
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffluent of biomass cooking with active oxygen and solid alkali (CAOSA): component separation, recovery and characterization†
Ning Dinga,
Huai Liua,
Xianhai Zeng *ab,
Yong Sunab,
Xing Tangab and
Lu Lin*ab
*ab,
Yong Sunab,
Xing Tangab and
Lu Lin*ab
aCollege of Energy, Xiamen University, Xiamen 361102, PR China. E-mail: xianhai.zeng@xmu.edu.cn; lulin@xmu.edu.cn; Fax: +86-592-2880701; Fax: +86-592-2880702; Tel: +86-592-2880701 Tel: +86-592-2880702
bFujian Engineering and Research Center of Clean and High-valued Technologies for Biomass, Xiamen Key Laboratory of Clean and High-valued Applications of Biomass, Xiamen University, Xiamen 361102, PR China
First published on 25th April 2020
Abstract
Effluent from biomass pretreatment industries affects a substantial proportion of industrial wastewater and can be troublesome to treat. In a new environmentally benign biomass pretreatment approach based on cooking with active oxygen and solid alkali (CAOSA), some precipitates were found to be produced while the effluent (yellow liquor) was treated with alcohol or acid. Based on the precipitation approaches, component contents in CAOSA yellow liquor were successfully made clear, consisting of 57.19 wt% low molecular weight acid salts, 15.47 wt% lignin, 4.30 wt% saccharides and 23.04 wt% inorganic salts. Different organic solvents were tested as the precipitants, and when ethanol was used, over 81 wt% of the compounds in the yellow liquor could be precipitated. Meanwhile, 89.2% of the solid alkali consumed in CAOSA pretreatment could be recovered, showing prospects for primary effluent treatment. Furthermore, CAOSA lignin (CL) was recovered by acid precipitation from yellow liquor. By characterization, CL was found to have lower molecular weight and higher β-O-4 bond (60.05%) ratio than milled wood lignin (MWL, 48.64%), and thus, it might be a more ideal feedstock for further valorization.
1. Introduction
Biomass refining, a concept that has arisen since the 1980s, has attracted increasing attention from practitioners and researchers.1 Countless chemicals are obtained from biomass, especially lignocellulosics.2,3 Due to the natural recalcitrance of biomass, employing efficient pretreatment technologies to separate their components is necessary for further utilization.4–6 However, effluent from the biorefinery industry, which makes quite a large proportion of industrial wastewater, has been particularly challenging to address because of its high total organic carbon (TOC) content, strong alkalinity and even toxicity.7–9 Current disposal methods mainly include coagulation, precipitation, biological aeration or the introduction of oxidizing agents such as Fenton's reagent, ozone and peroxide.10,11 By the method of oxygen delignification, 51, 57 and 43% of BOD3, COD and color were reduced of the soda-anthraquinone pulping effluent.12 And in Garg's work, with addition of H2O2 and FeCl3, the maximum lignin and TOC removals reached 85% and 48% from the Kraft pulping effluent.13 However, the complex disposal process will undoubtedly increase the treatment cost. Further, plenty of biomass resources are abandoned into effluent, which is used for supplying heat or discharged directly, thus also leading to the waste of biomass resources.10In current biomass pretreatment processes, lignin is often treated as a by-product in the effluent without high-value utilization.14,15 However, lignin is truly not waste without any application value; in contrast, it has great potential for use as feedstock to produce aromatic monomers.16 Lignin from different biomass species is considered to have different structures and application values.15 Lignin from gymnosperms is thought to lack a syringyl unit and, thus, shows a higher branching degree and a less active β-O-4 ether bond, resulting in more difficulties to be degraded.17 C-lignin has been found in plant seeds with a simple linear catechol structure; therefore, C-lignin is recognized as a better raw material for lignin valorization.18 Extraction methods can also influence the structure of lignin. Milled wood lignin (MWL) and cellulosic enzymatic lignin (CEL) are thought to have a structure that is most close to the structure of native lignin.14 Many different extraction methods have been put forward to obtain modified lignin, which is more suitable for further valorization.19 However, until now, technical lignin has only been applied to prepare low-value materials such as sulfonate, with industrial lignin use rarely reported for the large-scale production of lignin monomers.20–22
Previously, a new environmentally benign technology for biomass pretreatment based on cooking with active oxygen and solid alkali, short for CAOSA, has been exploited and developed by our group.23,24 By CAOSA, pulp with a desirable brightness and low kappa value can be obtained.25 Further, the viscosity, alkalinity, biochemical oxygen demand (BOD) and chemical oxygen demand (COD) of the yellow liquor are found to show lower values than black liquor in the traditional process, thus meaning easier treatment.26 However, even though the treatment difficulty of yellow liquor is much less than black liquor, there have also been some problems involving effluent treatment and compound re-utilization. Jiang and co-workers have tried to recover the solid alkali by calcium causticization. However, the calcium salts are always co-precipitated together with the magnesium alkali, resulting in the difficulty of the re-utilization of the solid alkali.24
To find an effective pathway for effluent organic separation and catalyst recovery, in this work, a precipitation approach utilizing alcohol and acid was proposed. Surprisingly, with such a precipitation method, qualitative and quantitative analysis of all constituents in yellow liquor was determined. This work provided a simple method to recover the solid alkali and reduce the TOC value of yellow liquor. Furthermore, CAOSA lignin, which was acid-precipitated from yellow liquor, was determined to contain a higher β-O-4 ether bond proportion (60.05%) compared with that of MWL (48.64%), showing great potential for further valorization to prepare vanillin or methyl vanillate. In a word, the developed precipitation approach can undoubtedly improve the efficiency of biomass utilization and provide preferable economic benefits for biomass refineries.
2. Experimental
2.1 Materials
Spruce wood was provided by YST Group Co. Ltd. (Dongguan, Guangdong, China). Deuterated water (D2O) and Deuterated dimethyl sulfoxide (DMSO-d6) with a purity of over 99.9% were purchased from Sigma–Aldrich (Shanghai, China). Other reagents were of analytical grade and purchased from Shanghai Aladdin Bio-Chem Technology Co. Ltd.2.2. Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) over 12 h, then dried and ball-milled for 48 h. The resulting powder was extracted with a mixture of dioxane and water (96:4) at 30 °C for over 24 h. This extraction process was repeated 3 times, and the extracts were combined and concentrated using a rotary evaporator. Then the concentrated liquid was dropped into ethanol solvent (10 times the volume of the liquid) to remove saccharides. The ethanol solvent was filtered and concentrated again. And the concentrated ethanol solvent was then dropped into a mixture (5 times the volume of the liquid) of acetic acid and water (9:1) to obtain the lignin precipitate. Finally, the precipitate was washed and lyophilized to obtain the MWL powder.:5, methanol, ethanol, acetone, 1,4-dioxane and tetrahydrofuran). Immediately the solid precipitate or the separated liquid phase was produced.
:1) over 12 h, then dried and ball-milled for 48 h. The resulting powder was extracted with a mixture of dioxane and water (96:4) at 30 °C for over 24 h. This extraction process was repeated 3 times, and the extracts were combined and concentrated using a rotary evaporator. Then the concentrated liquid was dropped into ethanol solvent (10 times the volume of the liquid) to remove saccharides. The ethanol solvent was filtered and concentrated again. And the concentrated ethanol solvent was then dropped into a mixture (5 times the volume of the liquid) of acetic acid and water (9:1) to obtain the lignin precipitate. Finally, the precipitate was washed and lyophilized to obtain the MWL powder.:5, methanol, ethanol, acetone, 1,4-dioxane and tetrahydrofuran). Immediately the solid precipitate or the separated liquid phase was produced.In a typical acid precipitation experiment, yellow liquor was acidified by HCl solution to pH about 1.0, and the precipitate was produced.
In two groups of cross precipitation experiments, ethanol was used as the organic precipitant. In the first group, YL was firstly added into ethanol solvent (v/v = 1:5). The obtained precipitate was separated by filtration and washed by ethanol, then stored as Pre-E. Afterward, the filtrate was evaporated to recover ethanol solvent. And the concentrated liquid was acidified by HCl solution to pH about 1.0. Precipitate in this step was then washed by deionized water and stored as Pre-E–A.
In the other group, the order of precipitants was exchanged. Yellow liquor was firstly added into HCl solution and Pre-A was obtained firstly. Subsequently, the liquid was evaporated to remove excess acid and then to be precipitated by ethanol (v/v = 1:5), the precipitate labeled as Pre-A–E.
2.3 Determination and characterization
Additionally, ash contents of YL dry matter and precipitates were determined by thermogravimetric analysis (TG) for 3 times according to the following program: 25–20 °C, 20 °C min−1; 120 °C, 10 min; 120 °C −800 °C, 10 °C min−1; 800 °C, 10 min. The average ash content and the representative thermo-gravimetric curve was displayed.
:1, v/v). 20 mg lignin (MWL or CL) was dissolved into 600 μL solvent A. 100 μL cyclohexanol solution (10.85 mg mL−1 in A) was added as an internal standard. 100 μL 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (TMDP) was added into above solution and the mixture was kept for 10 min. The final phosphorylated sample was transferred into a 5 mm NMR tube for subsequent determination. Semi-quantification was performed referred to the last item and all the proportion was normalized by the internal standard.3. Results and discussion
3.1 Yellow liquor precipitation by organic solvents
The yellow liquor used in this work was obtained from the CAOSA pretreatment process at 160 °C for 3 hours with spruce wood. Under such conditions, alkali or salts were dissolve in yellow liquor together with the organics. A lot of efforts had been put on recovering the components in YL, especially the solid alkali used in CAOSA process. In the meanwhile, once the alkali, salts and organics were recovered, the treatment difficulty of the effluent would also be reduced.Considering the low solubility of the salts and alkali in organic solvents, some organic solvents, ordered by their polarity, were selected as precipitants, including methanol, ethanol, acetone, 1,4-dioxane and tetrahydrofuran. Once the yellow liquor was added into methanol or ethanol, faint yellow sediment was immediately observed (Fig. 1). Compared with the original yellow liquor, the supernatant of the sample looked much more limpid and brighter, inferring that most of the color components were precipitated. However, when acetone, 1,4-dioxane or tetrahydrofuran was used as precipitant, the salt solution and organic solvents were separated into two liquid layers without any solid precipitate generated. The reason for the layer separation was perhaps that with the solvents' polarity decreased, the inter-solubility between the solvents and salt solution is not as good as that with pure water. Therefore, among the selected solvents, alcohols (methanol and ethanol) were verified as the better precipitants for yellow liquor.
 | ||
| Fig. 1 Yellow liquor (A) and its mixture with methanol (B)/ethanol (C)/acetone (D)/ 1,4-dioxane (E)/tetrahydrofuran (F). | ||
Interestingly, a larger precipitate ratio of 81.9 wt% (based on the total dry weight of yellow liquor, Table S1†) was achieved with ethanol precipitant than methanol (64.8 wt%, Table 1). When using ethanol as precipitant, a darker solid precipitate and brighter supernatant were obtained (Fig. 1). This difference indicated that the color compounds, such as oxidized lignin, had lower solubility in ethanol solvent. In Zhang's work, Kraft lignin was treated by a mixture of methanol and water and separated into the dissolved part and undissolved part.31 The undissolved Kraft lignin was proved with a higher molecular weight than the dissolved ones. The undissolved lignin occupied 47.51% of the total lignin sample while the methanol ratio was 70%. While the methanol ratio reached 90%, the undissolved lignin decreased to 37.73. Yet, in this work, ethanol, which has a lower polarity, could separate more lignin from yellow liquor than methanol, implying that the lignin derivates were not be precipitated as the lignin molecule. The ethanol precipitation scheme was similar to the YL alkali precipitation process described in Jiang's work.24 Therefore, in the organic solvent precipitation process, lignin was more likely to be co-precipitated from the CAOSA yellow liquor with magnesium ion as the salt form (Fig. 2).
| Componentsa | YL/% | Pre-ethanol/% | Pre-methanol/% | |
|---|---|---|---|---|
| a The acid contents were calculated in the corresponding acid form. | ||||
| Proportion to YL dry weight | — | 81.9 | 64.8 | |
| Ash content | 25.35 | 27.61 | 32.33 | |
| LMWA | Formic acid | 15.60 | 3.28 | 3.38 |
| Acetic acid | 12.49 | 3.75 | 4.14 | |
| Glycolic acid | 6.90 | 4.36 | 5.17 | |
| 3-Hydracrylic acid | 2.69 | 1.40 | 1.32 | |
| Oxalic acid | 4.27 | 4.86 | 5.38 | |
| Malonic acid | 1.21 | 1.44 | 1.73 | |
| Succinic acid | 3.09 | 3.79 | 4.39 | |
| Maleic acid | 1.32 | 1.43 | 1.73 | |
| Fumaric acid | 1.29 | 1.32 | 1.63 | |
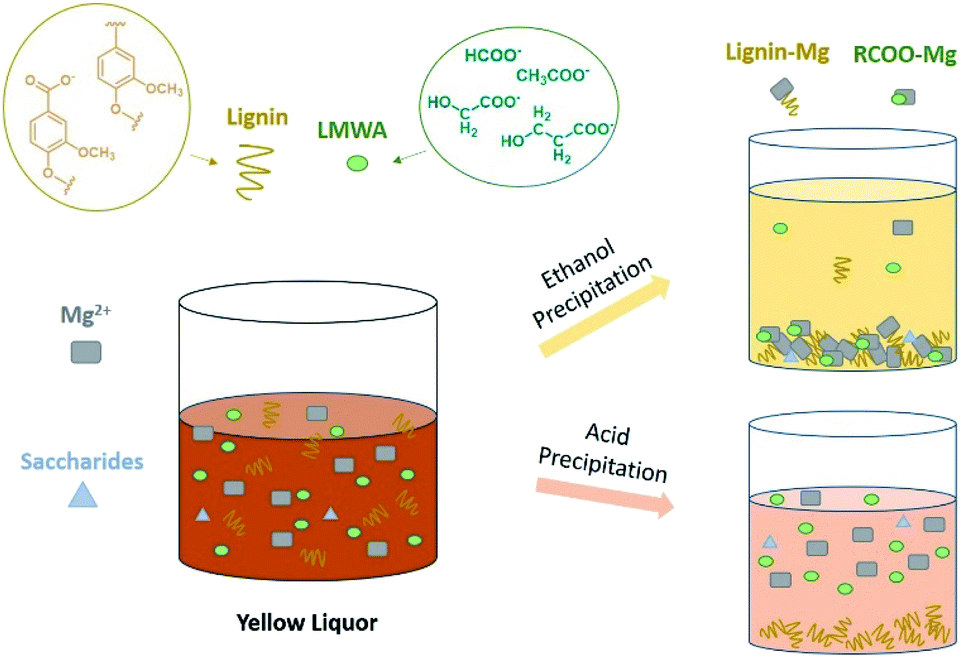 | ||
| Fig. 2 Schematic diagram about YL ethanol and acid precipitation. | ||
As revealed by all the precipitation cases, the precipitation ability of the precipitant could be concluded. The amount of precipitates would reduce both with the increment of the precipitant's magnesium salt solubility and decrement of its polarity. Based on the precipitation results, ethanol was suggested to be the best precipitant among the applied solvents and was therefore used in the following research.
3.2 Constituent distribution of yellow liquor
To clarify the constituents of yellow liquor, two different precipitation routes were designed, by which four kinds of precipitates were collected as Pre-A, Pre-A–E, Pre-E and Pre-E–A. In the CAOSA process, only biomass raw material, alkali, oxygen and water were brought into the pretreatment system, yellow liquor product could be reasonably classified into four parts: degraded lignin, degraded saccharides, low-molecular-weight degraded products and inorganic alkali/salts.Salts (organic and inorganic) in yellow liquor could be determined by TG analysis. The ash content of the dry component of yellow liquor was measured as an average of 25.34 wt% (Fig. 3, Table S2†). And the ash content of Pre-E was a little higher than YL and reached 27.61 wt%, meaning the magnesium salts were enriched into Pre-E during the ethanol precipitation process. Additionally, as for another three other kinds of precipitates (Pre-E–A, Pre-A and Pre-A–E), almost no solid residue was collected after the calcination at 800 °C, implying that these three precipitates were made up of organic compounds.
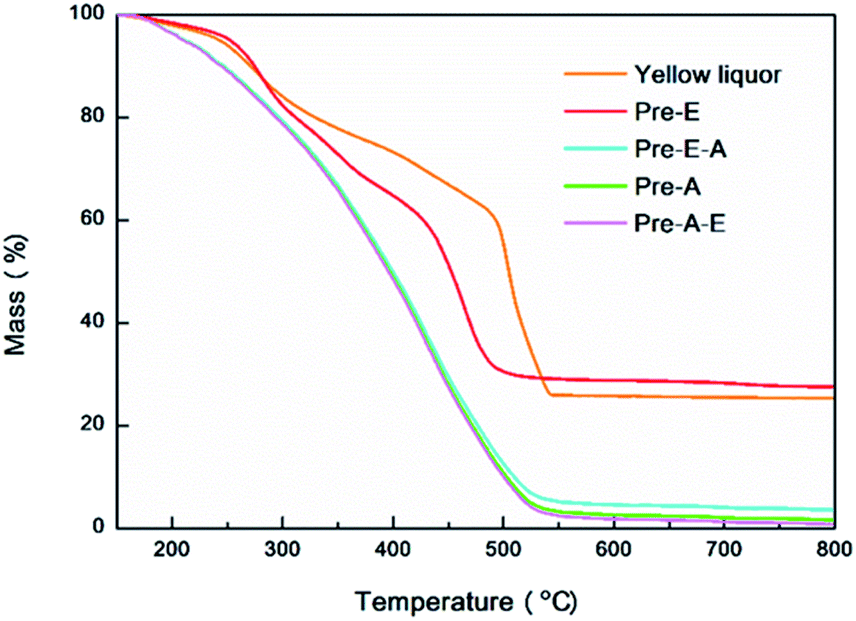 | ||
| Fig. 3 Typical TG curves of yellow liquor and precipitates. | ||
Considering the different ash contents and solubilities of the four precipitates, it was reasonable to assume that the precipitates were composed of different constituents. HSQC NMR was adopted to analyze the lignin and its linkage with saccharides, such as the lignin–carbohydrate complexes (LCC).14 Signals in the lignin NMR spectra were classified into two regions: side chain region and aromatic region.32
From the comparison of the HSQC NMR spectra, Pre-E–A and Pre-A were proven to show almost the same chemical shift, indicating that they had almost the same structure. Moreover, most of the signals in the Pre-E–A and Pre-A spectra were found in the MWL spectra for spruce wood. Thus, they were reasonably seen as the lignin fragment without complete degradation (Fig. 4). According to the signal assignment, some typical structures in lignin were disappeared after CAOSA pretreatment (Table S3,† Fig. 5). The regulation of the structural changes in spruce lignin was parallel with the bamboo lignin during the CAOSA process.26 The phenylcoumarans structure was degraded rapidly, while the β-O-4 structure and ferulates structure remained stable after the pretreatment process. Although both Pre-E–A and Pre-A showed the similarity to MWL, the amounts of precipitate from Pre-E–A (1.72 wt%) and Pre-A (15.47 wt%) were different. This difference could be explained by the fact that after CAOSA treatment, the oxygen-containing groups in lignin were oxidized. In alkaline conditions, salts could be formed once hydroxyl groups are oxidized to carboxyl groups, then the solubility of the salts in alcohol was decreased. Thus, after ethanol precipitation, most of the oxidized lignin was separated, and only a small amount of which was obtained in the following acid precipitation.
 | ||
| Fig. 4 HSQC NMR spectra of MWL and precipitates. | ||
 | ||
| Fig. 5 Main structure present in the bamboo lignin: (A) β-O-4 alkyl-aryl ethers; (A′) β-O-4 alkyl-aryl ethers with acylated γ-OH with p-coumaric acid; (B) resinols; (C) phenylcoumarans; (T) a likely incorporation of tricin into the lignin polymer through a G-type β-O-4 linkage; (PCE) p-coumarates; (FA) ferulates; (I) p-hydrocinnamyl alcohol end-groups (H) p-hydroxyphenyl units; (G) guaiacyl units; (S) syringyl units. | ||
It was worth mentioning that many signals in Pre-A–E could be assigned to saccharides and LCC linkage, indicating that Pre-A–E, which could be precipitated by alcohol and without any signal in aromatic region, was probably the product of incompletely degraded cellulose and hemicellulose.33
As reported previously, LMWA salts hold a majority part in yellow liquor.26 In an oxidation–delignification process such as CAOSA, aromatic rings are considered to be broken by active oxygen groups.34 LMWA products are mainly obtained from the ring-opening reaction of lignin aromatics. The LMWA salts, mainly including formate, acetate, glycolate and hydracrylate, were acidified and then determined by HPLC (Table 1). It was calculated that over half of the dry components of yellow liquor was made up of LMWA salts, evidencing the occurrence of severe delignification and a ring-opening reaction alongside the CAOSA process.
Pre-E had an ash content of 27.61 wt% (Table S2†), and the LMWA contents of Pre-E and Pre-Methanol were determined by HPLC (Table 1). By comparison of the contents, LWMA salts, especially the formic and acetic salts, showed a better solubility in methanol than ethanol. And with the polarity of the organic groups in the salts became lower, more proportion of which was precipitated, such as the salts combined with the oxidized lignin. Only a few proportions of the formic and acetic salts could be precipitated by ethanol or methanol, remaining the possibility to recover formic and acetic acids from YL for further fine utilizations.22 The amount of the precipitation proved that more LWMA salts would be separated with the polarity of the organic solvent decreased.
Apparently, the precipitates by organic solvent were not only composed of LMWA salts. Some lignin, saccharides and inorganic salts were perhaps precipitated together with the LMWA salts in the precipitation process. And the content of inorganic salts dissolved in yellow liquor could be calculated from the mass difference between the dry component of yellow liquor and other constituents. Inorganic salts were identified as magnesium salts including MgCO3, Mg(HCO3)2, Mg2OCO3, MgO and Mg(OH)2.24 These inorganic salts can be transformed again into solid alkali MgO by calcination, providing a potential means for catalyst recovery.
After analysis of the precipitates, the complete composition of the yellow liquor was finally made clear (Table 2).
3.3 Effluent treatment and catalyst recovery
By understanding the overall component distribution, the alcohol and acid precipitation approach can be substantially developed.Currently, alkali used in the traditional pulping process is usually recovered by evaporation-calcination or causticization by calcium oxide. However, different shortcomings have been exposed by such methods. In the evaporation–calcination process, effluent is firstly concentrated and evaporated, and then, the solid residue is collected to recover heat and alkali by calcination. The most obvious disadvantage of the evaporation–calcination method is the huge energy cost consumed by evaporation and the serious harm to equipment brought about by foaming and carbonization problems. For calcium oxide causticization method, effluent can be re-adjusted to a proper alkaline condition and re-used in the cooking process. However, while the calcium oxide is used to treat CAOSA yellow liquor, magnesium will be precipitated together with calcium. After that, separation of calcium and magnesium will be the subsequent difficulty.24 Removal of the organic compounds from yellow liquor is the other question. By usage of the activated sludge system, the COD removal is reported a decrease of 67–76%.35 And when nano-Fe2O3/H2O2/UV is used to treat the Kraft pulping effluent, the maximum aromatic compounds and COD reduction reach 68.4% and 89.8%.36 However, it's difficult to achieve the re-utilization of the recovered organic compounds from the effluent through those degradation processes.
Compared with the pathways mentioned above, alcohol precipitation, by which over 80% of the constituents can be simply separated, is indeed an effective approach for treating CAOSA yellow liquor (Fig. 6A). After alcohol precipitation, the secondary effluent would contain fewer organics and could be easier-treated. Especially, by this effluent treatment approach, the solid alkali could also be contemporarily recovered, avoiding an extra complicated catalyst recovery process. Based on the contents of each constituents, the solid alkali recovery, lignin removal and organics removal reached 89.2%, 88.9% and 76.2% respectively.
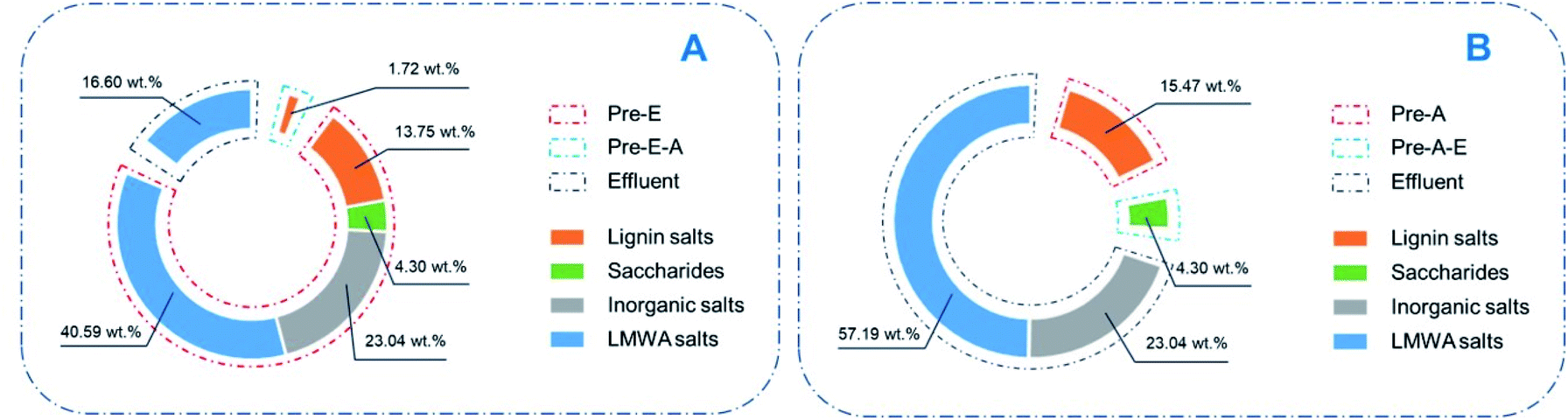 | ||
| Fig. 6 Components content in different precipitates (A: ethanol added first; B: acid added first). | ||
3.4 CL analysis
The acid precipitation approach affords more options for lignin utilization (Fig. 6B). Pre-A, the lignin obtained by acidification from yellow liquor, is named as CAOSA lignin, short for CL. Further structural analysis of CL will deepen the understanding of the CAOSA delignification mechanism and make better use of the undegraded component in biomass pretreatment effluent. | ||
| Fig. 7 GPC results of Pre-E–A and Pre-A. | ||
Additionally, GPC results for yellow liquor presented three peaks: 600, 750 and 1100 (Fig. S1†). By the acid precipitation method, it was found that the GPC result for Pre-A–E showed almost no signal at 750, and the signal of Mw 600 also decreased, meaning lignin was precipitated by the acid precipitation process.
| Entry | Structures | MWL | CL |
|---|---|---|---|
| 1 | β-O-4 ether bond (A) | 48.64 | 60.05 |
| 2 | Resinols (B) | 13.97 | 11.85 |
| 3 | Phenylcoumarans (C) | 20.90 | 26.42 |
| 4 | Cinnamyl alcohol end-groups (I) | 16.49 | 1.67 |
| 5 | S/G ratio | 0.89 | 0.74 |
| 6 | –OCH3 | 26.56 | 25.51 |
Notably, the proportion of β-O-4 linkages in the lignin side chain, which attracted great attention from researchers investigating lignin valorization, was increased from 48.64% to 60.05% after the CAOSA process. The increase of β-O-4 linkage was caused by its relatively lower reactivity under the weak alkaline conditions.26 Hence, CL has the potential to be a better feedstock to produce aromatic monomers through lignin valorization methods.
The other significant difference appeared around δC/δH 4.09/61.2 ppm, where belongs to the cinnamyl alcohol end-groups (I). The disappearance of that signal indicated the thorough degradation of the I structure. The conjugated Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cβ structure with aromatic ring was easy to be attacked by the electrophilic active oxygen groups, as a result, the CαCβ bond was then cleaved.23
Cβ structure with aromatic ring was easy to be attacked by the electrophilic active oxygen groups, as a result, the CαCβ bond was then cleaved.23
The content of methoxyl groups in CL was calculated to be 25.51%, which remained at the same level as the content in the MWL (26.56%). In one piece of our previous study, the aryl-ether bonds were found degraded under CAOSA conditions, including the methoxyl groups at lignin side chain. Cleavage of the aryl-ether bonds would decrease the methoxyl group content in lignin. But the hydroxyl group generated in the aryl bond cleavage process would further trigger the ring-opening reaction of the lignin aromatic ring. Because of that, the aromatic ring with a phenol hydroxyl group would be fast broken under CAOSA condition, and the methoxyl group was therefore remained at a similar value with the original one.
C bond between their α-C and β-C, such as cinnamyl alcohol end-groups (I), cinnamyl aldehyde (pCE) or ferulates (FA) structure, as shown in the HSQC NMR results obtained for spruce MWL and CL (Fig. 4).By semi-quantification with 31P NMR, content of α-OH was found decreased after CAOSA treatment, which was probably a result of Cα–Cβ bond cleavage under the oxidation conditions (Fig. 8, Table 4). In an alkaline condition, the phenol hydroxyl group tended to transform into the quinone structure, and then, β-C at the side chain became a stronger negative charge center. Followed by the charge center transformed, the β-C became easy to be attacked by active oxygen group (Scheme 1).23 Afterward, the other ending of the oxygen free radical was attracted by α-C, which had a lower electronegativity, and an extremely unstable tetra-atomic ring was formed. As the result of the crack of the unstable ring, the Cα–Cβ bond of the lignin side chain was broken and two molecules of carbonyl compounds were produced. Subsequently, benzoic structure and hydroxyl acid ester was generated and could be found out as the products in yellow liquor. Therefore, it is easy to understand that in 31P NMR spectra of CL, the signal of –COOH was much higher than that of MWL.
 | ||
| Fig. 8 31P NMR spectra of MWL and CL. | ||
| Entry | Signal range/ppm | Structures | MWL | CL |
|---|---|---|---|---|
| 1 | 136.0–135.0 | α-OH, E | 0.11 | 0 |
| 2 | 134.5–133.8 | α-OH, T | 0.32 | 0.04 |
| 3 | 133.8–133.3 | Internal standard | — | — |
| 4 | 133.3–132.0 | Primary –OH | 6.39 | 15.22 |
| 5 | 132.0–131.5 | S –OH | 0.33 | 0.99 |
| 6 | 131.5–129.5 | G –OH | 0.43 | 0.57 |
| 7 | 128.8–128.0 | pCE –OH | 0.58 | 0.75 |
| 8 | 128.0–126.0 | –COOH | 0.54 | 1.31 |
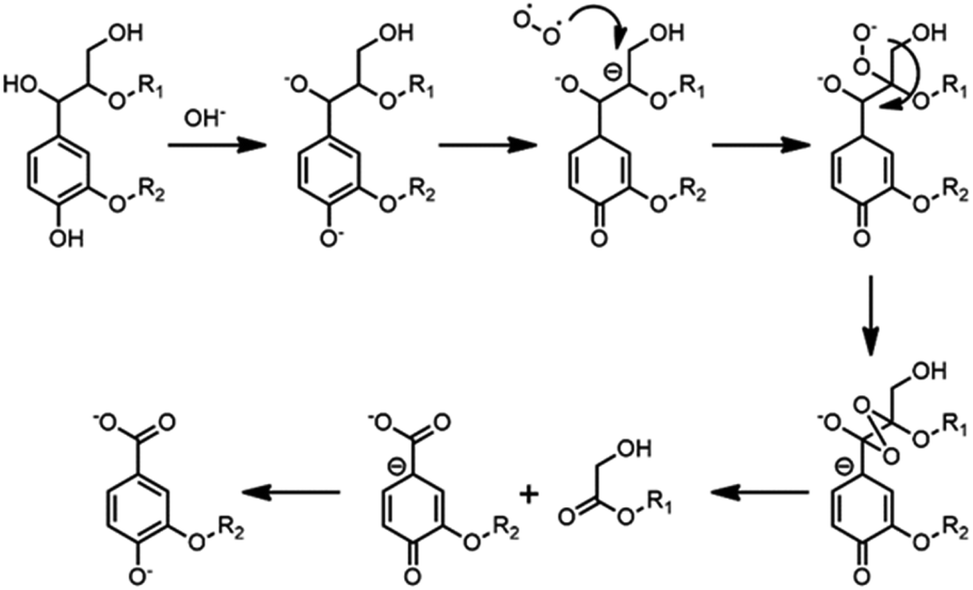 | ||
| Scheme 1 Possible routine of Cα–Cβ bond cleavage in CAOSA process. | ||
Moreover, as indicated by the difference in the α-OH content, the unconjugated Cα–Cβ bond was almost completely broken under CAOSA conditions. In contrast with the unconjugated bond, the conjugated Cα–Cβ structure in pCE and FA could be detected both before and after CAOSA. The relative stability of pCE and FA structure might because of that, in their conjugated structure, electronic density was diluted among the whole conjugated structure instead of being concentrated at the β-C. Therefore, the negative charge was no longer concentrated at the β-C. As a result, the β-C would not tend to attract active oxygen groups and the C–C bond would not be cleaved.
First of all, it should be noted that the main difference between MWL and CL was found for the peaks between 1750–1510 cm−1, which represented the stretching and vibration of C–O bonds, typically for the CO functional group. The band at 1710 cm−1, which was assigned to CO stretching in unconjugated ketone and carbonyl groups, was notably reduced after CAOSA pretreatment, indicating the severely broken of that structures in lignin during CAOSA process. Band around 1600 cm−1 was the signal for aromatic skeletal vibrations plus CO stretch. And in CL, the band around 1600 cm−1 was even stronger than MWL, meaning more CO bonds conjugated with the aromatic ring were detected in CL sample. This was consistent with the conclusion drawn from the 31P NMR results, which showed that under alkaline oxidation conditions, lots of carbonyl or carboxyl groups were generated.
Next, the decrease in the intensity of bands at 2925 cm−1 and 2850 cm−1, which were assigned to C–H stretching in methyl groups and methylene groups. Methyl and Methylene groups were mainly existing in lignin side chain structure, therefore, the decrease of that after CAOSA pretreatment might be caused by the degradation of lignin side chain structures, such as the Cα–Cβ bond cleavage. Furthermore, as the nature of spruce lignin, whether in MWL or CL samples, only the guaiacyl (G) units could be detected with the band range from 925–817 cm−1 (Fig. 9).
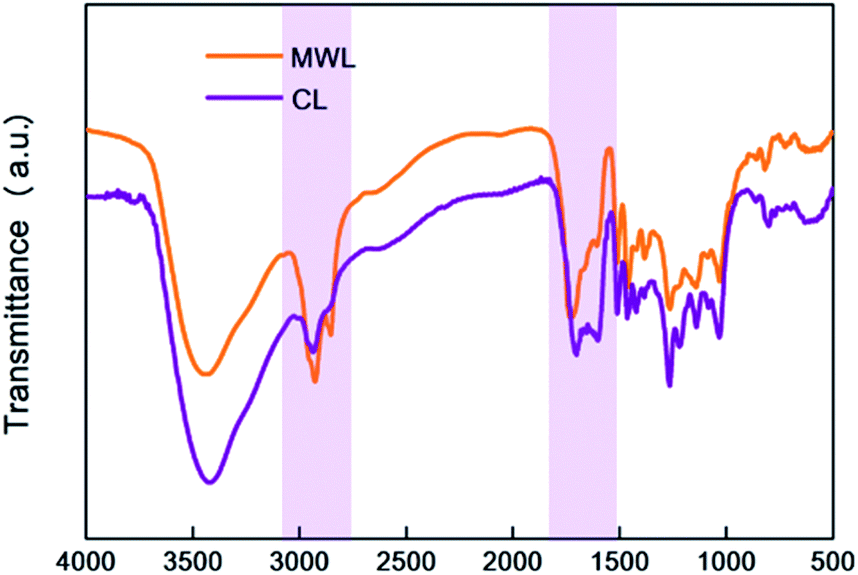 | ||
| Fig. 9 FTIR spectra of MWL and CL. | ||
Hence, CL, the acid precipitate of CAOSA yellow liquor with a higher β-O-4 bond ratio, shows better prospects for application on preparing substituted benzoic acid or its ester.
4. Conclusions
In summary, a preferable chemical precipitation method to treat yellow liquor, the effluent of the CAOSA process, was put forward. By use of this precipitation method, the content of the constituents in yellow liquor was determined. Alcohol precipitation showed a great potential to serve as an efficient approach to recover the solid alkali and reduce the TOC value of the CAOSA yellow liquor. While ethanol used as precipitant, over 81 wt% components could be separated from the liquid and solid alkali recovery and lignin removal reached 89.2% and 88.9% respectively. On the other hand, the structure of the acid precipitate, CL, was studied. CL has lower molecular weight and about a 10% higher ratio of β-O-4 than MWL, was regarded as a better feedstock for further lignin valorization. Thus, this precipitation method has remarkable application potential and is possibly an alternative technology for the treatment of effluent for biomass pretreatment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the financial support from the National Natural Science Foundation of China (Grant No. 21676223, 21978248), the Natural Science Foundation of Fujian Province of China (Grant No. 2019J06005).References
- H. R. Bungay, Science, 1982, 218, 643–646 CrossRef CAS PubMed.
- X. Tang, X. Zeng, Z. Li, L. Hu, Y. Sun, S. Liu, T. Lei and L. Lin, Renewable Sustainable Energy Rev., 2014, 40, 608–620 CrossRef CAS.
- M. Zuo, K. Le, Y. Feng, C. Xiong, Z. Li, X. Zeng, X. Tang, Y. Sun and L. Lin, Ind. Crops Prod., 2018, 112, 18–23 CrossRef CAS.
- M. Li, Y. Pu and A. J. Ragauskas, Front. Chem., 2016, 4, 1–8 Search PubMed.
- C. E. W. L. R. Lynd and T. U. Gerngross, Biotechnol. Prog., 1999, 15, 777–793 CrossRef PubMed.
- M. T. Rajeev Kumar, K. Karimi and I. S. Horvath, Biofuel Res. J., 2016, 9, 347–356 CrossRef.
- P. Asaithambi, A. R. A. Aziz, B. Sajjadi and W. Daud, Environ. Sci. Pollut. Res. Int., 2017, 24, 5168–5178 CrossRef CAS PubMed.
- G. V. Sjonkortekaas, Y. He, G. Lettinga and J. A. Field, J. Ferment. Bioeng., 1998, 86, 97–110 CrossRef.
- U. Patel and S. Suresh, Sep. Purif. Technol., 2008, 61, 115–122 CrossRef CAS.
- E. C. Catalkaya and F. Kargi, J. Hazard. Mater., 2007, 139, 244–253 CrossRef CAS PubMed.
- R. J. Stephenson and S. J. B. Duff, Water Res., 1996, 30, 781–792 CrossRef CAS.
- D. Kaur, N. K. Bhardwaj and R. K. Lohchab, Environ. Sci. Pollut. Res. Int., 2017, 24, 23488–23497 CrossRef CAS PubMed.
- K. Lal and A. Garg, Process Saf. Environ. Prot., 2017, 111, 766–774 CrossRef CAS.
- J. L. Wen, S. L. Sun, T. Q. Yuan, F. Xu and R. C. Sun, J. Agric. Food Chem., 2013, 61, 11067–11075 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 709–719 CrossRef CAS PubMed.
- N. Li, Y. Li, C. G. Yoo, X. Yang, X. Lin, J. Ralph and X. Pan, Green Chem., 2018, 20, 4224–4235 RSC.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed. Engl., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- F. Chen, Y. Tobimatsu, D. Havkin-Frenkel, R. A. Dixon and J. Ralph, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1772–1777 CrossRef CAS PubMed.
- X. Yang, N. Li, X. Lin, X. Pan and Y. Zhou, J. Agric. Food Chem., 2016, 64, 8379–8387 CrossRef CAS PubMed.
- R. Rinaldi, Joule, 2017, 1, 427–428 CrossRef CAS.
- Q. Mei, H. Liu, X. Shen, Q. Meng, H. Liu, J. Xiang and B. Han, Angew. Chem., Int. Ed. Engl., 2017, 56, 14868–14872 CrossRef CAS PubMed.
- X. Song, N. Ding, Y. Zai, X. Zeng, Y. Sun, X. Tang, T. Lei and L. Lin, J. Taiwan Inst. Chem. Eng., 2019, 96, 315–320 CrossRef CAS.
- Q. Yang, J. Shi and L. Lin, Energy Fuels, 2012, 26, 6999–7004 CrossRef CAS.
- Y. Jiang, N. Ding, B. Luo, Z. Li, X. Tang, X. Zeng, Y. Sun, S. Liu, T. Lei and L. Lin, ChemCatChem, 2017, 9, 2544–2549 CrossRef CAS.
- Y. Jiang, X. Zeng, R. Luque, X. Tang, Y. Sun, T. Lei, S. Liu and L. Lin, ChemSusChem, 2017, 10, 3982–3993 CrossRef CAS PubMed.
- N. Ding, X. Song, Y. Jiang, B. Luo, X. Zeng, Y. Sun, X. Tang, T. Lei and L. Lin, Sustainable Energy & Fuels, 2018, 2, 2206–2214 RSC.
- A. Björkman, Nature, 1954, 174, 1057–1058 CrossRef.
- J. L. Wen, S. L. Sun, B. L. Xue and R. C. Sun, Materials, 2013, 6, 359–391 CrossRef CAS PubMed.
- D. Argyropoulos, J. Wood Chem. Technol., 1994, 14, 45–63 CrossRef CAS.
- A. S. Jaaskelainen, Y. Sun, D. S. Argyropoulos, T. Tamminen and B. Hortling, Wood Sci. Technol., 2003, 37, 91–102 CrossRef.
- H. Zhang, Y. Bai, B. Yu, X. Liu and F. Chen, Green Chem., 2017, 19, 5152–5162 RSC.
- E. A. Capanema, M. Y. Balakshin and J. F. Kadla, J. Agric. Food Chem., 2004, 52, 1850–1860 CrossRef CAS PubMed.
- A. M. Socha, S. P. Plummer and V. Stavila, Biotechnol. Biofuels, 2013, 6, 61 CrossRef CAS PubMed.
- A. Kloekhorst, Y. Shen, Y. Yie, M. Fang and H. J. Heeres, Biomass Bioenergy, 2015, 80, 147–161 CrossRef CAS.
- G. Morales, S. Pesante and G. Vidal, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2015, 50, 639–645 CAS.
- V. Nogueira, I. Lopes, T. A. P. Rocha-Santos, F. Goncalves and R. Pereira, Environ. Technol., 2018, 39, 1586–1596 CrossRef CAS PubMed.
- M. Huang, J. Luo and Z. Fang, Chem. Ind. For. Prod., 2015, 35, 126–132 CAS.
- A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS.
- O. Faix, Holzforschung - International Journal of the Biology, Chemistry, Physics and Technology of Wood, 1991, 45, 21–28 CAS.
- L. Moghaddam, J. Rencoret, V. R. Maliger, D. W. Rackemann, M. D. Harrison, A. Gutiérrez, J. C. del Río and W. O. S. Doherty, ACS Sustainable Chem. Eng., 2017, 5, 4846–4855 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01225d |
| This journal is © The Royal Society of Chemistry 2020 |