
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphate-sensing with (di-(2-picolyl)amino)quinazolines based on a fluorescence on–off system†
Kazusa Aoki,
Ryuji Osako,
Jiahui Deng,
Takashi Hayashita ,
Takeshi Hashimoto and
Yumiko Suzuki*
,
Takeshi Hashimoto and
Yumiko Suzuki*
Department of Materials and Life Sciences, Faculty of Science and Technology, Sophia University, Kioi-cho 7-1, Chiyoda-ku, Tokyo 102-8554, Japan. E-mail: yumiko_suzuki@sophia.ac.jp
First published on 17th April 2020
Abstract
Detection and visualization of phosphates such as ATP in living organisms can facilitate the elucidation of various biological events. Although substantial efforts had been made in this area, present methods have disadvantages such as the need for specialized equipment and poor sensitivities. To address these limitations, novel fluorescent probes, (di-(2-picolyl)amino)quinazolines, were developed for application in ATP detection. They selectively recognized copper ions by fluorescence quenching, and their copper complexes displayed fluorescence enhancement in the presence of phosphoric acid derivatives. This fluorescence on–off system enabled highly sensitive fluorescence detection of ATP when combined with a phenyl boronic acid-modified γ-cyclodextrin through a plausible multipoint recognition system.
Introduction
Phosphates typified by adenosine 5′-triphosphate (ATP) are essential for life and are involved in various biochemical pathways. Therefore, real-time monitoring, visualization, or quantification of phosphate levels, can potentially aid in the elucidation of various biological events and disease mechanisms. The reported approaches for the detection of ATP require specialized and/or costly equipment such as HPLC,1 NMR,2 or micro-fluidic chips.3 To address these limitations, various fluorescent probes such as low molecular weight,4–6 polymer,7,8 and supramolecular sensors9 that enable simpler and efficient detection have been developed. However, these existing strategies have disadvantages such as the necessity for expensive supported metals or proteins, and also suffer from poor sensitivities and selectivities. To further address these challenges, we conducted a study for the development of novel strategies for fluorescence detection of phosphates using a combination of quinazoline fluorophores possessing di-2-(picolyl)amine (dpa) and cyclodextrins (CyDs).Dpa can complex with various metals, and the dpa-bound fluorophores display diverse fluorescence responses depending on the identity of the complexed metal species.10 Furthermore, the interaction between dpa–metal complexes and ATP weakens the dpa–metal coordination, which results in changes in the fluorescence of the molecule, which is critical for our design. In fact, dpa–metal complexes are well-known for their recognition of phosphoric acid derivatives, including ATP.11–16
CyDs are well-known water-soluble host molecules comprising nano-sized hydrophobic cavities that enable them to incorporate various organic compounds in aqueous solutions.17 In addition, the structural modifications of CyDs are straightforward, which allow the incorporation of a variety of functionalities for modulating their properties.18,19 These beneficial properties of CyDs have attracted significant attention for use in various avenues of science and technology, such as in molecular recognition systems,20,21 supramolecular chemistry,22,23 and drug delivery systems24 based on their host–guest interactions. We anticipated that the combination of CyDs and quinazoline fluorophores, recently discovered by our group,25 could potentially be employed for the development of a highly sensitive phosphate recognition system. We report herein, a novel recognition system for phosphate detection that is based on the (di-(2-picolyl)amino)quinazoline and phenylboronic acid-modified γ-cyclodextrin platforms.
The starting point for this study was the design and synthesis of the quinazoline derivatives dpa-QZ1 and dpa-QZ2, bearing a di-(2-picolyl)amine (dpa) moiety at position 2 of the quinazoline framework as probes for phosphate recognition (Scheme 1).
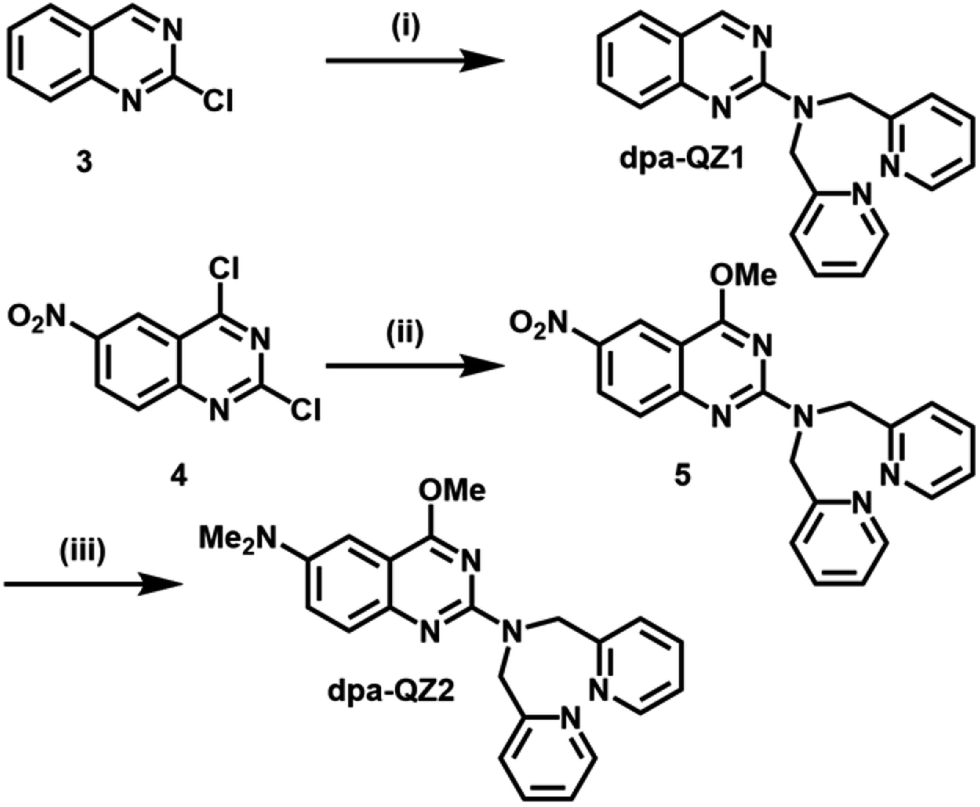 | ||
| Scheme 1 Synthesis of fluorescent quinazolines dpa-QZ1 and dpa-QZ2. (i) dpa, TEA, THF, reflux, 1 week, 70%. (ii) NaOMe, MeOH, 0 °C, 23 h; 20 °C, 3 h; dpa, reflux 4 h, 68%. (iii) H2, Pd/C (10 mol%), MeOH, r.t., 2 h; HCOOH, HCHO, reflux, 2 h, 29%. dpa = di-(2-picolyl)amine, TEA = trimethylamine. | ||
Experimental
Materials and instrument
Chemicals were supplied by WAKO Pure Chemical Industries, Ltd., Kanto Chemical Co. INC, Tokyo Chemical Industry Co., Ltd., or Sigma-Aldrich, and were used without purification. Column chromatography was performed on Merck Silica Gel 60 or Merck aluminium oxide using chloroform, ethyl acetate and n-hexane as eluents. TLC experiments were carried out on Merck Silica Gel 60 F254 plates for Merck Silica Gel 60 and on Merck aluminium oxide 60 F254 plates for Merck aluminium oxide. Structural identification of the synthesized compounds was confirmed by 1H-NMR, 13C-NMR, HRMS spectroscopy, and elemental analysis. The 1H or 13C nuclear magnetic resonance (NMR) spectra were recorded on a JEOL JNM-ECA500 NMR spectrometer (500 MHz) using TMS and CDCl3 or DMSO-d6 as internal standards. The 1H-NMR and 13C-NMR spectral data are reported as follows: chemical shift (ppm), integration, multiplicity (s. singlet; d, doublet; t, triplet; q, quartet, m, multiplet), coupling constants (J) in Hz. Mass spectra were recorded using a TOF (ESI) analyzer or a magnetic sector (EI and FAB) analyzer. Elemental analysis was conducted with a PerkinElmer PE2400 II apparatus. Melting points were measured using a ATM-02, AS ONE. UV-Vis absorption spectra in solution were recorded at room temperature with a Hitachi U-3900. Fluorescence spectra in solution were recorded at room temperature with a Hitachi F-7000. A quart cuvette with a 1 cm path length was used. Water was doubly distilled and deionized with a Mill-Q water system (WG222, Yamato Sci. Co. Ltd. and Autopure WR-600G, Millipore).Preparation of fluorescent quinazolines
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 n-hexane/ethyl acetate, then 1:2) to obtain the desired product (698 mg, 2.13 mmol, 70%) as a yellow solid.
:1 n-hexane/ethyl acetate, then 1:2) to obtain the desired product (698 mg, 2.13 mmol, 70%) as a yellow solid.Rf = 0.33 (2:1 n-hexane/ethyl acetate, Al2O3 plate).
Mp 118–120 °C.
Yellow needles, recrystallized from dichloromethane/hexane.
1H-NMR (CDCl3, 500 MHz): δ: 9.04 (1H, s), 8.54 (2H, m), 7.66 (2H, t, J = 9.2 Hz), 7.56–7.59 (3H, m), 7.30 (2H, s), 7.23 (1H, t, J = 6.5 Hz), 7.14 (2H, t, J = 6.5 Hz), 5.23 (4H, s).
13C-NMR (CDCl3, 500 MHz): δ: 161.8, 159.5, 158.7, 152.4, 149.4, 136.5, 134.0, 127.3, 126.0, 122.6, 121.9, 121.4, 121.3, 119.9, 52.6.
HRMS (FAB) m/z calcd for C20H17N5 (M − H)−: 328.1562, found: 328.1551.
Anal. calcd for C20H17N5: C, 73.37%; H, 5.23%; N, 21.39%, found: C, 73.46%; H, 5.26%; N, 21.35%.
:1 n-hexane/ethyl acetate) to obtain the desired product (1.68 g, 68%) as a yellow solid.Rf = 0.67 (1:1 n-hexane/ethyl acetate, Al2O3 plate).
Mp 149–150 °C.
Yellow needles, recrystallized from dichloromethane/hexane.
1H-NMR (500 MHz, CDCl3) δ: 8.84 (1H, d, J = 2.3 Hz), 8.56 (2H, s), 8.34 (1H, dd, J = 9.2, 2.3 Hz), 7.60–7.65 (2H, m), 7.50 (1H, d, J = 9.2 Hz), 7.40 (1H, d, J = 8.0 Hz), 7.16–7.23 (3H, m), 5.27 (2H, s), 5.16 (2H, s), 3.97 (3H, s).
13C-NMR (126 MHz, CDCl3) δ: 168.2, 160.5, 158.2, 157.7, 157.0, 149.4, 141.3, 136.6, 127.5, 126.1, 122.2, 122.1, 121.5, 120.9, 110.4, 54.4, 53.1, 52.9.
HRMS (FAB) m/z calcd for C21H19N6O3 (M + H)+: 403.1519, found: 403.1533.
:1 n-hexane/ethyl acetate) to obtain the desired product (145 mg, 29%) as a yellow solid.Rf = 0.35 (1:1 n-hexane/ethyl acetate, Al2O3 plate).
Mp 136–137 °C.
Yellow prisms, recrystallized from dichloromethane/hexane.
1H-NMR (500 MHz, CDCl3) δ: 8.53 (2H, d, J = 4.5 Hz), 7.57 (2H, td, J = 7.2, 1.5 Hz), 7.47 (1H, d, J = 9.2 Hz), 7.31 (3H, dd, J = 9.2, 2.9 Hz), 7.12 (2H, m), 7.06 (1H, d, J = 2.9 Hz), 5.16 (4H, s), 3.88 (3H, s), 2.97 (6H, s).
13C-NMR (126 MHz, CDCl3) δ: 166.5, 159.5, 156.8, 149.1, 146.8, 146.3, 136.4, 126.0, 123.3, 121.7, 121.3, 112.0, 103.1, 53.6, 52.8, 41.4.
HRMS (FAB) m/z calcd for C23H24N6O (M)+: 400.2012, found: 400.2017.
Anal. calcd for C23H24N6O: C, 68.98%; H, 6.04%; N, 20.99%, found: C, 68.97%; H, 5.79%; N, 20.87%.
1H-NMR (300 MHz, D2O) δ: 7.8 (t, 1H), 7.62 (d, 1H), 7.5 (d, 1H), 5.2–5.0 (m, 8H), 4.05–3.45 (m, 48H).
Anal. calcd for BC55H85FNO42·5.0H2O: C, 40.23%; H, 6.44%; N, 0.85%, found: C, 40.20%; H, 6.46%; N, 1.01%.
1H-NMR (300 MHz, D2O) δ: 8.2–7.2 (m, 3H), 5.25–4.9 (m, 3H), 4.4–3.2 (m, 42H).
Anal. calcd for BC49H75F NO37·6.5H2O: C, 41.53%; H, 6.26%; N, 0.99%, found: C, 41.48%; H, 6.06%; N, 1.01%.
Preparation of solutions for measurements
Results and discussion
First, the effect of acidity on the fluorescence of dpa-QZ1 and dpa-QZ2 were investigated, and both these probes exhibited weak fluorescence under acidic condition. The fluorescence intensities increased gradually with the continuous addition of the sodium hydroxide solution and reached a maximum around pH 7 (Fig. S1, ESI†). These observations can be attributed to the protonation of the nitrogen atoms of the dpa unit under acidic conditions, which cause the photo-induced electron transfer (PET) from the quinazoline fluorophore to the dpa moiety, leading to fluorescence quenching. As the pH increased, the nitrogen atom of the dpa unit underwent deprotonation, which resulted in the fluorescence recovery by PET suppression.Next, the responses of the probes to the presence of various metals were investigated by the addition of the metal ion—Cu2+, Co2+, Ni2+, Zn2+, Pb2+, and Cd2+ containing salts [metal (NO3)2] into the aqueous solutions of dpa-QZ1 and dpa-QZ2 at pH 7.4. Remarkably, the fluorescence of dpa-QZ1 and dpa-QZ2 was quenched by the addition of Ni2+ and Cu2+. Particularly, the fluorescence intensity decreased significantly with the addition of Cu2+ (Fig. 1). These results indicate that the fluorescence is quenched in the presence of the Ni2+ or Cu2+ ions due to the ligand-to-metal charge transfer (LMCT) from the excited state of the quinazoline ligands to metal ions. The difference between the fluorescence response to Ni2+ and Cu2+ could be attributed to the difference of the binding affinities of dpa to each metal ion due to difference in the ion size.
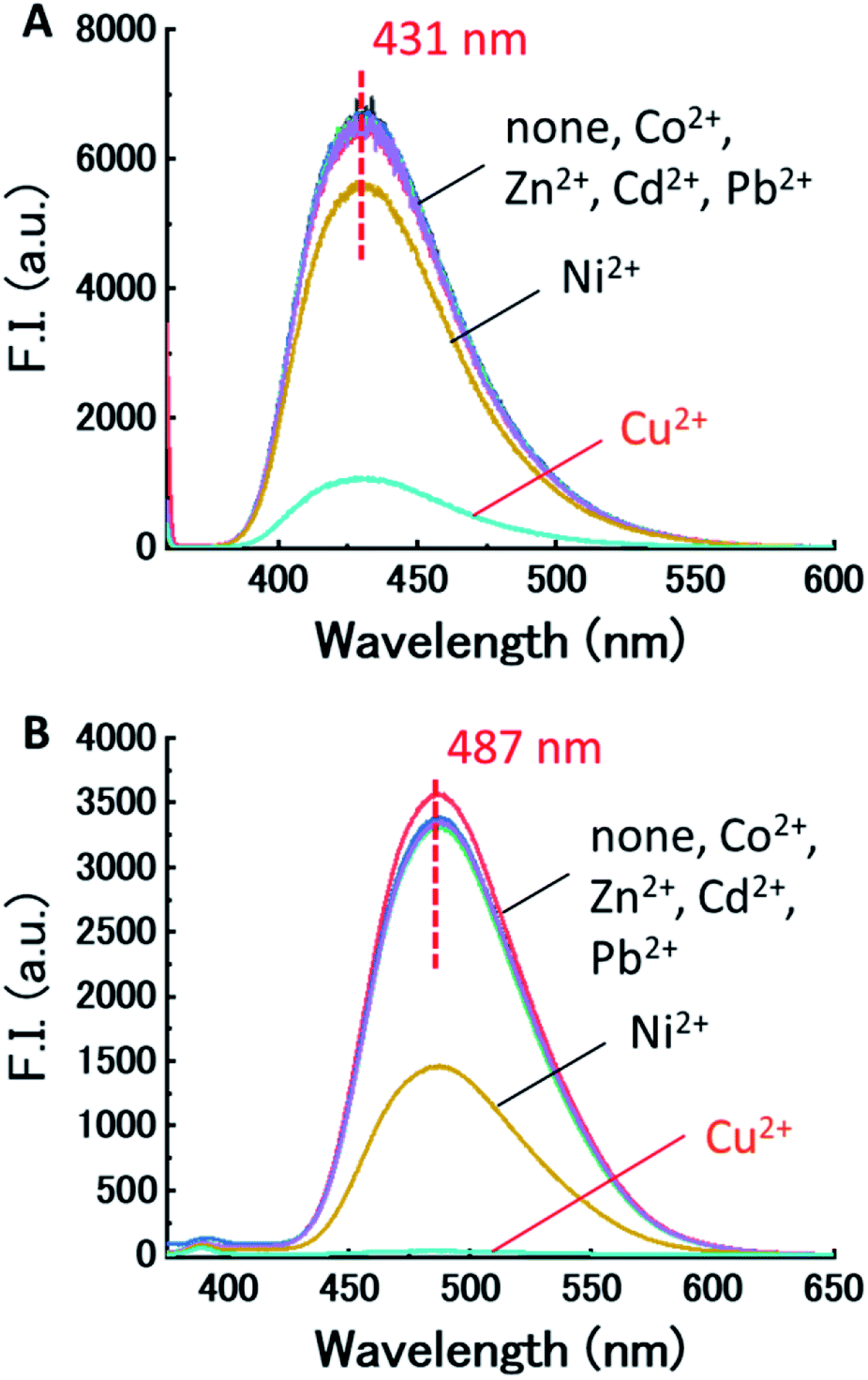 | ||
| Fig. 1 (A) Fluorescence spectra of (A) dpa-QZ1 and (B) dpa-QZ2 with various metals in 1% DMSO – 99% water (v/v), pH 7.4 adjusted using HEPES/NaOH buffer, at 25 °C (λ1ex = 355 nm, λdpa-QZ2ex = 345 nm). [probe] = 10 μM, [metal(NO3)2] = 20 μM, [NaNO3] = 0.10 M, [HEPES buffer] = 5.0 mM. | ||
The binding constants of the quinozaline probes to Cu2+ were calculated by the curve fitting method (5.7 × 105 M−1 for dpa-QZ1 and 1.1 × 107 M−1 for dpaQZ2; Fig. S2, ESI†), and indicated the formation of complexes in a 1:1 ratio with the copper ions. Because dpa-QZ2 has electron donating groups attaching to the quinazoline framework, methoxy group at the position 4 and di-methylamino group at the position 6, the binding capability of dpa-QZ2 to Cu2+ is higher than that of dpa-QZ1. The difference of the fluorescence response to Ni2+ between these two probes is also due to the difference of the electron density of dpa group. These results prompted us to investigate the use of these dpa–copper complexes (Cu-dpa-QZ1 and Cu-dpa-QZ2) for sensing phosphate anions.
We evaluated the recognition abilities of these two metal complexes in the presence of 100 equivalents of phosphate under investigation, and monophosphate (Pi), pyrophosphate (PPi), triphosphate (Tri), AMP, ADP, and ATP were assayed under these conditions. The fluorescence of Cu-dpa-QZ1 dramatically recovered upon addition of each of the phosphates, except AMP (Fig. 2). The fluorescence of Cu-dpa-QZ2 was fully recovered by the addition of PPi, Tri, and ATP, whereas, it recovered by half upon ADP addition. These results clearly indicate that the interactions between the copper ion and the phosphoric acid derivatives weakened the coordination of the dpa moieties to Cu2+, which caused the fluorescence recovery by the inhibition of LMCT from the excited quinazoline core to Cu2+. The binding constants (Kapp, M−1) of the copper complexes, obtained from the titration curves (Fig. S3 and S4, ESI†), indicated weak binding to AMP, moderate binding to Pi and ADP, and very strong binding to PPi, Tri, and ATP (Table 1). The binding constant of Cu-dpa-QZ1 to ATP (7.3 × 104 M−3) was comparable to that of Cu-dpa-QZ2 (1.2 × 104 M−3), whereas Cu-dpa-QZ1 exhibited a substantially higher binding constant to ADP (1.5 × 104 M−3) than Cu-dpa-QZ2 (4.7 × 103 M−3), indicating that dpa-QZ2 has higher ATP selectivity than dpa-QZ1.
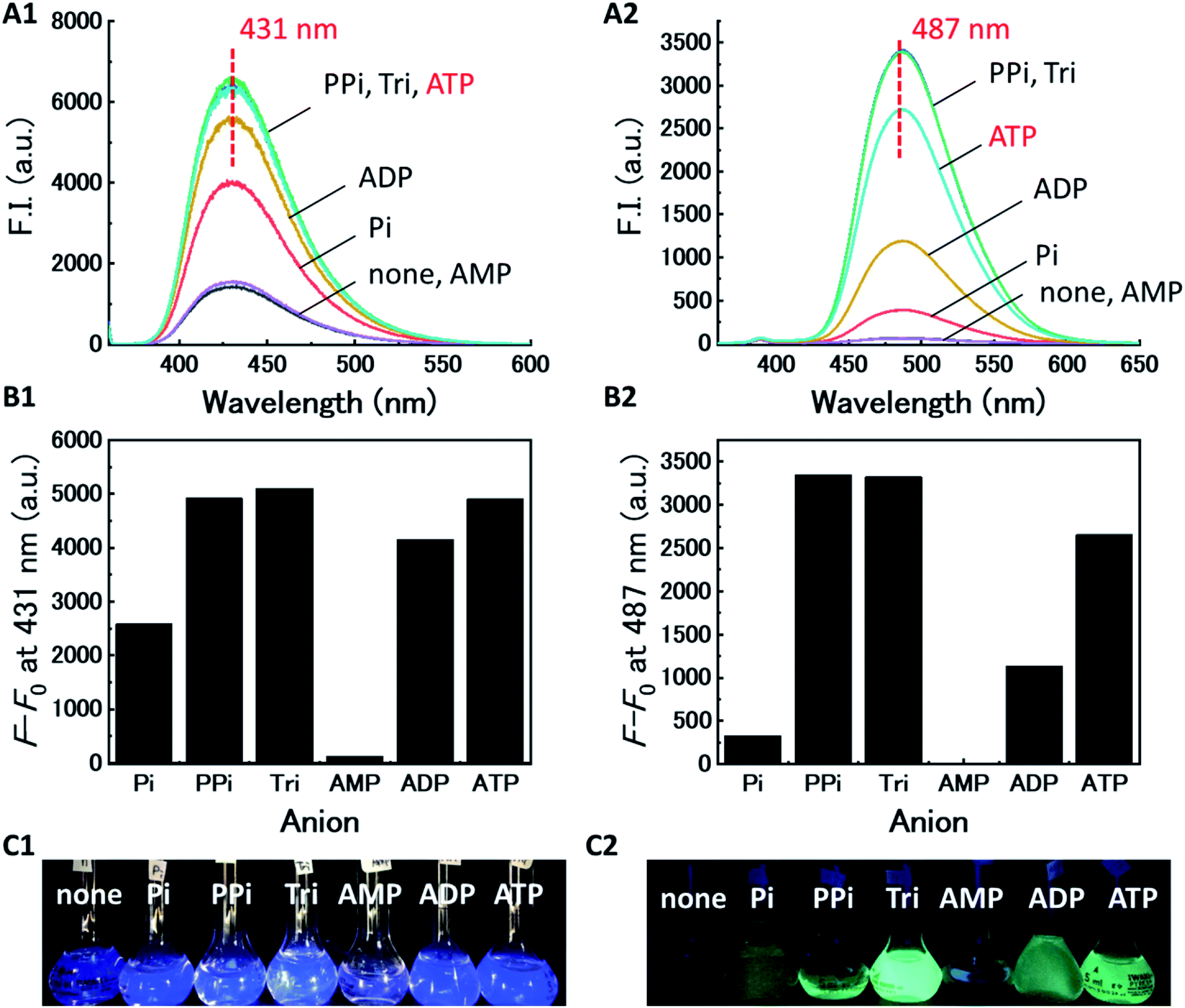 | ||
| Fig. 2 (A) Fluorescence spectra of Cu-dpa-QZ1 for (A1) and Cu-dpa-QZ2 for (A2) with phosphoric acid derivatives (Pi, PPi, Tri, AMP, ADP, and ATP) in 1% DMSO – 99% water (v/v), pH 7.4 adjusted by HEPES/NaOH buffer, at 25 °C (λdpa-QZ1ex = 355 nm, λdpa-QZ2ex = 345 nm). (B) F–F0 is the fluorescence intensity difference at fluorescence maximum wavelength of Cu-dpa-QZ1 for (B2) and Cu-dpa-QZ2 for (B2). [probe] = 10 μM, [Cu(NO3)2] = 20 μM, [phosphoric acid] = 1.0 mM, [HEPES buffer] = 5.0 mM. (C) UV irradiation images of Cu-dpa-QZ1 for (C1) and Cu-dpa-QZ2 for (C2) with phosphoric acid derivatives. | ||
| Phosphoric acid derivatives | Kapp [M−1] | |
|---|---|---|
| Cu-dpa-QZ1 | Cu-dpa-QZ2 | |
| a Fluorescence measurement for phosphoric acid titration curves were performed in 1% DMSO – 99% water (v/v), pH 7.4 adjusted by HEPES/NaOH buffer, at 25 °C (λdpa-QZ1ex = 355 nm, λdpa-QZ2ex = 345 nm). [probe] = 10 μM, [Cu(NO3)2] = 20 μM, [phosphoric acids] = 0–4.0 mM, [NaNO3] = 0.10 M, [HEPES buffer] = 5.0 mM.b n.d. = Not determined due to insufficient fluorescence enhancement even in the presence of 1.5 mM AMP. | ||
| Pi | 8.4 × 103 | 1.2 × 103 |
| PPi | 2.2 × 105 | 7.3 × 104 |
| Tri | 5.4 × 105 | 7.2 × 104 |
| AMP | n.d.b | n.d.b |
| ADP | 1.5 × 104 | 4.7 × 103 |
| ATP | 7.3 × 104 | 3.8 × 104 |
Having determined the binding abilities and the fluorescence behaviour of the developed probes, we turned to examine the use of cyclodextrins (CyDs) to improve the selectivity and sensitivity of the ATP sensing, based on a reported multipoint recognition system.28,29 It was assumed that the introduction of the dpa probe into the hydrophobic cavity of 3-fluorophenyl boronic acid (FPB)-modified CyD could likely allow the binding of ATP, wherein, the Cu–dpa recognizes the phosphoric acid unit of ATP, and the FPB recognizes the 1,2-cis-diol unit of the adenosine simultaneously (Fig. 3).
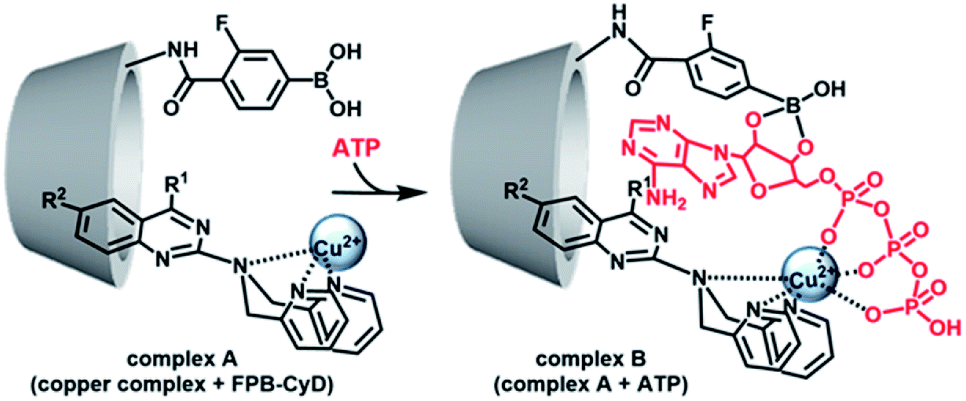 | ||
| Fig. 3 Proposed scheme for ATP-detection by the multipoint recognition system using copper complex and FPB-CyD. | ||
First, the fluorescence responses of dpa-QZ1 and dpa-QZ2 to β-CyD, γ-CyD, FPB-β-CyD, and FPB-γ-CyD were evaluated. Despite the lack of particular differences in the responses of dpa-QZ1 to the CyDs, the fluorescence intensity of dpa-QZ2 was enhanced in the presence of β-CyD, indicating the interaction of dpa-QZ2 with the hydrophobic cavity of β-CyD (Fig. 4A and B). Notably, the NOESY spectrum of the mixture of dpa-QZ2 and β-CyD exhibited interactions between the protons of the quinazoline and those of the CyD's concave side (Fig. 5), thus confirming the interaction of the probe with the CyD. Furthermore, the fluorescence intensity increased upon mixing dpa-QZ2 and β-CyD, and a blue shift in the fluorescence was observed in the presence of DMSO in a concentration-dependent manner (Fig. 4C and S5, ESI†). These observations support the incorporation of dpa-QZ2 into the cavity of the CyD and the stabilization by hydrophobic interactions.
 | ||
| Fig. 4 Fluorescence-response of (A) dpa-QZ1 and (B) dpa-QZ2 to CyDs and modified-CyDs with FPB in 1% DMSO – 99% water (v/v), pH 7.4 adjusted by HEPES/NaOH buffer, at 25 °C (λdpa-QZ1ex = 355 nm, λdpa-QZ2ex = 345 nm). [probe] = 10 μM, [CyD] = 5.0 mM, [FPB-CyD] = 0.50 mM, [NaNO3] = 0.10 M, [HEPES buffer] = 5.0 mM. Notably, for CyDs the concentration that exhibited the highest fluorescence response to the probe was adopted, whereas for FPB-CyDs the approximate saturation concentration was used. (C) Fluorescence-response of dpa-QZ2 to β-CyD (0–6.0 mM) in 1% DMSO – 99% water (v/v), and their wavelength shifts, pH 7.4 adjusted by HEPES/NaOH buffer, at 25 °C (λex = 345 nm). [dpa-QZ2] = 10 μM, [HEPES buffer] = 5.0 mM, [NaNO3] = 0.10 M. | ||
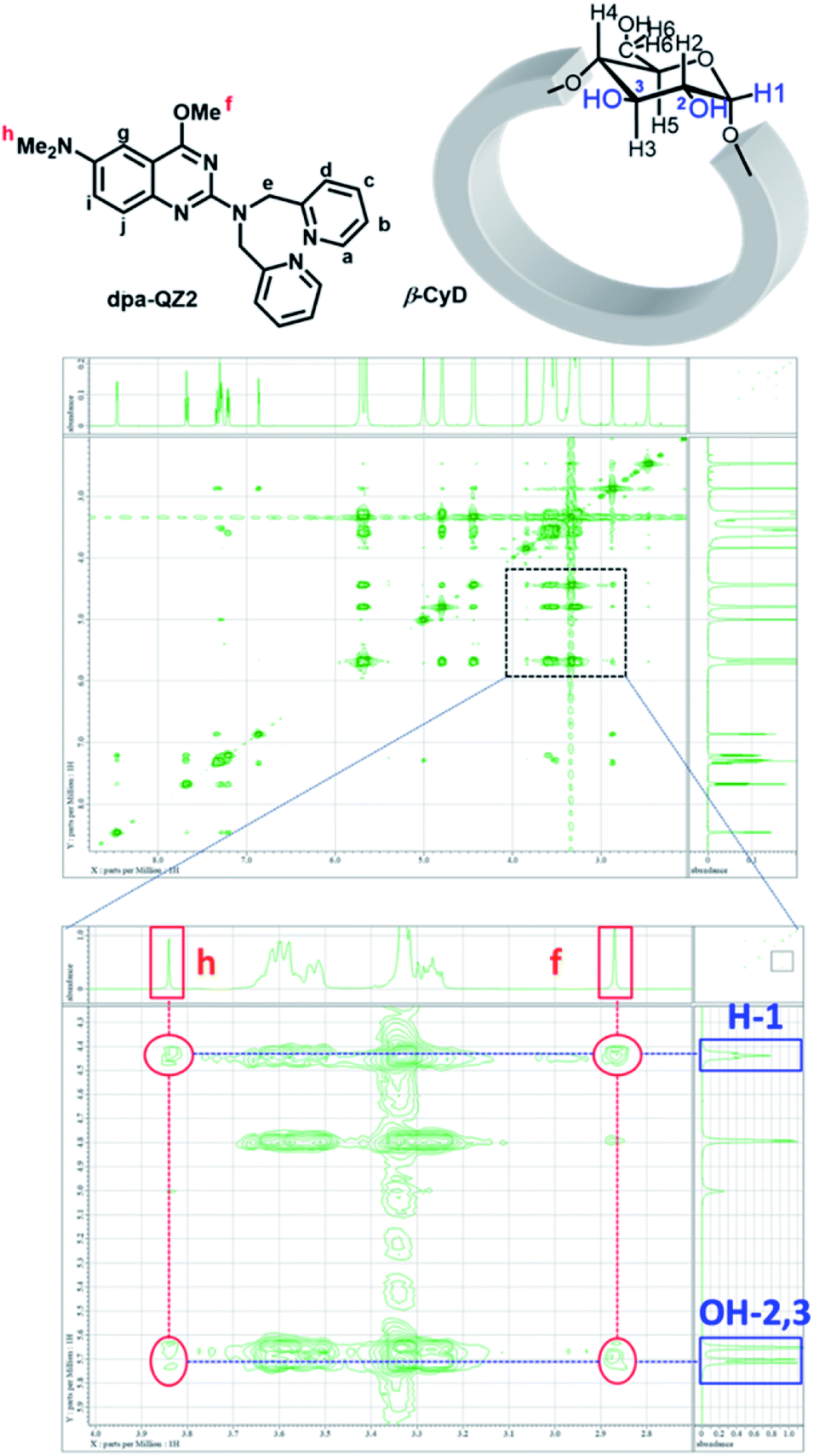 | ||
| Fig. 5 The NMR assignments and NOESY spectra of dpa-QZ2/β-CyD in 100% d6-DMSO.30 [dpa-QZ2] = 8 mM, [β-CyD] = 16 mM. | ||
The fluorescence responses of dpa-QZ1 and dpa-QZ2 to the metal ions in the presence of CyDs and the FPB-modified CyDs exhibited the same levels of responses as those in the absence of CyDs (Fig. S6 and S7 ESI†). The fluorescence recovery ratios (F/F0) calculated from the emission intensities of copper complexes with CyDs and the phosphoric acid derivatives were used to evaluate the selectivity and the sensitivity of the ATP sensing, where F is the relative fluorescence intensity (after the addition of phosphoric acids), and F0 is that of initial state (before the addition of phosphoric acids) (Fig. 6 and Table 2). In the case of Cu-dpa-QZ1, the presence of CyDs did not affect the fluorescence recovery ratios and sensitivities (Table 2 and Fig. S8, ESI†). In contrast, the sensitivities to PPi, Tri and ATP were increased by using Cu-dpa-QZ2 in the presence of FPB-γ-CyD (PPi: F/F0 from 55 to 133, Tri: F/F0 from 55 to 135, ATP: F/F0 from 44 to 69) and those to Pi, AMP, and ADP were not affected by the presence of FPB-γ-CyD (Pi: F/F0 6.2 and 2.9, AMP: F/F0 <1 and < 1, ADP: F/F0 19 and 18) (Table 2, Fig. 6 and S9, ESI†). However, the limit of detections (3 × standard deviation of the regression line/slope), calculated from the values of (F–F0) and the concentrations of ATP (Fig. S4F and S10, ESI†) did not exhibit any significant difference. They were found to be 1.50 and 0.32 μM in the presence and absence of FPB-γ-CyD, respectively. Further investigation using modified recognition systems with structurally-tuned quinazoline fluorophores and boronic acid-tethered CyDs is expected to further confirm the role of the boronic acid moiety.
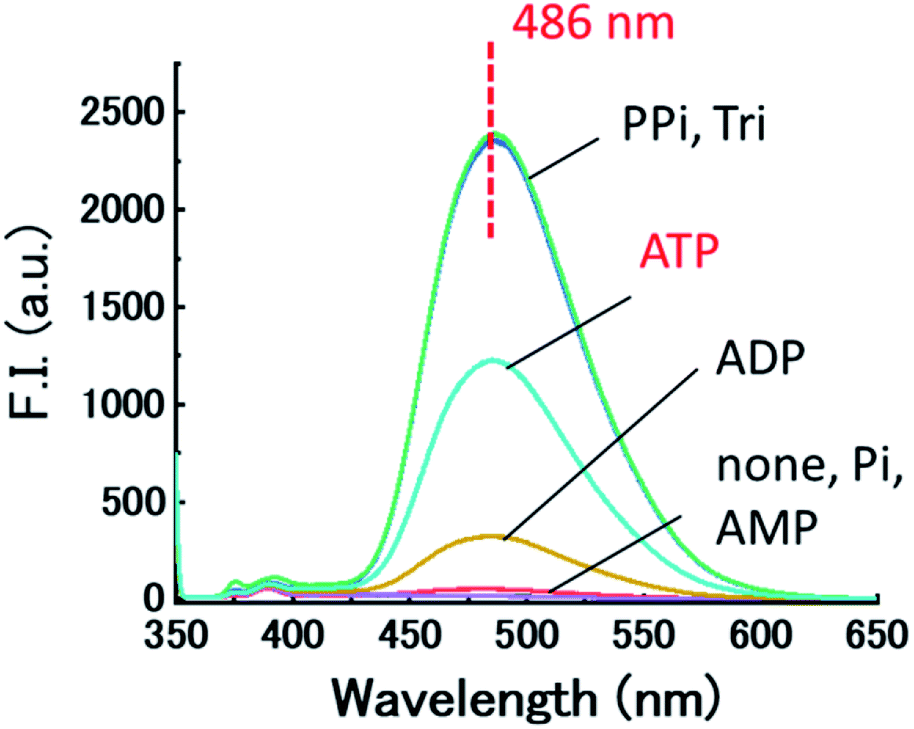 | ||
| Fig. 6 Fluorescence spectra of Cu-dpa-QZ2 with phosphoric acid derivatives (Pi, PPi, Tri, AMP, ADP and ATP) in the presence of FPB-γ-CyD in 1% DMSO – 99% water (v/v), pH 7.4 adjusted by HEPES/NaOH buffer, at 25 °C. F–F0 is the fluorescence intensity difference at fluorescence maximum wavelength (λex = 345 nm). [dpa-QZ2] = 10 μM, [Cu(NO3)2] = 20 μM, [FPB-γ-CyD] = 0.50 mM, [phosphoric acid] = 1.0 mM, [HEPES buffer] = 5.0 mM. | ||
| Phosphoric acid derivatives | Fluorescence recovery ratio (F/F0)b | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cu-dpa-QZ1 | Cu-dpa-QZ2 | |||||||||
| None | β-CyD | γ-CyD | FPB-β-CyD | FPB-γ-CyD | None | β-CyD | γ-CyD | FPB-β-CyD | FPB-γ-CyD | |
| a Fluorescence measurements for the selectivity of phosphoric acid derivatives were performed in 1% DMSO – 99% water (v/v), pH 7.4 adjusted by HEPES/NaOH buffer, at 25 °C (λdpa-QZ1ex = 355 nm, λdpa-QZ2ex = 345 nm). [probe] = 10 μM, [Cu(NO3)2] = 20 μM, [phosphoric acids] = 1.0 mM, [NaNO3] = 0.10 M, [HEPES buffer] = 5.0 mM.b Unless otherwise noted, F/F0 indicates the relative fluorescence intensity (F) at 431 nm of Cu-dpa-QZ1 and 486 nm of Cu-dpa-QZ2 in the presence of 1.0 mM of the phosphoric acid derivatives against that of the initial state (F0). | ||||||||||
| Pi | 2.8 | 2.5 | 1.9 | 3.1 | 2.4 | 6.2 | 2.5 | 2.1 | 4.3 | 2.9 |
| PPi | 4.5 | 3.9 | 2.6 | 8.7 | 5.5 | 55 | 14 | 14 | 65 | 133 |
| Tri | 4.6 | 3.9 | 2.7 | 8.6 | 5.3 | 55 | 14 | 14 | 64 | 135 |
| AMP | 1 | 1.2 | <1 | 1.5 | 1.3 | <1 | <1 | <1 | <1 | <1 |
| ADP | 4.0 | 3.6 | 2.5 | 6.6 | 4.5 | 19 | 8.3 | 6.2 | 16 | 18 |
| ATP | 4.5 | 3.7 | 2.6 | 8.1 | 5.2 | 44 | 12 | 12 | 47 | 69 |
Conclusions
In conclusion, novel quinazoline fluorescent probes, dpa-QZ1, and dpa-QZ2 were developed, which enabled the recognition of various phosphoric acid derivatives by fluorescence enhancement. While both probes showed similar levels of response to metal ions, their levels of response to phosphoric acid derivatives and CyDs were different. The ATP selectivity of dpa-QZ2 was better than that of dpa-QZ1, and in the presence of β-CyD, dpa-QZ2 showed fluorescence enhancement, indicating the binding of dpa-QZ2 to the hydrophobic cavity of β-CyD. The recognition system comprising dpa-QZ2 and FPB-modified γ-CyD demonstrated higher sensitivity to ATP (F/F0 = 69). Overall, 2-amioquinazolines display high fluorescence intensities and can serve as efficient probes or sensors for enabling sensitive, versatile, and wide-ranging applications, such as real-time monitoring of the intracellular ATP concentration, which facilitates to the elucidation of vital biological phenomena. Furthermore, ATP detection with higher sensitivity and selectivity can be achieved through structural modification of the quinazoline fluorophore and CyDs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (C) (18K06587) from the Japan Society for the Promotion of Science and MEXT, Japan.Notes and references
- F. Ozogul, T. Kda, P. C. Quantick and Y. Ozogul, Int. J. Food Sci. Technol., 2000, 35, 549 CrossRef CAS.
- Y. Lian, H. Jiang, J. Feng, X. Wang, X. Hou and P. Deng, talanta, 2016, 150, 485 CrossRef CAS PubMed.
- B. Liu, M. Ozaki, H. Hisamoto, Q. Luo, Y. Utsumi, T. Hattori and S. Terabe, Anal. Chem., 2005, 77, 573 CrossRef CAS PubMed.
- Y. Wu, J. Wen, H. Li, S. Sun and Y. Xu, Chin. Chem. Lett., 2017, 28, 1916 CrossRef CAS.
- A. Ojida, I. Takashima, T. Kohira, H. Nonaka and I. Hamachi, J. Am. Chem. Soc., 2008, 130, 12095 CrossRef CAS PubMed.
- L. Xiao, S. Sun, Z. Pei, Y. Pei, Y. Pang and Y. Xu, Biosens. Bioelectron., 2015, 65, 166 CrossRef CAS PubMed.
- H. Imamura, K. P. Huynh Nhat, H. Togawa, K. Saito, R. Iino, Y. Kato-Yamada, T. Nagai and H. Noji, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15651 CrossRef CAS PubMed.
- F. Li, X. Hu, F. Wang, B. Zheng, J. Du and D. Xiao, Talanta, 2018, 179, 285 CrossRef CAS PubMed.
- T. Hu, W. Na, X. Yan and X. Su, Talanta, 2017, 165, 194 CrossRef CAS PubMed.
- Y. Tsuchido, A. Yamasawa, T. Hashimoto and T. Hayashita, Anal. Sci., 2018, 34, 1125 CrossRef CAS PubMed.
- L. G. Pathberiya, N. Barlowa, T. Nguyen, B. Graham and K. L. Tuck, Tetrahedron, 2012, 68, 9435 CrossRef CAS.
- B. A. Wong, S. Friedle and S. J. Lippard, Inorg. Chem., 2009, 48, 7009 CrossRef CAS PubMed.
- A. Ojida, Y. Mito-oka, M. Inoue and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 6256 CrossRef CAS PubMed.
- A. Ojida, Y. Mito-oka, K. Sada and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 2454 CrossRef CAS PubMed.
- A. Ojida, Y. Miyahara, J. Wongkongkatep, S. Tamura, K. Sada and I. Hamachi, Chem.–Asian J., 2006, 1, 555 CrossRef CAS PubMed.
- J. B. Kim, Y. M. Lee, J. Ryu, W. J. Kim, G. Keum and E. K. Bang, Bioconjugate Chem., 2016, 27, 1850 CrossRef CAS PubMed.
- G. Crini, Chem. Rev., 2014, 114, 10940 CrossRef CAS PubMed.
- A. Harada, Y. Takashima and H. Yamaguchi, Chem. Soc. Rev., 2009, 38, 875 RSC.
- A. Antelo, A. Jover, L. Galantini, F. Meijide, M. A. Alcalde, N. V. Pavel and J. V. Tato, J. Inclusion Phenom. Macrocyclic Chem., 2011, 69, 245 CrossRef CAS.
- M. Kumai, S. Kozuka, M. Samizo, T. Hashimoto, I. Suzuki and T. Hayashita, Anal. Sci., 2012, 28, 121 CrossRef CAS PubMed.
- A. Ueno, T. Kuwabara, A. Nakamura and F. Toda, Nature, 1992, 356, 136 CrossRef CAS.
- G. Wenz, Angew. Chem., Int. Ed. Engl., 1994, 33, 803 CrossRef.
- D. Patra, H. Zhang, S. Sengupta and A. Sen, ACS Nano, 2013, 7, 7674 CrossRef CAS PubMed.
- R. Challa, A. Ahuja, J. Ali and R. K. Khar, AAPS PharmSciTech, 2005, 6, 329 CrossRef PubMed.
- Y. Suzuki, J. Sawada, P. Hibner, H. Ishii, K. Matsuno, M. Sato, B. Witulski and A. Asai, Dyes Pigm., 2017, 145, 233 CrossRef CAS.
- E. A. Raux, Master's thesis, Georgia State University, 2010.
- L. Zhu, J. Jin, C. Liu, C. Zhang, Y. Sun, Y. Guo, D. Fu, X. Chen and B. Xu, Bioorg. Med. Chem., 2011, 19, 2797 CrossRef CAS PubMed.
- Y. Tsuchido, S. Fujiwara, T. Hashimoto and T. Hayashita, Chem. Pharm. Bull., 2017, 65, 318 CrossRef CAS PubMed.
- T. Yamada, S. Fujiwara, K. Fujita, Y. Tsuchido, T. Hashimoto and T. Hayashita, Molecules, 2018, 23, 635 CrossRef PubMed.
- H. J. Schneider, F. Hacket, V. Rüdiger and H. Ikeda, Chem. Rev., 1998, 98, 1755 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01455a |
| This journal is © The Royal Society of Chemistry 2020 |