
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atomic dynamics of stress-induced lattice misalignment structures in a KDP subsurface
Y. Hua,
Z. Zhua,
H. Z. Shao c,
J. M. Xiaoa,
M. Xua,
L. Zhaob and
J. Zhuang*a
c,
J. M. Xiaoa,
M. Xua,
L. Zhaob and
J. Zhuang*a
aShanghai Engineering Research Center of Ultra-Precision Optical Manufacturing, Department of Optical Science and Engineering, Fudan University, Shanghai 200433, China. E-mail: junzhuang@fudan.edu.cn
bState Key Laboratory of Surface Physics, Department of Physics, Fudan University, Shanghai 200433, China. E-mail: lizhao@fudan.edu.cn
cCollege of Electrical and Electronic Engineering, Wenzhou University, Wenzhou, 325035, China
First published on 23rd June 2020
Abstract
We present an ab initio molecular dynamics study of the thermal stability and dynamics behaviors of lattice misalignment structures (LMSs) in the subsurface layers of KH2PO4 (KDP) crystals. The dehydration process at the atomic scale is observed in the LMS system, which is the same as that in a perfect KDP crystal. However, the paths entering the dehydration process are various. The interesting result is that compared with a perfect KDP crystal, many new paths appear in the LMS system, and even in the same paths, the dehydration is more likely to happen in the LMS system. This leads to a dramatic increase in the dehydration numbers in the LMS system, for which the reasons are given in terms of structural deformation and/or uneven distribution of protons. The results elucidate the underlying atomic mechanism of the effect of LMS defects on the thermal stability of KDP material.
Introduction
Potassium dihydrogen phosphate (KDP, KH2PO4) crystals have attracted intensive interest in the field of optical materials, due to their remarkable nonlinear electro-optical properties1 and capability of growing to large size,2–5 leading to many applications in various high-power large-aperture laser systems such as inertial confinement fusion reactors.6,7 However, the actual laser damage threshold (LDT) of a KDP crystal is usually far below its intrinsic damage threshold, which limits the output power of laser systems.8–13 It is widely accepted that various defects, including those in the surface/subsurface layers created during KDP lens surfacing, are the main reason for the decline of LDT of KDP.11–18 These defects may have an impact on the distribution of light and thus affect the LDT of KDP.19–24 Therefore, besides growing KDP crystals close to perfection, it is also important to obtain smoother surfaces and more damage-free subsurfaces after machining to increase the LDT of KDP.In recent years, with the improvement of machining technique, a smoother surface of KDP can be produced by different ultra-precision surface machining methods, such as single point diamond turning,25–27 ultra-precision grinding28,29 and magnetorheological finishing.30 Even so, after machining, the damage at a more microscopic level still cannot be avoided as expected. Recently, researchers evaluated more precisely the subsurface structure of a KDP crystal after ultra-precision machining by utilizing grazing incidence X-ray diffraction technique. The results show that a shallow subsurface layer of a machined KDP crystal has turned to a lattice misalignment structure (LMS).31,32 This means that, compared with other machining-induced defects,11,28 such as pits, cracks, and nicks, such stress-induced subsurface micro “LMS” defect seems impossible to avoid, even with a very advanced surface machining technique. Therefore, clarifying the characteristics of LMS, a kind of “intrinsic” defect or damage, and its effect on the LDT of KDP becomes more important. We have previously published an ab initio study of the lattice misalignment structures in KDP crystals. In this previous work, by utilizing the ab initio static calculations, we found out for the first time various stable lattice misalignment structures by applying stress to KDP crystals, and calculated their electronic band structures and optical absorption properties. The results show that the electronic band structure and the optical absorption of misalignment structure indeed have some differences compared to a perfect KDP crystal.33
Compared with the static study, the dynamic study on the evolution of the misalignment structure, however, is more important, since it directly determines the material thermal stability under the action of light and heat. This is particularly significant in large-aperture laser systems, where micro-defects may not hinder the device performance, but the growth and even decomposition of defects are catastrophic.34–37 In the present work, we picked two stable LMSs from our previous static calculations and studied the atomic dynamics behaviors in these misalignment structures by utilizing ab initio molecular dynamics (MD) simulations. We want to see the atomic-level dynamic evolution of LMS, the possible structural damage, the damage pathway and how difficult it is compared with the perfect structure of KDP, and so on. These results are helpful to evaluate the possible adverse effects of such stress-induced “intrinsic” misalignment structure during the working process of KDP components.
Model and method
As shown in Fig. 1(a), we give the unit cell of a perfect KDP crystal, which consists of four KH2PO4 molecules and contains two molecule layers. The perfect cell can also be seen in Fig. 1(d), i.e., the colored part, which differs from Fig. 1(a) only in the selection of unit cells; the grey parts are obtained by utilizing the periodic boundary conditions, a perfect bulk is thus formed. In our previous work,33 we presented a simple and effective method to obtain the atomic configuration of misalignment structure on the (200) plane. Specifically, we moved the grey atoms in the dashed box along the Z direction a small displacement az by changing the z-component of vector a, as shown in Fig. 1(d) and (e), the P–O bonds at the (200) boundary of the unit cell were thus slightly deformed [see Fig. 1(e)]. By doing so, we try to model the local strain induced by the stress resulting from the cutting tip. Then, the system was fully relaxed. Through relaxation, the stress in the deformed P–O bonds can be transferred to every layer in the system, during which the (200) LMS appears naturally. More details can be found from ref. 33. Following the similar procedures as the above, we simulated the LMS evolution and obtained lots of local stable LMSs, such as the two representative ones in Fig. 1(b) and (c). Their z-components of vector a change by 8.40 Å and 26.0 Å relative to the perfect KDP structure, as shown in Fig. 1(a)–(c). Different z-components of vector a mean different degrees of misalignment. Compared with the perfect structure (PS) of KDP, both of two LMSs produce obvious deformation [see Fig. 1(a)–(c)]. Specially, the LMS at 8.40 Å (LMS1) has new radicals of “[HPO4]2−” and “[H3PO4]” [see Fig. 1(b)], which is different from that in the PS and the LMS at 26.0 Å (LMS2).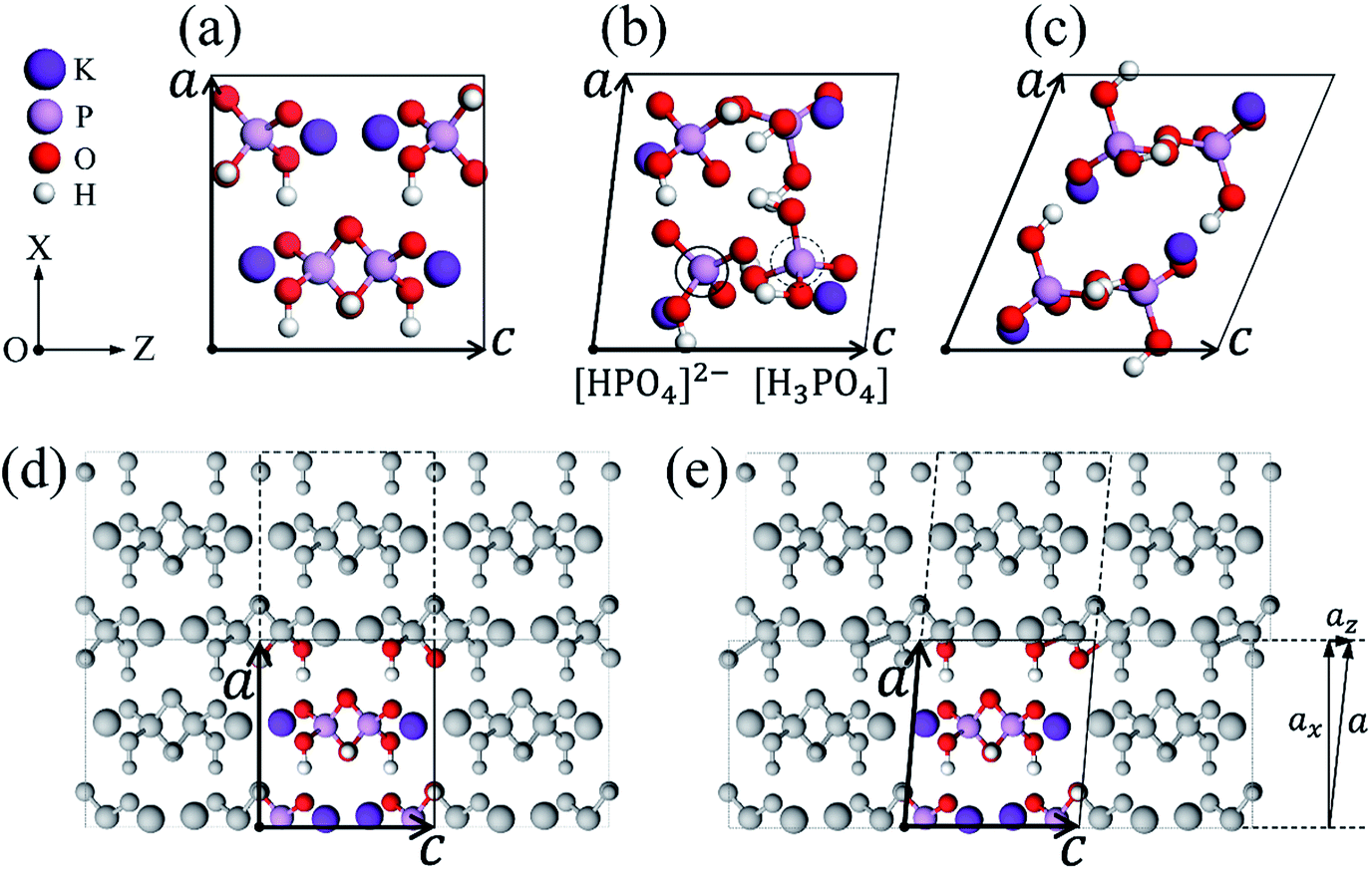 | ||
| Fig. 1 The unit cell of a perfect KDP crystal (a). Two stable LMSs with the z-component of vector a of 8.40 Å (b) and 26.0 Å (c). The radicals labeled by the solid and dashed circles in (b) denote radicals of [HPO4]2− and [H3PO4], respectively. The perfect KDP bulk system (d), by adjusting the z-component of vector a, the grey atoms in the dashed box are moved along the Z direction a small displacement az (e). The a and c indicate the basis vectors of the unit cell. The white, red, pink and purple balls denote H, O, P and K atoms, respectively, which are all the same in the following figures. | ||
In the present work, aiming at the two LMSs in Fig. 1(b) and (c), we use ab initio MD simulations to study their dynamics behaviors. For comparison, the perfect KDP structure is also studied under the same conditions. We first generate the 2 × 2 × 2 supercells of PS and two LMSs. Then, the three supercells including 256 atoms are fully relaxed. After that, the three optimized systems are heated at a constant temperature of 2000 K. It should be noted that an unreasonable high temperature adopted here for simulation is just to accelerate the chemical reaction, since the simulated time scale (ps) is too short for the chemical processes. The similar practice can be widely found in the literatures.38–42 A step size of 1.0 fs is used for all MD calculations with each simulation running for approximately 5000 steps.43 In all cases, dynamical equilibrium (based on the total system energy) appears to be reached in about 500 steps. After the systems reach to the dynamical equilibrium, we begin to trace the atomic trajectories. We focus on those steps with obvious atomic position and bond changes to find out the possible structural changes/damages. Then, we further extract the obvious changed structures and fully relax them to judge whether the changed structures can be stable. To compare the difficulty of the possible structural changes/damages in the LMS and PS systems, we performed 30 independent MD simulations for each system.
All calculations including the structural relaxations and MD simulations are performed based on the density function theory (DFT) utilizing the Vienna ab initio simulation program pack (VASP) package.44 The exchange–correlation potential is described by the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof functional.45 PAW potentials are used for electron–ion interactions. The kinetic energy cutoff for the plane-wave basis is set to be 680 eV, which leads to the converged result for the total system energy better than 1 meV per atom.21,46 A 2 × 2 × 2 supercell with a 3 × 3 × 3 Monkhorst–Pack k-point mesh is used for all calculations. The conjugate gradient algorithm is used to relax the ions and the residual force on each atom is taken as the convergence criterion which is smaller than 0.01 eV Å−1. With these parameters, the obtained lattice parameters of the relaxed perfect KDP crystal are a = b = 7.44 Å and c = 6.96 Å, which are in excellent agreement with the experimental values of a = b = 7.45 Å and c = 6.97 Å.47 Ab initio molecular dynamics (MD) calculations are achieved by the Verlet algorithm and the canonical ensemble (NVT) is adopted using the Nosé thermostat to control temperature.
Results and discussion
Based on the obtained MD trajectories, we first focus on the atomic dynamic evolution in the PS and LMS systems. It is known that in the static perfect KDP structure (paraelectric phase), each oxygen atom connects only one close (O–H) or far (O⋯H) proton through the H-bond (O–H⋯O).48–53 We will refer to the oxygen atom that connects the proton through the short distance as “Os” (O–H), and the other case as “Ol” (O⋯H), as shown in Fig. 2(a). In sharp contrast, in the dynamic evolution, as shown in Fig. 2(b)–(d), the oxygen atoms that connect two protons appear in the three systems because of the rotation and movement of PO4 tetrahedrons, the transfer of protons, and the change in the H-bond length. Here, we use the similar symbols like “Osl, Oss and Osm” to describe these new oxygen states; the subscript “m” (medium) indicates that the proton is approximately located at the midpoint position in the H-bond (O⋯H⋯O/O–H–O), as shown in Fig. 2(d). Another obvious dynamic phenomenon observed in the three systems, especially for the PS and LMS2 systems, is that the proton may escape from one PO4 tetrahedron to another, turning two [H2PO4]− radicals into one [HPO4]2− radical and one [H3PO4] radical. Lastly but most importantly, when the oxygen state Osl appears, the special dynamics behavior that probably results in a qualitative change in material usually occurs in the subsequent dynamic evolution. That is dehydration reaction by which some radicals will be decomposed. | ||
| Fig. 2 Schematic of atomic configuration of two [H2PO4]− radicals in the initial PS system (a). When the systems reach to the dynamical equilibrium, various configurations (b), (c) and (d) appear in any of the three systems. The oxygen atoms marked with black circles and Arabic numerals (1–6) denote five types of oxygen states: Os and Ol (a), Osl (b), Oss (c), and Osm (d). The subscripts “s” (short), “l” (long) and “m” (medium) describe the distance of proton relative to two oxygen atoms in the O–H⋯O or O⋯H⋯O (O–H–O) bonds, respectively. | ||
We find that the oxygen state Osl can appear in various possible combinations of PO4 tetrahedrons and protons, and they all have the potential to dehydrate. In Fig. 3, we give the three instances that correspond to three dehydration paths. Fig. 3(a) displays the PS system. Here, we take the colored atoms (2[H2PO4]−) as an example, which will be referred to as the “initial combination”. For clarity, we remove the grey atoms in the PS system and only display the initial combination [see Fig. 3(b)]. Through a series of dynamic evolution, as shown in Fig. 3(b) and (c), the initial combination of “2[H2PO4]−” turns into the combination of “[HPO4]2− + [H3PO4]”, owing to the transfer of protons; besides, the rotation and movement of PO4 tetrahedrons lead to the formation of the oxygen state “Osl” [see Fig. 3(c)]. The formation of oxygen state “Osl” is an important dynamic evolution result, since “Osl” may evolve into a water molecule in the subsequent evolution, which results in a qualitative change in KDP material [see Fig. 3(d)]. We will refer to the combination in Fig. 3(c) that is going to dehydrate as the “dehydrated combination”. Another two examples for the LMS1 and LMS2 systems are given in Fig. 3(e)–(h) and (i)–(l), respectively. Similarly, after a series of dynamic evolution, besides the obvious changes in structure and/or chemical composition, the oxygen state “Osl” appears in the dehydrated combinations [see Fig. 3(g) and (k)]. Significantly, the oxygen state “Osl” could also evolve into a water molecule in the subsequent evolution [see Fig. 3(h) and (l)], and the dynamics process from “Osl” to “H2O” are fully identical to that in the PS system.
 | ||
| Fig. 3 The supercells of PS (a), LMS1 (e), and LMS2 (i), where the colored atoms are referred to as the “initial combination”. For clarity, we only display the initial combinations in (b), (f), and (j). After a series of dynamic evolution, the initial combinations produce obvious changes in structure and/or chemical composition, and generate the oxygen state “Osl”; we will refer to such changed initial combinations as the “dehydrated combinations” (c), (g), and (k). In the subsequent evolution, the oxygen state “Osl” evolves into a water molecule (d), (h), and (l). The a and c denote the basis vectors of the unit cell. | ||
Certainly, in the MD simulations, there are more than three types of dehydration paths. In Table 1, we give all the dehydration paths observed in the three systems of PS, LMS1, and LMS2, which are numbered by the Arabic numeral “1–14”. The important result, as seen in the second column in Table 1, only three dehydration paths “1, 4, and 10” are in the PS system; the others are all in the LMS systems. In Table 1, we also give the corresponding initial combinations, which are labelled by the symbols of “A1, A2, A3, B1, …” (see the third column). In the subsequent dynamic evolution, the initial combinations evolve into the dehydrated combinations, in which “Osl” is formed. As seen in Table 1, some initial combinations evolve into the same dehydrated combination. For example, the initial combinations “A1, A2, and A3” all become into the dehydrated combination “A”. Therefore, there are only six types of dehydrated combinations, which are labeled by the capital letters from “A” to “F” in Table 1. Interestingly, after the formation of “Osl”, it is through the same dynamics process that the oxygen states “Osl” in the dehydrated combinations evolve into the water molecules. That is, from the six types of dehydrated combinations to the final dehydrated products, see the fourth to sixth columns in Table 1, the dynamics process from “Osl” to the “water molecule” are completely the same, indicated by s1–s5.
| Path | System | Initial combination (s0) | Dehydrated combination (s1) | Process | Products (s4, s5) |
|---|---|---|---|---|---|
| 1 | PS, LMS1, LMS2 | A1: 2[H2PO4]− | A: [HPO4]2− + [H3PO4] | s1–s5 | H2O, [PO3]−, [P2O7]4− |
| 2 | LMS1 | A2: [HPO4]2− + [H2PO4]− | |||
| 3 | LMS1 | A3: [H2PO4]− + [H3PO4] | |||
| 4 | PS, LMS2 | B1: 2[H2PO4]− | B: 2[H2PO4]− | ||
| 5 | LMS1 | B2: 2[H3PO4] | |||
| 6 | LMS1 | C1: [HPO4]2− + [H3PO4] | C: [H2PO4]− + [H3PO4] | ||
| 7 | LMS1 | C2: [HPO4]2− + [H2PO4]− | |||
| 8 | LMS1 | C3: [H2PO4]− + [H3PO4] | |||
| 9 | LMS2 | C4: 2[H2PO4]− | |||
| 10 | PS | D1: 2[H2PO4]− | D: 2[HPO4]2− | ||
| 11 | LMS2 | E1: 2[H2PO4]− | E: [HPO4]2− + [H2PO4]− | ||
| 12 | LMS1 | E2: [H2PO4]− + [H3PO4] | |||
| 13 | LMS1 | E3: [HPO4]2− + [H2PO4]− | |||
| 14 | LMS2 | F1: 3[H2PO4]− | F: [HPO4]2− + [H2P2O7]2− |
In Fig. 4, we display the atomic dynamic evolution in the dehydration process, step s1 to s5, where only the two protons that form water are given for clarity. In the “s1” step, the oxygen state Osl appears in the “dehydrated combination”. Then, in the s2 step, the H-bond gets shorter/longer, turning the O⋯H–O bond into the O–H–O/O⋯H⋯O bond; this results in the formation of oxygen state Osm. Next, in the s3 step, the O–H–O/O⋯H⋯O bond evolves into the O–H⋯O bond, during which the proton successfully escapes from one oxygen atom to the other, the oxygen state Oss thus formed. Finally, the most remarkable change occurs in the s4 step, that is, the newly formed oxygen state Oss is ejected from the PO4 tetrahedron as a water molecule and leaving behind a [PO3]− radical. By the way, the newly formed [PO3]− radical may interact with a nearest [PO4]3− radical to form a [P2O7]4− radical in the subsequent dynamic evolution [see Fig. 4(e)]. Indeed, researchers have detected the intermediate product [P2O7]4− radical in the direct heating of KDP crystals above 500 K.54,55 Furthermore, we extract the structures in the s4 and s5 steps from the MD trajectories and fully relax them, to judge whether the water molecules can exist stably; the results show that the water molecules in both two steps still exist stably after relaxation. In summary, despite the paths (1–14) entering the dehydration process are various, their dehydration processes from “Osl” to the “water molecule” are the same; that is, after the formation of “Osl”, it first evolves into “Osm”, then turns into “Oss”, eventually the oxygen state “Oss” liberates from the PO4 tetrahedron in the form of a water molecule. If only the two protons that form water are considered, the dehydration process can be described as the following equations, which are consistent with the experimental observations.54,55
 | (1) |
 | ||
| Fig. 4 The atomic dynamic evolution from the dehydrated combination (s1) to the final products (s4, s5), where only the two protons that form water are given. In the s1 step, Osl appears in the dehydrated combination (a); then in the s2 (b) and s3 (c) steps, Osl evolves into Osm and then Oss; the oxygen states are marked by the grey dashed circles; finally, in the s4 step (d), “Oss” liberates from the PO4 tetrahedron in the form of a water molecule and leaving behind a [PO3]− radical. In the s5 step, the [PO3]− radical interacts with an adjacent [PO4]3− radical to form a [P2O7]4− radical (e). | ||
To compare the decomposition difficulty of LMS and PS systems, as shown in Fig. 5, we give the statistical results for the dehydrated numbers of each system in the 30 MD simulations. From Fig. 5, we can see that, compared with the PS system, the dehydration paths in the LMS systems increase significantly, especially in the LMS1 system. Specifically, only three dehydration paths are in the PS system (paths 1, 4, and 10); in sharp contrast, there are nine and five dehydration paths in the LMS1 and LMS2 systems, respectively (see Fig. 5). For the LMS1 system, this result should be mainly attributed to its initial chemical composition. Indeed, in the three initial systems, only the LMS1 system has different radicals: [HPO4]2−, [H2PO4]−, and [H3PO4]. These different radicals result in the formation of some unique dehydration paths. For example, in the LMS1 system, there are some dehydration paths that start from the initial combinations which consist of two different radicals, such as “[HPO4]2− + [H2PO4]−”, “[H2PO4]− + [H3PO4]”, etc. (see Table 1); while in the PS and LMS2 systems, there are no such dehydration paths. For the LMS2 system, the increased dehydration paths should be caused by the deformed structure. As shown in Fig. 1(a)–(c), the H-bonds in the LMSs are obviously stretched compared with that in the PS. The stretched H-bonds decrease the attraction between the adjacent PO4 tetrahedrons, making it easier for the PO4 tetrahedrons to move and rotate. This catalyzes the scission of the H-bond network, making the consequence that the protons in the LMS systems have more transfer degrees of freedom, thus generating more types of dehydration paths in the LMS2 system.
 | ||
| Fig. 5 Statistical results for the dehydrated numbers in the PS (red rectangles), LMS1 (blue stars), and LMS2 (green triangles) systems. The abscissas (1–14) denote fourteen dehydration paths observed in the three systems (see Table 1). Inset: the total dehydrated numbers for the three systems. | ||
Another interesting result in Fig. 5 is that, in the same dehydration paths, such as path 1 and path 4, the dehydrated numbers for the LMS systems are obviously greater than that of the PS system. There are two possible reasons: on one hand, the PO4 tetrahedrons in the LMS systems are easier to move and rotate due to the stretched H-bonds, so that the LMS systems are easier to generate the oxygen state “Osl” compared with the PS system. On the other hand, the protons are unevenly distributed in the initial LMS1 system. The uneven distribution of protons results in the formation of some radicals in local area with charged states. The radical in the positively state, such as [KH3PO4]+, is apt to emit a proton outward, while the radical in the negatively state, such as [KHPO4]−, tends to contribute one [OH]−. That is, the uneven distribution of protons catalyzes the occurrence of dehydration.
The above facts show that, compared with the PS system, not only do many new dehydration paths appear in the LMS systems, but also the dehydrated numbers of LMS systems increase significantly in the same dehydration paths (see Fig. 5). Therefore, the total dehydrated numbers for both LMS systems are much greater than that of the PS system. As shown in the inset in Fig. 5, for the LMS1 and LMS2 systems, there are 78 and 65 dehydrations in the 30 MD simulations with each running for 5000 steps; this is in sharp contrast with the 21 dehydrations in the PS system.
The statistical results in Fig. 5 show that the dehydration paths in the three systems are various; but interestingly, they all contain path “1”, also the numbers of radicals dehydrated through this path account for the largest proportion (see Fig. 5). Thus, we take the path “1” as an example; we calculate the formation energy of the corresponding configurations in dehydration path 1, step s0 to s4, in which all the atoms are fully relaxed. The aim is to compare the decomposition difficulty of the same dehydration path in the three systems, and thus to further understand the statistical results. In Fig. 6(a), we display the energy level comparison diagram for the PS (red lines), LMS1 (blue lines), and LMS2 (green lines) systems; the abscissas “s0” and “s1” correspond to the initial combination “A1” and the dehydrated combination “A”, respectively (see Table 1); “s2–s4” correspond to the corresponding configurations in dehydration path 1; the ordinate denotes the formation energy. The formation energy is calculated by the following equation:56–58
| Ef = En − En−x | (2) |
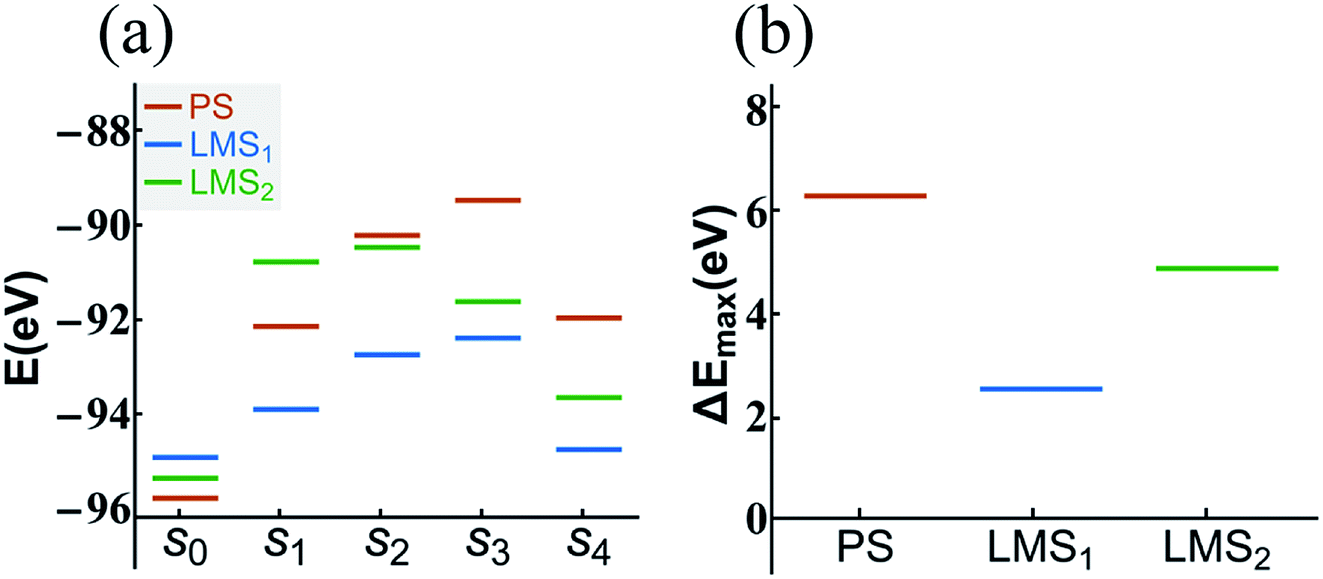 | ||
| Fig. 6 The energy level comparison diagram (a) of dehydration path 1 in the PS (red lines), LMS1 (blue lines), and LMS2 (green lines) systems, where the abscissas “s0” and “s1” correspond to the initial combination “2[H2PO4]−” and the dehydrated combination “[HPO4]2− + [H3PO4]”, respectively (see Table 1); “s2–s4” correspond to the corresponding configurations in dehydration path 1. For clarity, the energy difference comparison diagram (ΔEmax = Emax − Es0) is shown in (b). | ||
In summary, compared with the PS system, the LMS systems have more dehydration paths, and even in the same dehydration paths, the LMS systems have lower energy barrier to dehydrate. The results reveal that the LMS defect decomposes more easily than the perfect KDP structure, suggesting that the thermal stability of KDP material would be much affected by such stress-induced subsurface micro LMS defect. Indeed, it has been discovered that the laser-induced or thermal-induced decomposition reactions in general begin with the local reactions in the surface/subsurface layers of KDP.37,54,55,59 We believe that the LMS defect may play an important role in the decomposition of the surface/subsurface layers of KDP components, and also may be one of the possible reasons for the formation of the laser-induced damages in the use of KDP.
Conclusion
The LMS, which appears in the machining process of KDP, is investigated regarding the dynamics behaviors by utilizing the ab initio MD simulations. The details of atomic dynamic evolution in the three systems of PS, LMS1 and LMS2 are obtained. Especially, the dehydration phenomenon is observed, and its process is the same in the three systems. The dehydration process includes the formation of “Osl”, its transformation, and ejection in the form of a water molecule. However, the paths entering the dehydration process are various. Fourteen dehydration paths are observed in the three systems. The interesting result is that compared with the PS system, a lot of new dehydration paths appear in the LMS systems, and even in the same dehydration paths, the dehydration is more likely to occur in the LMS systems. This leads to the consequence that the total dehydrated numbers in the LMS systems are much greater than that in the PS system, which can be attributed to the deformed structure and/or the uneven distribution of protons. These results suggest that once the LMS defect resulting from processing appears in KDP crystals, the possibility of material decomposition will be greatly increased, which will much affect the damage threshold of KDP components.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Open Research Fund of Key Laboratory of Neutron Physics, CAEP (No. 2014BB07).References
- D. Eimerl, Electro-optic, linear, and nonlinear optical properties of KDP and its isomorphs, Ferroelectrics, 1987, 72, 95–139 CrossRef CAS.
- R. Hawey-Fedder, H. Robey, T. Biesiada, M. Dehaven, R. Floyd and A. Burnham, Rapid Growth of Very Large KDP and KD*P Crystals in Support of the National Ignition Facility, Proc. SPIE, 2000, 4102, 152–161 CrossRef.
- N. P. Zaitseva, J. J. De Yoreo, M. R. Dehaven, R. L. Vital, K. E. Montgomery, M. Richardson and L. J. Atherton, Rapid growth of large-scale (40–55 cm) KH2PO4 crystals, J. Cryst. Growth, 1997, 180, 255–262 CrossRef CAS.
- C. Maunier, P. Bouchut, S. Bouillet, H. Cabane, R. Courchinoux, P. Defossez, J.-C. Poncetta and N. Ferriou-Daurios, Growth and characterization of large KDP crystals for high power lasers, Opt. Mater., 2007, 30, 88–90 CrossRef CAS.
- N. Zaitseva and L. Carman, Rapid Growth of KDP-type Crystals, Prog. Cryst. Growth Charact., 2001, 43, 1–118 CrossRef CAS.
- J. J. De Yoreo, A. K. Burnham and P. K. Whitman, Developing KH2PO4 and KD2PO4 crystals for the world's most power laser, Int. Mater. Rev., 2002, 47, 113–152 CrossRef CAS.
- R. Hawley-Fedder, P. Geraghty, S. Locke, M. McBurney, M. Runkel, T. Suratwala, S. Thompson, P. Wegner and P. Whitman, NIF Pockels Cell and Frequency Conversion Crystals, Proc. SPIE, 2004, 5341, 121–126 CrossRef CAS.
- A. Yokotani, T. Sasaki, K. Yoshida, T. Yamanaka and C. Yamanaka, Improvement of the bulk laser damage threshold of potassium dihydrogen phosphate crystals by ultraviolet irradiation, Appl. Phys. Lett., 1986, 48, 1030–1032 CrossRef CAS.
- M. F. Singleton, J. F. Cooper, B. D. Andresen and F. P. Milanovich, Laser-induced bulk damage in potassium dihydrogen phosphate crystal, Appl. Phys. Lett., 1988, 52, 857–859 CrossRef CAS.
- J. Swain, S. Stokowski, D. Milam and F. Rainer, Improving the bulk laser damage resistance of potassium dihydrogen phosphate crystals by pulsed laser irradiation, Appl. Phys. Lett., 1982, 40, 350–352 CrossRef CAS.
- J. Cheng, M. J. Chen, W. Liao, H. J. Wang, J. H. Wang, Y. Xiao and M. Q. Li, Influence of surface cracks on laser-induced damage resistance of brittle KH2PO4 crystal, Opt. Express, 2014, 22, 28740–28755 CrossRef PubMed.
- S. F. Wang, J. Wang, Q. Xu, X. Y. Lei, Z. C. Liu and J. F. Zhang, Influences of surface defects on the laser-induced damage performances of KDP crystal, Appl. Opt., 2018, 57, 2638–2646 CrossRef CAS PubMed.
- S. G. Demos and M. Staggs, Application of fluorescence microscopy for noninvasive detection of surface contamination and precursors to laser-induced damage, Appl. Opt., 2002, 41, 1977–1983 CrossRef CAS PubMed.
- C. W. Carr, H. B. Radousky and S. G. Demos, Wavelength Dependence of Laser-Induced Damage: Determining the Damage Initiation Mechanisms, Phys. Rev. Lett., 2003, 91(1–4), 127402 CrossRef CAS PubMed.
- S. G. Demos, M. Yan, M. Staggs, J. J. De Yoreo and H. B. Radousky, Raman scattering investigation of KH2PO4 subsequent to high fluence laser irradiation, Appl. Phys. Lett., 1998, 72, 2367–2369 CrossRef CAS.
- M. J. Chen, M. Q. Li, C. H. An, L. Zhou, J. Cheng, Y. Xiao and W. Jiang, Influence of Light Intensification by Subsurface Cracks on KH2PO4 Crystal's Laser Induced Damage Threshold, Jpn. J. Appl. Phys., 2013, 52(1–8), 032701 CrossRef.
- M. Q. Li, M. J. Chen, J. Cheng, Y. Xiao and W. Jiang, Two important mechanisms damaging KH2PO4 crystal processed by ultraprecision fly cutting and their relationships with cutting parameters, Appl. Opt., 2013, 52, 3451–3460 CrossRef CAS PubMed.
- Y. Zhang, L. C. Zhang, M. Liu, F. H. Zhang, K. Mylvaganam and W. D. Liu, Revealing the mechanical properties of potassium dihydrogen phosphate crystals by nanoindentation, J. Mater. Res., 2016, 31, 1056–1064 CrossRef CAS.
- T. T. Sui, Y. F. Lian, M. X. Xu, L. S. Zhang, Y. L. Li, X. Zhao, X. G. Xu and X. Sun, Comparison of hydrogen vacancies in KDP and ADP crystals: a combination of density functional theory calculations and experiment, Phys. Chem. Chem. Phys., 2019, 21, 6186–6197 RSC.
- X. Chen, Q. Q. Zhao, X. C. Wang, J. Chen and X. Ju, Linear optical properties of defective KDP with oxygen vacancy: first-principles calculations, Chin. Phys. B, 2015, 24(1–6), 077802 CrossRef.
- C. S. Liu, N. Kioussis, S. G. Demos and H. B. Radousky, Electron- or Hole-Assisted Reactions of H Defects in Hydrogen-Bonded KDP, Phys. Rev. Lett., 2003, 91(1–4), 015505 CrossRef CAS PubMed.
- C. S. Liu, Q. Zhang, N. Kioussis, S. G. Demos and H. B. Radousky, Electronic structure calculations of intrinsic and extrinsic hydrogen point defects in KH2PO4, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68(1–11), 224107 CrossRef.
- C. S. Liu, C. J. Hou, N. Kioussis, S. G. Demos and H. B. Radousky, Electronic structure calculations of an oxygen vacancy in KH2PO4, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72(1–6), 134110 CrossRef.
- C. D. Marshall, S. A. Payne, M. A. Henesian, J. A. Speth and H. T. Powell, Ultraviolet-induced transient absorption in potassium dihydrogen phosphate and its influence on frequency conversion, J. Opt. Soc. Am. B, 1994, 11, 774–785 CrossRef CAS.
- Q. Xu and J. Wang, Analysis of the influence of vacuum chucking on the distortion of the KDP crystal, Proc. SPIE, 2000, 4231, 464–468 CrossRef.
- Q. Xu, J. Wang, W. Li, X. Zeng and S. Y. Jing, Defects of KDP Crystal Fabricated by Single Point Diamond Turning, Proc. SPIE, 1999, 3862, 236–239 CrossRef CAS.
- P. Lahaye, C. Chomont and P. Dumont, Using a design of experiment method to improve KDP crystals machining process, Proc. SPIE, 1999, 3492, 814–820 CrossRef CAS.
- D. J. Wu, B. Wang, H. Gao, R. K. Kang and X. S. Cao, Influence of Grinding to the Surface and Subsurface Quality of KDP crystal, Adv. Mater. Res., 2008, 53–54, 203–208 CAS.
- Y. Namba, K. Yoshida, H. Yoshida and S. Nakai, Ultraprecision grinding of optical materials for high power lasers, Proc. SPIE, 1997, 3244, 320–330 CrossRef.
- J. Menapace, P. Ehrmann and R. Bickel, Magnetorheological finishing (MRF) of potassium dihydrogen phosphate (KDP) crystals: nonaqueous fluids development, optical finish, and laser damage performance at 1064 nm and 532 nm, Proc. SPIE, 2009, 7504(1–12), 750414 Search PubMed.
- N. Hou, Y. Zhang, L. C. Zhang and F. H. Zhang, Assessing microstructure changes in potassium dihydrogen phosphate crystals induced by mechanical stresses, Scr. Mater., 2016, 113, 48–50 CrossRef CAS.
- N. Hou, Y. Zhang, L. C. Zhang, K. Mylvaganam, J. Q. Wei and F. H. Zhang, An investigation on the surface/subsurface defects in potassium dihydrogen phosphate crystals after fly-cutting, Proc. SPIE, 2016, 9683(1–4), 968309 Search PubMed.
- Y. Hu, Z. Zhu, J. M. Xiao, H. Z. Shao, L. Zhao, M. Xu and J. Zhuang, Atomic scale study of stress-induced misaligned subsurface layers in KDP crystals, Sci. Rep., 2019, 9(1–9), 10399 CrossRef PubMed.
- R. A. Negres, S. O. Kucheyev, P. Demange, C. Bostedt, T. Van Buuren, A. J. Nelson and S. G. Demos, Decomposition of KH2PO4 crystals during laser-induced breakdown, Appl. Phys. Lett., 2005, 86(1–3), 171107 CrossRef.
- A. Surmin, S. Lambert, F. Guillet, B. Minot, D. Damiani and F. Gervais, Thermal mechanism of laser induced damages in KDP crystals, Proc. SPIE, 2007, 6720(1–5), 672007 CrossRef.
- T. Tsujibayashi, M. Ichimiya, K. Toyoda and N. Ohno, Molecular-Chain Formation Induced by Infrared-Laser Light Observed in L-Cysteine and Potassium Dihydrogenphosphate, Jpn. J. Appl. Phys., 2011, 50(1–4), 101601 CrossRef.
- S. G. Demos, M. Staggs and M. R. Kozlowski, Investigation of processes leading to damage growth in optical materials for large-aperture lasers, Appl. Opt., 2002, 41, 3628–3633 CrossRef CAS PubMed.
- C. F. Li, Z. Mei, F. Q. Zhao, S. Y. Xu and X. H. Ju, Molecular dynamic simulation for thermal decomposition of RDX with nano-AlH3 particles, Phys. Chem. Chem. Phys., 2018, 20, 14192–14199 RSC.
- C. Deng, X. G. Xue, Y. Chi, H. Z. Li, X. P. Long and C. Y. Zhang, Nature of the Enhanced Self-Heating Ability of Imperfect Energetic Crystals Relative to Perfect Ones, J. Phys. Chem. C, 2017, 121, 12101–12109 CrossRef CAS.
- S. I. Stoliarov, R. E. Lyon and M. R. Nyden, A reactive molecular dynamics model of thermal decomposition in polymers. II. Polyisobutylene, Polymer, 2004, 45, 8613–8621 CrossRef CAS.
- I. V. Schweigert, Quantum mechanical simulations of condensed-phase decomposition dynamics in molten RDX, J. Phys.: Conf. Ser., 2014, 500(1–6), 052039 CrossRef.
- K. Zhong, J. Liu, L. Y. Wang and C. Y. Zhang, Influence of Atmospheres on the Initial Thermal Decomposition of 1,3,5-Trinitro-1,3,5-triazinane: Reactive Molecular Dynamics Simulation, J. Phys. Chem. C, 2019, 123, 1483–1493 CrossRef CAS.
- D. J. Carter and A. L. Rohl, Ab initio Molecular Dynamics Simulations of (101) Surfaces of Potassium Dihydrogenphosphate, J. Chem. Theory Comput., 2011, 7, 1604–1609 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- Q. Zhang, F. Chen, N. Kioussis, S. G. Demos and H. B. Radousky, Ab initio study of the electronic and structural properties of the ferroelectric transition, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 65(1–10), 024108 Search PubMed.
- R. J. Nelmes, Structural studies of KDP and the KDP-type transition by neutron and x-ray diffraction: 1970-1985, Ferroelectrics, 1987, 71, 87–123 CrossRef CAS.
- G. Beutier, S. P. Collins, G. Nisbet, K. A. Akimova, E. N. Ovchinnikova, A. P. Oreshko and V. E. Dmitrienko, Proton configurations in the hydrogen bonds of KH2PO4 as seen by resonant x-ray diffraction, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92(1–11), 214116 CrossRef.
- V. H. Schmidt, Review of order-disorder models for KDP-family crystals, Ferroelectrics, 1987, 72, 157–173 CrossRef CAS.
- A. A. Levin and S. P. Dolin, Molecular Models and Calculations in Microscopic Theory of Order-Disorder Structural Phase Transitions: Application to KH2PO4 and Related Compounds, J. Phys. Chem., 1996, 100, 6258–6261 CrossRef CAS.
- R. J. Nelmes, Z. Tun and W. F. Kuhs, A compilation of accurate structural parameters for KDP and DKDP, and a users guide to their crystal structures, Ferroelectrics, 1987, 71, 125–141 CrossRef CAS.
- R. J. Nelmes, Recent structural studies of the KDP-type transition: A review, Ferroelectrics, 1984, 53, 207–214 CrossRef CAS.
- S. Endo, T. Chino, S. Tsuboi and K. Koto, Pressure-induced transition of the hydrogen bond in the ferroelectric compounds KH2PO4 and KD2PO4, Nature, 1989, 340, 452–455 CrossRef CAS.
- B. Choi, High temperature phase transitions and thermal decomposition of KH2PO4 crytsals, J. Phys. Chem. Solids, 1995, 56, 1023–1030 CrossRef CAS.
- K. Lee, Hidden nature of the high-temperature phase transitions in crystals of KH2PO4-type: is it a physical change?, J. Phys. Chem. Solids, 1996, 57, 333–342 CrossRef CAS.
- H. Z. Shao, Y. Li, J. H. Zhang, B. Y. Ning, W. X. Zhang, X. J. Ning, L. Zhao and J. Zhuang, Physical mechanisms for the unique optical properties of chalcogen-hyperdoped silicon, Europhys. Lett., 2012, 99, 46005 CrossRef.
- Z. Zhu, H. Shao, X. Dong, N. Li, B. Y. Ning, X. J. Ning, L. Zhao and J. Zhuang, Electronic Band Structure and Sub-band-gap Absorption of Nitrogen Hyperdoped Silicon, Sci. Rep., 2015, 5(1–9), 10513 CrossRef PubMed.
- H. Sawada and K. Kawakami, First-principles calculation of the interaction between nitrogen atoms and vacancies in silicon, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 1851–1858 CrossRef CAS.
- R. H. Chen, C. C. Yen, C. S. Shern and T. Fukami, Impedance spectroscopy and dielectric analysis in KH2PO4 single crystal, Solid State Ionics, 2006, 177, 2857–2864 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |