the case of oxidants such as KMnO4, NBS, Na2O2 (entry 1) and iodine reagents like PIDA, PIFA, IBX or NaIO4 (entry 2) the reaction failed to deliver the expected product 3a. Interestingly, with K2S2O8 (1.0 equiv., entry 3) both oxidation of HQ to quinone as well as C–S bond-forming reaction took place to afford the product 3a in 10% isolated yield. Based on this encouraging result, we tried the same reaction using (NH4)2S2O8 (1.0 equiv., entry 4) and CrO3 (1.0 equiv., entry 5), to get product 3a in 20% and 30% respectively. Further, when 3.0 equiv. of CrO3 (entry 5) was used the yield improved to 75%. However, with higher equivalents (3.0 equiv.) of K2S2O8 (35%) and (NH4)2S2O8 (48%) the product 3a was obtained in less isolated yield as compared with CrO3 (entries 3–5). By decreasing (2.0 equiv., entry 6) or increasing (4.0 equiv., entry 6) the quantity of CrO3 the yield did not improve. Additionally, when the reaction was carried out with only DCM (no reaction) or H2O (12%, entry 7), the result was not encouraging. This proves that solvent combinations are more favorable and water is essential for this reaction. Further, when the reaction was carried out in a combination of H2O with various solvents such as DCE, DMF and ACN (Table 1, entries 8–10) the reaction yield diminished significantly. Finally, the effects of different acids as additive was evaluated.
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Additive | Solvent | Yieldb (%) |
| a Reaction conditions: hydroquinone 1a (1.0 mmol), thiol 2a (1.5 mmol) oxidant (1 equiv.), additive (1 equiv.), solvent 1:1 ratio (4.0 mL) at rt for 1 h.b Isolate yield.c Oxidant (3.0 equiv.).d Oxidant (2.0 equiv.).e Oxidant (4.0 equiv.).f CrO3 (0.1 equiv.) + (NH4)2S2O8 (3.0 equiv.); NR = no reaction. |
||||
| 1 | KMnO4/NBS/Na2O2 | — | DCM:H2O |
NR |
| 2 | PIDA/PIFA/IBX/NaIO4 | — | DCM:H2O |
NR |
| 3 | K2S2O8 | — | DCM:H2O |
10/35c |
| 4 | (NH4)2S2O8 | — | DCM:H2O |
20/48c |
| 5 | CrO3 | — | DCM:H2O |
30/75c |
| 6 | CrO3 | — | DCM:H2O |
68d/72e |
| 7 | CrO3 | — | DCM/H2O | NRc/12c |
| 8 | CrO3 | — | DCE:H2O |
45c |
| 9 | CrO3 | — | ACN:H2O |
NRc |
| 10 | CrO3 | — | DMF:H2O |
NRc |
| 11 | CrO3 | TFA | DCM:H2O |
25c |
| 12 | CrO3 | p-TSA | DCM:H2O |
38c |
| 13 | CrO3 | H2SO4/HCl | DCM:H2O |
NRc |
| 14 | CrO3–(NH4)2S2O8 | — | DCM:H2O |
52f |
When TFA (25%, entry 11) or p-TSA (38%, entry 12) was used the product 3a was obtained only in very less yield. Furthermore, when HCl and H2SO4 were used as an additive, hydroquinone (1a) was simply converted into p-bezoquinone (6a) (entry 13). Finally, a catalytic amount of CrO3 (0.1 equiv.) with (NH4)2S2O8 (3.0 equiv.) reaction was performed (entry 14), under this condition only 52% of desired product was obtained. The systematic screening study revealed that 1a (1.0 equiv.), 2a (1.5 equiv.) and CrO3 (3.0 equiv.) in DCM:H2O (1:1 v/v, 4 mL) at room temperature could be the optimum condition for oxidative direct C–S bond formation and synthesis of quinonyl thioethers from hydroquinone.
With the optimum condition in hand, substrate scope of various alkyl and aryl thiols was examined under the optimized reaction condition and the results are depicted in Scheme 2. The reaction of a series of linear long-chain (C1–C5) mercaptopropanoates (2b–2e) with HQ afforded corresponding thioethers 3b–3e in good to moderate yield (75% to 48%). As the chain length of thioesters increased, the reactivity decreased. Mercaptopropanoate (2f) containing isopropyl group provided product 3f in best isolated yield (85%). Similarly, mercaptopropanoates 2g–2h, containing substituents such as 2-methoxyethyl (2g), benzyl (2h) and 3-phenylpropyl (2i) esters provided corresponding products 3g–3i in 1 h in good to moderate (82–40%). As noted earlier, with an increase in chain length, the yield decreased. In the case of free acids, such as 3-mercaptopropanonic acid (2j), amino-3-sulfhydrylpropanoic acid (2k) the reaction failed to deliver the expected product 3j and 3k. Furthermore, simple thiols like butanethiol (2l) containing no ester group also reacted quickly with HQ to offer product 3l, albeit in moderate yield (45%).
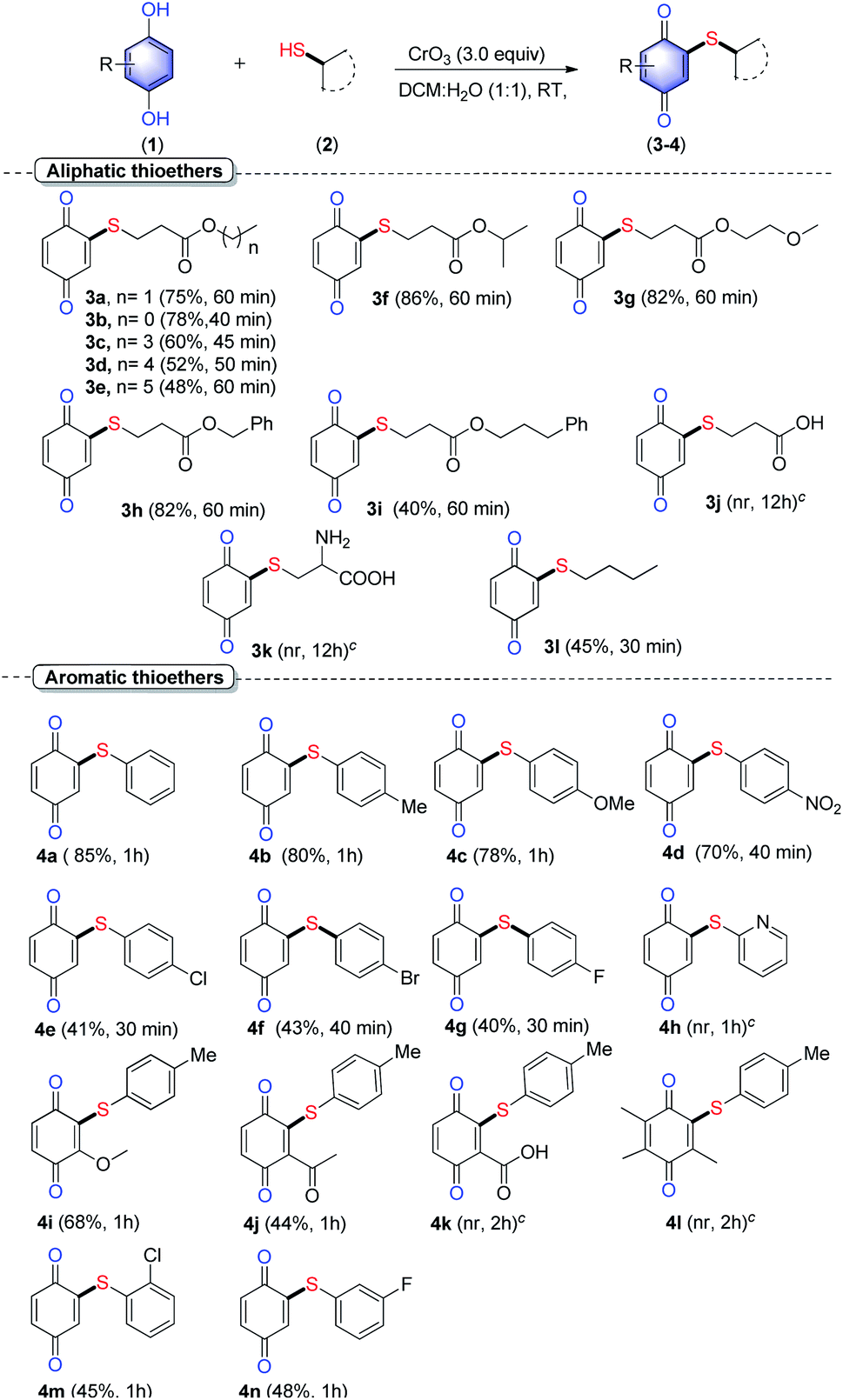 | ||
| Scheme 2 Scope of C–H/S–H cross-coupling between HQ and thiols.a,b aReaction conditions: hydroquinone (1.0 equiv., 100 mg), thiol (1.5 equiv.), CrO3 (3.0 equiv.), DCM:H2O (4 mL, 1:1) at rt, bisolated yield; cexpected product. | ||
As the next part of our study, the suitability of various aryl thiols was examined. The unsubstituted thiophenol (2m) gave aryl thioether 4a in a very good yield (85%). Similarly, thiophenols bearing electron-donating groups such as 4-Me (2n) and 4-OMe (2o) showed good reactivity and yielded corresponding aryl thioethers 4b (80%) and 4c (78%) in very good yield. The thiophenol 2p containing electron-withdrawing 4-NO2 group provided product 4d in moderate yield 70%. However, thiophenols 2q–s, substituted with deactivating substituents like 4-Cl, 4-Br, 4-F gave low to the moderate yield of desired products 4e–g (40–43%). However, 2-mercaptopyridine (2t) failed to deliver the expected product 4h. Further, the reaction of substituted hydroquinones with 4-methylthiophenol (2n) was examined. The 2-methoxy HQ (1b) and 2-acelyl HQ (1c) underwent reaction very smoothly to afford desired products in 68% (4i) and 44% (4j) respectively. HQ 1d containing deactivating –COOH group and HQ 1e containing highly substituted, 2,3,5-trimethyl failed to deliver the expected product (4k–l). Finally, the reactivity of ortho and meta substituted aryl thiol was also evaluated. Accordingly, 2-chlorothiophenol (2u) and 3-fluorothiophenol (2v) underwent reaction smoothly and the desired products 4m and 4n were obtained in 45% and 48% respectively. This result when combined with the result obtained from compounds 4e–f reveals that the position of the substituent at o/m/p did not influence the reactivity of aromatic thiols much under this oxidative condition.
Having demonstrated the synthesis of mono derivatives of quinonyl thioether in one pot using 1.5 equiv. of thiols, the effect of using a super stoichiometric quantity of thiols was examined (Scheme 3). Interestingly, when the reaction was carried out under standard condition using 3.0 equiv. aryl thiol 2n, bis-thiolated product 4o was obtained in 60% yield.
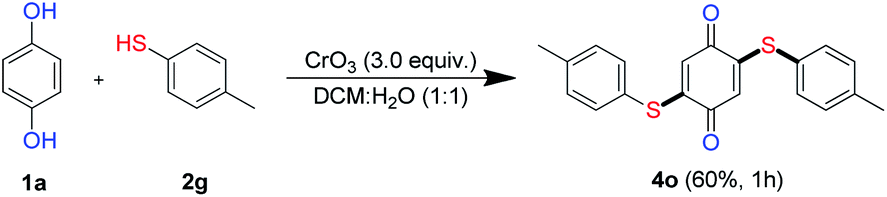 | ||
| Scheme 3 One-pot C–H difunctionalization of HQ. | ||
Vitamin K family consists of 2-methyl-1,4-naphthoquinone derivatives substituted with a long alkyl chain at 3-position. Thioether analogs of vitamin K were found to inhibit protein-tyrosine phosphatases and induce protein-tyrosine phosphorylation in a human hepatoma cell line (Hep3B).24 Utility of our method for the synthesis of Hep3B inhibitor was examined (Scheme 4). Accordingly, under the optimized condition, mercaptoethanol underwent reaction with menadione (6b) to provide corresponding product 7a, a Hep3B inhibitor, in 56% (Scheme 4).24
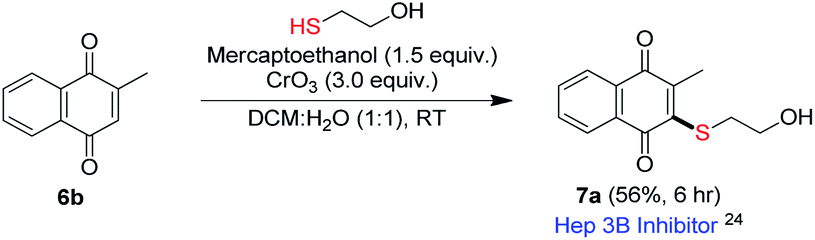 | ||
| Scheme 4 Synthesis of Hep3B inhibitor. | ||
As the next part of our study, we examined the reactivity of thiophenols with an additional functional group. Interestingly, under the optimal condition, when HQ was treated with 2-amino thiophenol derivatives 2w and 2x, we observed the formation of biologically important phenothiazines 5a (–Cl) and 5b (–CF3) through tandem C–S and C–N bond formation and oxidation of hydroquinone to quinone in 80% and 70% yield respectively (Scheme 5). However, reaction with sterically substituted HQ (1f) provides the expected phenothiazines 5c only in trace quantity.
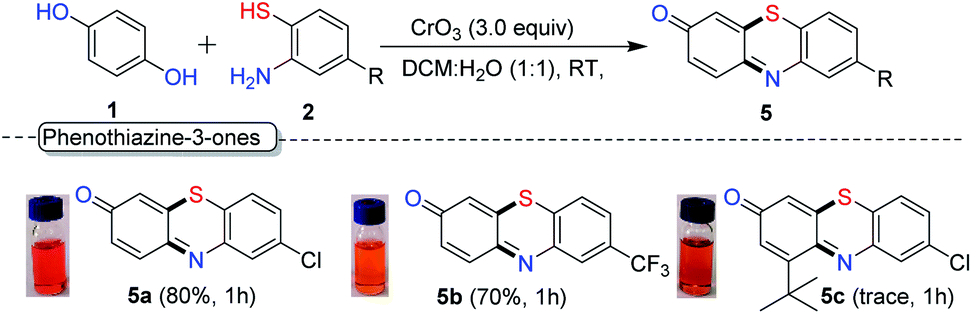 | ||
| Scheme 5 Domino reaction for phenothiazine-3-one. Reaction conditions: hydroquinone (1.0 equiv., 100 mg), 2-aminothiolphenol (1.5 equiv.), CrO3 (3.0 equiv.), DCM:H2O (4 mL, 1:1) at rt, isolated yield. | ||
In general, phenothiazines are excellent chromogenic molecules that find application as coloring agents.25a Moreover, this moiety is used as a common building block for the synthesis of pharmaceuticals including promazine drugs, antitubercular agents and cholinesterase inhibitors etc.25b The computer modeling of Absorption, Distribution, Metabolism, and Excretion (ADME) properties of compounds provides an idea about structure–property relationships and drug metabolism and pharmacokinetics (DMPK) properties based on the compound structure. In silico ADME-PK is applied at an early phase of the drug development process, in order to remove molecules with poor ADME-PK properties and leads to significant savings in research and development costs.
In this contest, we tried to understand ADME-PK properties of phenothiazine-3-one derivatives 5a–c in silicon study was carried out using Swiss-ADME,26 web tool (see ESI†). Results revealed that all compounds showed high gastrointestinal (GI) absorption, good blood–brain barrier (BBB) permeability and also compounds do not have P-glycoprotein (P-gp) permeability. The high lipophilicity (logPo/w) and less skin permeation (logKp) of compounds were observed in the range of 3.11 to 3.61 and −5.08 to −5.96 cm s−1 respectively. Furthermore, compounds 5a–c showed inhibition of cytochrome P450 isomers such as CYP1A2, CYP2C19 and CYP2C9. In addition topological polar surface area (TPSA) of 5a–c was found to 58.20 Å (≤140 Å), indicating that compounds have appropriate oral bioavailability (0.55). All compounds 5a–c meet the criteria of drug-likeness assessment based on Lipinski, Ghose and Veber rules. The drug lead-likeness shows, compound 5b with trifluromethyl (–CF3) is the most druggable substance without any violation. A combination of fragment contributions and a complexity penalty of 5b indicated good synthetic accessibility (2.81).
Further, to establish interaction mode with Alzheimer's disease (AD) associated AChE (Fig. 2A) target (PDB ID:1EVE) compound 5b was selected and in silico docking studies of 5a was performed by using Glide 10.7 Schrödinger27 software (see ESI†). Compound 5b exhibited G-score −7.30 kcal mol−1 and glide model score −44.18. Moreover, compound 5b showed strong hydrophobic interaction (Fig. 2B and C) with the amino acid residue Tyr121. These results reveal that the compound 5b strongly binds with the AChE active site. Thus compound 5b has good potential to show AChE inhibition.
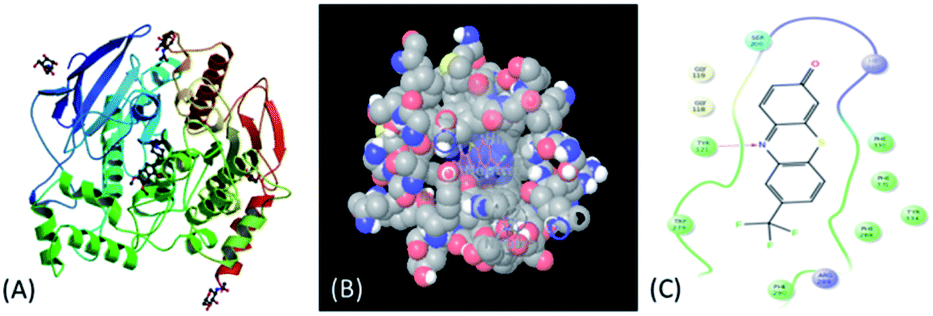 | ||
| Fig. 2 (A) Acetylcholinesterase target; (B) 3D structure of protein–ligand interaction; (C) 2D structure of protein–ligand interaction. | ||
Next, we examined the conversion efficiency of hydroquinone to benzoquinone under optimized reaction conditions. The tert-butyl benzoquinone (6b) obtained in excellent yield 91%. It reveals that the optimized condition was suitable for the conversion of HQ to BQ (Scheme 6).
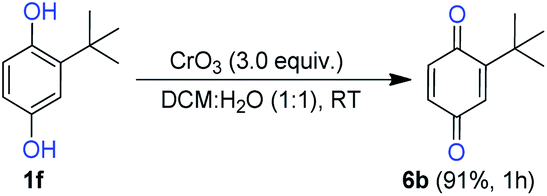 | ||
| Scheme 6 Conversion of HQ to BQ. | ||
To gain an understanding of the plausible mechanism for the C–S coupling reaction, a series of control experiments were conducted (Scheme 7). The reaction of isopropyl 3-mercaptopropanoate (2f) with benzoquinone (6a) under optimal condition provided only product 3f in low yield (36% equiv. 1), which is much less than the yield (86%, Scheme 2) obtained with hydroquinone (1a). This result indicated the benzoquinone may not the intermediate in the formation of product 3f and a sequential oxidative C–H/S–H cross-coupling between HQ and thiol should have taken place. Furthermore, the reaction carried out in the absence of CrO3 failed to deliver the desired product 3f (equiv. 2), which revealed that, CrO3 plays the role of oxidant in the oxidative cross-coupling reaction.
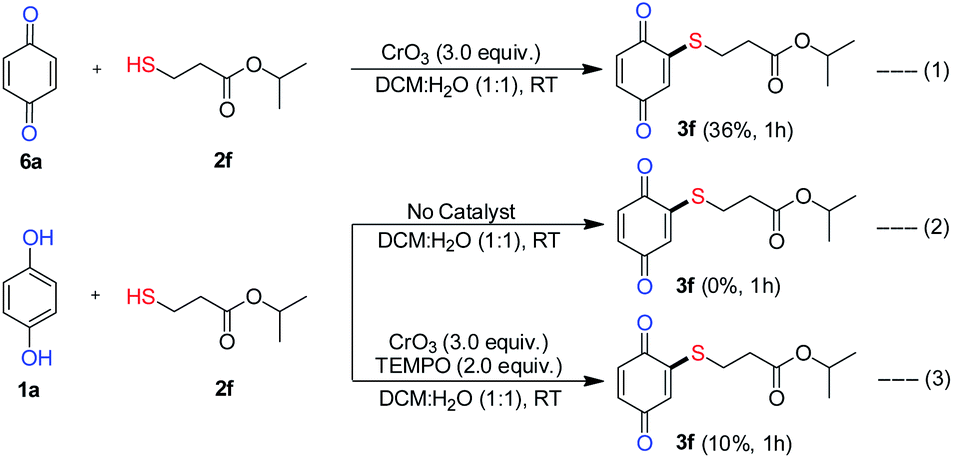 | ||
| Scheme 7 Verification experiments for the mechanism. | ||
Additionally, to determine whether radical intermediate was involved, the reaction was carried out between 1a and 2f in the presence of radical scavenger TEMPO (2.0 equiv.). The yield of product 3f decreased from 86% to 10% which clearly indicated that radical intermediates are involved in the reaction. Further, unlike the formation of poly thioalklylated quinones18,19 in nucleophilic addition of thiol to quinones, under our reaction conditions, only mono thioalkylated quinones were obtained. If it is radical cross-coupling the steric effect did not impact severely on the reactivity. From this view of point, we carried out the reaction with o/m substituted aryl thiophenol (Scheme 2, entry 4m–4n) and desired products obtained in moderately. This result reveals that the steric hindrance did not much influence the reactivity. This clearly shows that no conjugate addition of thiol to quinone. The formation of thiyl radicals from thiol is known.28 As it was mentioned earlier, without the presence of water (Table 1, entries 6 and 7) no reaction took place. Based on the verification experiments and background information, a plausible mechanism is proposed as shown in Scheme 8.
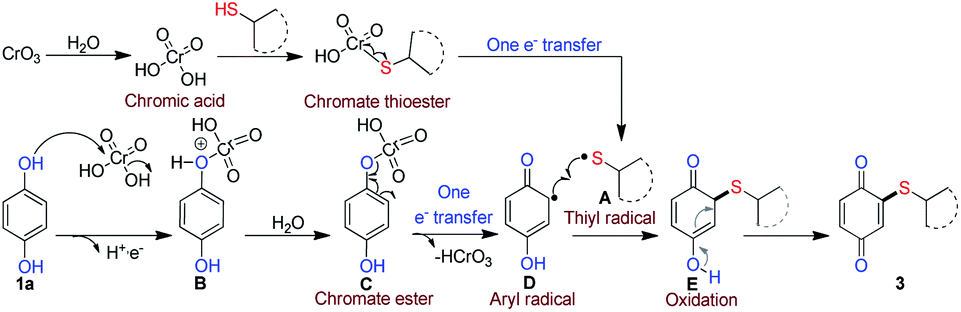 | ||
| Scheme 8 A Plausible mechanism. | ||
CrO3 on reaction with water is expected to produce chromic acid, a powerful oxidant used in controlled oxidation of organic compounds. After the formation of chromate thioester by the reaction between thiol and chromic acid, it is expected to undergo one-electron transfer reaction to afford thiyl radical A. Similarly, hydroquinone is expected to form corresponding chromate ester C via deprotonation of intermediate B, which is then transformed into a phenoxy followed by semi-quinone radical D via oxidation. Further, thiyl radical A and semi-quinone radical D might undergo radical cross-coupling to form intermediate E, which reoxidizes to form more stable quinonyl thioether.
Conclusion
A simple and efficient method for oxidative radical C–H/S–H cross-coupling of hydroquinones with alkyl thiols/thiophenol/amino thiophenol to form quinonyl thioethers in mono-selective fashion under mild condition is demonstrated. A series of quinonyl alkyl/aryl thioethers and phenothiazines-3-ones were obtained in moderate to good yield at rt. To the best of knowledge, this is the first example of C–S bond formation using a chromium reagent such as H2CrO4. Furthermore, in silico analysis of ADME-PK properties of phenothiazines established that compound 5b meets the criteria of drug and lead likeness and also posses good bioavailability. Moreover, it was also observed that compound 5b strongly binds with Alzheimer's disease (AD) associated AChE target.Experimental section
General information
All the reagents were purchased commercially and used without further purification. 1H NMR (400 MHz) and 13C NMR (100 MHz) were recorded with Bruker 400 MHz spectrometer in CDCl3 with tetramethylsilane (TMS) as the internal standard. Multiplicities are reported using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, sep = septet, br = broad resonance. All the NMR spectra were acquired at ambient temperature. Analytical thin-layer chromatography (TLC) was performed using Silica Gel 60 Å F254 pre-coated plates (0.25 mm thickness). Visualization was accomplished by irradiation with a UV lamp and staining with I2 on silica gel. High-resolution mass spectra (HRMS) were recorded on the Thermo Executive Plus spectrometer.General method-A
To a slowly stirred solution of hydroquinone (1.0 equiv.) and a suitable thiols (1.5 equiv.) in DCM:H2O (1:1, 4 mL), followed by which CrO3 (3.0 equiv.) was added portion wise at room temperature. The progress of the reaction was monitored by TLC. Upon the complete consumption of starting materials it was diluted with ethyl acetate and water. The organic phase was separated, extracted with two or more times with ethyl acetate, dried over Na2SO4, filtered and concentrated. The crude product was purified by silica gel column chromatography using hexane/ethyl acetate as eluent to get product. The disposal of chromic acid starts with adding H2SO4 or CaCO3 to the aqueous phase (pH = 1). Further, solid Na2S2O3 was added while stirring until the solution turns blue and cloudy. For MSDS information of CrO3 (http://www.labchem.com/tools/msds/msds/LC13090.pdf).
:10 to get the product as an orange yellow solid, 162.0 mg, 75.0% yield, mp 82–84 °C; 1H NMR (400 MHz, CDCl3): δ = 6.71 (d, J = 10.0 Hz, 1H), 6.63 (d, J = 10.0 Hz, 1H) 6.33 (s, 1H), 4.08–4.05 (q, 2H), 2.99–2.96 (t, 2H), 2.64–2.60 (t, 2H); 1.17–1.14 (s, 3H), 13C NMR (100 MHz, CDCl3): δ = 183.8, 183.6, 170.6, 151.8, 137.4, 136.1, 124.9, 61.0, 32.1, 24.9, 14.1; HRMS (ESI): m/z [M + H]+ calcd for C11H12NaO4S: 263.0354; found: 263.0348.:10 to get the product as an orange yellow solid, 160.2 mg, in 78.0% yield, mp 84–86 °C; 1H NMR (400 MHz, CDCl3): δ = 6.73 (d, J = 10.0 Hz 1H), 6.65 (d, J = 10.0 Hz, 1H) 6.35 (s, 1H), 3.64 (s, 3H), 3.01–2.98 (t, 2H), 2.68–2.64 (t, 2H); 13C NMR (100 MHz, CDCl3): δ = 183.8, 183.6, 171.2, 151.8, 137.4, 136.1, 124.9, 52.1, 31.9, 24.9; HRMS (ESI): m/z [M + H]+ calcd for C10H10NaO4S: 249.0197; found: 249.0200.:10 to get the product as an orange yellow solid, 144.8 mg, 60.0% yield, mp 87–89 °C; 1H NMR (400 MHz, CDCl3): δ = 6.75 (d, J = 10.0 Hz, 1H), 6.68–6.65 (dd, J1 = 10.0 Hz, J2 = 10.0 Hz, 1H) 6.37 (s, 1H), 4.07–4.04 (t, 2H), 3.02–3.00 (t, 2H), 2.68–2.65 (t, 2H), 1.59–1.52 (m, 2H), 1.35–1.26 (m, 2H), 0.88–0.85 (t, 3H); 13C NMR (100 MHz, CDCl3): δ = 183.8, 183.6, 170.8, 151.9, 137.4, 136.1, 124.9, 65.4, 32.2, 30.5, 25.0, 19.0, 13.7; HRMS (ESI): m/z [M + H]+ calcd for C13H16O4S: 268.0269; found: 268.0774.:10 to get the product as an orange yellow solid, 132.0 mg, 52.0% yield, mp 86–88 °C; 1H NMR (400 MHz, CDCl3): δ = 6.76 (d, J = 10.0 Hz, 1H), 6.76 (d, J = 10.0 Hz, 1H), 6.38 (d, J = 1.6 Hz, 1H), 4.07–4.04 (t, 2H), 3.04–3.00 (t, 2H), 2.69–2.65 (t, 2H), 1.58 (s, 2H), 1.28 (d, 4H), 0.89 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 183.8, 183.6, 170.7, 151.9, 137.4, 136.1, 124.9, 65.4, 32.2, 28.2, 27.9, 25.1, 22.2, 13.9; HRMS (ESI): m/z [M + H]+ calcd for C14H18NaO4S: 305.0823; found: 305.0824.:10 to get the product as an orange yellow solid, 127.9 mg, 48.0% yield, mp 87–89 °C; 1H NMR (400 MHz, CDCl3): δ = 6.79 (d, J = 10.0 Hz, 1H), 6.72–6.69 (dd, J1 = 10.0 Hz, J2 = 10.0 Hz, 1H), 6.40 (s, 1H), 4.10–4.07 (t, 2H), 3.06–3.02 (t, 2H), 2.72–2.68 (t, 2H), 1.64–1.57 (m, 2H), 1.29 (s, 6H), 0.87–0.84 (t, 3H); 13C NMR (100 MHz, CDCl3): δ = 183.8, 183.6, 170.8, 152.0, 137.4, 136.1, 124.9, 65.4, 32.2, 31.4, 28.5, 25.5, 25.1, 22.5, 13.9; HRMS (ESI): m/z [M + H]+ calcd for C15H20NaO4S: 319.0980; found: 319.0973.:10 to get the product as an orange yellow solid, 194.4 mg, 85.0% yield, mp 81–83 °C; 1H NMR (400 MHz, CDCl3): δ = 7.10 (d, J = 10.0 Hz, 1H), 7.01 (d, J = 10.0 Hz, 1H), 6.71 (s, 1H), 5.36–5.27 (m, 1H), 3.36–3.33 (t, 2H), 2.99–2.96 (t, 2H), 1.53–1.52 (d, 6H); 13C NMR (100 MHz, CDCl3): δ = 183.8, 183.6, 170.2, 151.9, 137.4, 136.1, 124.9, 32.4, 29.6, 25.0, 21.7; HRMS (ESI): m/z [M + H]+ calcd for C12H14NaO4S: 277.0510; found: 277.0502.:10 to get the product as an orange yellow solid, 199.5 mg, 82.0% yield, mp 76–78 °C; 1H NMR (400 MHz, CDCl3): δ = 6.59 (d, J = 10.0 Hz, 1H), 6.50 (d, J = 10.0 Hz, 1H), 6.21 (s, 1H), 4.03–4.00 (t, 2H), 3.36–3.34 (t, 2H), 3.11 (s, 3H), 2.87–2.84 (t, 2H), 2.56–2.53 (m, 2H); 13C NMR (100 MHz, CDCl3): δ = 183.7, 183.6, 170.7, 151.6, 137.2, 136.0, 124.9, 70.0, 63.8, 58.6, 31.8, 24.7; HRMS (ESI): m/z [M + H]+ calcd for C12H14NaO4S: 293.0460; found: 293.0451.:10 to get the product as an orange yellow solid, 223.0 mg, 82.0% yield, mp 82–84 °C; 1H NMR (400 MHz, CDCl3): δ = 7.24–7.21 (m, 5H), 6.66 (d, J = 10.0 Hz, 1H), 6.58 (d, J = 10.0 Hz, 1H), 6.30 (s, 1H), 5.03 (s, 2H), 2.97–2.93 (t, 2H), 2.67–2.64 (t, 2H); 13C NMR (100 MHz, CDCl3): δ = 183.8, 183.7, 170.7, 151.8, 137.4, 136.1, 135.5, 128.7, 128.5, 128.4, 66.9, 32.2, 24.9; HRMS (ESI): m/z [M + H]+ calcd for C16H14NaO4S: 325.0510; found: 325.0514.:10 to get the product as an orange yellow semi solid, 118.8 mg, 40.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.29–7.16 (m, 5H), 6.80 (d, J = 10.0 Hz, 1H), 6.71 (d, J = 10.0 Hz, 1H), 6.41 (s, 1H), 4.16–4.13 (t, 2H), 3.05–3.01 (t, 2H), 2.71–2.66 (q, 4H), 1.99–1.96 (t, 2H); 13C NMR (100 MHz, CDCl3): δ = 183.8, 183.7, 170.7, 151.9, 141.0, 137.4, 136.2, 128.5, 126.1, 124.9, 64.6, 32.2, 32.1, 30.0, 25.0; HRMS (ESI): m/z [M + H]+ calcd for C18H18NaO4S: 353.0823; found: 353.0816.:10 to get the product as an orange yellow solid, 79.5 mg, 45.0% yield, mp 80–82 °C; 1H NMR (400 MHz, CDCl3): δ = 7.11 (d, J = 10.0 Hz, 1H), 7.05–7.02 (dd, J1 = 10.0 Hz, J2 = 10.0 Hz, 1H), 6.69 (s, 1H), 3.10–3.06 (t, 2H), 2.06–1.99 (q, 2H), 1.85–1.77 (q, 2H), 1.28–1.25 (t, 3H); 13C NMR (100 MHz, CDCl3): δ = 183.9, 183.8, 153.1, 137.4, 136.1, 124.7, 30.1, 29.2, 22.1, 13.5.:10, orange red solid, 150.0 mg, 85.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.50 (s, 5H), 6.83 (d, J = 10.4 Hz, 1H), 6.70–6.66 (dd, J1 = 10.4 Hz, J2 = 10.4 Hz, 1H), 5.88 (d, J = 2.4 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 184.5, 183.9, 154.5, 137.5, 135.8, 135.6, 130.6, 130.4, 126.9, 125.9. The spectral data of the compound 4a was complies with the values reported in the literature.29:10 to get the product as an orange red solid, 165.6 mg, 80.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.38 (d, J = 8.4 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 6.81 (d, J = 10.0 Hz, 1H), 6.68–6.65 (dd, J1 = 10.0 Hz, J2 = 10.0 Hz, 1H), 5.87 (d, J = 2.4 Hz, 1H), 2.4 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 184.5, 184.1, 154.9, 141.1, 137.5, 135.5, 131.2, 125.8, 123.2, 21.4. The spectral data of the compound 4b was complies with the values reported in the literature.29:10 to get the product as an orange red solid, 168.3 mg, 76.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.37 (d, J = 8.8 Hz, 2H), 6.98 (d, J = 8.8 Hz, 2H), 6.78 (d, J = 10.0 Hz, 1H), 6.66–6.63 (dd, J1 = 10.0 Hz, J2 = 10.0 Hz, 1H), 5.84 (d, J = 2.4 Hz, 1H), 3.84 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 184.5, 184.1, 161.5, 155.3, 137.5, 137.1, 135.9, 125.8, 116.9, 116.0, 55.5. The spectral data of the compound 4c was complies with the values reported in the literature.29:10 to get the product as a yellow solid, 165.0 mg, 70.0% yield; 1H NMR (400 MHz, CDCl3): δ = 8.34 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 6.88 (d, J = 10.0 Hz, 1H), 6.74 (d, J = 10.0 Hz, 1H), 5.95 (s, 1H); 13C NMR (100 MHz, CDCl3): δ = 184.0, 183.2, 152.3, 149.0, 137.5, 136.3, 135.9, 135.7, 126.7, 125.3, 124.1. The spectral data of the compound 4d was complies with the values reported in the literature.29:10 to get the product as an yellow solid, 92.3 mg, 41.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.50–7.43 (dd, J1 = 8.4 Hz, J2 = 8.4 Hz, 2H), 6.85 (d, J = 10.0 Hz, 2H), 6.71–6.68 (dd, J1 = 10.0 Hz, J2 = 10.0 Hz, 1H), 5.87 (d, J = 2.0 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 184.3, 183.8, 153.9, 137.5, 137.3, 136.9, 135.8, 130.7, 126.0, 125.3. The spectral data of the compound 4e was complies with the values reported in the literature.29:10 to get the product as an orange solid, 113.8 mg, 43.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.65–7.61 (m, 2H), 7.39–7.36 (m, 2H), 6.84 (d, J = 10.0 Hz, 1H), 6.72–6.68 (dd, J1 = 10.0 Hz, J2 = 10.0 Hz, 1H), 5.88 (d, J = 2.4 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 184.3, 183.7, 153.8, 137.5, 137.2, 135.9, 133.7, 126.1, 126.0, 125.9, 125.6. The spectral data of the compound 4f was complies with the values reported in the literature.29:10 to get the product as an orange solid, 84.3 mg, 40.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.51–7.48 (q, 2H), 7.22–7.18 (t, 2H), 6.83 (d, J = 10.0 Hz, 1H), 6.71–6.68 (dd, J1 = 10.0 Hz, J2 = 10.0 Hz, 1H), 5.85 (d, J = 2.4 Hz, 1H); 13C NMR (100 MHz, CDCl3): δ = 184.4, 183.9, 165.9, 162.9, 154.4, 137.9, 137.8, 137.5, 125.9, 117.9, 117.7. The spectral data of the compound 4g was complies with the values reported in the literature.29:10 to get the product as an orange red solid, 125.8 mg, 68.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.36 (d, J = 8.0, 2H), 7.28 (d, J = 8.0, 2H), 5.97 (s, 1H), 5.78 (s, 1H), 3.84 (s, 3H), 2.41 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 184.8, 184.0, 178.8, 159.6, 157.0. 141.1, 135.5, 135.4, 131.2, 123.4, 107.6, 106.7, 56.6, 21.4.:10 to get the product as an orange red solid, 77.8 mg, 44.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.33 (d, J = 8.0, 2H), 7.15 (d, J = 8.0, 2H), 6.79 (d, J = 8.0 1H), 6.72 (d, J = 8.0, 1H), 2.35 (s, 3H), 2.12 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 198.8, 183.5, 183.1, 145.4, 142.6, 139.9, 136.7, 136.5, 134.3, 130.3, 126.3, 31.3, 21.4.:10 to get the product as an orange yellow solid, 101.3 mg, 45.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.52–7.41 (m, 4H), 6.84 (d, J = 10, 1H), 6.72–6.69 (dd, J1 = 2.4, J2 = 2.4, 1H), 5.90 (d, J1 = 2.4, 1H); 13C NMR (100 MHz, CDCl3): δ = 184.3, 183.6, 153.6, 137.5, 135.9, 135.3, 133.8, 131.9, 130.9, 128.8, 126.1.:10 to get the product as an orange yellow solid, 112.5 mg, 48.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.55–7.51 (m, 2H), 7.29–7.23 (m, 2H), 6.85 (d, J = 10, 1H), 6.72–6.69 (dd, J1 = 2.4, J2 = 2.4, 1H), 5.88 (q, 1H); 13C NMR (100 MHz, CDCl3): δ = 184.3, 183.7, 163.9, 161.4, 151.7, 137.5, 137.4, 135.9, 133.5, 133.4, 126.0, 125.9, 125.8, 117.2, 116.9, 114.2, 114.0.:10, to get the product as an orange red solid, 190.1 mg, 60.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.36 (d, J = 8.0, 4H), 7.27–7.25 (t, 4H), 5.57 (s, 2H), 2.39 (s, 6H); 13C NMR (100 MHz, CDCl3): δ = 182.4, 181.3, 154.0, 141.1, 135.5, 131.2, 126.5, 123.5, 21.4.:10 to get the product as a red solid, 177.8 mg, 80.0% yield; 1H NMR (400 MHz, CDCl3): δ = 7.90 (d, 1H), 7.60 (d, 1H), 7.44–7.37 (m, 2H), 6.95–6.75 (m, 1H), 6.74 (s, 1H); 13C NMR (100 MHz, CDCl3): δ = 182.4, 147.5, 139.9, 139.7, 135.6, 134.6, 133.4, 133.1, 131.0, 125.9, 121.9, 120.5.:10 to get the product as a red solid, 177.0 mg, 70.0% yield, mp 76–78 °C; 1H NMR (400 MHz, CDCl3): δ = 8.13 (s, 1H), 7.67–7.54 (m, 3H), 6.97–6.76 (m, 1H), 6.76 (s, 1H); 13C NMR (100 MHz, CDCl3): δ = 182.4, 147.8, 139.8, 139.1, 135.8, 134.1, 130.8, 130.7, 127.5, 126.9, 126.8, 125.8, 121.1.:10, orange red solid, trace yield; 1H NMR (400 MHz, CDCl3): δ = 7.85 (d, 1H), 7.50 (s, 1H), 7.14–7.33 (m, 2H), 6.66 (s, 1H), 1.38 (s, 9H).:10 to get the product as a yellow solid, 90.0 mg, 91.0% yield; 1H NMR (400 MHz, CDCl3): δ = 6.65 (s, 2H), 6.56 (2, 1H), 1.25 (s, 9H); 13C NMR (100 MHz, CDCl3): δ = 188.4, 187.4, 156.0, 138.7, 135.0, 131.5, 35.3, 29.2, 29.0.:10 to get the product as a yellow solid, 80.5 mg, 56.0% yield; 1H NMR (400 MHz, CDCl3): δ = 8.08–8.04 (m, 2H), 7.70–7.67 (t, 2H), 3.80–3.78 (t, 2H), 3.35–3.32 (t, 2H), 2.39 (s, 3H); 13C NMR (100 MHz, CDCl3): δ = 182.3, 181.6, 148.3, 145.7, 133.8, 133.5, 132.7, 132.0, 126.9, 126.7, 62.0, 37.3, 15.5. The spectral data of the compound 7a was complies with the values reported in the literature.24Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
AKTP and SP thanks UGC-RFSMS, New Delhi for the award of the fellowship. We thank DST-FIST and DST-PURSE New Delhi, India for NMR and HRMS facilities at School of Chemistry, Bharathidasan University, Tiruchirappalli, India.References
- (a) G. A. Kraus and J. Mengwasser, Molecules, 2009, 14, 2857–2861 CrossRef CAS PubMed; (b) A. E. Wendlandt and S. S. Stahl, Angew. Chem., Int. Ed., 2015, 54, 14638–14658 CrossRef CAS PubMed; (c) B. Hosamani, M. F. Ribeiro, E. N. da Silva Junior and I. N. N. Namboothiri, Org. Biomol. Chem., 2016, 14, 6913–6931 RSC; (d) C. Song, X. Dong, H. Yi, C. W. Chiang and A. Lei, ACS Catal., 2018, 8, 2195–2199 CrossRef CAS.
- (a) P. R. Dandawate, A. C. Vyas, S. B. Padhye, M. W. Singh and J. B. Baruah, Mini-Rev. Med. Chem., 2010, 10, 436–454 CrossRef CAS PubMed; (b) M. K. Hadden, S. A. Hill, J. Davenport, R. L. Matts and B. S. J. Blagg, Bioorg. Med. Chem., 2009, 17, 634–640 CrossRef CAS PubMed.
- (a) J. Madeo, A. Zubair and F. Marianne, SpringerPlus, 2013, 2(139), 1–8 Search PubMed; (b) S. Park, E. Yun, I. H. Hwanf, S. Yoon, D. E. Kim, M. Na, G. Y. Song and S. Oh, Mar. Drugs, 2014, 12, 3231–3244 CrossRef CAS PubMed; (c) M. Arai, T. Kawachi, H. Sato, A. Setiawan and M. Kobayashi, Bioorg. Med. Chem. Lett., 2014, 24, 3155–3157 CrossRef CAS PubMed; (d) J. L. Bolton and T. Dunlap, Chem. Res. Toxicol., 2017, 30, 13–37 Search PubMed; (e) P. A. Garcia, A. P. Hernández, A. S. Feliciano and M. A. Castro, Mar. Drugs, 2018, 16(292), 1–51 Search PubMed.
- (a) V. J. Litchfield, R. B. Smith, A. M. Franklin and J. Davis, Synth. Commun., 2008, 38, 3447–3455 CrossRef CAS; (b) S. Er, C. Suh, M. P. Marshaka and A. A. Guzik, Chem. Sci., 2015, 6, 885–893 RSC; (c) E. J. Son, J. H. Kim, K. Kim and C. B. Park, J. Mater. Chem. A, 2016, 4, 11179–11202 RSC; (d) Y. Ding, Y. Li and G. Yu, Chem, 2016, 1, 790–801 CrossRef CAS; (e) O. Taran, Front. Chem., 2017, 5(49), 1–13 Search PubMed.
- (a) D. Sladic and M. J. Gasic, Molecules, 2006, 11(1), 1–33 CrossRef CAS PubMed; (b) N. E. Najjar, H. G. Muhtasib, R. A. Ketola, P. Vuorela, A. Urtti and H. Vuorela, Phytochem. Rev., 2011, 10, 353–370 CrossRef; (c) P. S. Guin, S. Das and P. C. Mandal, Int. J. Electrochem., 2011, 816202, 1–22 Search PubMed.
- (a) J. Fourie, C. J. Oleschuk, F. Guziec Jr, L. Guziec, D. J. Filterman, C. Monterrosa and A. Begleiter, Canc. Chemother. Pharmacol., 2002, 49, 101–110 CrossRef CAS PubMed; (b) C. Frontana, A. V. Mayagoitia, J. Garza, R. Vargas and I. Gonzalez, J. Phys. Chem. A, 2006, 110, 9411–9419 CrossRef CAS PubMed; (c) L. Zhou, Y. Chen, X. Yang, Y. Su, W. Zhang and J. Xu, Catal. Lett., 2008, 125, 154–159 CrossRef CAS; (d) X. Yang, Y. Wang, C. Zhang, T. Fang, L. Zhou, W. Zhang and J. Xu, J. Phys. Org. Chem., 2011, 24, 693–697 CrossRef CAS.
- W. Yu, P. Hjerrild, K. M. Jacobsen, H. N. Tobiesen, L. Clemmensen and T. B. Poulsen, Angew. Chem., Int. Ed., 2018, 57, 9805–9809 CrossRef CAS PubMed and references cited therein.
- (a) K. Mozaina, C. L. Cantrell, A. B. Mims, A. R. Lax, M. R. Tellez and W. L. A. Osbrink, J. Agric. Food Chem., 2008, 56, 4021–4026 CrossRef CAS PubMed; (b) C. K. Ryu, S. Y. Lee, N. Y. Kim, J. A. Hong, J. H. Yoon and A. Kim, Bioorg. Med. Chem. Lett., 2011, 21, 427–430 CrossRef CAS PubMed; (c) V. K. Tandon, S. Kumar, N. N. Mishra and P. K. Shukla, Eur. J. Med. Chem., 2012, 56, 375–386 CrossRef CAS PubMed; (d) A. Kacmaz, E. T. Acar, G. Atun, K. Kaya, B. D. Sigirci and F. Bagcigil, ChemistrySelect, 2018, 3, 8615–8623 CrossRef CAS; (e) H. Yıldırım, N. Bayrak, M. Yıldız, E. M. Kara, B. O. Celik and A. F. Tuyun, J. Mol. Struct., 2019, 1195, 681–688 CrossRef.
- Z. Wang, E. C. Southwick, M. Wang, S. Kar, K. S. Rosi, C. S. Wilcox, J. S. Lazo and B. I. Car, Cancer Res., 2001, 61, 7211–7216 CAS.
- (a) H. O. Sjögren, M. Isaksson, D. Willner, I. Hellström, K. E. Hellström and P. A. Cancer, Res, 1997, 57, 4530–4536 Search PubMed; (b) A. M. Keith Walker, A. C. Braemer, S. Hitt, R. E. Jones and T. R. Matthews, J. Med. Chem., 1978, 21, 840–842 CrossRef PubMed; (c) A. J. Sedman, Am. J. Med., 1984, 76, 109–114 CrossRef CAS PubMed; (d) D. M. Livermore, A. M. Sefton and G. M. Scott, J. Antimicrob. Chemother., 2003, 52, 331–344 CrossRef CAS PubMed; (e) X. Y. Zhang, G. S. Liu and J. Yuan, Chin. J. Clin. Rehabil., 2013, 7(24), 3340–3342 Search PubMed; (f) T. F. Luscher, S. Taddei, J. C. Kaski, J. W. Jukema, D. Kallend, T. Munzel, J. J. P. Kastelein and J. E. Deanfield, Eur. Heart J., 2012, 33, 857–865 CrossRef PubMed.
- (a) A. S. Thigpen, S. T. Nestor, R. A. O'Brien, S. Minkowicz, Y. Sheng, J. H. Davis Jr, K. N. West and A. Mirjafari, New J. Chem., 2017, 41, 1625–1630 RSC; (b) A. Bhadani and S. Singh, Langmuir, 2011, 27(23), 14033–14044 CrossRef CAS PubMed.
- (a) T. Kondo and T. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS PubMed and references cited therein; ; (b) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. Andy Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC and references cited therein; For latest papers-; (c) W. H. Bao, C. Wu, J. T. Wang, W. Xia, P. Chen, Z. T. X. Xu and W. M. He, Org. Biomol. Chem., 2018, 16, 8403–8407 RSC; (d) G. S. Sorabad and M. R. Maddani, Asian J. Org. Chem., 2019, 8, 1–9 CrossRef; (e) W. H. Bao, Z. Wang, X. Tang, Y. F. Zhang, J. X. Tana, Q. Zhu, Z. Cao, Y. W. Lin and W. M. He, Chin. Chem. Lett., 2019, 30, 2259–2262 CrossRef CAS; (f) L. Y. Xie, Y. L. Chen, L. Qin, Y. Wen, J. W. Xie, J. X. Tan, Y. Huang, Z. Cao and W. M. He, Org. Chem. Front., 2019, 6, 3950–3955 RSC.
- (a) X. Yu, Q. Wu, H. Wan, Z. Xu, X. Xu and D. Wang, RSC Adv., 2016, 6, 62298–62301 RSC; (b) C. Zhang, J. Mc-Clure and C. J. Chou, J. Org. Chem., 2015, 80, 4919–4927 CrossRef CAS PubMed.
- (a) D. Wang, X. Yu, L. Wang, W. Yao, Z. Xu and H. Wan, Tetrahedron Lett., 2016, 57, 5211–5214 CrossRef CAS; (b) D. Wang, X. Yu, W. Yao, W. Hu, C. Ge and X. Shi, Chem. - Eur. J., 2016, 22, 5543–5546 CrossRef CAS PubMed.
- S. Rajasekar, T. P. A. Krishna, N. Tharmalingam, A. Ilangovan and E. Mylonakis, ChemistrySelect, 2019, 4, 2281–2287 CrossRef CAS.
- F. L. Zeng, X. L. Chen, S. Q. He, K. Sun, Y. Liu, R. Fu, L. B. Qu, Y. F. Zhao and B. Yu, Org. Chem. Front., 2019, 6, 1476–1480 RSC.
- (a) V. K. Tandon and H. K. Maurya, Tetrahedron Lett., 2009, 50, 5896–5902 CrossRef CAS; (b) V. K. Tandon, H. K. Maurya, S. Kumar, A. Rashid and D. Panda, RSC Adv., 2014, 4, 12441–12447 RSC.
- (a) I. H. Spinner, W. D. Raper and W. Metanomski, Can. J. Chem., 1963, 41, 483–494 CrossRef; (b) A. R. Katritzky, D. Fedoseyenkoa, P. P. Mohapatraa and P. J. Steel, Synthesis, 2008, 5, 777–787 CrossRef; (c) V. K. Tandona, D. B. Yadava, R. V. Singh, M. Vaisha, A. K. Chaturvedi and P. K. Shukla, Bioorg. Med. Chem. Lett., 2005, 15, 3463–3466 CrossRef PubMed; (d) D. E. Allgeier, S. A. Herbert, R. Nee, K. D. Schlecht and K. T. Finley, J. Org. Chem., 2003, 68, 4988–4990 CrossRef CAS PubMed.
- (a) Y. Lu, Y. Zhao, S. Wang, X. Wang, Z. Ge and R. Li, RSC Adv., 2016, 6, 11378–11381 RSC; (b) K. W. Wellington, G. E. Gordon, L. A. Ndlovu and P. Steenkamp, ChemCatChem, 2013, 5, 1570–1577 CrossRef CAS; (c) K. W. Wellington, R. Bokako, N. Raseroka and P. Steenkamp, Green Chem., 2012, 14, 2567–2576 RSC; (d) M. Schlippert, A. Mikolasch, V. Hahn and F. Schauer, J. Mol. Catal. B Enzym., 2016, 126, 106–114 CrossRef CAS.
- (a) X. Zeng and X. Cong, Org. Chem. Front., 2015, 2, 69–72 RSC; (b) M. Yadava and X. Qiang, Chem. Commun., 2013, 49, 3327–3329 RSC; (c) A. Levina, L. Zhang and P. A. Lay, J. Am. Chem. Soc., 2010, 132, 8720–8731 CrossRef CAS PubMed; (d) M. Juaristi, J. M. Aizpurua, B. Lecea and C. Palomo, Can. J. Chem., 1984, 62, 2941–2944 CrossRef CAS.
- (a) A. Ilangovan, S. Saravanakumar and S. Malayappasamy, Org. Lett., 2013, 15(19), 4968–4971 CrossRef CAS PubMed; (b) A. Ilangovan, A. Polu and G. Satish, Org. Chem. Front., 2015, 12, 1616–1620 RSC; (c) A. Polu and A. Ilangovan, Tetrahedron Lett., 2018, 59, 438–441 CrossRef; (d) T. P. A. Krishna, P. Sakthivel and A. Ilangovan, Org. Chem. Front., 2019, 6, 3244–3251 RSC; (e) P. Sakthivel, T. P. A. Krishna and A. Ilangovan, Org. Biomol. Chem., 2020, 18, 3027–3031 RSC.
- Y. Hong, G. Zhang, H. Wang, Z. Huang, J. Wang, A. K. Singh and A. Lei, Chem. Rev., 2017, 117, 9016–9085 CrossRef PubMed and references cited therein.
- Z. Huang, D. Zhang, X. Qi, Z. Yan, M. Wang, H. Yan and A. Lei, Org. Lett., 2016, 18, 2351–2354 CrossRef CAS PubMed.
- Y. Nishikawa, Z. Wang, J. Kerns, C. S. Wilcox and B. I. Carr, J. Biol. Chem., 1999, 274, 34803–34810 CrossRef CAS PubMed.
- (a) V. Venkatraman, M. Foscato, V. R. Jensenb and B. K. Alsberg, J. Mater. Chem. A, 2015, 3, 9851–9860 RSC; (b) Y. Liaoa, P. Jianga, S. Chena, F. Xiaoa and G. J. Deng, RSC Adv., 2013, 3, 18605–18608 RSC.
- (a) A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed; (b) T. Sander, J. Freyss, M. Von-Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed; (c) A. Daina and V. Zoete, Curr. Med. Chem., 2016, 11(11), 1117–1121 CAS.
- J. R. Greenwood, D. Calkins, A. P. Sullivan and J. C. Shelley, J. Comput. Aided Mol. Des., 2010, 24, 591–604 CrossRef CAS PubMed.
- F. Denes, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 CrossRef CAS PubMed and cited therein.
- D. Wang, X. Yu, L. Wang, W. Yao, Z. Xu and H. Wan, Tetrahedron Lett., 2016, 57, 5211–5214 CrossRef CAS and references cited therein.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, spectral data and copies of spectra. See DOI: 10.1039/d0ra01519a |
| This journal is © The Royal Society of Chemistry 2020 |