
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltra-fine platinum species supported on niobium pentoxide for CO oxidation†
Miao-Miao Wangad,
Jing Yuc,
Dao-Lei Wange and
Rui Si *ab
*ab
aShanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai, 201204, Shanghai, China. E-mail: sirui@sinap.ac.cn
bShanghai Synchrotron Radiation Facility, Zhangjiang Laboratory, Shanghai, 201204, Shanghai, China
cShanghai Institute of Measurement and Testing Technology, Shanghai 200233, China
dUniversity of Chinese Academy of Science, Beijing 100049, China
eDivision of China, TILON Group Technology Limited, Shanghai 200090, China
First published on 26th March 2020
Abstract
Platinum oxide supported on a Lewis acid niobium oxide (Nb2O5) support has been used for various heterogeneous and homogeneous catalysts. In this work, we used urea as a precipitating agent to obtain crystallized Nb2O5 with high surface area via a hydrothermal route. Nb2O5-supported Pt catalysts were subsequently synthesized via an incipient wetness impregnation approach. Multiple characterizations including X-ray diffraction (XRD), high-resolution transmission electron microscopy (HRTEM) and nitrogen adsorption/desorption confirmed the identical structural and textural properties of the Nb2O5 support before and after the impregnation process. Furthermore, the X-ray absorption fine structure technique (XAFS) results with related data analysis indicate that the platinum species in the fresh and H2-pretreated samples were in the form of single atoms or ultrafine clusters. In addition, the decrease in coordination number (CN) of the first-shell Pt–O bond, as well as the formation of Pt–Pt contribution with very low CN, after H2-pretreatment was verified, which corresponds to the decrease of oxidation state for Pt species on the surface of supports. Thus, the ultrafine-clustered metallic Pt species are considered to be more active than the oxidized Pt single ions. The current results will be of great significance in controllable synthesis of active Pt-based catalysts for other catalytic oxidation reactions.
1 Introduction
Platinum-based catalysts have been widely used in industry to catalyse a large number of important chemical reactions because of their superior activity and stability.1–4 On the other hand, highly dispersed metal atoms on redox supports normally show excellent catalytic reactivity in various reactions by means of maximizing the efficiency of active metal atoms.5–7 Therefore, more and more research groups have shown that sub-nanosized clusters or single atoms exhibit better catalytic performance than nanosized metal or metal oxide particles.8–14 Judai et al. confirmed that the cluster catalysts consisting of only a few Pd atoms are active at much lower temperature than the bulk analogues for the NO reduction by CO.9 The Zhang group synthesized isolated single Pt atoms anchored onto iron oxide (FeOx), which exhibited very high activity and stability for both CO oxidation and preferential oxidation (PROX) of CO.11Other research results have shown that increasing the surface area of the supports can effectively improve dispersion to optimize activity.15 The pretreatment of the catalysts by specific gas can also disperse the active metal particles thereby increasing catalytic activity. For instance, The Qin group used HCl steam to re-disperse Pt nanoparticles in process of propane dehydrogenation for better catalytic activity.13 Original large-sized Mo oxide on carbon support was calcined in an H2 atmosphere for obtaining small-sized Mo species.16 After vacuum calcination and H2 reduction, thermal treatment of 0.2 wt% Pt/SiO2 was used to synthesize Pt particles less than 1 nm that are uniformly dispersed on the SiO2.17 At present, some researchers on the evolution of single-atom catalysts (SACs) during activation and catalytic reactions show that ultrafine cluster is more active than pure single atoms. Recently, Piccolo Group demonstrated the importance of Pt clusters for CO oxidation reaction by means of in situ characterization methods.18 Pt–O–Pt sites, represented by a Pt8O14 model cluster were built on ceria for low-temperature CO oxidation reaction, which is 100–1000 times more active than their single atom Pt1/CeO2 parent.19
Many oxides (Al2O3,18,20–22 Fe3O4,23,24 Co3O4,25,26 ZrO2,22,27 and, MnO2,28,29 etc.) were applied to support Pt for various heterogeneous catalytic reactions. Among them, niobium oxide (Nb2O5) has been used as a solid acid catalyst with the Lewis acid sites on its surface for various heterogeneous or homogeneous catalysis.15,30–38 Oxygen vacancies were introduced into Nb2O5 via thermal treatment to obtain defective Nb2O5-supported Pt catalysts for CO oxidation.36 So, niobium pentoxide supported platinum metal catalyst may be a promising catalyst. However, present published results had limited research about Pt-based Nb2O5 catalysts for heterogeneous catalysis, especially for CO oxidation reactions.
In this work, we used urea as a precipitating agent to obtain pure-phase Nb2O5 with high specific surface area by hydrothermal method using niobium(V) oxalate hydrate as a precursor and loaded different amounts of Pt species via incipient wetness impregnation subsequently. The catalytic activity of catalysts were tested via the CO oxidation reaction which is a typical benchmark reaction for estimating catalysts and plays a pivotal role in catalysis for both fundamental research and industrial applications,34,39–41 including purification of polluted gases, preferential removal of CO in proton exchange membrane fuel cells under hydrogen-rich conditions, CO gas sensors and CO elimination of automotive exhaust gases. Our results indicate that the method of preparing Nb2O5 can effectively disperse and anchor Pt atoms due to high specific surface area (127 m2 g−1) of the Nb2O5 compared to that of other reports (10 m2 g−1–65 m2 g−1).32–34,36 In the case of Pt-based catalysts, the pretreatment with H2 of the catalysts was to reduce the platinum species for obtaining reduced platinum which is active species for CO oxidation. But X-ray absorption fine structure (XAFS) analysis results of this work indicated the increase of catalyst activity after hydrogen pretreatment is not only due to the decrease of the oxidation state for Pt species but also to the appearance of Pt fine-clusters on the surface of supports.
2 Materials and methods
2.1 Materials
All the chemicals are analytical grade and were used without further purified and modified. Urea (CO(NH2)2, ≥ 99.0%) was purchased from Sinopharm Chemical Reagent Co., Ltd. Tetraammineplatinum(II) nitrate (Pt(NH3)4(NO3)2, 99.99%) and niobium(V) oxalate hydrate powder was purchased from Alfa Aesar Chemical Reagent Co., Ltd.2.2 Catalyst preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Nb = 10:1) was dissolved in 20 mL of deionized and 60 mL of deionized water at room temperature, respectively. The mixed solution was transferred to a Teflon-lined stainless-steel autoclave (inner volume: 100 mL) and then heated at 80 °C for 8 hours in vessel. And the temperature was sequentially elevated up to 200 °C and kept for two days. After the hydrothermal process finished and the vessel was cooled down to room temperature, the resulting precipitate was isolated by centrifugation, washed repeatedly with deionized water until neutral. The as-washed products were dried at 80 °C overnight and then calcined at 400 °C for 4 h at muffle furnace. After grinding and sieving (100 mesh) the calcined samples, the Nb2O5 fresh powder was obtained with the white colour.
:Nb = 10:1) was dissolved in 20 mL of deionized and 60 mL of deionized water at room temperature, respectively. The mixed solution was transferred to a Teflon-lined stainless-steel autoclave (inner volume: 100 mL) and then heated at 80 °C for 8 hours in vessel. And the temperature was sequentially elevated up to 200 °C and kept for two days. After the hydrothermal process finished and the vessel was cooled down to room temperature, the resulting precipitate was isolated by centrifugation, washed repeatedly with deionized water until neutral. The as-washed products were dried at 80 °C overnight and then calcined at 400 °C for 4 h at muffle furnace. After grinding and sieving (100 mesh) the calcined samples, the Nb2O5 fresh powder was obtained with the white colour.2.3 Characterization
564.0 eV) were performed at BL14W1 beamline of the Shanghai Synchrotron Radiation Facility (SSRF) operated at 3.5 GeV under “top-up” mode with a constant current of 220 mA. The sample mixing LiF uniformly was pressed into a sheet, then sealed with Kapton tape before the XAFS test. The XAFS data were recorded under fluorescence mode with a standard Lytle ion chamber. The energy was calibrated with Pt foil as a reference. Athena and Artemis codes were used to extract the data and fit the profiles. For the X-ray absorption near-edge spectroscopy (XANES) part, the experimental absorption coefficients as function of energies μ(E) were processed by background subtraction and normalization procedures and reported as “normalized absorption”. Based on the normalized XANES profiles, both the molar proportion of Pt4+/Pt0 and the oxidation state of Pt can be determined by the linear combination fit with the help of various references (Pt foil for Pt0 and PtO2 for Pt4+). For the extended X-ray absorption fine structure (EXAFS) part, the Fourier transformed (FT) data in R space were analysed by applying PtO2 and metallic Pt model for Pt–O and Pt–Pt contributions. The passive electron factors, S02, were determined by fitting the experimental data on Pt foils and setting the coordination number (CN) of Pt–Pt to be 12, and then fixed for further analysis of the measured samples. The parameters describing the electronic properties (e.g., correction to the photoelectron energy origin, E0) and local structure environment including CN, bond distance (R), and Debye–Waller factor around the absorbing atoms (σ2) were allowed to vary during the fit process. The fitted ranges for K and R spaces were selected to be k = 2.5–11.8 Å−1, 2.5–10.5 Å−1, 2.5–8.0 Å−1, 2.5–12.0 Å−1, 2.5–8.5 Å−1 or 2.5–12.0 Å−1 for 1 Pt/Nb2O5-fresh, 1 Pt/Nb2O5–H2, 1 Pt/Nb2O5-used, 0.3 Pt/Nb2O5-fresh, 0.3 Pt/Nb2O5–H2, 0.3 Pt/Nb2O5-used, respectively.2.4 Catalytic test
CO oxidation reactions were carried out in micromeritics AutoChem 2920. The detection processes of catalyst activity were as follows: The sieved sample (40–60 mesh, 50 mg) was loaded into a U-shaped quartz tube, and then preactivated at 5% H2/95% He or 5% O2/95% N2 for 30 min with a flow rate of 50 mL min−1 before the reaction. After the reactor cooled to room temperature, pure He gas was used to purge H2 or O2 for 20 minutes. Afterwards, CO oxidation activities of Pt/Nb2O5 catalysts were measured in a stoichiometric gas mixture of 2% CO/1% O2/97% He at a flow rate of 30 mL min−1, corresponding to a gas hourly space velocity (GHSV) of 36000 mL h−1 gcat−1. The reactant temperature began from 30 °C to 300 °C with the heating rate of 10 °C min−1 and the outlet gas compositions of CO and CO2 were recorded on-line at 30 °C per step by Micro GC fusion gas chromatography (INFICON company). The CO conversion of CO oxidation was calculated according to the following equation: CO conversion (%) = (COin − COout)/COin × 100. The experimental error is within 3%.
3 Results and discussion
3.1 Catalytic performance of Pt/Nb2O5 catalysts for CO oxidation reaction
CO oxidation reaction, as model catalysis reaction, was used to evaluate the catalytic performance of platinum-doped Nb2O5 catalysts. Fig. 1a and b show the “light-off” profiles of H2-pretreated and O2-pretreated samples, respectively, which display that all the Pt/Nb2O5 samples exhibited CO conversions. In addition, this figure shows that CO conversion increases with elevating Pt content and the catalysts pretreated with H2 exhibit improved activity whereas the ones pretreated with O2 exhibit worse activity. At 50% conversion of the same component, the conversion temperature of the samples after H2-pretreatment is about 40 °C to 50 °C lower than the conversion temperature of the oxygen pretreated samples (Table 1). For example, the temperature of 50% CO conversion is 231 °C for O2-pretreated 1% Pt catalysts, but 189 °C for H2-pretreated 1% Pt catalysts. Similarly, the temperature of 50% CO conversion is 265 °C for O2-pretreated 0.3% Pt catalysts, but 215 °C for H2-pretreated 0.3% Pt catalysts. This result implies that Pt species with low valence are more effective than Pt species with high valence for CO oxidation.19,42 Further reasons are explored in the following characterization. Recently, Tran et al. reported about 50% CO oxidation at 160 °C under 60000 mL g−1 h−1 over 4 wt% Pt/Nb2O5 under 2% CO/5% O2/He.36 CO oxidation activities of Pt/Nb2O5 catalysts were measured in a stoichiometric gas mixture of 2% CO/1% O2/97% He corresponding to a gas hourly space velocity (GHSV) of 36000 mL h−1 gcat−1 in this work. Considering the Pt content and the oxygen enrichment conditions in that work, the reactivity of our catalyst is competitive.
 | ||
| Fig. 1 CO oxidation conversion over Pt–Nb2O5 samples for “light-off” experiments from 30 to 300 °C of catalysts. | ||
| Sample | Pt loadinga (at%) | SBETb (m2 g−1) | Vpb (cm3 g−1) | T1/2c (°C) |
|---|---|---|---|---|
| a Determined by ICP.b From N2 adsorption/desorption.c Temperature for 50% CO conversion.d After O2-pretreatment.e After H2-pretreatment. | ||||
| Nb2O5 | — | 127 ± 1 | 0.296 ± 0.002 | — |
| 1 Pt–Nb2O5 | 0.8 | 121 ± 1 | 0.280 ± 0.002 | 231d |
| 189e | ||||
| 0.5 Pt–Nb2O5 | 0.4 | 120 ± 1 | 0.287 ± 0.003 | 258d |
| 209e | ||||
| 0.3 Pt–Nb2O5 | 0.3 | 121 ± 1 | 0.290 ± 0.002 | 265d |
| 215e | ||||
| 0.1 Pt–Nb2O5 | 0.1 | 119 ± 1 | 0.289 ± 0.002 | 282d |
| 233e | ||||
| 0.05 Pt–Nb2O5 | 0.07 | 114 ± 1 | 0.280 ± 0.003 | >300d |
| 244e | ||||
3.2 Structural and textural properties of Pt/Nb2O5 catalysts
XRD was performed to determine the crystal structure of platinum-doped niobium oxide catalysts. The XRD patterns in Fig. S1a† identify that Pt-free Nb2O5 sample presented a pseudo-hexagonal structure (JCPDS card no: 28-317) with TT phase.43 In addition, no Pt-containing peaks were identified for all fresh catalysts shown in Fig. S1a.† 1 Pt and 0.3 Pt catalysts were selected for hydrogen pretreatment and further XRD testing (Fig. S1b†). There are no Pt peaks on H2-pretreated catalysts, which preliminarily indicates the high dispersion or small size of Pt species. As for used samples, no Pt species are found for all samples (see Fig. S2a and b†). The average crystallite size calculated by the Scherrer equation for pure Nb2O5 is 11.9 nm (see Table 2). Crystallite sizes of niobium pentoxide with hexagonal structure synthesized using solvent evaporation method were 61.4 nm in Ziolek' work.32 Table 2 shows that the average crystallite size is between 10.4 and 14.9 nm for all the investigated fresh catalysts, H2-pretreated and used samples after CO oxidation according to the results of XRD calculated by the Scherrer equation, which indicating that contents of Pt and pretreatment conditions have no clear effect for the Nb2O5 support. (001) and (100) peaks of the XRD of the used samples were selected to plot the intensity seen in Fig. S3.† There is no obvious evidence to prove the regular development of materials with hydrogen pretreatment or oxygen pretreatment, as mentioned above. A series of Pt/Nb2O5 samples prepared using NbCl5 as a precursor shown crystal size of 45–54 nm reported by Tran et al.36 These comparative data are indication that the successful precipitation of niobium ions via urea to obtain small nanosized Nb2O5 support in this work.| Sample | DXRDa (nm) | DTEMb (nm) | ||||
|---|---|---|---|---|---|---|
| Freshc | H2d | Usede | Freshc | H2d | Usede | |
| a Calculated from XRD patterns by Scherrer equation.b Statistic data on the basis of TEM images.c Fresh samples.d After H2-pretreatment.e Used samples after CO oxidation.f After CO oxidation with H2-pretreatment.g After CO oxidation with O2-pretreatment. | ||||||
| Nb2O5 | 11.9 | — | — | 11.3 ± 2.4 | — | — |
| 1 Pt–Nb2O5 | 14.9 | 12.5 | 13.5f | 12.4 ± 2.6 | 12.1 ± 1.9 | 13.1 ± 2.3f |
| 10.8g | ||||||
| 0.5 Pt–Nb2O5 | 11.7 | — | 9.6f | — | — | — |
| 13.7g | ||||||
| 0.3 Pt–Nb2O5 | 13.3 | 12.5 | 10.8f | 11.2 ± 2.8 | 11.7 ± 2.5 | 11.1 ± 3.1f |
| 11.4g | ||||||
| 0.1 Pt–Nb2O5 | 12.6 | — | 13.4f | — | — | — |
| 12.4g | ||||||
| 0.05 Pt–Nb2O5 | 10.4 | — | 10.7f | — | — | — |
| 10.7g | ||||||
Summarizing the results of XRD analysis, the fresh catalysts, the H2-pretreated samples and the used samples are not observed peaks related to platinum structure due to the ultrafine sizes or high dispersion of platinum species.
The ICP-AES results (see Table 1) show that the experimental Pt content is close to the target value for each catalyst. N2 adsorption–desorption results are present in Fig. S4† and summarized in Table 1. Both Nb2O5 support and Pt–Nb2O5 catalysts show type IV adsorption isotherm and H1-type hysteresis loop with the BJH pore volumes (Vp) between 0.280–0.296 cm3 g−1. Also, the surface area was almost the same after Pt deposition (114–121 m2 g−1), indicating the good support of nanosized Nb2O5 for platinum. Furthermore, a particularly high BET specific surface area (127 m2 g−1) was determined for Nb2O5, which provides a matrix for Pt loading in this work. All these data indicate that the introduction of Pt species has no significant effect on the textural properties of Nb2O5, and Pt–Nb2O5 catalysts were stable after air-calcination at 400 °C.
For further analysis, the Nb2O5, 0.3 Pt/Nb2O5 and 1 Pt/Nb2O5 samples were subjected to TEM characterization. The TEM images of pure Nb2O5 support in Fig. 2a show a worm-like aggregation with a primary particle size of about 11 nm (see Table 2), which is consistent with the XRD results. The HRTEM image in Fig. 2b clearly displays an interplanar spacing of 0.392 nm, corresponding to the (001) lattice fringe of pseudo-hexagonal Nb2O5. No metallic Pt particles but the only structure of Nb2O5 was observed in HRTEM for both 0.3 Pt/Nb2O5 (Fig. 3a) and 1 Pt/Nb2O5 (Fig. 3b). Similarly, the HRTEM images of the H2-pretreated samples and used samples shown in Fig. 3c–f also show no Pt species. Clearly visible lattice fringes appearing on all images belong to the (001) crystal plane of Nb2O5. STEM-EDS mapping of fresh 1 Pt/Nb2O5 and used 0.3 Pt–Nb2O5 with H2-pretreatment are shown in Fig. S5,† which indicates that Pt is uniformly dispersed on the support, but size and form of Pt are still unclear. Consulting other groups' work about Pt/Nb2O5, Pt species were loaded on Nb2O5 with size of particles of 2.7–17.9 nm,30,31,36,44 while the Pt species of calcined samples in this work cannot be identified on HRTEM images, suggesting that ultrafine Pt species supported in Nb2O5 obtained by the urea-based deposition–precipitation approach, proving the superiority of nanosized Nb2O5 in this work.
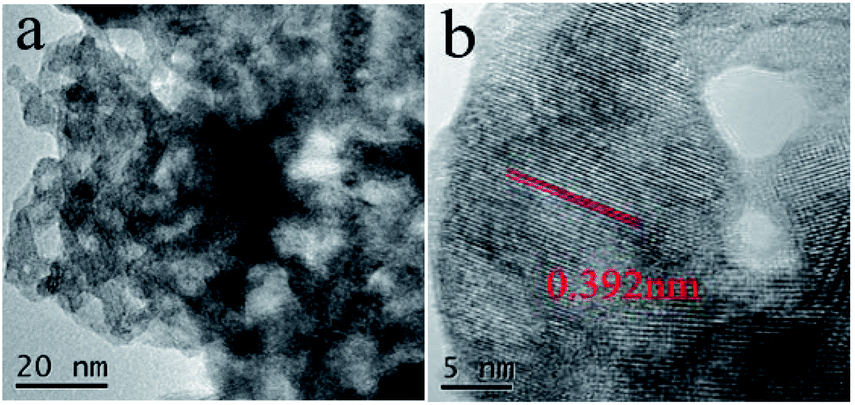 | ||
| Fig. 2 TEM images of Nb2O5 samples. | ||
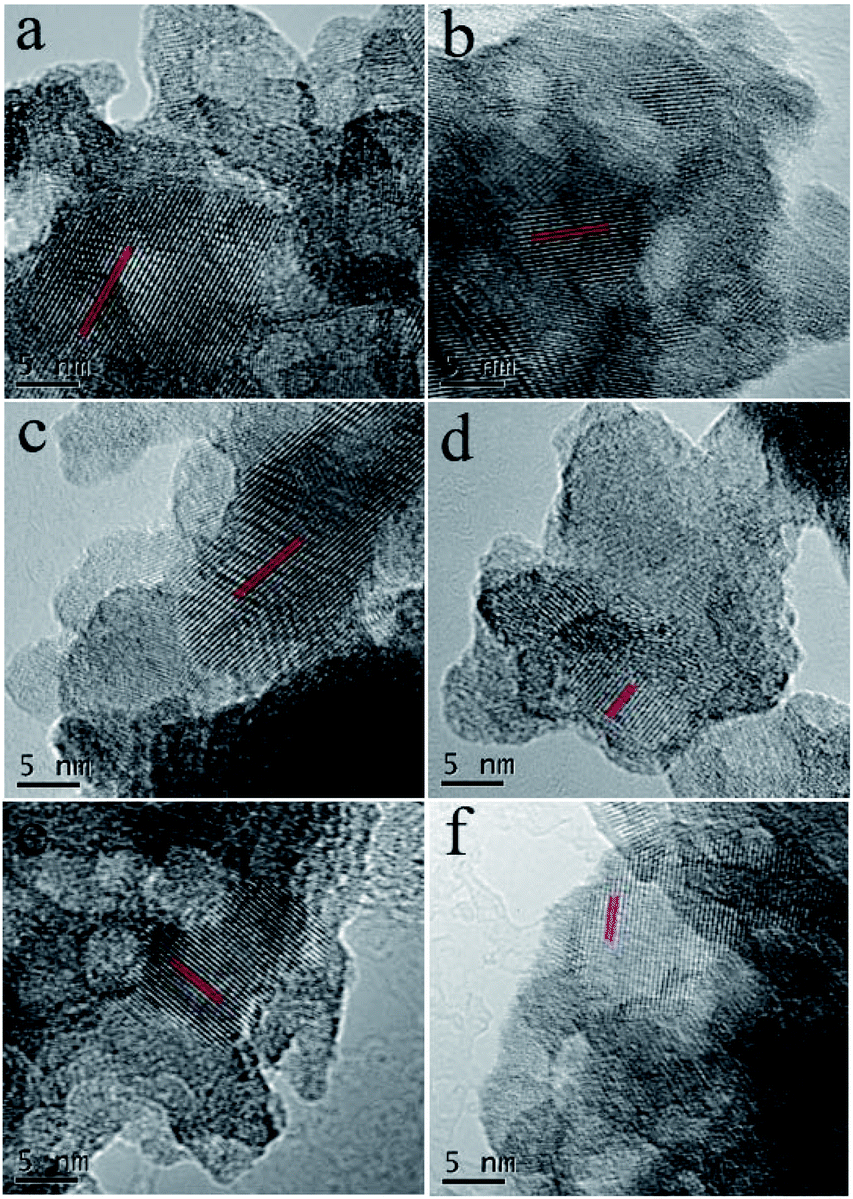 | ||
| Fig. 3 HRTEM images of (a, c and e) 0.3 Pt/Nb2O5 and (b, d and f) 1 Pt/Nb2O5; (a and b) fresh samples; (c and d) after H2-pretreatment samples; (e and f) used samples with H2-pretreatment samples. | ||
The results of this part are briefly summarized: Nb2O5 nanocrystalline obtained by the urea-based deposition–precipitation approach is thermally stable. XRD and HRTEM characterization revealed that the deposited Pt species on Nb2O5 are extremely small.
3.3 Reducibility and active species of Pt/Nb2O5 catalysts
Fig. 4 shows the H2-TPR profiles of the as-calcined samples. On the one hand, the TPR of the Nb2O5 support results in no reduction peak between 30 and 500 °C, confirming that no surface reduction of the support takes place without Pt. On the other hand, the two small reduction peaks for Nb2O5-supported Pt samples were observed with the peaks at 61 °C and 331 °C. The former reduction peak suggested a reduction of surface Pt oxide to metallic platinum.36,44 And the latter reduction peak showed that the reduction of surface lattice oxygen related to the metal oxide support interacting with Pt species.36,45,46 | ||
| Fig. 4 H2-TPR analysis of the synthesized catalysts. | ||
Since no peaks of Pt species could be observed from XRD and about the size and dispersion of Pt species could not be obtained from HRTEM. For further research, XAFS was used to study the electronic and the local coordination structure of the 1 Pt/Nb2O5 and 0.3 Pt/Nb2O5. The XANES data for Pt L3-edge in Fig. 5a and b clearly demonstrate that the white line peaks of samples are all between Pt foil and PtO2 but closer to Pt foil, indicating that the low valence state of Pt in fresh samples and H2-pretreatment samples. By means of the linear combination Pt/Nb2O5 fresh sample and the 1 Pt/Nb2O5 fresh sample are 1.82 (Table 3). However, the valence states of Pt in the H2-pretreated samples is reduced, that for 0.3 Pt/Nb2O5 sample and the 1 Pt/Nb2O5 sample are 1.20 and 1.13, respectively. Obviously, the supported Pt were reduced partially by the hydrogen. As for used samples with H2-pretreatment, the valence of Pt is further reduced compared to the H2-pretreated samples for both of 0.3 Pt/Nb2O5 and 1 Pt/Nb2O5, which evidently shown in Fig. 5a, b and Table 3.
 | ||
| Fig. 5 Pt L3-edge XANES profiles (a and b) and EXAFS fitting results in R space (c and d) of Pt–Nb2O5 samples: (a and c) 0.3 Pt–Nb2O5 and (b and d) 1 Pt–Nb2O5. | ||
| Sample | δ | Pt–O | Pt–Pt | σ2 (Å2) | ΔE0 (eV) | ||
|---|---|---|---|---|---|---|---|
| R (Å) | CN | R (Å) | CN | ||||
| 1 Pt–Nb2O5-fresh | 1.82 ± 0.04 | 1.98 ± 0.01 | 4.1 ± 0.2 | — | — | 0.003(O)0.005 ± 0.001(Pt) | 12.1 ± 0.7 |
| 1 Pt–Nb2O5–H2 | 1.20 ± 0.02 | 2.00 ± 0.01 | 2.2 ± 0.2 | — | — | 10.2 ± 2.5 | |
| 1 Pt–Nb2O5-used | 1.01 ± 0.02 | 1.97 ± 0.01 | 1.9 ± 0.2 | 2.70 ± 0.02 | 1.8 ± 0.6 | 8.3 ± 1.4 | |
| 0.3 Pt–Nb2O5-fresh | 1.84 ± 0.02 | 1.98 ± 0.01 | 4.1 ± 0.3 | — | — | 12.1 ± 0.7 | |
| 0.3 Pt–Nb2O5–H2 | 1.13 ± 0.02 | 2.00 ± 0.02 | 2.3 ± 0.5 | — | — | 10.2 ± 2.5 | |
| 0.3 Pt–Nb2O5-used | 0.72 ± 0.02 | 1.97 ± 0.01 | 1.7 ± 0.3 | 2.74 ± 0.01 | 4.9 ± 1.2 | 8.3 ± 1.4 | |
The related EXAFS spectra of Pt L3-edge exhibit a single prominent peak at ca. 1.98 Å originating from the first shell of Pt–O contribution with a coordination number (CN) of around 4.1 (see Table 3) for both of 0.3 Pt/Nb2O5 fresh sample (Fig. 5c) and 1 Pt/Nb2O5 fresh sample (Fig. 5d), which strongly indicate that Pt species exist with the form of single atoms for calcined samples. After hydrogen pretreatment, according to the results of the fitting, only the Pt–O shell was found without the peak of the Pt–Pt shell because the second shell is too small to fitted. But the existence of ultrafine clusters cannot be denied and neglected because there are very small peak corresponding to the Pt–Pt shell in Fig. 5c and d for 0.3 Pt/Nb2O5 and 1 Pt/Nb2O5. It is worth noting that the intensity of Pt–O peak of H2-pretreated catalysts significantly decline compared to fresh samples, accompanied by considerable decrease of coordination number (4.1 to 2.2 for 1 Pt and 4.1 to 2.3 for 0.3 Pt) and slight extension of Pt–O bond (1.98 Å to 2.00 Å for both of them), and the second shell Pt–Pt is very inconspicuous, which indicates that some single atoms formed to ultrafine clusters during H2-pretreatment. In other words, H2-pretreatment promotes a few of single atoms to form ultrafine clusters on the surface of the Nb2O5 resulting in higher activity for CO oxidation than O2-pretreatment catalysts. For the used samples, the very obvious Pt–Pt shell (2.70 Å for 1 Pt and 2.74 Å for 0.3 Pt) appeared with high CN that is 1.8 and 4.9 for 1 Pt and 0.3 Pt respectively besides the Pt–O shell seen Fig. 5c and d, which show that bigger Pt clusters were formed on used samples.
3.4 Discussion
There are no characteristic peaks of Pt species on the XRD patterns (Fig. S1 and S2†) of the all Pt-based catalysts. 0.3 Pt–Nb2O5 and 1 Pt–Nb2O5 samples after CO oxidation with H2-pretreatment were chosen to be characterized by TEM. The TEM images are not available for obtaining the information of Pt species of the used samples because the size of Pt species is less than the limit of detection of the device, which indicates that no Pt nanoparticles were formed after the reaction. TEM also shows no structural transformation on supports for H2-pretreatment and used catalysts compared with fresh samples. All the above analysis shows that catalyst supports have no obvious deformation or aggregation during reaction test, so the catalysts are very stable during the CO reaction.H2-TPR confirmed the interaction between the Pt species and Nb2O5 because the pure support did not exhibit the reduction peak of the surface lattice oxygen in 331 °C in the test temperature range, while it appeared after the loading of Pt. It should be noted that since the low-content catalysts do not easily exhibit peaks in H2-TPR, only the spectrum of the 1 Pt catalyst is shown here.
The most important information about explanation of the above phenomenon is derived from XAFS results undoubtedly. Comparing the XAFS spectrum and analytical results of the H2-pretreated catalysts and the as-prepared catalysts, the H2-pretreated samples possess a tiny peak belonging to the Pt–Pt shell that is absent for fresh catalysts. The only one Pt–O shell for fresh catalysts sufficiently indicate that Pt species exist in the form of single atoms, which illustrates the success of the synthetic method of Nb2O5. The lower valence of Pt after H2-pretreatment accounts for higher activity of CO conversion corresponding to the “light-off” curves of the CO oxidation reaction, which confirmed by the works of other research groups.19,42 Then, decrease of coordination number and slight extension of Pt–O bond after H2-pretreatment and the appearance of Pt–Pt shell, which indicates H2-pretreatment process promotes a few of single atoms to form ultrafine Pt clusters on the Nb2O5 surface resulting in higher activity than O2-pretreatment catalysts. We hypothesize the similar local coordination structure for O2-pretreated catalysts and the fresh catalysts because the fresh catalysts were obtained by calcination in air. Therefore, the above conclusions obtained by analysing and comparing the XAFS results of fresh catalysts and H2-pretreated catalysts are reliable and reasonable. XAFS reveals more details about the used catalysts. The used samples produce Pt–O shell and Pt–Pt shell according to the fitting results (see Fig. 5 and Table 3). At the same time, the coordination number of Pt–O shell for used samples decreased (3.9–1.2 Å for 1 Pt–Nb2O5 and 4.2–1.6 Å for 0.3 Pt–Nb2O5) compared to H2-pretreat samples. It indicates that the coordination environment of Pt species undergone dramatic changes during the reaction. In other words, ultrafine Pt clusters form bigger Pt clusters during CO oxidation reaction. The structural transformation of Pt/N2O5 is shown in Scheme 1.
 | ||
| Scheme 1 Schematic illustration of the structural transformation of Pt/N2O5. | ||
4 Conclusions
Nb2O5 with a high surface area was obtained by the hydrothermal method using urea as a precipitating agent and different content Pt/Nb2O5 catalysts were synthesized via incipient wetness impregnation. XAFS, a powerful tool for exploring the relationship between catalyst structure and structure-activity, indicates that Pt species present in fresh and H2-pretreated catalysts with the form of single atoms and fine clusters even when the content of Pt is 1%. The reasons for higher catalyst activity after H2-pretreated catalysts than O2-pretreated is not only due to the reduction of the valence state of Pt species but also to the appearance of Pt fine-clusters on the surface of supports according to XAFS spectrum and its corresponding analysis results. The current results will be of great significance in controllable synthesis of active ultrafine clusters or single atom Pt-based catalysts for other catalytic oxidation reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial supported from the National Science Foundation of China (NSFC) (grant no. 21773288) and the National Key Basic Research Program of China (2017YFA0403402).Notes and references
- P. Gélin and M. Primet, Appl. Catal., B, 2002, 39, 1–37 CrossRef.
- L. V. Mattos and F. B. Noronha, J. Power Sources, 2005, 145, 10–15 CrossRef CAS.
- C. Zhou, H. Wang, F. Peng, J. Liang, H. Yu and J. Yang, Langmuir, 2009, 25, 7711–7717 CrossRef CAS PubMed.
- K. An, S. Alayoglu, N. Musselwhite, S. Plamthottam, G. Melaet, A. E. Lindeman and G. A. Somorjai, J. Am. Chem. Soc., 2013, 135, 16689–16696 CrossRef CAS PubMed.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlogl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef CAS PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernandez, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- M. Valden, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- K. Judai, S. Abbet, A. S. Wörz, U. Heiz and C. R. Henry, J. Am. Chem. Soc., 2004, 126, 2732–2737 CrossRef CAS PubMed.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981–983 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- L. Wang, H. Li, W. Zhang, X. Zhao, J. Qiu, A. Li, X. Zheng, Z. Hu, R. Si and J. Zeng, Angew. Chem., 2017, 129, 4790–4796 CrossRef.
- J. Liu, C. Liu, A. Ma, J. Rong, Z. Da, A. Zheng and L. Qin, Appl. Surf. Sci., 2016, 368, 233–240 CrossRef CAS.
- M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS PubMed.
- T. Murayama, W. Ueda and M. Haruta, ChemCatChem, 2016, 8, 2620–2624 CrossRef CAS.
- J. Chen, H. Wang, Z. Wang, S. Mao, J. Yu, Y. Wang and Y. Wang, ACS Catal., 2019, 9, 5302–5307 CrossRef CAS.
- H. Wang, J.-X. Liu, L. F. Allard, S. Lee, J. Liu, H. Li, J. Wang, J. Wang, S. H. Oh, W. Li, M. Flytzani-Stephanopoulos, M. Shen, B. R. Goldsmith and M. Yang, Nat. Commun., 2019, 10, 3808 CrossRef PubMed.
- C. Dessal, T. Len, F. Morfin, J.-L. Rousset, M. Aouine, P. Afanasiev and L. Piccolo, ACS Catal., 2019, 9, 5752–5759 CrossRef CAS.
- H.-C. Wu, T.-C. Chen, Y.-C. Chen, J.-F. Lee and C.-S. Chen, J. Catal., 2017, 355, 87–100 CrossRef CAS.
- A. S. Chellappa, C. M. Fischer and W. J. Thomson, Appl. Catal., A, 2002, 227, 231–240 CrossRef CAS.
- A. S. Ivanova, E. M. Slavinskaya, R. V. Gulyaev, V. I. Zaikovskii, O. A. Stonkus, I. G. Danilova, L. M. Plyasova, I. A. Polukhina and A. I. Boronin, Appl. Catal., B, 2010, 97, 57–71 CrossRef CAS.
- F. Pompeo, N. N. Nichio, M. M. V. M. Souza, D. V. Cesar, O. A. Ferretti and M. Schmal, Appl. Catal., A, 2007, 316, 175–183 CrossRef CAS.
- P. Zhang, R. Li, Y. Huang and Q. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 2671–2678 CrossRef CAS PubMed.
- C. Wang, H. Daimon and S. Sun, Nano Lett., 2009, 9, 1493–1496 CrossRef CAS PubMed.
- Z. Jiang, X. Feng, J. Deng, C. He, M. Douthwaite, Y. Yu, J. Liu, Z. Hao and Z. Zhao, Adv. Funct. Mater., 2019, 29, 1902041 CrossRef.
- Z. Yan, Z. Xu, B. Cheng and C. Jiang, Appl. Surf. Sci., 2017, 404, 426–434 CrossRef CAS.
- D.-W. Jeong, H. S. Potdar, J.-O. Shim, W.-J. Jang and H.-S. Roh, Int. J. Hydrogen Energy, 2013, 38, 4502–4507 CrossRef CAS.
- Y. Chen, J. He, H. Tian, D. Wang and Q. Yang, J. Colloid Interface Sci., 2014, 428, 1–7 CrossRef CAS PubMed.
- S. Furukawa, D. Tsukio, T. Shishido, K. Teramura and T. Tanaka, J. Phys. Chem. C, 2012, 116, 12181–12186 CrossRef CAS.
- J. C. Matsubu, S. Zhang, L. DeRita, N. S. Marinkovic, J. G. Chen, G. W. Graham, X. Pan and P. Christopher, Nat. Chem., 2017, 9, 120–127 CrossRef CAS.
- V. M. Benitez, S. P. de Lima, M. do Carmo Rangel, D. Ruiz, P. Reyes and C. L. Pieck, Catal. Today, 2017, 289, 53–61 CrossRef CAS.
- L. Wolski, I. Sobczak and M. Ziolek, J. Catal., 2017, 354, 100–112 CrossRef CAS.
- D. A. G. Aranda, A. L. D. Ramos, F. B. Passos and M. Schmal, Catal. Today, 1996, 28, 119–125 CrossRef CAS.
- S. Guerrero, J. T. Miller and E. E. Wolf, Appl. Catal., A, 2007, 328, 27–34 CrossRef CAS.
- B. Liu, H. Wang, Y. Chen, J. Wang, L. Peng and L. Li, J. Alloys Compd., 2016, 682, 584–589 CrossRef CAS.
- S. B. T. Tran, H. Choi, S. Oh and J. Y. Park, J. Catal., 2019, 375, 124–134 CrossRef CAS.
- Y. Liu, M. Tursun, H. Yu and X. Wang, Mol. Catal., 2019, 464, 22–28 CrossRef CAS.
- P. Justin, P. Hari Krishna Charan and G. Ranga Rao, Appl. Catal., B, 2010, 100, 510–515 CrossRef CAS.
- P.-P. Du, X.-C. Hu, X. Wang, C. Ma, M. Du, J. Zeng, C.-J. Jia, Y.-Y. Huang and R. Si, Inorg. Chem. Front., 2017, 4, 668–674 RSC.
- Y. Yuan, A. P. Kozlova, K. Asakura, H. Wan, K. Tsai and Y. Iwasawa, J. Catal., 1997, 170, 191–199 CrossRef CAS.
- P.-P. Du, W.-W. Wang, C.-J. Jia, Q.-S. Song, Y.-Y. Huang and R. Si, Appl. Catal., A, 2016, 518, 87–101 CrossRef CAS.
- R. M. Rioux, H. Song, J. D. Hoefelmeyer, P. Yang and G. A. Somorjai, J. Phys. Chem. B, 2005, 109, 2192–2202 CrossRef CAS PubMed.
- I. Nowak and M. Ziolek, Chem. Rev., 1999, 99, 3603–3624 CrossRef CAS PubMed.
- S. Y. Moon, RSC Adv., 2016, 6, 18198–18203 RSC.
- E. O. Jardim, S. Rico-Francés, F. Coloma, J. A. Anderson, E. V. Ramos-Fernandez, J. Silvestre-Albero and A. Sepúlveda-Escribano, Appl. Catal., A, 2015, 492, 201–211 CrossRef CAS.
- T. S. Mozer and F. B. Passos, Int. J. Hydrogen Energy, 2011, 36, 13369–13378 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01252a |
| This journal is © The Royal Society of Chemistry 2020 |