
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot copper-catalyzed three-component reaction: a modular approach to functionalized 2-quinolones†
Ah Reum Kimab and
Hee Nam Lim *a
*a
aEco-Friendly New Materials Research Center, Therapeutics & Biotechnology Division, Korea Research Institute of Chemical Technology (KRICT), 141 Gajeong-ro, Yuseong-gu, Daejeon 34114, Republic of Korea. E-mail: heenam@krict.re.kr
bDepartment of Chemistry, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
First published on 24th February 2020
Abstract
A copper-catalyzed three-component annulation for the synthesis of functionalized 2-quinolones was developed. Three reactions including an SN2, a Knoevenagel, and finally C–N bond formation are involved in the designed cascade reaction using 2-bromoacylarenes, 2-iodoacetamide, and nucleophiles as the three components. A new catalytic system was discovered during the study and this modular approach is highly efficient to access functionalized 2-quinolone derivatives, compatible with a broad range of functional groups, scalable, and step-economic. Further derivatization of the obtained product demonstrates the synthetic utility of this method.
Introduction
2-Quinolones are widely used in the medicinal arena and material science owing to their versatile biological activities1 and utility as biomaterials2 with interesting functions. They are also found as core structures of various natural products.3 For 2-quinolone synthesis, two traditional approaches have been well established: the acid-assisted (Knorr)4 or base-promoted synthesis (Friedländer).5 However, the harsh acidic conditions in the Knorr type synthesis limits the widespread application of this reaction. In the complementary Friedländer type reaction, 2-aminoacylarenes are common motifs that couple with malonates; this has been a widely used protocol to provide substituted 2-quinolones. In spite of the practicality and scalability, it often requires multi-step pre-functionalization to introduce the amine moiety of arenes or it needs a long process to install functional groups at the target position of quinolones.6As alternative methods, a number of synthetic procedures have been recently demonstrated via intermolecular coupling reaction with transition-metal (TM) complexes.7–10 Involving C–N bond and/or C–C bond forming reaction, each protocol has provided orthogonal bond formation and broad functional group compatibility for the classical methods. In 2004, Kadnikov and Larock developed Pd-catalyzed three-component annulation using N-substituted o-iodoanilines, internal alkynes and CO.7a Alper disclosed in 2014 the preparation of 4-substituted 2-quinolones by the Pd-catalyzed oxidative cyclocarbonylation of N-monosubstituted 2-vinylanilines.7b More recently, Jiao and coworkers found a novel Rh-catalyzed carbonylation and annulation of N-alkyl anilines with CO and internal alkyne through N–H and C–H activation.7c Consequently, the Wu group succeeded in applying the same strategy with an Ir catalyst, which could now tolerate halogen groups.7d Very recently, Das developed a method to afford 3-substituted 2-quinolones with terminal alkynes and oxalic acid as a CO source under supported Pd-catalysis.7e
In addition to utilizing CO gas or its equivalent, two-component reactions via C–H bond activation have also been of much interests. In 2014, Jeganmohan reported a Ru-catalyzed cyclization of anilides with acrylates or propiolates that delivers unsubstituted 2-quinolones or 4-alkyl substituted 2-quinolones, respectively;8a the method involves amide-directed C–H alkenylation followed by an intramolecular amidation. Liu also used acetanilide as a directing group in a Pd-catalyzed cascade reaction, but the directing group was installed from a free aniline with acetic anhydride and acetyl was subsequently removed during the reaction.8b In a similar manner, Maiti and coworkers disclosed a straightforward methodology to afford N-aryl-4-substituted quinolones by a Pd-catalyzed dehydrogenative coupling reaction with simple diarylamines and 3-substituted acrylic acids.8c More recently, Yu explored a metal-free/base-promoted lactamization with carbon dioxide and the 2-alkenylanilines.8d In 2015, Rong and Dong developed a unique method using a Rh-catalyzed C–C bond activation strategy to prepare 3,4-disubstituted 2-quinolones from isatins containing various directing groups.9 Besides these strategies, benzynes have also been adopted for Pd-catalyzed annulation: the Wang group used α-carbamoyl ketene dithioacetals as coupling partners for C–S/N–H activation10a while the Xu group chose N-methoxy acrylamides for C–H/N–H activation.10b Each protocol provides distinct mechanistic insights and useful substrate scope. Nevertheless, introducing various functional groups at the 3 and/or 4-position of 2-quinolones in one-pot from readily available reagents is still an unmet synthetic challenge.
Our interest was to find a convenient and practical protocol for the controlled synthesis of 2-quinolones containing various functional groups with inexpensive copper catalysts and simple starting materials. One viable way is a one-pot coupling reaction between 2-haloacylarenes with pre-functionalized acetamides. Several methods with Pd or Cu metals have been developed in this fashion, but the reported procedures are subject to the substituents at C2 position of the acetamides.11 In this view, a unified strategy to access 3- and/or 4-substituted 2-quinolones in one-pot remains elusive.
Herein, we suggest a one-pot copper-catalyzed three-component reaction12 as a new strategy for the functionalized 2-quinolone synthesis (Scheme 1). We envisioned that 2-substituted acetamides can be prepared in situ from amide 2 and nucleophile 3, which then react with 2-haloacylarenes 1 in the same pot; following this sequence, the unprecedented three-component reaction involving SN2, Knoevenagel and copper-catalyzed C–N bond coupling reactions can offer 3,4-difunctionalized 2-quinolones by forming three new C–X bonds (X = carbon or heteroatoms). There are many possible reaction pathways to give the desired product 4, however, the actual reaction path is likely dependent on the rate of each reaction. In principle, this modular approach will allow a diverse set of 2-quinolones to be prepared by introducing various nucleophiles. However, the challenge lies in securing high compatibility between three independent reactions in which many reactants might hamper the operation of each reaction or disable the catalytic activity.13
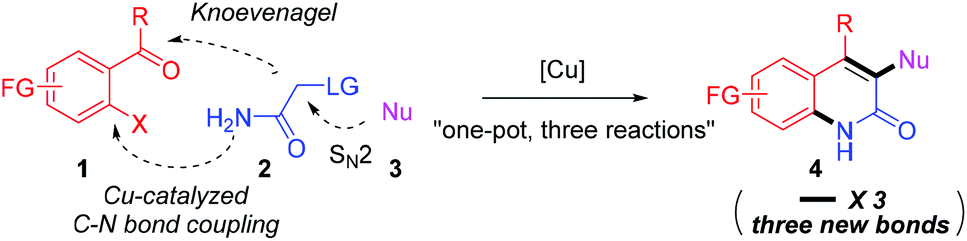 | ||
| Scheme 1 Proposed three-component approach for 2-quinolone synthesis. | ||
Results and discussion
To realize the proposed multicomponent reaction, the commercially available substrates 1a, 2, and 3a were chosen for the model reaction. We initially focused on a catalytic system with CuI and diamine ligands;14 gratifyingly, it was effective, although the desired product 4aa was given in only low to moderate yields (30–50%). Despite much efforts, the catalytic system employing this frequently used copper salt was not further improved by adopting various ligands, solvents, nor even by targeting other copper salts with different counter anions. Inspired by a previous study,11c we next screened copper powder-based catalytic conditions. After significant efforts to optimize the reaction, the use of copper powder (60–80 nm) with 2-picolinic acid (L1) and K2CO3 was found to be most effective with high reproducibility. It is notable that the combination of copper powder with ligand are rarely reported for synthesis of nitrogen-incorporated small molecules. The selected control experiments during optimization studies are summarized in Table 1.
|
||
|---|---|---|
| Entry | Variations from standard conditions | Yielda |
| a Determined by 1H NMR using 1,3,5-trimethoxybenzene as the internal standard.b Isolated yield. | ||
| 1 | None | 55% (52)b |
| 2 | w/o Cu powder | Trace |
| 3 | w/o L1 | 30% |
| 4 | w/o K2CO3 | Trace |
| 5 | 90 °C instead of 110 °C | 21% |
| 6 | Other nonpolar solvents instead of DMF | 10–40% |
| 7 | 10 mol% Cu powder and L1 instead of 20% | 44% |
| 8 | L2 instead of L1 | 27% |
| 9 | L3 instead of L1 | 48% |
| 10 | L4 instead of L1 | 41% |
| 11 | L5 instead of L1 | 23% |
| 12 | L6 instead of L1 | 14% |
| 13 | L7 instead of L1 | 44% |
| 14 | L8 instead of L1 | 34% |
 |
||
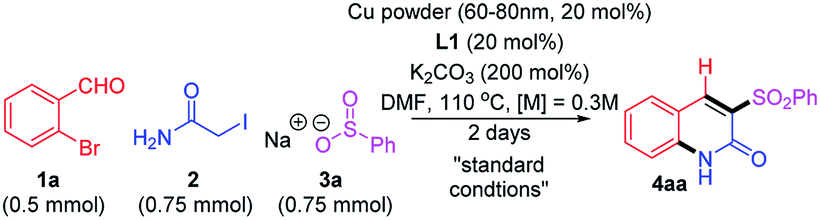
It was not surprising to observe no conversion without copper powder (entry 2); however, the reaction proceeded without L1 to give 4aa in 30% yield (entry 3) and the base was essential (entry 4) with only trace product observed when the base was omitted. This reaction was operative at even lower temperatures, although the efficacy was highly reduced (entry 5). While other less polar solvents such as tetrahydrofuran, chloroform, and 1,4-dioxane were less productive (10–20%), moderate yield (40%) was observed using toluene (entry 6). Although the amount of Cu catalyst can be reduced to 10%, the yield was decreased (entry 7). The choice of ligand proved to be very important for further reaction optimization.15 A monodentate ligand such as 2,6-lutidine (L2) was not as good as L1 (entry 8). Phenanthroline and bipyridine ligands (L3 and L4) were comparable to L1 (entry 9 and 10). However, N, O-ligands (L5, L6) were much less efficient (entry 11 and 12).16 A diamine ligand (DMEDA, L7) was also compatible with the copper powder with slightly lower yield (entry 13). It was interesting to observe that the reaction was still working to give a reasonable yield in the presence of an anionic O,O-ligand (L8) that contains an active methylene (entry 14).17
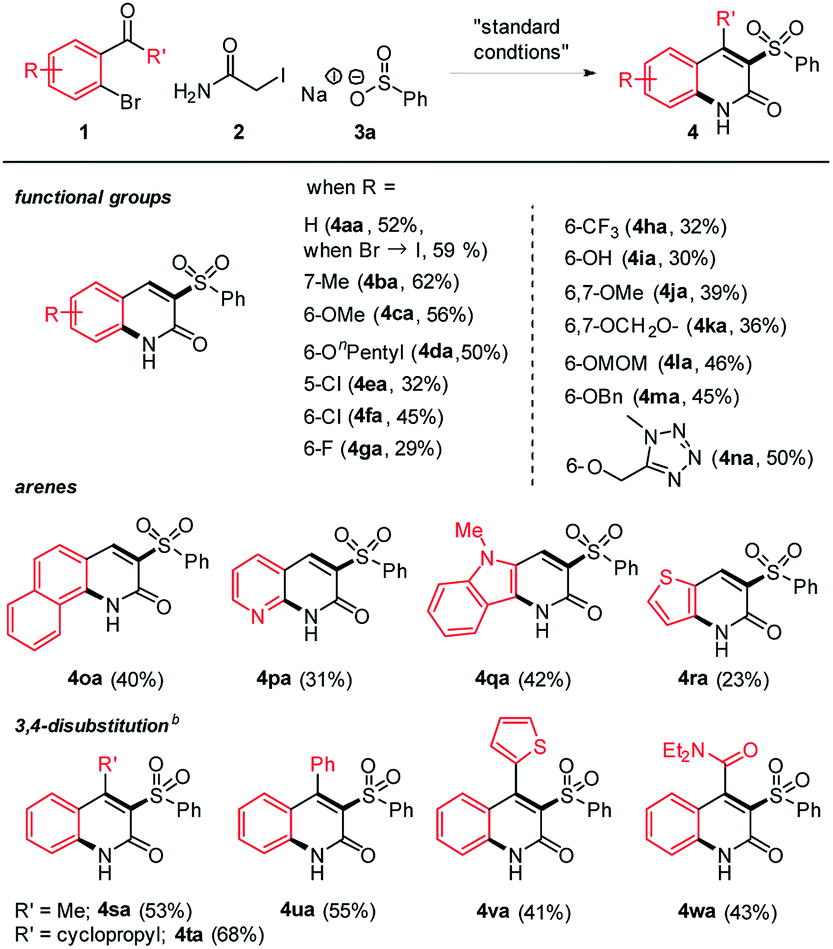
With the optimized conditions in hand, we first tested the functional group compatibility. Gratifyingly, a range of functional groups are well suited in this reaction (Table 2). For example, alkyl and alkoxy substituents worked smoothly, giving moderate to good yields (4ba–4da). In the case of halides, chloride (4ea–4fa) showed better yield than fluoride (4ga) and a trifluoromethyl group (4ha). It is notable that a free hydroxy group was tolerated in the reaction to give a reasonable yield (4ia). Electron rich functional groups such as dialkoxy and methylenedioxy were also compatible (4ja–4ka). The acid-sensitive MOM protecting group (4la) and redox-unstable benzyl protecting group (4ma) were tolerated in this transformation. A heterocycle such as tetrazole also participated in the three-component reaction. While testing the arene scope, the efficiency of other arylaldehydes containing naphthalene, pyridine, thiophene, and biologically more relevant indole was relatively low (4oa–4ra), but the products were still obtained in synthetically useful yields. Under the standard conditions, ketones were much less reactive, likely due to inefficient Knoevenagel condensation. In this case, additional use of Ca(OH)2 was discovered to promote the reaction while the driving force is unclear. The optimization process for the ketone substrate is summarized (see the page S3 in the ESI†). In this way, 3,4-disubstituted 2-quinolones were also accessible, offering the facile entry to densely functionalized derivatives (4sa–4wa).
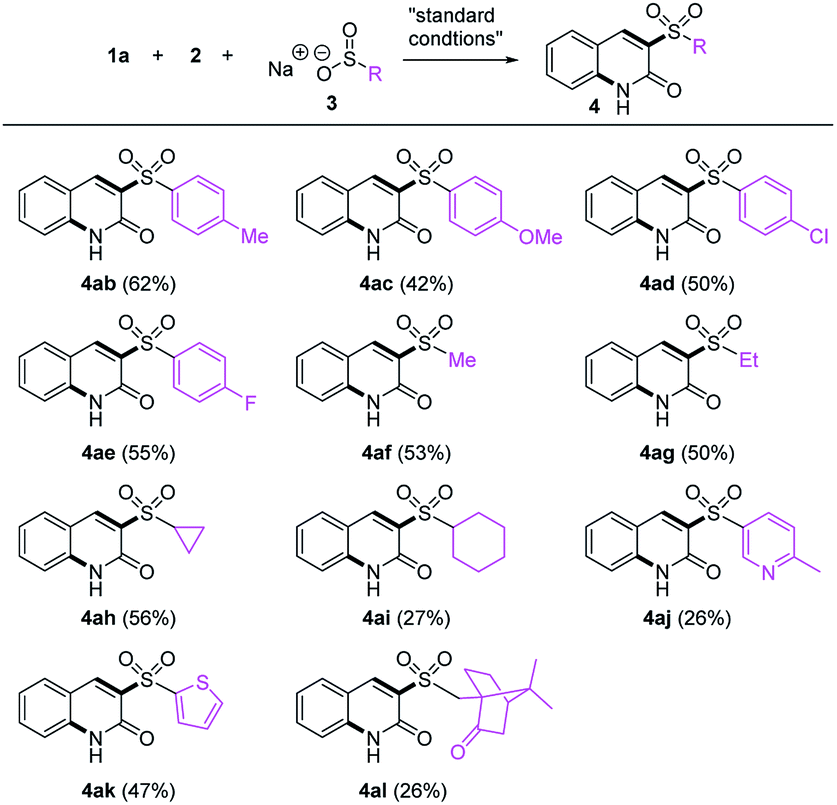
Next, the reaction scope with respect to sulfinates was surveyed (Table 3). Various arylsulfinic acid sodium salts18 containing both electron-donating and -withdrawing groups were examined (4ab–4ae), all of which were amenable to the reaction. When alkyl sulfinates containing methyl, ethyl, cyclopropyl, and cyclohexyl groups (4af–4ai) were employed, more liphophilic quinolones were obtained. The scope was further extended to other heterocycles: both picoline and thiophene rings (4aj–4ak) survived to give the corresponding products albeit in low yield for the picoline substrate. To our delight, the reaction with 10-camphorsulfinic acid sodium salt proceeded smoothly to provide 4al.
 |
Given the success using sulfinates, other nucleophiles were next examined (Scheme 2a). When sodium thiolate was employed in the standard condition, 3-phenylthio-2-quinolone 5 was obtained in only 30% isolated yield; however, the yield was improved by adding Ca(OH)2 (55%). In addition to sulfur(II), oxygen-based nucleophiles such as sodium phenoxides also worked in the presence of a stronger base, thus affording 3-aryloxy-2-quinolones 6. Notably, the use of nitrogen-based nucleophile, N-methylphenylamine, resulted in the desired transformation using ketone substrate 1r, giving the 2-quinolone 7 in 44% yield.19 However, the use of other amine nucleophiles such as aniline and dialkylamines was unsuccessful. In addition, the application of the carbon-based soft nucleophiles such as malonates remains unresolved.
 | ||
| Scheme 2 Further scope and applications. (a) Use of other nucleophiles. (b) Gram scale. (c) Further applications. | ||
The protocol was readily scalable (Scheme 2b); in the gram scale reaction 4aa was obtained with improved efficiency (52% → 62%). Next, we demonstrated the synthetic utility of the prepared quinolones (Scheme 2c). As 4aa holds many useful functional groups, it could be further functionalized. For instance, it was successfully transformed to 2-chloroquinoline 8. N-Benzyl quinolone 9 was obtained by the selective N-alkylation in good yield, and 9 could be further functionalized to 3,4-disubstituted hydroquinolone 10 by Grignard-initiated Michael addition.20 Meanwhile, the sulfonyl group can be converted to organotin compound 1121 which is a key intermediate for the synthesis of 3-halogenated quinolones 1222 and 1323 and is expected as a useful platform for coupling reactions.24 The versatile conversion of the product obtained by our method is believed to have many implications in synthetic and medicinal chemistry.
Possible mechanistic pathways to give 4aa are illustrated in Scheme 3a. Depending on the reaction order, they can be classified into three ways. The first route (path A) is deconstructive as the resulting intermediate I cannot afford 4aa. The second route is firstly forming the intermediate II by C–N coupling reaction: paths B and C are conceivable to give 4aa. The third route is initiated by a SN2 reaction between 2 and 3a, which provides the intermediate V. The intermediate V can form 4aa by reacting with 1a via the intermolecular C–N bond formation followed by the Knoevenagel condensation (path D) or vise versa (path E).
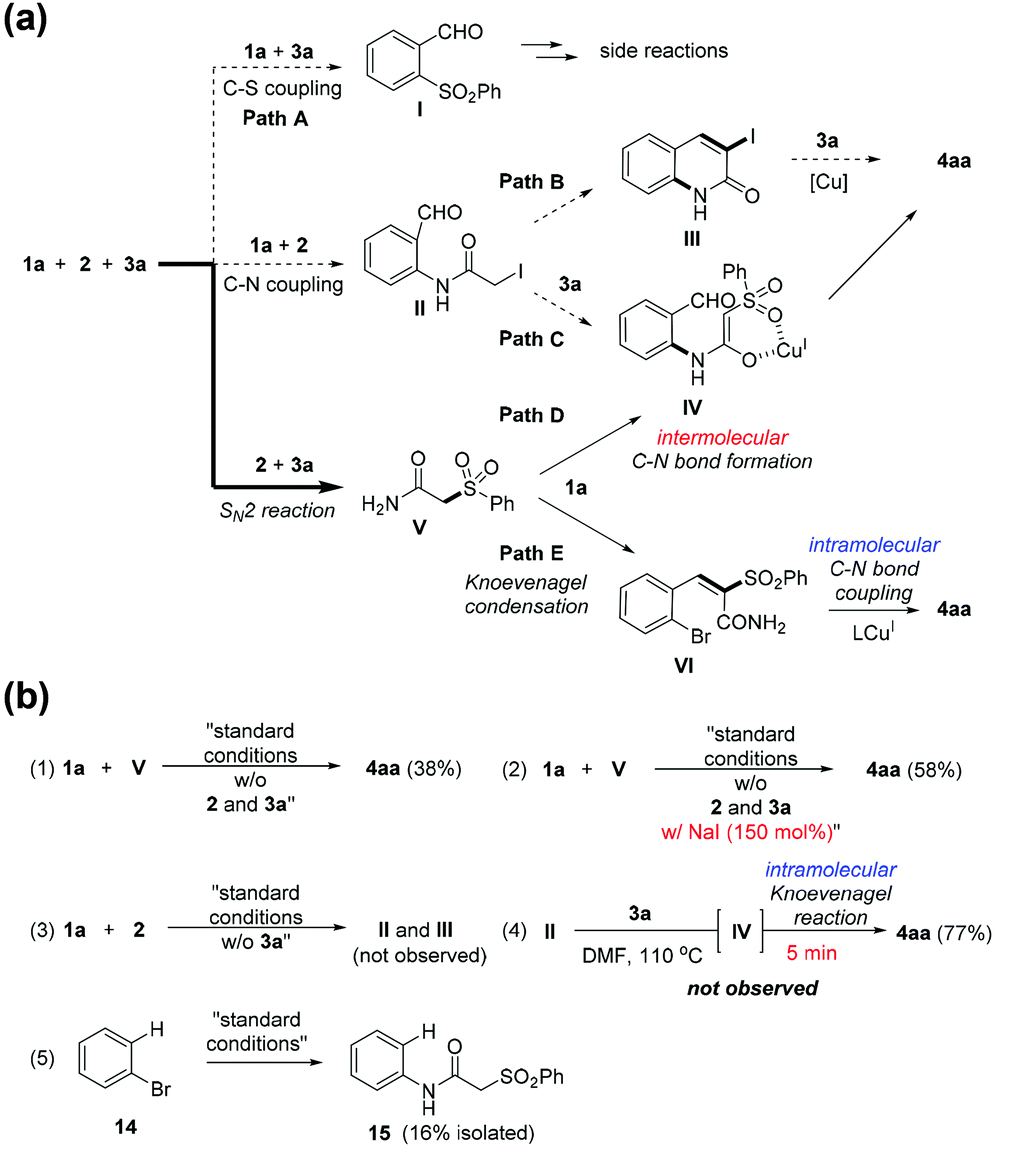 | ||
| Scheme 3 A proposed mechanism. (a) Possible reaction pathways. (b) Control experiments. | ||
The reaction with 1a, 2, and 3a was monitored by checking tlc, tracking LC-MS and 1H nmr experiment; we found no LC traces corresponding to I, II, III, and IV. On the other hand, the intermediate V was clearly identified while 2 was fully consumed within 1 hour. This indicates that the reaction quickly proceeds into a two-component reaction (1a and V). In addition, the intermediate VI was found as a major resting intermediate in the LC-trace and tlc during the entire course of the reaction. Although the intermediate IV was not detected in the LC-trace, a possibility cannot be excluded that the intramolecular Knoevenagel reaction rapidly occurs, thus not allowing VI itself to stay on.
To gain insights into the mechanism, we first conducted a control experiment using 1a and V under the standard conditions [Scheme 3b-(1)]. Interestingly, in this two-component reaction, the yield of 4aa was even lower than that of the three-component reaction. The only difference was the presence of NaI which is a by-product by SN2 reaction between 2 and 3a; NaI was indeed an important promoter of the three-component reaction [Scheme 3b-(2)].25 As expected, the compounds II and III were not formed in the control experiment [Scheme 3b-(3)]. The intermediate IV was highly reactive species. We attempted to isolate the intermediate IV by treating II with 3a. However, a isolated product was 4aa and IV was not traceable [Scheme 3b-(4)]. This observation, in part, supports feasibility of path D if C–N bod forming reaction is facile. The formation of 15 in the Scheme 3b-(5) further supports our postulate about the path D while the efficiency for C–N bond formation was low. Although not completely conclusive, at this point, path E (major contribution) and D (minor contribution) are plausible pathways to 4aa.
Conclusions
In conclusion, under the conceptually simple one-pot operation, three separate reagents reacted to synergistically form the functionalized 2-quinolones. This was enabled by three cascade reactions of SN2, Knoevenagel condensation, and C–N coupling with good control over reactive intermediates. During reaction optimization, a new catalytic system employing copper power with 2-picolinic acid was discovered and this method highlights streamlined preparation of biologically important 2-quinolone derivatives. Studies to further extend the reaction scope of acyl groups, arenes and nucleophiles is underway in our laboratory.Experimental section
General remarks
All reactions were carried out in 5 mL capped vials. N,N-Dimethylformamide (DMF) was directly used from SPS system (Hansen, Puresolve MD) without further purification. Thin layer chromatography (TLC) analysis was run on silica gel plates. Spots were visualized by exposure to ultraviolet (UV) light (254 nm). High-resolution mass spectra were reported for the molecular ion [M]+. Melting points were determined with the Mettler Toledo MP50 and the values were uncorrected. Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker (400 MHz for 1H, 100 MHz for 13C and 376 MHz for 19F). Chemical shifts for proton NMR spectra are reported in parts per million (ppm) relative to the singlet at 7.26 ppm for chloroform-d and to the quintet at 2.50 ppm for dimethylsulfoxide-d6. Chemical shifts for carbon NMR spectra are reported in 77.2 ppm with the center line of triplet for chloroform-d, in 39.5 ppm with the center line of the septet for dimethylsulfoxide-d6 and in 116.6 ppm, 164.2 ppm with the center line of the quartet for trifluoroacetic acid-d. Data for 1H NMR were presented as following: chemical shifts (δ, ppm), multiplicity (br = broad, s = singlet, d = doublet, t = triplet, dd = doublet of doublets, td = triplet of doublet, m = multiplet), coupling constant (Hz), and integration. The chemical shifts of peaks found were reported for 13C NMR spectra. Infrared spectra were recorded with a Smith Detection ATR-FTIR.2-Bromo-5-((1-methyl-1H-tetrazol-5-yl)methoxy)benzaldehy-de (1n)
A mixture of 2-bromo-5-hydroxybenzaldehyde (1.00 g, 5.0 mmol), K2CO3 (1.04 g, 7.5 mmol) and 5-(chloromethyl)-1-methyl-1H-tetrazole (0.79 g, 6.0 mmol) in DMSO (10 mL) was stirred at 50 °C for 4 h. Then, the reaction mixture was cooled to room temperature, quenched with saturated NH4Cl solution, and extracted with EtOAc (30 × 3 mL). The organic solution was dried over MgSO4, concentrated, and subjected to column chromatography (Hex![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 3:1) to afford the benzaldehyde 1n (1.02 g, 69%) as white solid, mp 66–67 °C.
:EtOAc = 3:1) to afford the benzaldehyde 1n (1.02 g, 69%) as white solid, mp 66–67 °C.
Rf = 0.2 (Hex:EtOAc = 3:1); 1H NMR (400 MHz, CDCl3-d) δ 4.37 (s, 3H), 5.35 (s, 2H), 7.15 (dd, J = 8.8 Hz and 3.1 Hz, 1H), 7.54–7.56 (m, 2H), 10.29 (s, 1H). 13C NMR (100 MHz, CDCl3-d) δ 39.8, 61.2, 114.1, 119.0, 123.7, 134.2, 135.0, 157.7, 162.1, 191.6. IR (neat) νmax 1010, 1162, 1224, 1684, 2876, 3066 cm−1. HRMS[EI+] calcd for C10H9BrN4O2 [M]+ 295.9909, found 295.9904.
General procedure for 4aa–4ra (Table 1) and 4ab–4al (Table 2)
A 5 mL vial was charged with 2-bromobenzaldehyde 1 (0.50 mmol), 2-iodoacetamide 2 (138.7 mg, 0.75 mmol), a corresponding sodium sulfinate 3 (0.75 mmol), Cu powder (60–80 nm, 6.4 mg, 0.10 mmol), 2-picolinic acid (12.3 mg, 0.10 mmol), K2CO3 (138.2 mg, 1.00 mmol) in dry DMF (1.5 mL). The vial was sealed with the cap and heated at 110 °C under stirring for 48 h. After cooling to room temperature, the reaction mixture was diluted with saturated NH4Cl solution and extracted with ethyl acetate (20 × 5 mL). The combined organic solution was dried over MgSO4 and concentrated. The residue was subjected to silica gel column chromatography (DCM:MeOH = 97:3 to 95:5 or Hex:EtOAc = 1:1) to afford the 2-quinolones.
Purification process for 4qa (Table 1)
After cooling to room temperature, the resulting brownish suspension was filtered and washed with saturated aq. NH4Cl solution. The dark yellow solid was then washed with H2O, MeOH, and DCM to afford the compound 4qa (light yellow solid).General procedure for 4sa–4wa (ketone substrates in Table 1)
A 5 mL vial was charged with ketone 1s–1w (0.50 mmol), 2-iodoacetamide 2 (185.0 mg, 1.00 mmol), sodium benzenesulfinate 3a (164.2 mg, 1.00 mmol), Cu powder (60–80 nm, 6.4 mg, 0.10 mmol), 2-picolinic acid (12.3 mg, 0.10 mmol), K2CO3 (172.8 mg, 1.25 mmol), and Ca(OH)2 (92.6 mg, 1.25 mmol) in dry DMF (1.5 mL). The vial was sealed with the cap and heated at 110 °C under stirring for 48 h. After cooling to room temperature, the reaction mixture was diluted with saturated aq. NH4Cl solution and extracted with ethyl acetate (20 × 5 mL). The combined organic solution was dried over MgSO4 and concentrated, and subjected to silica gel column chromatography (DCM:MeOH = 97:3 to 95:5 or Hex:EtOAc = 1:1) to afford the 2-quinolones.
3-(Phenylsulfonyl)quinolin-2(1H)-one (4aa)
Yield = 52%; white solid; 1H NMR (400 MHz, DMSO-d6) δ 7.30 (t, J = 7.6 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.59–7.63 (m, 2H), 7.66–7.73 (m, 2H), 7.99–8.04 (m, 3H), 8.97 (s, 1H), 12.17 (s, 1H). This spectral data is in agreement with the reported ref. 11c.7-Methyl-3-(phenylsulfonyl)quinolin-2(1H)-one (4ba)
Yield = 62%; white solid; mp ≥ 300 °C; Rf = 0.3 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 2.40 (s, 3H), 7.13–7.15 (m, 2H), 7.58–7.62 (m, 2H), 7.70 (m, 1H), 7.90 (m, 1H), 7.98–8.01 (m, 2H), 8.91 (s, 1H), 12.18 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 21.7, 114.9, 115.1, 124.4, 128.3, 128.9, 129.4, 130.4, 133.6, 139.8, 141.2, 144.4, 145.0, 156.5. IR (neat) νmax 1155, 1308, 1478, 1603, 1649, 3144 cm−1. HRMS[EI+] calcd for C16H13NO3S [M]+ 299.0616, found 299.0626.
6-Methoxy-3-(phenylsulfonyl)quinolin-2(1H)-one (4ca)
Yield = 56%; pale yellow solid; mp ≥ 300 °C; Rf = 0.25 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 3.81 (s, 3H), 7.28–7.36 (m, 2H), 7.59–7.63 (m, 3H), 7.70 (m, 1H), 7.99–8.01 (m, 2H), 8.92 (s, 1H), 12.16 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 55.6, 110.9, 116.8, 117.7, 124.0, 128.4, 128.9, 130.9, 133.6, 135.8, 139.6, 144.0, 154.6, 155.9. IR (neat) νmax 1151, 1291, 1493, 1618, 1649, 3401 cm−1. HRMS[EI+] calcd for C16H13NO4S [M]+ 315.0565, found 315.0570.
6-(Pentyloxy)-3-(phenylsulfonyl)quinolin-2(1H)-one (4da)
Yield = 50%; pale yellow solid; mp 279–280 °C; Rf = 0.25 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 0.90 (t, J = 6.9 Hz, 3H), 1.30–1.44 (m, 4H), 1.71–1.77 (m, 2H), 4.00 (t, J = 6.5 Hz, 2H), 7.27–7.35 (m, 2H), 7.58–7.63 (m, 3H), 7.70 (t, J = 7.4 Hz, 1H), 8.00 (d, J = 7.8 Hz, 2H), 8.90 (s, 1H), 12.14 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 13.9, 21.9, 27.7, 28.2, 68.0, 111.5, 116.7, 117.8, 124.3, 128.4, 128.9, 130.9, 133.6, 135.7, 139.7, 144.0, 153.9, 155.9. IR (neat) νmax 1150, 1289, 1474, 1619, 1648, 3085 cm−1. HRMS[EI+] calcd for C20H21NO4S [M]+ 371.1191, found 371.1187.
5-Chloro-3-(phenylsulfonyl)quinolin-2(1H)-one (4ea)
Yield = 32%; pale yellow solid; mp ≥ 300 °C; Rf = 0.4 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 7.33 (d, J = 8.5 Hz, 1H), 7.46 (d, J = 7.8 Hz, 1H), 7.60–7.74 (m, 4H), 8.03–8.05 (m, 2H), 8.95 (s, 1H), 12.54 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 114.8, 115.1, 123.3, 128.7, 129.0, 131.9, 133.1, 134.0, 134.7, 139.0, 139.7, 142.5, 156.0. IR (neat) νmax 1155, 1319, 1444, 1571, 1612, 3078 m−1. HRMS[EI+] calcd for C15H10ClNO3S [M]+ 319.0070, found 319.0065.
6-Chloro-3-(phenylsulfonyl)quinolin-2(1H)-one (4fa)
Yield = 45%; ivory solid; mp ≥ 300 °C; Rf = 0.25 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 7.35 (d, J = 8.9 Hz, 1H), 7.59–7.63 (t, J = 7.6 Hz, 2H), 7.69–7.73 (m, 2H), 8.00 (d, J = 7.6 Hz, 2H), 8.16 (d, J = 1.8 Hz, 1H), 8.97 (s, 1H), 12.36 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 117.4, 118.3, 126.5, 128.5, 129.0, 129.3, 131.8, 133.7, 133.8, 139.3, 139.8, 143.7, 156.2. IR (neat) νmax 1158, 1312, 1477, 1617, 1649, 3455 cm−1. HRMS[EI+] calcd for C15H10ClNO3S [M]+ 319.0070, found 318.9950.
6-Fluoro-3-(phenylsulfonyl)quinolin-2(1H)-one (4ga)
Yield = 29%; ivory solid; mp ≥ 300 °C; Rf = 0.25 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 7.38 (dd, J = 9.1 and 4.6 Hz, 1H), 7.58–7.63 (m, 3H), 7.71 (m, 1H), 7.93 (dd, J = 8.9 and 2.8 Hz, 1H), 8.00 (d, J = 7.5 Hz, 2H), 8.98 (s, 1H), 12.32 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 114.9 (d, JC,F = 23 Hz), 117.5 (d, JC,F = 8 Hz), 117.8 (d, JC,F = 10 Hz), 122.4 (d, JC,F = 25 Hz), 128.5, 129.0, 131.9, 133.8, 137.9, 139.4, 143.9 (d, JC,F = 4 Hz), 156.1, 157.1 (d, JC,F = 238 Hz). 19F NMR (376 MHz, DMSO-d6) δ −119.7 IR (neat) νmax 1158, 1312, 1498, 1628, 1650, 3073 cm−1. HRMS[EI+] calcd for C15H10FNO3S [M]+ 303.0365, found 303.0356.
3-(Phenylsulfonyl)-6-(trifluoromethyl)quinolin-2(1H)-one (4ha)
Yield = 32%; white solid; mp ≥ 300 °C; Rf = 0.3 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 7.50 (d, J = 8.7 Hz, 1H), 7.60–7.64 (m, 2H), 7.72 (m, 1H), 7.96–8.05 (m, 3H), 8.54 (s, 1H), 9.13 (s, 1H), 12.55 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 116.6, 116.8, 123.0 (q, JC,F = 32 Hz), 124.0 (q, JC,F = 270 Hz), 128.4 (q, JC,F = 4 Hz), 128.5, 129.0, 129.7 (q, JC,F = 3 Hz) 132.1, 133.9, 139.3, 143.3, 144.6, 156.5. 19F NMR (376 MHz, DMSO-d6) δ −60.4 IR (neat) νmax 1126, 1324, 1482, 1631, 1654, 3157 cm−1. HRMS[EI+] calcd for C16H10F3NO3S [M]+ 353.0333, found 353.0315.
6-Hydroxy-3-(phenylsulfonyl)quinolin-2(1H)-one (4ia)
Yield = 30%; yellow solid; mp ≥ 300 °C; Rf = 0.15 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 7.18–7.24 (m, 2H), 7.30 (s, 1H), 7.58–7.62 (m, 2H), 7.69 (t, J = 7.4 Hz, 1H), 7.99 (d, J = 7.8 Hz, 2H), 8.84 (s, 1H), 9.74 (s, 1H), 12.04 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 113.2, 116.6, 118.0, 124.1, 128.4, 128.9, 130.7, 133.6, 134.6, 139.8, 143.9, 152.7, 155.8. IR (neat) νmax 1151, 1302, 1413, 1627, 1658, 3433 cm−1. HRMS[EI+] calcd for C15H11NO4S [M]+ 301.0409, found 301.0410.
7-Dimethoxy-3-(phenylsulfonyl)quinolin-2(1H)-one (4ja)
Yield = 39%; pale yellow solid; mp ≥ 300 °C; Rf = 0.15 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 3.81 (s, 3H), 3.84 (s, 3H), 6.85 (s, 1H), 7.54 (s, 1H), 7.57–7.61 (m, 2H), 7.68 (m, 1H), 7.97–8.00 (m, 2H), 8.80 (s, 1H), 12.04 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 55.8, 55.9, 97.1, 110.1, 110.6, 126.9, 128.2, 128.8, 133.4, 138.1, 140.1, 143.4, 145.6, 155.1, 156.3. IR (neat) νmax 1148, 1252, 1413, 1617, 1649, 3015 cm−1. HRMS[EI+] calcd for C17H15NO5S [M]+ 345.0671, found 345.0688.
7-(Phenylsulfonyl)-[1,3]dioxolo[4,5-g]quinolin-6(5H)-one (4ka)
Yield = 36%; pale brown solid; mp ≥ 300 °C; Rf = 0.2 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 6.17 (s, 2H), 6.82 (s, 1H), 7.49 (s, 1H), 7.57–7.61 (m, 2H), 7.68 (t, J = 7.3 Hz, 1H), 7.98 (d, J = 7.4 Hz, 2H), 8.78 (s, 1H), 12.15 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 94.8, 102.6, 106.6, 111.8, 126.9, 128.2, 128.8, 133.4, 139.6, 140.0, 143.6, 144.1, 153.3, 156.3. IR (neat) νmax 1152, 1245, 1444, 1629, 1650, 2933 cm−1. HRMS[EI+] calcd for C16H11NO5S [M]+ 329.0358, found 329.0357.
6-(Methoxymethoxy)-3-(phenylsulfonyl)quinolin-2(1H)-one (4la)
Yield = 46%; pale yellow solid; mp 239–240 °C; Rf = 0.2 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 3.39 (s, 3H), 5.23 (s, 2H), 7.31 (d, J = 9.0 Hz, 1H), 7.42 (dd, J = 9.0 and 2.3 Hz, 1H), 7.58–7.62 (m, 2H), 7.67–7.71 (m, 2H), 8.00 (d, J = 7.8 Hz, 2H), 8.91 (s, 1H), 12.18 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 55.7, 94.3, 114.9, 116.7, 117.7, 124.9, 128.4, 128.9, 131.1, 133.7, 136.4, 139.6, 144.1, 151.8, 156.0. IR (neat) νmax 1152, 1312, 1495, 1625, 1649, 3144 cm−1. HRMS[EI+] calcd for C17H15NO5S [M]+ 345.0671, found 345.0671.
6-(Benzyloxy)-3-(phenylsulfonyl)quinolin-2(1H)-one (4ma)
Yield = 45%; yellow solid; mp ≥ 300 °C; Rf = 0.2 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 5.15 (s, 2H), 7.29–7.36 (m, 2H), 7.39–7.44 (m, 3H), 7.48–7.50 (m, 2H), 7.59–7.62 (m, 2H), 7.68–7.72 (m, 2H), 8.00 (d, J = 7.8 Hz, 2H), 8.90 (s, 1H), 12.18 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 69.8, 112.2, 116.8, 117.7, 124.4, 127.9, 128.0, 128.4, 128.5, 128.9, 131.0, 133.7, 135.9, 136.6, 139.6, 143.9, 153.6, 156.0. IR (neat) νmax 1149, 1293, 1498, 1622, 1654, 3149 cm−1. HRMS[EI+] calcd for C22H17NO4S [M]+ 391.0878, found 391.0870.
6-((1-Methyl-1H-tetrazol-5-yl)methoxy)-3-(phenylsulfony-l)quino lin-2(1H)-one (4na)
Yield = 50%; pale yellow solid; mp 274–275 °C; Rf = 0.1 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 4.40 (s, 3H), 5.42 (s, 2H), 7.32 (d, J = 9.1 Hz, 1H), 7.44 (dd, J = 9.1 and 2.8 Hz, 1H), 7.59–7.63 (m, 2H), 7.68–7.75 (m, 2H), 8.01 (d, J = 7.8 Hz, 2H), 8.89 (s, 1H), 12.20 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 39.6, 60.7, 112.6, 116.9, 117.6, 124.1, 128.4, 128.9, 131.1, 133.7, 136.3, 139.6, 143.9, 152.9, 156.0, 161.8. IR (neat) νmax 1153, 1290, 1493, 1624, 1658, 3015 cm−1. HRMS[EI+] calcd for C18H15N5O4S [M]+ 397.0845, found 397.0852.
3-(Phenylsulfonyl)benzo[h]quinolin-2(1H)-one (4oa)
Yield = 40%; pale brown solid; mp ≥ 300 °C; Rf = 0.3 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 7.61–7.65 (m, 3H), 7.69–7.74 (m, 3H), 7.92 (d, J = 8.6 Hz, 1H), 7.98 (d, J = 8.0 Hz, 1H), 8.07 (d, J = 7.7 Hz, 2H), 8.84 (d, J = 8.3 Hz, 1H), 9.04 (s, 1H), 12.59 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 113.7, 120.9, 123.2, 123.3, 126.2, 127.0, 128.4, 128.6, 128.9, 129.6, 129.7, 133.7, 135.1, 139.5, 139.7, 145.0, 157.1. IR (neat) νmax 1150, 1308, 1508, 1627, 1644, 3059 cm−1. HRMS[EI+] calcd for C19H13NO3S [M]+ 335.0616, found 335.0614.
3-(Phenylsulfonyl)-1,8-naphthyridin-2(1H)-one (4pa)
Yield = 31%; pale yellow solid; mp ≥ 300 °C; Rf = 0.2 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 7.37 (m, 1H), 7.60–7.64 (m, 2H), 7.72 (t, J = 7.4 Hz, 1H), 8.00–8.02 (m, 2H), 8.48 (m, 1H), 8.67 (m, 1H), 9.02 (s, 1H), 12.71 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 112.6, 119.3, 128.5, 129.0, 131.8, 133.8, 139.3, 139.5, 144.2, 151.2, 154.1, 157.2. IR (neat) νmax 1147, 1304, 1470, 1608, 1646, 3020 cm−1. HRMS[EI+] calcd for C14H10N2O3S [M]+ 286.0412, found 286.0396.
9-Methyl-3-(phenylsulfonyl)-1,9-dihydro-2H-pyrido[2,3-b]indol-2-one (4qa)
Yield = 42%; yellow solid; mp ≥ 300 °C; Rf = 0.2 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 3.92 (s, 3H), 7.14 (t, J = 7.5 Hz, 1H), 7.50 (t, J = 7.8 Hz, 1H), 7.57–7.62 (m, 3H), 7.67 (t, J = 7.3 Hz, 1H) 8.02 (d, J = 7.7 Hz, 2H), 8.08 (d, J = 8.2 Hz, 1H), 8.76 (s, 1H), 12.99 (s, 1H). 13C NMR (100 MHz, TFA-d) δ 30.8, 112.8, 115.4, 123.7, 124.9, 130.1, 130.3, 130.8, 131.9, 133.7, 135.7, 137.7, 140.0, 147.4, 156.8. IR (neat) νmax 1090, 1154, 1306, 1555 1616, 1641 cm−1. HRMS[EI+] calcd for C18H14N2O3S [M]+ 338.0725, found 338.0732.
6-(Phenylsulfonyl)thieno[3,2-b]pyridin-5(4H)-one (4ra)
Yield = 23%; pale brown solid; mp 259–260 °C; Rf = 0.1 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 7.04 (d, J = 5.4 Hz, 1H), 7.56–7.60 (m, 2H), 7.67 (t, J = 7.3 Hz, 1H), 7.98 (d, J = 7.7 Hz, 2H), 8.23 (d, J = 5.4 Hz, 1H), 9.04 (s, 1H), 12.72 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 115.9, 116.6, 125.3, 128.2, 128.8, 133.3, 139.2, 139.3, 140.1, 147.6, 156.8. IR (neat) νmax 1152, 1312, 1441, 1631, 1641, 3102 cm−1. HRMS[EI+] calcd for C13H9NO3S2 [M]+ 291.0024, found 291.0022.
4-Methyl-3-(phenylsulfonyl)quinolin-2(1H)-one (4sa)
Yield = 53%; white solid; mp 282–283 °C; Rf = 0.25 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 3.11 (s, 3H), 7.29–7.32 (m, 2H), 7.54–7.58 (m, 2H), 7.63–7.67 (m, 2H), 7.94–7.96 (m, 2H), 8.09 (m, 1H), 11.98 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 14.8, 115.7, 119.0, 122.6, 126.9, 127.4, 128.5, 128.9, 132.9, 133.3, 139.3, 142.4, 154.6, 156.7. IR (neat) νmax 1143, 1302, 1497, 1636, 1653, 2840 cm−1. HRMS[EI+] calcd for C16H13NO3S [M]+ 299.0616, found 299.0613.
4-Cyclopropyl-3-(phenylsulfonyl)quinolin-2(1H)-one (4ta)
Yield = 68%; pale yellow soild; mp 261–262 °C; Rf = 0.3 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 0.75–0.79 (m, 2H), 1.32–1.37 (m, 2H), 2.46 (m, 1H), 7.26–7.32 (m, 2H), 7.51–7.55 (m, 2H), 7.60–7.64 (m, 2H), 7.91 (d, J = 7.6 Hz, 2H), 8.35 (d, J = 8.3 Hz, 1H), 11.99 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 10.2, 12.8, 115.7, 119.9, 122.3, 127.6, 127.9, 128.3, 132.1, 132.6, 132.8, 139.2, 143.0, 156.8, 158.6. IR (neat) νmax 1159, 1314, 1593, 1637, 2847, 2995 cm−1. HRMS[EI+] calcd for C18H15NO3S [M]+ 325.0773, found 352.0776.
4-Phenyl-3-(phenylsulfonyl)quinolin-2(1H)-one (4ua)
Yield = 55%; pale white solid; mp 287–288 °C; Rf = 0.35 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 6.87 (d, J = 8.3 Hz, 1H), 7.13 (t, J = 7.7 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H), 7.40–7.42 (m, 2H), 7.50–7.57 (m, 5H), 7.61–7.66 (m, 2H), 7.89 (d, J = 7.6 Hz, 2H), 12.18 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 115.5, 119.5, 122.6, 127.7, 127.9, 128.1, 128.5, 128.6, 128.8, 133.0, 133.5, 134.4, 139.9, 141.6, 155.9, 156.7. IR (neat) νmax 1149, 1306, 1653, 2851, 2876, 2982 cm−1. HRMS[EI+] calcd for C21H15NO3S [M]+ 361.0773, found 361.0791.
3-(Phenylsulfonyl)-4-(thiophen-2-yl)quinolin-2(1H)-one (4va)
Eluent used for the chromatography (Hex:EtOAc = 1:1); yield = 41%; gray solid; mp 250–251 °C; Rf = 0.3 (Hex:EtOAc = 1:1); 1H NMR (400 MHz, CDCl3-d) δ 7.19–7.22 (m, 2H), 7.23–7.27 (m, 2H), 7.36 (d, J = 8.2 Hz, 1H), 7.46–7.49 (m, 2H), 7.54 (t, J = 7.2 Hz, 1H), 7.62–7.65 (m, 2H), 8.11 (d, J = 7.7 Hz, 2H), 12.78 (s, 1H). 13C NMR (100 MHz, CDCl3-d) δ 116.3, 121.2, 123.7, 127.2, 127.5, 128.4, 128.9, 129.2, 129.2, 131.2, 132.7, 133.2, 133.7, 139.2, 141.7, 150.8, 159.4. IR (neat) νmax 1145, 1309, 1637, 1653, 2845, 3109 cm−1. HRMS[EI+] calcd for C19H13NO3S2 [M]+ 367.0337, found 367.0339.
N,N-Diethyl-2-oxo-3-(phenylsulfonyl)-1,2-dihydroquinoline-4-carboxamide (4wa)
Eluent for the chromatography (Hex:EtOAc = 2:1 to 3:7); yield = 43%; pale white solid; mp 214–215 °C; Rf = 0.3 (Hex:EtOAc = 1:2); 1H NMR (400 MHz, CDCl3-d) δ 1.12 (t, J = 7.1 Hz, 3H), 1.42 (t, J = 7.1 Hz, 3H), 3.27–3.40 (m, 2H), 3.65 (m, 1H), 3.86 (m, 1H), 7.26–7.31 (m, 2H), 7.45–7.49 (m, 2H), 7.54 (t, J = 7.3 Hz, 1H), 7.62–7.66 (m, 2H), 8.23–8.25 (m, 2H), 12.65 (s, 1H). 13C NMR (100 MHz, CDCl3-d) δ 12.2, 13.2, 39.0, 43.5, 116.3, 116.8, 124.1, 125.9, 127.8, 128.6, 129.4, 133.7, 134.2, 140.0, 140.6, 151.4, 159.4, 164.5. IR (neat) νmax 1151, 1309, 1459, 1616, 1674, 2991 cm−1. HRMS[EI+] calcd for C20H20N2O4S [M]+ 384.1144, found 384.1155.
3-Tosylquinolin-2(1H)-one (4ab)
Yield = 62%; white solid; mp ≥ 300 °C; Rf = 0.1 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 2.37 (s, 3H), 7.28 (m, 1H), 7.34 (d, J = 8.3 Hz, 1H), 7.40 (d, J = 8.2 Hz, 2H), 7.66 (m, 1H), 7.88–7.90 (m, 2H), 8.00 (d, J = 7.5 Hz, 1H), 8.94 (s, 1H), 12.22 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 21.1, 115.3, 117.2, 122.7, 128.5, 129.4, 130.5, 131.0, 133.9, 136.7, 141.0, 144.2, 144.3, 156.4. IR (neat) νmax 1149, 1315, 1556, 1620, 1657, 2830 cm−1. HRMS[EI+] calcd for C16H13NO3S [M]+ 299.0616, found 299.0603.
3-((4-Methoxyphenyl)sulfonyl)quinolin-2(1H)-one (4ac)
Yield = 42%; pale yellow solid; mp 285–286 °C; Rf = 0.1 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 3.84 (s, 3H), 7.10–7.13 (m, 2H), 7.29 (t, J = 7.5 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.67 (m, 1H), 7.93–7.95 (m, 2H), 8.00 (d, J = 8.0 Hz, 1H), 8.91 (s, 1H), 12.15 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 55.8, 114.1, 115.3, 117.2, 122.7, 130.5, 130.9, 131.0, 131.4, 133.8, 140.9, 143.8, 156.4, 163.3. IR (neat) νmax 1150, 1263, 1496, 1619, 1648, 2998 cm−1. HRMS[EI+] calcd for C16H13NO4S [M]+ 315.0565, found 315.0569.
3-((4-Chlorophenyl)sulfonyl)quinolin-2(1H)-one (4ad)
Yield = 50%; off-white solid; mp 264–265 °C; Rf = 0.2 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 7.29 (t, J = 7.6 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.66–7.70 (m, 3H), 8.02 (d, J = 8.2 Hz, 3H), 8.97 (s, 1H), 12.27 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 115.4, 117.2, 122.8, 129.1, 130.2, 130.5, 130.7, 134.1, 138.4, 138.8, 141.1, 144.9, 156.4. IR (neat) νmax 1162, 1321, 1476, 1619, 1651, 2828 cm−1. HRMS[EI+] calcd for C15H10ClNO3S [M]+ 319.0070, found 319.0057.
3-((4-Fluorophenyl)sulfonyl)quinolin-2(1H)-one (4ae)
Yield = 55%; white solid; mp ≥ 300 °C; Rf = 0.2 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 7.29 (t, J = 7.5 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 7.43–7.48 (m, 2H), 7.68 (m, 1H), 8.02 (d, J = 7.8 Hz, 1H), 8.07–8.12 (m, 2H), 8.96 (s, 1H), 12.27 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 115.4, 116.1 (d, JC,F = 23 Hz), 117.2, 122.8, 130.5, 130.6, 131.8 (d, JC,F = 10 Hz), 134.0, 135.8 (d, JC,F = 3 Hz), 141.0, 144.7, 156.4, 165.0 (d, JC,F = 251 Hz). 19F NMR (376 MHz, DMSO-d6) δ −104.9 IR (neat) νmax 1147, 1319, 1488, 1621, 1657, 2829 cm−1. HRMS[EI+] calcd for C15H10FNO3S [M]+ 303.0365, found 303.0361.
3-(Methylsulfonyl)quinolin-2(1H)-one (4af)
Yield = 53%; pale white solid; mp 277–278 °C; Rf = 0.2 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 3.33 (s, 3H), 7.30 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.69 (m, 1H), 7.99 (m, 1H), 8.71 (s, 1H), 12.46 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 41.4, 115.4, 117.2, 122.8, 130.4, 131.1, 133.8, 140.6, 143.2, 157.4. IR (neat) νmax 1142, 1292, 1557, 1619, 1648, 2824 cm−1. HRMS[EI+] calcd for C10H9NO3S [M]+ 223.0303, found 223.0320.
3-(Ethylsulfonyl)quinolin-2(1H)-one (4ag)
Yield = 50%; off-white solid; mp 242–243 °C; Rf = 0.15 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 1.15 (t, J = 7.4 Hz, 3H), 3.52 (q, J = 7.4 Hz, 2H), 7.30 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.70 (m, 1H), 7.99 (d, J = 7.9 Hz, 1H), 8.72 (s, 1H), 12.46 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 6.7, 46.8, 115.5, 117.3, 122.8, 128.9, 130.4, 133.9, 140.7, 144.6, 157.4. IR (neat) νmax 1127, 1289, 1555, 1618, 1648, 2839 cm−1. HRMS[EI+] calcd for C11H11NO3S [M]+ 237.0460, found 237.0456.
3-(Cyclopropylsulfonyl)quinolin-2(1H)-one (4ah)
Yield = 56%; pale yellow solid; mp ≥ 300 °C; Rf = 0.25 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 1.03–1.11 (m, 4H), 3.22 (m, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.96 (d, J = 7.9 Hz, 1H), 8.61 (s, 1H), 12.44 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 5.1, 30.0, 115.4, 117.1, 122.8, 130.4, 130.7, 133.7, 140.6, 142.8, 157.4. IR (neat) νmax 1142, 1287, 1618, 1654, 2867, 3006 cm−1. HRMS[EI+] calcd for C12H11NO3S [M]+ 249.0460, found 249.0457.
3-(Cyclohexylsulfonyl)quinolin-2(1H)-one (4ai)
Yield = 27%; pale yellow solid; mp 268–269 °C; Rf = 0.2 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 1.12–1.29 (m, 3H), 1.35–1.44 (m, 2H), 1.62 (d, J = 12.0 Hz, 1H), 1.79 (d, J = 12.6 Hz, 2H), 1.89 (d, J = 11.5 Hz, 2H), 3.72 (m, 1H), 7.29 (t, J = 7.5 Hz, 1H), 7.40 (d, J = 8.3 Hz, 1H), 7.69 (m, 1H), 7.98 (d, J = 7.9 Hz, 1H), 8.69 (s, 1H), 12.42 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 24.2, 24.4, 24.9, 58.8, 115.4, 117.3, 122.8, 128.2, 130.3, 133.9, 140.7, 145.1, 157.4. IR (neat) νmax 1127, 1303, 1480, 1620, 1642, 2927 cm−1. HRMS[EI+] calcd for C15H17NO3S [M]+ 291.0929, found 291.0923.
3-((6-Methylpyridin-2-yl)sulfonyl)quinolin-2(1H)-one (4aj)
Yield = 26%; pale yellow solid; mp 275–276 °C; Rf = 0.2 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 2.43 (s, 3H), 7.32 (t, J = 7.6 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H), 7.55 (m, 1H), 7.70 (m, 1H), 8.03–8.08 (m, 3H), 9.01 (s, 1H), 12.24 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 23.7, 115.5, 117.2, 120.2, 122.9, 127.4, 129.1, 130.5, 134.1, 138.6, 141.0, 145.6, 156.4, 156.6, 159.0. IR (neat) νmax 1117, 1313, 1452, 1619, 1649, 2840 cm−1. HRMS[EI+] calcd for C15H12N2O3S [M]+ 300.0569, found 300.0556.
3-(Thiophen-2-ylsulfonyl)quinolin-2(1H)-one (4ak)
Yield = 47%; off-white solid; mp ≥ 300 °C; Rf = 0.2 (DCM:MeOH = 93:7) 1H NMR (400 MHz, DMSO-d6) δ 7.22 (t, J = 4.4 Hz, 1H), 7.29 (t, J = 7.6 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.92 (d, J = 3.8 Hz, 1H), 8.01 (d, J = 8.0 Hz, 1H), 8.08 (d, J = 5.0 Hz, 1H), 8.92 (s, 1H), 12.35 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 115.4, 117.1, 122.8, 127.8, 130.6, 131.1, 134.1, 135.4, 135.8, 140.2, 140.9, 143.9, 156.4. IR (neat) νmax 1147, 1313, 1480, 1620, 1655, 2833 cm−1. HRMS[EI+] calcd for C13H9NO3S2 [M]+ 291.0024, found 291.0024.
3-(((7,7-Dimethyl-2-oxobicyclo[2.2.1]heptan-1yl)methyl)sulfo-nyl)quino-lin-2(1H)-one (4al)
Eluent for the chromatography (Hex:EtOAc = 1:1); yield = 26%; white solid; mp 260–261 °C; Rf = 0.2 (Hex:EtOAc = 1:1); 1H NMR (400 MHz, DMSO-d6) δ 0.81 (s, 3H), 1.01 (s, 3H), 1.38 (m, 1H), 1.57 (m, 1H), 1.86 (d, J = 18.4 Hz, 1H), 1.93 (m, 1H), 2.04 (t, J = 4.3 Hz, 1H), 2.26–2.32 (m, 2H), 3.45 (d, J = 15.2 Hz, 1H), 3.96 (d, J = 15.2 Hz, 1H), 7.29 (t, J = 7.5 Hz, 1H), 7.41 (d, J = 8.3 Hz, 1H), 7.69 (m, 1H), 7.98 (d, J = 7.8 Hz, 1H), 8.65 (s, 1H), 12.40 (s, 1H). 13C NMR (100 MHz, DMSO-d6) d 19.3, 19.4, 24.7, 26.4, 41.8, 42.0, 48.0, 49.7, 58.4, 115.4, 117.3, 122.7, 130.4, 131.8, 133.7, 140.7, 143.1, 157.6, 214.1. IR (neat) νmax 1138, 1314, 1626, 1646, 1746, 2954 cm−1. HRMS[EI+] calcd for C19H21NO4S [M]+ 359.1191, found 359.1187.
3-(Phenylthio)quinolin-2(1H)-one (5)
A 5 mL vial was charged with 2-bromobenzaldehyde 1a (92.5 mg, 0.50 mmol), 2-iodoacetamide 2 (185.0 mg, 1.00 mmol), sodium thiophenoxide (90%, 146.8 mg, 1.00 mmol), Cu powder (60–80 nm, 6.4 mg, 0.10 mmol), 2-picolinic acid (12.3 mg, 0.10 mmol), K2CO3 (172.8 mg, 1.25 mmol) and Ca(OH)2 (92.6 mg, 1.25 mmol) in dry DMF (1.5 mL). The vial was sealed with the cap and heated at 110 °C under stirring for 48 h. After cooling to room temperature, the reaction mixture was diluted with saturated NH4Cl solution and extracted with ethyl acetate (20 × 5 mL). The combined organic solution was dried over MgSO4 and concentrated, and subjected to silica gel column chromatography (DCM:MeOH = 97:3 to 95:5) to afford the quinolone 5 (69.7 mg, 55%).
Off-white solid; mp 241–242 °C; Rf = 0.3 (DCM:MeOH = 93:7); 1H NMR (400 MHz, DMSO-d6) δ 7.11 (t, J = 7.5 Hz, 1H), 7.21 (s, 1H), 7.31 (d, J = 8.2 Hz, 1H), 7.41–7.45 (m, 2H), 7.48–7.56 (m, 5H), 12.14 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 115.0, 119.3, 122.2, 126.8, 129.0, 129.5, 130.0, 130.7, 132.3, 133.4, 133.7, 136.9, 159.1. IR (neat) νmax 1424, 1551, 1638, 1655, 2882, 2989 cm−1. HRMS[EI+] calcd for C15H11NOS [M]+ 253.0561, found 253.0561.
General procedure for 6a–c
A 5 mL vial was charged with 2-bromobenzaldehyde 1a (92.5 mg, 0.50 mmol), 2-iodoacetamide 2 (185.0 mg, 1.00 mmol), a corresponding sodium phenoxide (1.00 mmol), Cu powder (60–80 nm, 6.4 mg, 0.10 mmol), 2-picolinic acid (12.3 mg, 0.10 mmol) and K3PO4 (265.3 mg, 1.25 mmol) in dry DMF (1.5 mL). The vial was sealed with the cap and heated at 110 °C under stirring for 48 h. After cooling to room temperature, the reaction mixture was diluted with saturated NH4Cl solution and extracted with ethyl acetate (20 × 5 mL). The combined organic solution was dried over MgSO4 and concentrated, and subjected to silica gel column chromatography (Hex:EtOAc = 1:2 to 1:1) to afford the 2-quinolones.
3-Phenoxyquinolin-2(1H)-one (6a)
Yield = 37%; off-white solid; mp 209–210 °C; Rf = 0.4 (Hex:EtOAc = 1:1); 1H NMR (400 MHz, DMSO-d6) δ 7.03–7.05 (m, 2H), 7.12 (t, J = 7.4 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.33–7.39 (m, 3H), 7.45 (m, 1H), 7.53 (s, 1H), 7.61 (d, J = 7.8 Hz, 1H), 12.13 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 114.8, 117.3, 119.0, 122.1, 123.2, 123.7, 127.3, 128.9, 129.8, 136.1, 145.1, 156.4, 157.5. IR (neat) νmax 1228, 1571, 1638, 1655, 2862, 3011 cm−1. HRMS[EI+] calcd for C15H11NO2 [M]+ 237.0790, found 237.0802.
3-(4-Chlorophenoxy)quinolin-2(1H)-one (6b)
Yield = 51%; white solid; mp 220–221 °C; Rf = 0.4 (Hex:EtOAc = 1:1); 1H NMR (400 MHz, DMSO-d6) δ 7.05–7.07 (m, 2H), 7.19 (m, 1H), 7.34–7.40 (m, 3H), 7.46 (m, 1H), 7.62–7.64 (m, 2H), 12.19 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 114.9, 118.9, 118.9, 122.2, 124.8, 126.8, 127.5, 129.1, 129.6, 136.3, 144.5, 155.5, 157.3. IR (neat) νmax 1226, 1430, 1479, 1654, 2854, 3013 cm−1. HRMS[EI+] calcd for C15H10ClNO2 [M]+ 271.0400, found 271.0395.
3-(4-Methoxyphenoxy)quinolin-2(1H)-one (6c)
Yield = 28%; pale brown solid; mp 184–185 °C; Rf = 0.2 (Hex:EtOAc = 1:1); 1H NMR (400 MHz, DMSO-d6) δ 3.75 (s, 3H), 6.95–6.98 (m, 2H), 7.03–7.06 (m, 2H), 7.13 (m, 1H), 7.22 (s, 1H), 7.31 (m, 1H), 7.40 (m, 1H), 7.54 (d, J = 7.9 Hz, 1H), 12.11 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 55.4, 114.7, 115.0, 119.1, 119.8, 120.0, 122.1, 127.0, 128.3, 135.4, 147.0, 149.0, 155.7, 157.4. IR (neat) νmax 1224, 1497, 1569, 1654, 2847, 2995 cm−1. HRMS[EI+] calcd for C16H13NO3 [M]+ 267.0895, found 267.0895.
4-Methyl-3-(methyl(phenyl)amino)quinolin-2(1H)-one (7)
A 5 mL vial was charged with 2′-bromoacetophenone 1s (99.5 mg, 0.50 mmol), 2-iodoacetamide 2 (138.7 mg, 0.75 mmol), N-methylaniline (0.08 mL, 0.75 mmol), CuI (19.0 mg, 0.10 mmol), 2-picolinic acid (6.2 mg, 0.05 mmol), K2CO3 (138.2 mg, 1.00 mmol) and Ca(OH)2 (74.1 mg, 1.00 mmol) in dry DMF (1.5 mL). The vial was sealed with the cap and heated at 110 °C under stirring for 48 h. After cooling to room temperature, the reaction mixture was diluted with saturated NH4Cl solution and extracted with ethyl acetate (20 × 5 mL). The combined organic solution was dried over MgSO4 and concentrated, and subjected to silica gel column chromatography (Hex:EtOAc = 2:1 to 1:1) to afford the quinolone 7 (58.1 mg, 44%).
Pale yellow solid; mp 250–251 °C; Rf = 0.3 (Hex:EtOAc = 1:1); 1H NMR (400 MHz, DMSO-d6) δ 2.31 (s, 3H), 3.12 (s, 3H), 6.51 (d, J = 8.1 Hz, 2H), 6.64 (t, J = 7.2 Hz, 1H), 7.10–7.14 (m, 2H), 7.23 (t, J = 7.6 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.51 (t, J = 7.7 Hz, 1H), 7.77 (d, J = 8.1 Hz, 1H), 11.85 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 13.6, 37.4, 111.8, 115.2, 116.5, 119.7, 121.8, 125.4, 128.9, 129.9, 134.6, 137.4, 144.9, 148.2, 159.4. IR (neat) νmax 1338, 1496, 1560, 1636, 2811, 2938 cm−1. HRMS[EI+] calcd for C17H16N2O [M]+ 264.1263, found 264.1270.
2-Chloro-3-(phenylsulfonyl)quinoline (8)
A mixture of compound 4aa (42.8 mg, 0.15 mmol) in POCl3 (0.35 mL, 3.75 mmol) was stirred at reflux for 1 h. Then, the reaction mixture was cooled to room temperature and quenched with ice–water. The mixture was neutralized with 1 N NaOH (15 mL) at 0 °C and extracted with EtOAc (10 × 3 mL). The combined organic solution was dried over MgSO4, and concentrated. The residue was subjected to column chromatography (Hex:EtOAc = 6:1) to afford the quinoline 8 (43.4 mg, 95%) as a white solid.
Mp 150–151 °C; Rf = 0.4 (Hex:EtOAc = 5:1); 1H NMR (400 MHz, CDCl3-d) δ 7.52–7.56 (m, 2H), 7.64 (t, J = 7.4 Hz, 1H), 7.71 (t, J = 7.6 Hz, 1H), 7.91 (m, 1H), 8.00–8.06 (m, 4H), 9.23 (s, 1H). 13C NMR (100 MHz, CDCl3-d) δ 126.0, 128.6, 128.7, 129.0, 129.2, 129.4, 133.1, 133.9, 134.1, 139.3, 142.2, 145.4, 149.2. IR (neat) νmax 1140, 1319, 1444, 1571, 2927, 3078 cm−1. HRMS[EI+] calcd for C15H10ClNO2S [M]+ 303.0121, found 303.0119.
1-Benzyl-3-(phenylsulfonyl)quinolin-2(1H)-one (9)
To a stirred solution of the compound 4aa (285.3 mg, 1.00 mmol) in THF (10 mL) was added NaH (60% dispersed in mineral oil, 52.0 mg, 1.30 mmol) at 0 °C under N2. After 10 min, benzyl bromide (0.14 mL, 1.20 mmol) was added at 0 °C and the reaction mixture was stirred at room temperature for 1 h. Then, the reaction mixture was quenched with water and extracted with ethyl acetate. The combined organic solution was dried over MgSO4 and concentrated. The residue was subjected to column chromatography (Hex:EtOAc = 4:1 to 3:2) to afford the product 9 (274 mg, 73%) as a white solid.
Mp 200–201 °C; Rf = 0.65 (Hex:EtOAc = 1:1); 1H NMR (400 MHz, DMSO-d6) δ 5.44 (s, 2H), 7.08 (d, J = 7.4 Hz, 2H), 7.18–7.27 (m, 3H), 7.36 (t, J = 7.5 Hz, 1H), 7.47 (d, J = 8.7 Hz, 1H), 7.60–7.64 (m, 2H), 7.68–7.73 (m, 2H), 8.04 (d, J = 7.7 Hz, 2H), 8.14 (d, J = 7.8 Hz, 1H), 9.09 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 44.9, 115.5, 118.2, 123.1, 126.4, 127.2, 128.4, 128.6, 128.9, 130.0, 132.0, 133.7, 134.4, 136.0, 139.5, 140.9, 144.30, 156.2. IR (neat) νmax 1143, 1285, 1440, 1559, 1636, 3066 cm−1. HRMS[EI+] calcd for C22H17NO3S [M]+ 375.0929, found 375.0940.
4-Allyl-1-benzyl-3-(phenylsulfonyl)-3,4-dihydroquinolin-2(1H)-one (10)
To a stirred solution of the compound 9 (75.1 mg, 0.20 mmol) in THF (2 mL) was added allyl magnesium bromide (1.0 M in ether, 0.30 mL, 0.30 mmol) at room temperature. After 1 h, the reaction mixture was quenched with water and extracted with EtOAc (5 × 3 mL). The combined organic solution was dried over MgSO4, and concentrated. The residue was subjected to column chromatography (Hex:EtOAc = 4:1) to afford the product 10 (76.8 mg, 92%) as a white solid.
Mp 77–78 °C; Rf = 0.1 (Hex:EtOAc = 5:1); 1H NMR (400 MHz, CDCl3-d) δ 2.28 (m, 1H), 2.40 (m, 1H), 3.83 (m, 1H), 4.24 (s, 1H), 4.99 (d, J = 8.2 Hz, 1H) 5.03 (d, J = 9.2 Hz, 1H), 5.12 (d, J = 10.2 Hz, 1H), 5.22 (d, J = 16.1 Hz, 1H), 5.70 (m, 1H), 6.80 (d, J = 8.1 Hz, 1H), 7.04 (t, J = 7.3 Hz, 1H), 7.11 (m, 1H), 7.21 (m, 1H), 7.27 (m, 1H), 7.32–7.40 (m, 6H), 7.55 (t, J = 7.5 Hz, 1H), 7.70 (d, J = 7.8 Hz, 2H). 13C NMR (100 MHz, CDCl3-d) δ 37.2, 40.0, 47.0, 70.3, 116.0, 119.6, 124.1, 125.4, 126.9, 127.5, 128.4, 128.9, 128.9, 129.0, 133.2, 134.1, 136.3, 137.8, 138.2, 161.1. IR (neat) νmax 1147, 1308, 1601, 1665, 2927, 3069 cm−1. HRMS[EI+] calcd for C25H23NO3S [M]+ 417.1399, found 417.1403.
3-(Tributylstannyl)quinolin-2(1H)-one (11)
A mixture of compound 4aa (42.8 mg, 0.15 mmol), AIBN (2.5 mg, 15 μmol), SnBu3H (0.06 mL, 0.23 mmol) in dioxane (1.5 mL) was stirred at reflux for 30 h. Then, the reaction mixture was cooled to room temperature and concentrated. The residue was subjected to a short pad of silica gel (silica gel was pretreated with 3% NEt3 in DCM) and rinsed with DCM to afford the compound 11 (35.1 mg, 54%) as a white solid.Mp 69–70 °C; Rf = 0.4 (DCM); 1H NMR (400 MHz, CDCl3-d) δ 0.89–0.92 (m, 10H), 1.15–1.19 (m, 5H), 1.33–1.42 (m, 6H), 1.58–1.66 (m, 6H), 7.17 (t, J = 7.5 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.44 (m, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.89 (m, 1H), 12.39 (s, 1H). 13C NMR (100 MHz, CDCl3-d) δ 10.1, 13.9, 27.6, 29.3, 116.0, 120.8, 122.1, 127.2, 130.0, 139.1, 139.4, 149.2, 167.8. IR (neat) νmax 1424, 1611, 1626, 2849, 2914, 2952 cm−1.
3-Chloroquinolin-2(1H)-one (12)
To a stirred solution of the compound 11 (43.4 mg, 0.10 mmol) in THF (1 mL) was added CuCl2 (33.6 mg, 0.25 mmol) and 2,6-lutidine (10 μL, 0.10 mmol) at room temperature. After 1 h, the reaction mixture was quenched with saturated NaHCO3 solution and extracted with EtOAc (5 × 3 mL). The combined organic solution was dried over MgSO4, and concentrated. The residue was subjected to column chromatography (DCM:MeOH = 98:2) to afford the product 12 (16.0 mg, 89%) as a white solid.
Mp 252–253 °C; Rf = 0.1 (DCM); 1H NMR (400 MHz, DMSO-d6) δ 7.22 (t, J = 7.5 Hz, 1H), 7.34 (d, J = 8.3 Hz, 1H), 7.53 (t, J = 7.7 Hz, 1H), 7.67 (d, J = 7.9 Hz, 1H), 8.30 (s, 1H), 12.29 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 115.2, 118.8, 122.4, 125.7, 127.4, 130.6, 137.7, 137.7, 157.6. IR (neat) νmax 1203, 1429, 1637, 1654, 2921, 3006 cm−1. HRMS[EI+] calcd for C9H6ClNO [M]+ 179.0138, found 179.0136.
3-Iodoquinolin-2(1H)-one (13)
A mixture of compound 11 (43.4 mg, 0.10 mmol), NaI (16.5 mg, 0.11 mmol), FeCl3 (35.7 mg, 0.22 mmol) in THF/H2O (0.2 mL/0.2 mL) was stirred at rt for 1 h. Then, the reaction mixture was diluted with saturated NaHSO3 solution and extracted with ethyl acetate (5 × 3 mL). The combined organic solution was dried over MgSO4, and concentrated. The residue was subjected to column chromatography (DCM:MeOH = 98:2 to 97:3) to afford the product 12 (21.7 mg, 80%) as a white solid.
Mp 259–260 °C; Rf = 0.4 (Hex:EtOAc = 1:1); 1H NMR (400 MHz, DMSO-d6) δ 7.18 (t, J = 7.5 Hz, 1H), 7.31 (d, J = 8.3 Hz, 1H), 7.53 (t, J = 7.7 Hz, 1H), 7.64 (d, J = 7.9 Hz, 1H), 8.70 (s, 1H), 12.11 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 95.9, 115.2, 120.3, 122.1, 127.0, 130.8, 138.8, 148.8, 158.8. IR (neat) νmax 1210, 1421, 1627, 1637, 2922, 3122 cm−1. HRMS[EI+] calcd for C9H6INO [M]+ 270.9494, found 270.9495.
N-(2-Formylphenyl)-2-iodoacetamide (II)
To a stirred solution of 2-iodoacetic acid (1.95 g, 10.5 mmol) in ethyl acetate (25 mL) was added 2-aminobenzaldehyde (1.21 g, 10.0 mmol) at 0 °C. Subsequently, N,N′-dicyclohexylcarbo-diimide (2.06 g, 10.0 mmol) in EA (10 mL) was added dropwise to the mixture. After 30 min, the reaction mixture stirred at room temperature for 10 h. Then, the resulting slurry was filtered through a pad of Celite. The filtrate was concentrated and was subjected to column chromatography (Hex:EtOAc = 7:1) to afford the product II (1.38 g, 48%) as a pale yellow solid.
Mp 110–111 °C; Rf = 0.38 (Hex:EtOAc = 5:1); 1H NMR (400 MHz, CDCl3-d) δ 3.90 (s, 2H), 7.28 (m, 1H), 7.64 (t, J = 8.0 Hz, 1H), 7.71 (d, J = 7.7 Hz, 1H), 8.68 (d, J = 8.5 Hz, 1H), 9.94 (s, 1H), 11.52 (s, 1H). 13C NMR (100 MHz, CDCl3-d) δ −0.1, 120.0, 122.0, 123.7, 136.1, 136.3, 140.5, 167.0, 195.6. IR (neat) νmax 1074, 1192, 1290, 1444, 1653, 3231 cm−1.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the KRICT, Republic of Korea (SI1935-10) and the National Research Foundation of Korea (NRF-2019R1C1C1004970) are greatly appreciated. Prof. Michael Young (The University of Toledo) is acknowledged for proofreading the manuscript.Notes and references
- (a) B. Joseph, F. Darro, A. Behard, B. Lesur, F. Collignon, C. Decaestecker, A. Frydman, G. Guillaumet and R. Kiss, J. Med. Chem., 2002, 45, 2543 CrossRef CAS; (b) J. Shi, Z. Xiao, M. A. Ihnat, C. Kamat, B. Pandit, Z. Hu and P. K. Li, Bioorg. Med. Chem. Lett., 2003, 6, 1187 CrossRef; (c) S. Pasquini, M. De Rosa, V. Pedani, C. Mugnaini, F. Guida, L. Luongo, M. De Chiaro, S. Maione, S. Dragoni, M. Frosini, A. Ligresti, V. Di Marzo and F. Corelli, J. Med. Chem., 2011, 54, 5444 CrossRef CAS; (d) A. A. Al-Amiery, R. I. H. Al-Bayati, K. Y. Saour and M. F. Radi, Res. Chem. Intermed., 2012, 38, 559 CrossRef CAS; (e) Y. Zhang, Y. Fang, H. Liang, H. Wang, K. Hu, X. Liu, X. Yi and Y. Peng, Bioorg. Med. Chem. Lett., 2013, 23, 107 CrossRef CAS; (f) S. K. Suthar, V. Jaiswal, S. Lohan, S. Bansal, C. Chaudhary, A. Tiwari, A. T. Alex and A. Joseph, Eur. J. Med. Chem., 2013, 63, 589 CrossRef CAS; (g) T. Tashima, Bioorg. Med. Chem. Lett., 2015, 25, 3415 CrossRef CAS; (h) T. Shiro, T. Fukaya and M. Tobe, Eur. J. Med. Chem., 2015, 97, 397 CrossRef CAS PubMed.
- (a) W. M. F. Fabian, K. S. Niederreiter, G. Uray and W. Stadlbauer, J. Mol. Struct., 1999, 477, 209 CrossRef CAS; (b) N. S. Badgujar, M. Pazicky, P. Traar, A. Terec, G. Uray and W. Stadlbauer, Eur. J. Org. Chem., 2006, 2715 CrossRef CAS; (c) T. L. Micotto, A. S. Brown and J. N. Wilson, Chem. Commun., 2009, 7548 RSC; (d) R. S. Singh, A. Kumar, S. Mukhopadhyay, G. Sharma, B. Koch and D. S. Pandey, J. Phys. Chem. C, 2016, 120, 22605 CrossRef CAS; (e) A. Joliton, J.-M. Plancher and E. M. Carreira, Angew. Chem., Int. Ed., 2016, 55, 2113 CrossRef CAS PubMed; (f) A. S. Murugan, N. Vidhyalakshmi, U. Yamesh and J. Annaraj, J. Mater. Chem. B, 2017, 5, 3195 RSC.
- (a) M. F. Grundon, in The Alkaloids: Quinoline Alkaloids Related to Anthranilic Acid, Academic Press, London, 1988, vol. 32, p. 341 Search PubMed; (b) J. P. Michael, Nat. Prod. Rep., 1995, 12, 465 RSC; (c) S. D. Sarker and P. G. Waterman, J. Nat. Prod., 1995, 58, 574 CrossRef CAS; (d) C. Ito, M. Itoigawa, A. Furukawa, T. Hirano, T. Murata, N. Kaneda, Y. Hisada, K. Okuda and H. Furukawa, J. Nat. Prod., 2004, 67, 1800 CrossRef CAS PubMed.
- (a) B. Staskun, J. Org. Chem., 1964, 29, 1153 CrossRef CAS; (b) K. Kumar, S. Sai, T. M. Gilbert and D. Klumpp, J. Org. Chem., 2007, 72, 9761 CrossRef.
- C.-C. Cheng and S.-J. Yan, Organic Reactions, John Wiley and Sons, New York, 1982, vol. 28, p. 37 Search PubMed.
- (a) K. H. Raitio, J. R. Savinainen, J. Vepsäläinen, J. T. Laitinen, A. Poso, T. Järvinen and T. J. Nevalainen, J. Med. Chem., 2006, 49, 2022 CrossRef CAS PubMed; (b) B. F. McGuinness, K.-K. Ho, T. M. Stauffer, L. L. Rokosz, N. Mannava, S. G. Kultgen, K. Saionz, A. Klon, W. Chen, H. Desai, W. L. Rogers, M. Webb, J. Yin, Y. Jiang, T. Li, H. Yan, K. King, S. Zahng, K. K. Majmadar, V. Srivastava and S. Saha, Bioorg. Med. Chem. Lett., 2010, 20, 7414 CrossRef CAS PubMed.
- (a) D. V. Kadnikov and R. C. Larock, J. Org. Chem., 2004, 69, 6772 CrossRef CAS PubMed; (b) J. Ferguson, F. Zeng, N. Alwis and H. Alper, Org. Lett., 2013, 15, 1998 CrossRef CAS PubMed; (c) X. Li, X. Li and N. Jiao, J. Am. Chem. Soc., 2015, 137, 9246 CrossRef CAS; (d) F. Zhu, Y. Li, Z. Wang and X.-F. Wu, Adv. Synth. Catal., 2016, 358, 3350 CrossRef CAS; (e) V. Thakur, A. Sharma, Yamini, N. Sharma and P. Das, Adv. Synth. Catal., 2019, 361, 426 CrossRef CAS.
- (a) R. Manikandan and M. Jeganmohan, Org. Lett., 2014, 16, 3568 CrossRef CAS; (b) J. Wu, S. Xiang, J. Zeng, M. Leow and X.-W. Liu, Org. Lett., 2015, 17, 222 CrossRef CAS PubMed; (c) R. Kancherla, T. Naveen and D. Maiti, Chem.–Eur. J., 2015, 21, 8360 CrossRef CAS; (d) Z. Zhang, L.-L. Liao, S.-S. Yan, L. Wang, Y.-Q. He, J.-H. Ye, J. Li, Y.-G. Zhi and D.-G. Yu, Angew. Chem., Int. Ed., 2016, 55, 7068 CrossRef CAS PubMed.
- R. Zeng and G. Dong, J. Am. Chem. Soc., 2015, 137, 1408 CrossRef CAS PubMed.
- (a) Y. Dong, B. Liu, P. Chen, Q. Liu and M. Wang, Angew. Chem., Int. Ed., 2014, 53, 3442 CrossRef CAS PubMed; (b) W. Wang, X. Peng, X. Qin, X. Zhao, C. Ma, C.-H. Tung and Z. Xu, J. Org. Chem., 2015, 80, 2835 CrossRef CAS.
- For use of pre-functionalized acetamides, (a) P. J. Manley and M. T. Bilodeau, Org. Lett., 2004, 6, 2433 CrossRef CAS; (b) L. Fu, X. Huang, D. Wang, P. Zhao and K. Ding, Synthesis, 2011, 10, 1547 Search PubMed; (c) B. H. Ahn, I. Y. Lee and H. N. Lim, Org. Biomol. Chem., 2018, 16, 7851 RSC.
- For recent reviews for the copper catalyzed multicomponent reaction, see: (a) S. Hassan and T. J. J. Mueller, Adv. Synth. Catal., 2015, 357, 617 CrossRef CAS; (b) N. K. Ojha, G. V. Zyryanov, A. Majee, V. N. Charushin, O. N. Chupakhin and S. Santra, Coord. Chem. Rev., 2017, 353, 1 CrossRef CAS; (c) S. B. Sing, Curr. Catal., 2018, 7, 80 CrossRef; (d) R. Rossi, G. Angelici, G. Casotti, C. Manzini and M. Lessi, Adv. Synth. Catal., 2019, 361, 2737 CrossRef CAS; (e) A. Cherian, N. A. Harry, S. Saranya, K. R. Rohit and G. Anilkumar, Asian J. Org. Chem., 2019, 8, 197 CrossRef.
- M. Nallagangula and K. Namitharan, Org. Lett., 2017, 19, 3536 CrossRef CAS.
- (a) A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421 CrossRef CAS PubMed; (b) J. C. Antilla, A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 11684 CrossRef CAS PubMed; (c) J. C. Antilla, J. M. Baskin, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2004, 69, 6514 CrossRef CAS; (d) E. R. Strieter, D. G. Blackmond and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4120 CrossRef CAS PubMed; (e) D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13 RSC.
- (a) S. Ley and A. W. Thomas, Angew. Chem., Int. Ed., 2003, 42, 5400 CrossRef CAS PubMed; (b) I. P. Beletskaya and A. V. Cheprakov, Coord. Chem. Rev., 2004, 248, 2337 CrossRef CAS; (c) A. Shafir, P. Lichtor and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3490 CrossRef CAS PubMed; (d) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2008, 47, 3096 CrossRef CAS PubMed; (e) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed; (f) A. Casita and X. Ribas, Chem. Sci., 2013, 4, 2301 RSC; (g) C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525 RSC.
- The exact reason for the low yields in comparison with the control (entry 3) is not clear at this point. However, it cannot be excluded that the ligands L5 and L6 react with the substrates 1a and 2, which leads to the formation of many intractable side products.
- No condensation product between 1a and L8 was detected, presumably due to the steric hindrance of t-butyl group.
- (a) A. U. Meyer, K. Straková, T. Slanina and B. König, Chem.–Eur. J., 2016, 22, 8694 CrossRef CAS PubMed; (b) T. Markovic, B. N. Rocke, D. C. Blakemore, V. Mascitti and M. C. Willis, Org. Lett., 2017, 19, 6033 CrossRef CAS PubMed; (c) G. Bogonda, D. V. Patil, H. Y. Kim and K. Oh, Org. Lett., 2019, 21, 3774 CrossRef CAS PubMed.
- When 1a was used, many intractable side-products were observed. Gratifyingly, the ketone substrate turned out to be better suited to the secondary amine than aldehyde. In addition, use of CuI (20 mol%) and picolinic acid (10 mol%) was more efficient than that of copper powder (29%).
- T.-H. Tsai, W.-H. Chung, J.-K. Chang, R.-T. Hsu and N.-C. Chang, Tetrahedron, 2007, 63, 9825 CrossRef CAS.
- S. Caddick and S. Khan, Tetrahedron Lett., 1993, 34, 7469 CrossRef CAS.
- M. Heinrich, J. J. Murphy, M. K. Ilg, A. Letort, J. Flasz, P. Philipps and A. Fürstner, Angew. Chem., Int. Ed., 2018, 57, 13575 CrossRef CAS PubMed.
- D. W. Blevins, M.-L. Yao, L. Yong and G. W. Kabalka, Tetrahedron Lett., 2015, 56, 3130 CrossRef CAS.
- (a) V. J. K. Stille, Angew. Chem., 1986, 98, 504 CrossRef; (b) M. S. Holt, W. L. Wilson and J. H. Nelson, Chem. Rev., 1989, 89, 11 CrossRef CAS.
- Halogen exchange with NaI in aryl bromides under the Cu(I) catalyst was reported, see: A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 14844 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01352h |
| This journal is © The Royal Society of Chemistry 2020 |