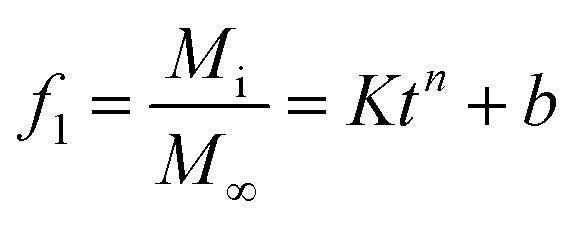DOI:
10.1039/D0RA01215G
(Paper)
RSC Adv., 2020,
10, 13174-13184
Release kinetics study and anti-corrosion behaviour of a pH-responsive ionic liquid-loaded halloysite nanotube-doped epoxy coating†
Received
8th February 2020
, Accepted 4th March 2020
First published on 1st April 2020
Abstract
This study focuses on the release kinetics of inhibitor-loaded nanocontainers and the anti-corrosive properties of epoxy coatings doped and undoped with the nanocontainers. In this work, 1-butyl-3-methylimidazolium chloride [Bmim][Cl] was loaded into halloysite nanotubes (HNTs), and the loaded HNTs were encapsulated with polyethyleneimine (PEI)/polyacrylic acid (PAA) and poly(diallyldimethylammonium chloride) (PDADMAC)/polyacrylic acid (PAA) to allow controlled release upon pH stimuli. The polyelectrolyte layer deposition was characterized using zeta potential analysis, and the release profiles were evaluated in neutral, acidic, and alkaline media. The release kinetics was studied and found to conform to the Ritger–Peppas and Korsmeyer–Peppas model, and the results proved that the combination of weak polyelectrolytes (PEI and PAA) provided a good response for up to 50% release of [Bmim][Cl] in acidic and alkaline media after 72 hours. The loaded HNTs encapsulated with the PEI/PAA combination were incorporated into an epoxy coating matrix and applied on an X52 steel substrate. The corrosion resistance of the coated and uncoated substrates was evaluated using electrochemical impedance spectroscopy (EIS) after immersion in a 3.5 wt% NaCl solution up to 72 hours. An artificial defect was created on the coating prior to immersion to evaluate the active corrosion inhibition ability. The coating doped with the smart pH-responsive halloysite nanotubes showed promising results in corrosion protectiveness even after 72 hours of exposure to a salt solution through EIS and SEM.
Introduction
Protective coatings are generally applied on metallic substrates to prevent direct exposure to weathering and attack from acids, bases, and particulates, which cause corrosion and lead to economic loss. These coatings, however, are susceptible to various degradation processes; this results in coating failure, where defects in the coatings allow the propagation of corrosion agents on the surface of the metallic structure.1 In practice, this damage, especially micron-size cracking, can be challenging to detect and in most cases, repair is almost impossible.2 In industrial applications, corrosion inhibitors are applied directly in the coating matrix to help suppress the corrosion rate, thus prolonging the service life of the coating. However, the direct addition of corrosion inhibitors results in several drawbacks: free inhibitors may react with the elements in the coatings, resulting in coating matrix disruption;3 the inhibitors are susceptible to losses due to washing, which reduces the functionality of the corrosion inhibitors; microporous coatings, which allow the corrosion agents to diffuse through, are potential threats to the environment. One way to prevent these drawbacks is by incorporating self-healing ability into a coating via the intercalation or encapsulation of a corrosion inhibitor in a micro/nano-scaled container.4
Technological advancement has led to the discovery of many types of nanomaterials (nanotubes, nanoribbons, etc.), which can act as miniature reactors for chemical interactions, such as CO2 reduction5–7 and CO2 oxidation;8,9 they are efficient nanocontainers2,10,11 and present the possibility of surface modification (PEMs,12,13 grafting,14etc.) owing to their large available surface areas. Many types of nanocontainers have been explored by researchers to encapsulate inhibitors, such as carbon nanotubes,15 halloysite nanotubes,4,16 polymeric capsules,17,18 and layered double hydroxides.19 Among these, halloysite nanotubes are one of the most cost-effective because they are found naturally, inexpensive, and highly abundant nanostructures with a unique hollow lumen structure that is capable of high loading capacity.20 Halloysite nanotubes possess a unique multilayer structure with a chemical formula of Al2Si2O5(OH)4, where the silica layer is on the exterior and the alumina layer is on the interior. Due to the different compositions of the exterior and interior of the tubular structure, halloysite possesses different surface charges, where the exterior (silica) is negatively charged and the interior (alumina) is positively charged. With this unique property, HNTs can be loaded with various types of functional compounds possessing dissimilar charges and has been proved to provide prolonged release of material. Abdullayev and his coworkers successfully loaded an azole-type corrosion inhibitor into halloysite nanotubes; the HNTs were capable of prolonging release up to 20 to 50 hours.21 Loading of a functional compound into HNTs effectively avoids direct interaction of the functional compound with the coating matrix and external environment while maintaining the coating continuity due to the small size of the HNTs.11
An ionic liquid is a molten salt typically composed entirely of ions that exist in a liquid state below 100 °C.22 It is generally considered to be a green solvent due to advantages such as non-flammable nature, low vapor pressure, and excellent stability over a wide range of temperatures; thus, it an excellent corrosion inhibitor to be used in extreme corrosion conditions.23 A highlight of ionic liquid is that it is a “designable” material, where the chemistry of the cation and anion can be altered to achieve desirable properties. Among the family of ionic liquids, imidazolium compounds have been proved to be effective mixed-type corrosion inhibitors for mild steel due to the presence of –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– groups.24 The anion counterpart of imidazolium ILs serves as an electron donating group which transfers its electrons to the empty d orbitals of iron in mild steel via chemisorption. On the other hand, the positively charged imidazolium structure will be attracted towards the negatively charged steel surface via electrostatic interaction, resulting in physisorption and formation of a protective monolayer.24,25 Due to this interaction, imidazolium-based ILs are highly suitable for inhibition of corrosion in mild steel.26 However, while the ionic liquid is considered to be an effective corrosion inhibitor, it has only been explored to be mixed in chemical process streams such as acidizing treatment and completion fluid in the oil and gas industry, acid cleaning, acid descaling, acid etching, and acid pickling.27 Applying these techniques allows the usage of a lower grade of steel and improves cost-effectiveness. Direct addition of ionic liquid can cause undesirable changes to coating properties; in the case of epoxy resin, the ionic liquid acts as a latent hardener or curing agent, resulting in loss of corrosion inhibition functionality.28,29 Loading of ionic liquids into nanocontainers unlocks the possibility to incorporate them into coating formulations, providing active corrosion protection, enhanced mechanical properties, and increased service life of the coating.
N– groups.24 The anion counterpart of imidazolium ILs serves as an electron donating group which transfers its electrons to the empty d orbitals of iron in mild steel via chemisorption. On the other hand, the positively charged imidazolium structure will be attracted towards the negatively charged steel surface via electrostatic interaction, resulting in physisorption and formation of a protective monolayer.24,25 Due to this interaction, imidazolium-based ILs are highly suitable for inhibition of corrosion in mild steel.26 However, while the ionic liquid is considered to be an effective corrosion inhibitor, it has only been explored to be mixed in chemical process streams such as acidizing treatment and completion fluid in the oil and gas industry, acid cleaning, acid descaling, acid etching, and acid pickling.27 Applying these techniques allows the usage of a lower grade of steel and improves cost-effectiveness. Direct addition of ionic liquid can cause undesirable changes to coating properties; in the case of epoxy resin, the ionic liquid acts as a latent hardener or curing agent, resulting in loss of corrosion inhibition functionality.28,29 Loading of ionic liquids into nanocontainers unlocks the possibility to incorporate them into coating formulations, providing active corrosion protection, enhanced mechanical properties, and increased service life of the coating.
The release of the loaded corrosion inhibitor can be designed based on a desired stimulus, and the release rate can be tuned accordingly with the modification of PEMs, thus achieving smart release with prolonged storage capability. Polyelectrolyte multilayers (PEMs) with defined characteristics and tuneable properties can be fabricated via the layer-by-layer (LBL) technique. The LBL technique enables the self-assembled sequential build-up of polymeric layers via deposition of charged polymers alternating between cationic and anionic onto a charged substrate. The deposition through electrostatic attraction can be applied with a wide variety of building blocks, allowing the incorporation of a broad range of functional units.30 PEMs have been constructed that possess distinctive properties based on their individual polyelectrolytes,30,31 molecular weight,32 salt concentration,33,34 and pH.35 It has also been reported that the ultimate properties of PEMs are highly dependent on the outermost assembled layer.36 PEMs fabricated via the LBL technique can be engineered to respond to various stimuli, such as pH level, humidity, temperature, and conductivity.37 Incorporation of pH-responsive PEMs enables the corrosion inhibitor to be contained without deterioration and release when triggered by local changes in pH at the cathodic and anodic areas of corrosion initiation. Yu et al. proved that the deposition of polyelectrolyte layers on HNTs loaded with 2-mercaptobenthiazole can result in more controlled and sustained release.11
In this research, an imidazolium-based ionic liquid was loaded into HNTs. 1-Butyl-3-methylimidazolium chloride [Bmim][Cl] was used because it has been proven to be an effective mixed-type corrosion inhibitor.38 Surface encapsulation of the loaded HNTs with polyethyleneimine (PEI), polydiallyldimethylammonium chloride (PDADMAC), and polyacrylic acid (PAA) via the LBL technique was conducted to achieve smart release upon pH stimulus. The release was analysed in neutral, acidic, and alkaline mediums to observe the effects of pH on the release profile. Finally, the corrosion inhibition of the synthesized smart pH-responsive halloysite nanotubes was evaluated after they were incorporated in an epoxy matrix and coated on a steel substrate.
Experimental
Materials
Halloysite nanotubes (HNTs) with a molecular weight of 294.19 were purchased from Sigma Aldrich. The corrosion inhibitor (CI) used was 1-butyl-3-methylimidazolium chloride [[Bmim][Cl]] 98% from Acros Organic. Hydrochloric acid, 37%, and sodium hydroxide ACS reagent pellets, ≥97%, were obtained from Sigma Aldrich. Polyethylenimine (PEI) with Mw ∼ 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 by LS and an average Mn ∼ 10000 by GPC, polyacrylic acid (PAA) with average Mw ∼ 1800, and poly(diallyldimethylammonium chloride) (PDADMAC, 35 wt%) in H2O with Mw < 100000 were purchased from Sigma Aldrich and were used as surface encapsulation materials for the HNTs. The coating materials used were Epolam 2015 resin (bisphenol-A-epichlorohydrin, MV < 700) and Epolam 2014 hardener. All materials were used without further purification.
000 by LS and an average Mn ∼ 10000 by GPC, polyacrylic acid (PAA) with average Mw ∼ 1800, and poly(diallyldimethylammonium chloride) (PDADMAC, 35 wt%) in H2O with Mw < 100000 were purchased from Sigma Aldrich and were used as surface encapsulation materials for the HNTs. The coating materials used were Epolam 2015 resin (bisphenol-A-epichlorohydrin, MV < 700) and Epolam 2014 hardener. All materials were used without further purification.
Loading of [Bmim][Cl] into HNTs
As shown in Scheme 1, 1-butyl-3-methylimidazolium chloride [Bmim][Cl] solution was prepared by a adding minimal amount of water to [Bmim][Cl] (1:12 in wt%) to create a saturated solution. Halloysite nanotubes (HNTs) were added to the solution and subjected to sonication for 30 minutes. The solution was transferred to a vacuum jar to be subjected to vacuum conditions for 6 hours. The solution was reverted to atmospheric pressure after every hour up to 6 cycles. Finally, the mixture was centrifuged at 5000 rpm for 5 minutes, washed with water and dried in an oven at 50 °C.
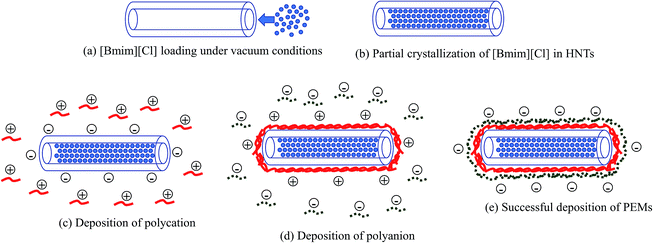 |
| | Scheme 1 Schematic of (a and b) the loading process of [Bmim][Cl] into the HNTs and (c–e) polyelectrolyte deposition via the LBL technique. | |
Characterization of HNT–[Bmim][Cl]
The surface morphology and inner lumen of the halloysite nanotubes (HNTs) were analyzed via field emission scanning electron microscope (FESEM 55VP, Zeiss supra) and transmission electron microscope (Zeiss Libra 200). Structural analysis of HNT was confirmed using Fourier transform infrared (FTIR) spectroscopy (Spectrum One, PerkinElmer). The loading efficiency of the corrosion inhibitor in the HNTs was characterized using a thermogravimetric analyzer (TGA) from 0 °C to 600 °C at 10 °C min−1 under nitrogen atmosphere (STA 6000 from PerkinElmer).
Surface encapsulation of HNT–[Bmim][Cl]
Polyethyleneimine (PEI), polydiallyldimethylammonium chloride (PDADMAC) and polyacrylic acid (PAA) solutions were prepared at 5 mg ml−1 concentration in 0.5 M aqueous NaCl. HNT–[Bmim][Cl] was mixed with a polycation, either PEI or PDADMAC solution, and the incubation time was 10 minutes. Each mixture was then subjected to centrifugation at 3000 rpm to separate the nanoparticles from the polyelectrolyte solution. The resulting nanoparticles were washed with water to remove the unreacted polyelectrolyte molecules. The process was repeated with PAA for subsequent layer deposition.
Release kinetics study
The release kinetics of HNT–[Bmim][Cl] was measured via UV-Vis spectroscopy (UV-1800 Shimadzu Scientific Instruments). 25 mg of sample were suspended in 100 ml of water adjusted to a pH value of 4.0, 7.0, or 10 using 1 M HCl and NaOH. The suspension of water and HNT–[Bmim][Cl] was constantly stirred with a magnetic stirrer at 360 rpm to achieve equilibrium of the mixture. 2 ml of the suspension was collected at several intervals up to 72 hours and replaced with the same volume of distilled solution. The collected samples were analyzed by UV-Vis spectroscopy by monitoring the absorption peak at 211 nm to determine the amount of [Bmim][Cl] released. Finally, the samples were subjected to sonication for 30 minutes to achieve total release of the loaded content and were measured.
The release profiles of all the samples were fitted using the Ritger–Peppas and Korsmeyer–Peppas model (power law)39 with a modification of the initial drug release (burst effect) proposed by Kim and Fassihi:40
| | 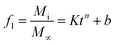 | (1) |
where
f1 represents the released amount of loaded content,
M∞ represents the amount of loaded content at equilibrium,
Mi represents the released amount of loaded content at time
t, and
K represents the release rate constant, taking into consideration the structural modification; the geometrical characteristic
n represents the exponent of release, indicating the release mechanism, and
b represents the burst effect.
Corrosion studies
Preparation of substrates for corrosion tests.
X52 mild steels were used for electrochemical impedance spectroscopy (EIS). Epoxy-moulded rectangular steel samples with a single side surface area of 6 cm2 were prepared. Prior to the conducting tests, surface finishing was performed on the steel samples by mechanical grinding with silicon carbide (SiC) paper with grades of 80, 100, 320, 600 and 1000 to ensure even surfaces and lack of corrosion products. The steel samples were then rinsed with water, degreased with acetone and finally air-dried. Epoxy resin and hardener were mixed in 100:32 ratios, stirred well and subjected to vacuum conditions for up to 30 minutes to eliminate bubbles prior to coating the steel surface. 3 different samples were prepared: bare steel (referred to as BS), steel coated with epoxy (referred to as SE) and steel coated with epoxy with the addition of 5 wt% HNT–[Bmim][Cl] (PEI, PAA) selected from the release kinetics study (referred to as SH).
Electrochemical impedance spectroscopy (EIS).
The corrosion mitigation performance was evaluated using electrochemical impedance spectroscopy (EIS) (WonaTech WEIS510). The test was conducted over a frequency range of 100 kHz to 0.01 Hz using an AC signal of 10 mV amplitude in a conventional three-electrode electrochemical cell which included a counter electrode made of graphite, a reference electrode of Ag/AgCl with saturated KCl aqueous solution, and the steel sample as the working electrode. Prior to the initiation of the EIS test, an artificial defect with a length of 0.5 cm was created on the SE and SH samples with a cutter. The test was performed at room temperature, and EIS readings were obtained after 1, 24, and 72 hours of immersion in 3.5 wt% NaCl solution. Scanning electron microscope (SEM) (ZEISS EVO LS15) and energy dispersive X-ray spectroscopy (EDS) analysis were conducted on the SE and SH samples before and after immersion in salt solution at the artificial defect area to observe changes.
Results and discussion
Blank HNTs characterization
The structural morphology and transmission electron microscopy (TEM) images of the blank HNTs are shown in Fig. 1. The HNTs are composed of bilayers of aluminosilicates with a tubular structure. The outer diameters of the HNTs ranged from 60 to 112 nm, the inner diameters ranged from 15 to 18 nm and the lengths ranged from 393.7 to 814.3 nm. The diameter of the HNTs was in the overall diameter range suggested by Abdullayev and Lvov,10 which varies between 50 and 200 nm, the inner diameter was within the suggested lumen size range of 10–50 nm, and the length was slightly shorter than the suggested overall length range of 0.5–2 microns.
 |
| | Fig. 1 FESEM and TEM images of the blank HNTs. | |
Loading efficiency of [Bmim][Cl] into the HNTs
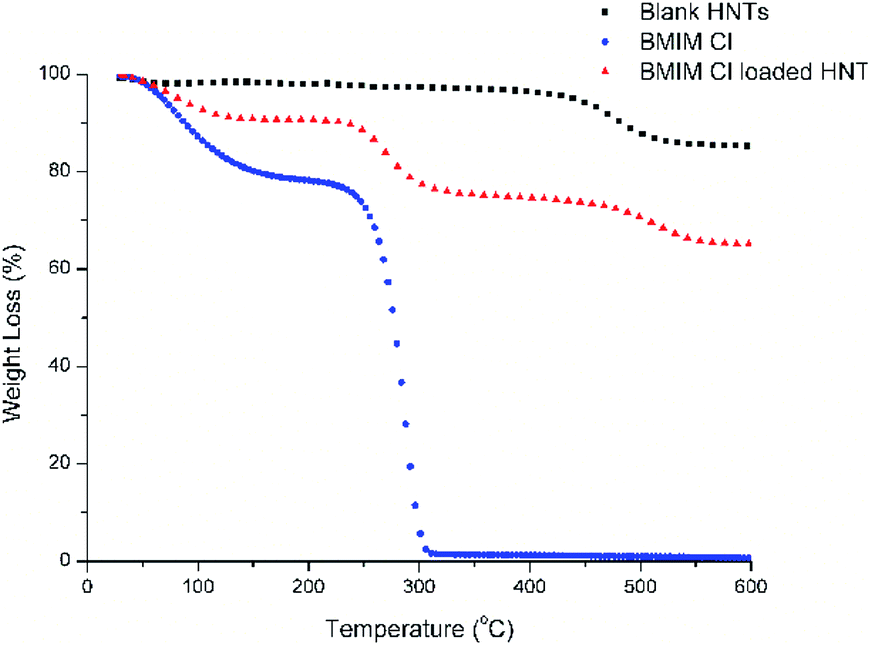
HNTs possess surface charges; therefore, water with a high dielectric constant can stabilize the solution and aid the loading of charged molecules onto the HNTs.20 Slight fizzling of bubbles could be observed immediately after the solution was subjected to vacuum conditions, which indicates that the air trapped within the HNTs was evacuated. The solution was subjected to several cycles of changes in pressure from vacuum conditions back to atmospheric pressure to ensure that the solution enters the HNTs and partial crystallization of the [Bmim][Cl] can take place.10 The thermogravimetry analysis (TGA) results of the blank HNTs, [Bmim][Cl], and HNT–[Bmim][Cl] were obtained and are shown in Fig. 2. From the figure, it can be identified that decomposition of the blank HNTs occurred around 400 °C to 550 °C. The decomposition of [Bmim][Cl], on the other hand, occurred between 231.25 °C and 321.85 °C; this was proved in ref. 41 to begin at 240.3 °C and end at around 317.4 °C. The initial drop of pure [Bmim][Cl] from 0 °C to 100 °C may be due to evaporation of water, mainly due to the hygroscopic nature of [Bmim][Cl], which tends to absorb moisture from the surrounding environment. From the results of HNT–[Bmim][Cl], the presence of the first drop which occurred at around 25 °C to 100 °C indicated weight loss due to water content. The second drop may be due to the decomposition of [Bmim][Cl], which occurred in the range of 231.25–321.85 °C. The loading efficiency obtained was 13.40%. The loading efficiency of [Bmim][Cl] was slightly lower compared to the loading of benzotriazole as reported by Yi He and her coworkers, which was 22.6%;16 however, it falls within the range described by Abdullayev and Lvov.10
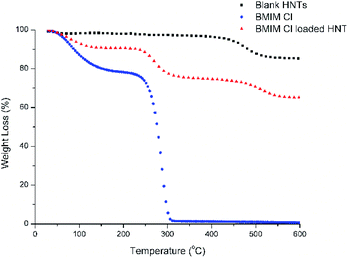 |
| | Fig. 2 TGA analysis of blank HNTs, [Bmim][Cl] and HNT–[Bmim][Cl]. | |
Chemical structure confirmation
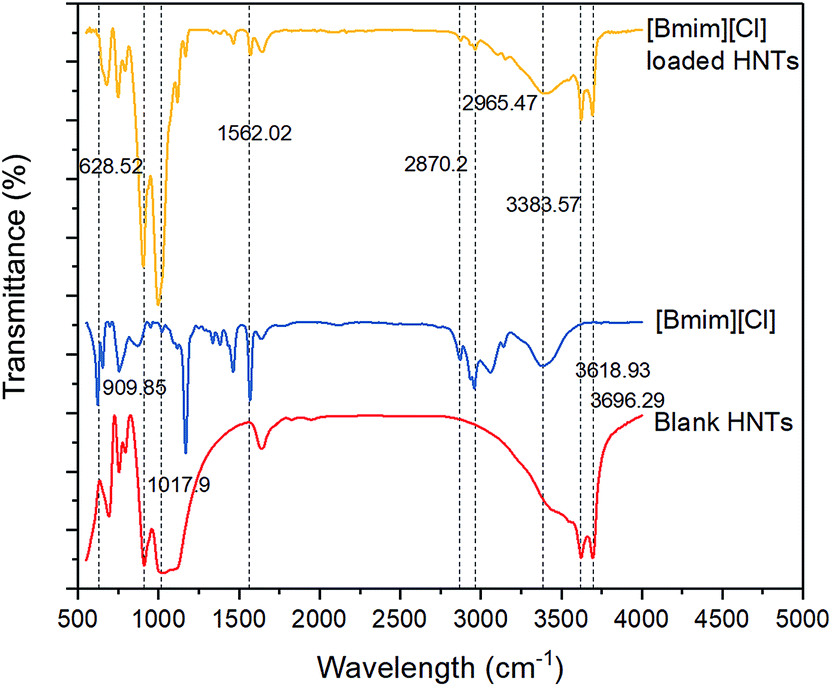
The FTIR spectra of the three different samples, including HNTs, [Bmim][Cl], and HNT–[Bmim][Cl], are illustrated in Fig. 3. In the FTIR spectrum of the HNTs, peaks at 3696.29 cm−1 and 3618.93 cm−1, which signify hydroxyl vibration, were clearly observed. The asymmetrical stretch vibration absorption of Si–O–Si can be attributed to the presence of a peak at 1017.90 cm−1.42 The peak at 909.85 cm−1 accounts for the Al–OH bending band. In terms of the [Bmim][Cl] FTIR spectrum, the peaks at 2965.47 cm−1 and 2870.20 cm−1 represent the aliphatic asymmetric and symmetric C–H stretching of the methyl group.41 A small but broad characteristic peak around 3300 to 3500 cm−1 may be related to the formation of a quaternary amine salt with chlorine. The peak at 1562.02 cm−1 indicates CC stretching. Peaks ranging from 628.52 to 1562.02 cm−1 represent the stretching of C–C and C–N bonds. After loading [Bmim][Cl] into the HNTs, all the characteristic peaks of blank HNTs, such as 3696.29 cm−1, 3618.93 cm−1 and 1017.90 cm−1, and the characteristic peaks of [Bmim][Cl], such as 2965.47 cm−1, 2870.20 cm−1 and 3300 to 3500 cm−1, can be seen clearly. This phenomenon confirms the existence of both HNTs and [Bmim][Cl], and the loading process occurred via physical diffusion without involving chemical bonding.
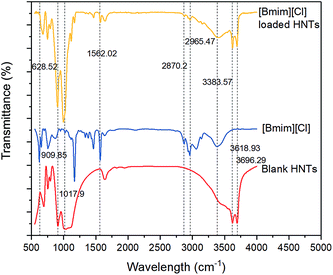 |
| | Fig. 3 FTIR spectra of the HNTs, [Bmim][Cl], and HNT–[Bmim][Cl]. | |
Confirmation of surface encapsulation
The polyelectrolyte multilayer encapsulation was performed using the layer-by-layer (LBL) electrostatic assembly technique. Hence, to confirm the deposition of layers, zeta potential analysis was performed because this technique allows determination of electrical charges on the surface, enabling the monitoring of stepwise growth of the PEMs.43Table 1 depicts the zeta potentials of blank HNTs and HNTs after deposition of two weak polyelectrolyte layers (polyethyleneimine/polyacrylic acid) and strong/weak polyelectrolyte layers (polydiallyldimethylammonium chloride/polyacrylic acid). The halloysite nanotubes possessed an initial charge of −32.4 mV. In PEM formation, the deposition of polyelectrolyte will always result in neutralization of the surface charge of the outermost layer, while the overcompensation of charge becomes the driving force for subsequent deposition of the alternating polyelectrolyte.30,44 The zeta potential reversed to −11.39 mV after deposition of the second PAA layer. These zeta potential values were similar to results reported by Abdullayev and his coworkers, who observed a range from +15 to 32 mV for the first layer of PEI deposition but a slightly lower range for the PAA layer of around −15 to −30 mV.12 After deposition of the strong polycation PDADMAC onto the HNTs, it was observed that the initial surface charge of the HNTs of −32.46 mV was reversed and reached 33.30 mV. Further deposition of a PAA layer reversed the charge to −15.56 mV. All these results showed successful deposition of the polyelectrolyte layer for neutralization and that overcompensation of the surface charge occurred after each layer deposition, as indicated by the positive and negative zeta potential values.
Table 1 Zeta potentials of layer-by-layer deposition on the HNTs
| Sample |
Zeta potential (mV) |
| Blank HNTs |
−32.47 |
| HNT–[Bmim][Cl] (PEI) |
+28.93 |
| HNT–[Bmim][Cl] (PEI, PAA) |
−11.39 |
| HNT–[Bmim][Cl] (PDA) |
+33.30 |
| HNT–[Bmim][Cl] (PDA, PAA) |
−15.57 |
Release profile of [Bmim][Cl]
The release profiles of [Bmim][Cl] in acidic (pH 4), neutral (pH 7) and alkaline (pH 10) mediums from unencapsulated HNT–[Bmim][Cl] are displayed in Fig. 4. From (Fig. 4), it can be observed that the initial release of 50–55% occurred within the first 10 minutes for all three mediums. Xing et al. reported the same situation, where complete release occurred within seconds due to the fast dissolution of loaded content stored on the surface and within the HNTs.31,45. After 72 hours, the ultimate release of all three mediums was achieved between 80% and 90% in neutral (86.50%), pH 4 (83.18%) and pH 10 (89.59%) mediums. The ultimate release of [Bmim][Cl] in alkaline medium is slightly higher compared to the other two mediums; this is mainly because the surface charges of the exterior are negatively charged above pH 1.5, while the interior has a positive charge of up to pH 8.5.10 When exposed to alkaline medium, an abundance of OH− ion neutralizes the interior surface charge, which can affect the release rate of the [Bmim][Cl] adsorbed on the surface of the HNTs. This phenomenon, however, increases the release only slightly compared to neutral medium and acidic medium, mainly due to the small and negligible amount of [Bmim][Cl] that remained adsorbed on the interior surface of the HNTs after 72 hours.
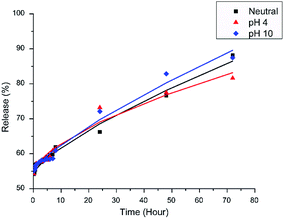 |
| | Fig. 4 The release profile of [Bmim][Cl] from unencapsulated HNT–[Bmim][Cl] subjected to neutral, acidic and alkaline medium for up to 72 hours (black squares represent neutral medium, red triangles represent pH 4 medium, and blue diamonds represent pH 10 medium). | |
In this research, planar thin films are assumed because the HNTs possess openings at both ends of the tubes, while the radial surface is impenetrable. The Ritger–Peppas and Korsmeyer–Peppas model adequately describes the mass transport phenomena of a porous polymer system containing active compounds. In this research, the polyelectrolyte layer forms a hydrophilic slab; exposure of the PEMs to water causes several changes, including changes in mobility and relaxation of macromolecular chains, leading to changes in the shape and size of the pore distribution.
The fitted curves and the corresponding parameters are displayed in Fig. 5 and Table 2. The release of [Bmim][Cl] with n < 0.5 indicates that the release was controlled by its low diffusivity in the polyelectrolyte layer as the inner content swells. From Table 2, most of the samples fall within this category, mainly because the encapsulation restricted the release of [Bmim][Cl]. The release exponent of n = 0.5 indicates Fickian diffusion (Case I), where the release of inner content is governed by diffusion across the concentration gradient. The release exponent within the range of 0.5 < n < 1 indicates that the release of [Bmim][Cl] is controlled by the viscoelasticity of the relaxation of polyelectrolyte layers during solvent penetration, causing the anomalous time-dependent effect. Lastly, n = 1 indicates that the release follows zero-order kinetics, in which the [Bmim][Cl] was released at a constant rate.
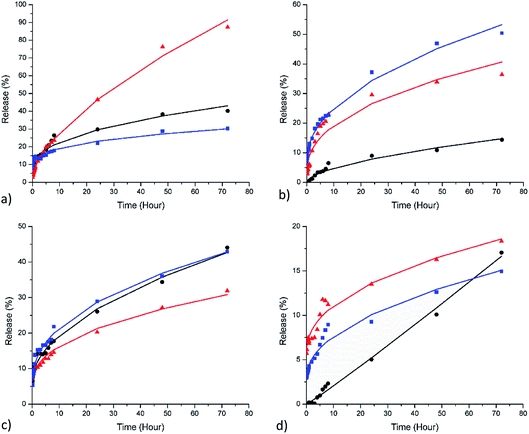 |
| | Fig. 5 The release profiles of [Bmim][Cl] from (a) HNT–[Bmim][Cl] (PEI), (b) HNT–[Bmim][Cl] (PEI, PAA), (c) HNT–[Bmim][Cl] (PDA), and (d) HNT–[Bmim][Cl] (PDA, PAA) subjected to neutral, acidic and alkaline medium for up to 72 hours (black circles represent neutral medium, red triangles represent pH 4 medium, and blue squares represent pH 10 medium). | |
Table 2 Parameters of the curves fitted with the Ritger–Peppas and Korsmeyer–Peppas model (power law), where n represents the release exponent (release mechanism) and k represents the release rate constant
| Sample |
Exposed medium |
n
|
k
|
R
2
|
| HNT–[Bmim][Cl] |
Neutral |
0.74 |
1.39 |
0.9863 |
| HNT–[Bmim][Cl] |
pH 4 |
0.61 |
2.15 |
0.9718 |
| HNT–[Bmim][Cl] |
pH 10 |
0.78 |
1.24 |
0.9827 |
| HNT–[Bmim][Cl] (PEI) |
Neutral |
0.42 |
6.01 |
0.9463 |
| HNT–[Bmim][Cl] (PEI) |
pH 4 |
0.64 |
5.72 |
0.9956 |
| HNT–[Bmim][Cl] (PEI) |
pH 10 |
0.34 |
5.09 |
0.9481 |
| HNT–[Bmim][Cl] (PEI, PAA) |
Neutral |
0.57 |
1.28 |
0.9572 |
| HNT–[Bmim][Cl] (PEI, PAA) |
pH 4 |
0.42 |
6.23 |
0.9290 |
| HNT–[Bmim][Cl] (PEI, PAA) |
pH 10 |
0.48 |
5.95 |
0.9895 |
| HNT–[Bmim][Cl] (PDA) |
Neutral |
0.52 |
3.89 |
0.9901 |
| HNT–[Bmim][Cl] (PDA) |
pH 4 |
0.43 |
4.15 |
0.9767 |
| HNT–[Bmim][Cl] (PDA) |
pH 10 |
0.43 |
5.97 |
0.9787 |
| HNT–[Bmim][Cl] (PDA, PAA) |
Neutral |
1.06 |
0.18 |
0.9940 |
| HNT–[Bmim][Cl] (PDA, PAA) |
pH 4 |
0.44 |
1.94 |
0.9527 |
| HNT–[Bmim][Cl] (PDA, PAA) |
pH 10 |
0.48 |
1.54 |
0.9596 |
In the HNT–[Bmim][Cl] (PEI) sample, PEI is a weak acid; therefore, pH changes significantly affect the porosity of the layer. PEI is an attractive cationic polyelectrolyte that is often used for nanoparticle engineering because of its weak-base buffering properties.46 In acidic medium, PEI has greater positive charge density owing to the excess H+ ion protonating the secondary and tertiary amines in the backbone.47 The highly charged PEI displays high intrachain repulsion, resulting in more linear backbone extension.46 When the HNTs encapsulated with PEI were exposed to acidic medium, the highly charged PEI with more linear expansion resulted in a more porous polycationic layer, thus allowing the [Bmim][Cl] with small molecular size to escape. This phenomenon can be observed by the change in the release exponent from 0.42 in neutral medium to 0.64 in the acidic medium of HNT–[Bmim][Cl] (PEI). In alkaline medium, PEI experiences deprotonation due to the lack of H+ ions and loses charge, resulting in low intrachain repulsion; this eventually causes aggregation. The shrinkage of PEI can reduce the permeation of loaded content, resulting in an even smaller release exponent of 0.34. The difference in the release mechanism resulted in very different ultimate releases of 43.10% (neutral), 95.99% (pH 4), and 30.15% (pH 10) after 72 hours, although the release rate constants for all three conditions are similar. With the addition of the PAA layer to the HNT–[Bmim][Cl] (PEI, PAA) sample, the responsiveness of its release in both acidic and alkaline mediums is more pronounced. PAA is a weak polyanion with increasing charge at an elevated pH level. When the pH drops, PAA tends to shrink and prevent PEI from further expansion, restricting the pathway for the loaded content to pass through. On the other hand, PAA is highly charged, resulting in a more linear expansion in the alkaline medium. PAA also possesses unique characteristics of capacity to absorb water.48 When exposed to alkaline medium, the PAA layers swell due to the highly charged linear expansion and absorption of water. This phenomenon causes Fickian diffusion of [Bmim][Cl] in neutral medium, with an n value equal to 0.57; here, the partially charged PEI and PAA act as porous watery shells that enable diffusion based on concentration. Subjecting the material to acidic and alkaline medium causes the release exponent to fall below 0.5. However, the release rate constants in acidic medium and alkaline medium differ considerably from that in neutral medium, namely 6.23 (pH 4), 5.95 (pH 10) and 1.28 (neutral); this is directly reflected in the actual ultimate releases of 14.74% (neutral), 40.66% (pH 4) and 53.27% (pH 10).
On the other hand, the strong/weak combination of PDADMAC and PAA, in which PDADMAC is a strong polycation, will show a lower response to pH changes.31 The deposition of a single layer of PDADMAC in the HNT–[Bmim][Cl] (PDA) sample showed a release exponent of 0.52; this exhibits release near to Fickian diffusion, where the release depends on the concentration gradient. Exposure to both acidic and alkaline medium changes the release mechanism to 0.43, which exhibits restricted release, while the inner content swells with solvent penetration. The ultimate release of HNT–[Bmim][Cl] (PDA) reached 42.70% (neutral), 30.89% (pH 4) and 42.86% (pH 10). The addition of another layer of PAA further restricts the release of [Bmim][Cl]. This effect can be seen in HNT–[Bmim][Cl] (PDA, PAA) subjected to neutral medium, where the release exponent was 1.06 but the release rate constant was only 0.18. The encapsulation of PDADMAC and PAA creates a porous watery outer shell caused by high association of water molecules with the carboxylate groups; this allows constant rate diffusion but a significantly low release rate due to restriction by the organic environment of PDADMAC, which does not favour water.31 When subjected to acidic medium, the restriction increases as the PAA layer loses charge and shrinks, resulting in a release exponent of only 0.44. In alkaline medium, the PAA layer swells, causing the release to be near to Fickian diffusion. All three mediums afforded similar ultimate releases of 16.65% (neutral), 18.60% (pH 4) and 15.07% (pH 10) at 72 hours.
Corrosion initiation or an incoming external solution can cause pH changes to affect the stability of a complex, triggering release of the inner content.49 To develop an effective smart responsive nanocontainer containing a corrosion inhibitor, the PEMs should be tuned to release when triggered by pH changes while remaining stable in neutral medium. From the release kinetics of the samples, it can be observed that the weak polyelectrolyte displayed high response towards pH changes, while the strong polyelectrolyte retained its stability throughout the pH variation. Responsiveness towards either acidic or alkaline medium is highly dependent on the types of polyelectrolytes deposited and their configuration. This effect can be seen clearly in the HNT–[Bmim][Cl] (PEI) sample, where the loaded HNTs were deposited with single layer PEI. Due to the nature of PEI, the release responsiveness of the layer favoured acidic medium while providing a lower release rate in alkaline medium. The HNT–[Bmim][Cl] (PEI, PAA) sample appears to be an excellent candidate as an effective smart-responsive nanocontainer because it can respond to both acidic and alkali mediums; it was selected for evaluation of its anti-corrosion performance.
Electrochemical impedance spectroscopy (EIS)
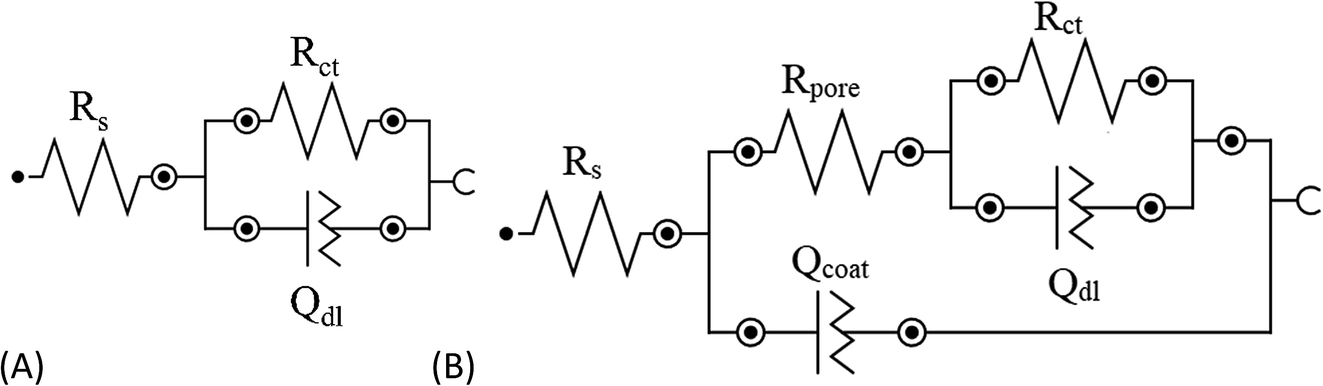
To evaluate the anticorrosion behavior of the selected SH particles, electrochemical impedance spectroscopy (EIS) was conducted on steel with and without the presence of HNT–[Bmim][Cl] (PEI, PAA). The obtained data for all the samples were fitted using several different equivalent circuit models (ECMs), as shown in Fig. 6.
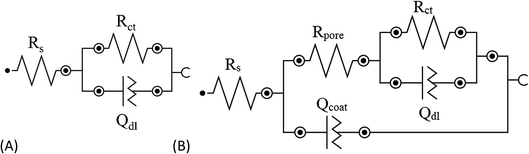 |
| | Fig. 6 (A) Simple randles circuit (R(RQ)) for the BS sample; (B) Randles circuit (R(R(RQ)Q)) for the SE and SH samples. | |
For the bare steel (BS) sample, a simple Randles circuit of R(QR) was employed which involves the resistance of the solution (Rs), double layer capacitance (Qdl) and charge transfer resistance (Rct). The Nyquist diagram did not show the perfect semicircle of a pure capacitor, which can be attributed to two reasons: frequency dispersion due to interfacial porosity or roughness and the kinetic dispersion effect.50,51 Hence, to model the imperfect capacitor, the constant phase element (Q) was preferred. The impedance of the constant phase element can be computed from the formula below:
| | 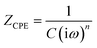 | (2) |
where
C indicates the capacitance,
ω indicates the frequency, and
n indicates the frequency dispersion parameter resulting from the surface morphology of the electrode.
For steel coated with bare epoxy (SE) and epoxy with the addition of smart pH-responsive halloysite nanotubes loaded with the [Bmim][Cl] (SH) sample, a Randles circuit ECM of R(R(QR)Q) was used to fit the data, as shown in Fig. 6. In the high-frequency range, the electric current was distributed uniformly throughout the coating area, presenting as a semicircle fitted with a parallel circuit (Rpore, Qcoat), corresponding to the pore resistance and dielectric behavior of the coating. When the frequency range moves to the lower range, the electrical current is focused at the active corrosion site, which occurs where the artificial defect was created. This phenomenon was fitted with another parallel circuit of (Rct, Qdl) in series with Rpore, corresponding to charge transfer resistance and double layer capacitance, respectively (Fig. 6).
From the analysis of the Nyquist plot as shown in Fig. 7 and Table 3, the bare steel (BS) sample showed an increasing trend in Rct from 129.78 Ω cm−2 at the 1st hour of exposure to 201.07 Ω cm−2 after 72 hours of exposure owing to the formation of stable oxide films as a result of corrosion, which offers slight resistance towards the propagation of the current. The decrease of the Rct value from 24 hours to 72 hours may be due to detachment of the thick accumulation of oxide layer. When an artificial defect was created on the epoxy-coated mild steel sample, ions in the surrounding solution came into contact with the metal substrate, leading to corrosion initiation without hindrance. The formation of thick oxide layers, as shown in the SEM images in Fig. 8b, provides a certain limitation of the corrosion at the defective coating area, leading to an increase in charge transfer resistance from 7337 Ω cm−2 at the 1st hour of exposure to 12677 Ω cm−2 after 72 hours of exposure. When the coating continuity of the SH sample was damaged due to the artificially induced crack, the embedded SH nanoparticles exposed to the surrounding environment experienced pH changes due to localized corrosion. The inner content was released as the polyelectrolyte layer experienced changes due to pH changes in the surroundings. The released [Bmim][Cl] was able to undergo both chemisorption and physisorption onto the negatively charged mild steel surface, thus forming a protective layer that provided a significantly higher charge transfer resistance of 170210 Ω cm−2 at the 1st hour of exposure.25 The charge transfer resistance further increased to 310760 Ω cm−2 at 72 hours of exposure with the continuous release of [Bmim][Cl] from the embedded SH nanoparticles. The Rpore values in the SE and SH samples showed a decreasing trend with increasing immersion time. As the immersion time increases up to 72 hours, the decreasing trend of Rpore may be caused by the increase in the exposed area.52 This may be due to propagation of the corrosion site, which leads to slight delamination of the coating. The decrease of Rpore, however, could be compensated with the increasing Rct as a result of the inhibition effect of [Bmim][Cl]. Comparing the SEM images from Fig. 8 for both the SE and SH samples after immersion, one can observe a distinct visual difference in the amount of corrosion product above the metal substrate. This phenomenon was in agreement with the EDS results as shown in Table 4, where the oxygen weight percent of the SE sample increased from 0.5 wt% to 21.2 wt% and that of the SH sample increased from 1.2 wt% to 10 wt%. The presence of nitrogen and chloride in the scratched area of the SH sample proved the release of [Bmim][Cl] from the nanotubes, and the inhibition of [Bmim][Cl] results in lower formation of corrosion product, as indicated by the oxygen weight percentage.
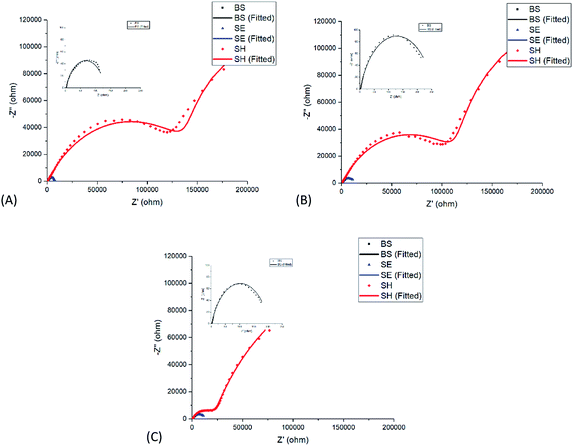 |
| | Fig. 7 Nyquist plots for the SE and SH samples after (A) 1 h, (B) 24 h, and (C) 72 h. | |
Table 3 Values of the fitted ECMs for the BS, SE and SH samples
| Sample |
Immersion time (Hr) |
R
s (Ω cm2) |
R
pore (Ω cm2) |
R
ct (Ω cm2) |
| BS |
1 |
2.9 |
— |
129.78 |
| 24 |
2.43 |
— |
252.73 |
| 72 |
2.19 |
— |
201.07 |
| SE |
1 |
105.03 |
1035.5 |
7337 |
| 24 |
105.31 |
858.4 |
12921 |
| 72 |
114.15 |
978.15 |
12677 |
| SH |
1 |
407.11 |
163290 |
170210 |
| 24 |
456.38 |
132220 |
222460 |
| 72 |
550.84 |
23148 |
310760 |
 |
| | Fig. 8 SEM images of SE (a) before and (b) after and of SH (c) before and (d) after 72 hours of immersion in 3.5 wt% NaCl solution. | |
Table 4 EDS analysis of SE and SH samples before and after 72 hours of immersion in 3.5 wt% NaCl solution
| Sample |
Immersion |
% weight percent ratio of elements |
| Fe |
C |
O |
N |
Cl |
Na |
| SE |
Before |
90.8 |
8.3 |
0.5 |
— |
— |
— |
| After |
70.9 |
7 |
21.2 |
— |
0.4 |
0.4 |
| SH |
Before |
83.3 |
14.9 |
1.2 |
— |
— |
— |
| After |
79.2 |
8.5 |
10 |
0.8 |
0.2 |
0.1 |
Conclusions
[Bmim][Cl] was successfully loaded into HNTs with a loading efficiency of 13.40 wt%, as confirmed using TGA. HNT–[Bmim][Cl] was characterized by FTIR, which confirmed that no chemical interaction of [Bmim][Cl] with the chemical structure of the HNTs occurred. Zeta potential analysis proved the successful deposition of polyelectrolyte. The release profile of HNT–[Bmim][Cl] was studied via UV-Vis, and it was found that the initial release of 50–55% occurred within the first 10 minutes and achieved an ultimate release of approximately 90%. Loading of [Bmim][Cl] into the HNTs provides extended release over 72 hours compared to direct addition into the coating matrix, which causes immediate release. When the loaded HNTs were encapsulated with PEMs, the release of [Bmim][Cl] was controlled and displayed pH responsiveness in its release profile. A weak polyelectrolyte provides rapid response towards pH variation, while a strong polyelectrolyte is more resistant and stable to pH variation. The PEI/PAA multilayers are an excellent candidate for effective smart pH-responsive encapsulation of corrosion inhibitor-loaded nanocontainers. The addition of the smart pH-responsive halloysite nanotubes to the epoxy coating could provide responsive release of [Bmim][Cl] when an artificial defect was induced on the coating surface and significantly increased corrosion protection of the steel, as confirmed by the EIS corrosion study and SEM. Therefore, this study provided a novel encapsulation method of [Bmim][Cl] and produced smart pH-responsive nanoparticles that can be readily incorporated into an epoxy matrix for combined passive and active corrosion protection of mild steel.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Great appreciation is offered to Universiti Teknologi PETRONAS for the facilities and research fund, Yayasan Universiti Teknologi PETRONAS (YUTP) 0153AA-H34.
References
- M. Zheludkevich, J. Tedim and M. Ferreira, Electrochim. Acta, 2012, 82, 314–323 CrossRef CAS.
- K. A. Zahidah, S. Kakooei, M. C. Ismail and P. B. Raja, Prog. Org. Coat., 2017, 111, 175–185 CrossRef CAS.
- E. Abdullayev, R. Price, D. Shchukin and Y. Lvov, ACS Appl. Mater. Interfaces, 2009, 1, 1437–1443 CrossRef CAS PubMed.
- E. Shchukina, D. Shchukin and D. Grigoriev, Prog. Org. Coat., 2017, 102, 60–65 CrossRef CAS.
- J.-H. Liu, L.-M. Yang and E. Ganz, ACS Sustainable Chem. Eng., 2018, 6, 15494–15502 CrossRef CAS.
- J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 3805–3814 RSC.
- J.-H. Liu, L.-M. Yang and E. Ganz, RSC Adv., 2019, 9, 27710–27719 RSC.
- L. Xu, L.-M. Yang and E. Ganz, Theor. Chem. Acc., 2018, 137, 98 Search PubMed.
- Y. Liu, L.-M. Yang and E. Ganz, Condens. Matter, 2019, 4, 65 Search PubMed.
- E. Abdullayev and Y. Lvov, J. Nanosci. Nanotechnol., 2011, 11, 10007–10026 CrossRef CAS PubMed.
- D. Yu, J. Wang, W. Hu and R. Guo, Mater. Des., 2017, 129, 103–110 CrossRef CAS.
- E. Abdullayev, D. Shchukin and Y. Lvov, Mater. Sci. Eng., 2008, 99, 331–332 CAS.
- S. C. Olugebefola, A. R. Hamilton, D. J. Fairfield, N. R. Sottos and S. R. White, Soft Matter, 2013, 10, 544–548 RSC.
- J. Zhang, D. Zhang, A. Zhang, Z. Jia and D. Jia, Iran. Polym. J., 2013, 22, 501–510 CrossRef CAS.
- H. Ye, D. Chen, N. Li, Q. Xu, H. Li, J. He and J. Lu, Environ. Sci.: Nano, 2019, 6, 1259–1266 RSC.
- Y. He, W. Xu, R. Tang, C. Zhang and Q. Yang, RSC Adv., 2015, 5, 90609–90620 RSC.
- H. Choi, Y. K. Song, K. Y. Kim and J. M. Park, Surf. Coat. Technol., 2012, 206, 2354–2362 CrossRef CAS.
- N. Pirhady Tavandashti, M. Ghorbani, A. Shojaei, J. M. C. Mol, H. Terryn, K. Baert and Y. Gonzalez-Garcia, Corros. Sci., 2016, 112, 138–149 CrossRef CAS.
- J. Tedim, M. L. Zheludkevich, A. C. Bastos, A. N. Salak, A. D. Lisenkov and M. G. S. Ferreira, Electrochim. Acta, 2014, 117, 164–171 CrossRef CAS.
- Y. M. Lvov, D. G. Shchukin, H. Mohwald and R. R. Price, ACS Nano, 2008, 2, 814–820 CrossRef CAS PubMed.
- E. Abdullayev, V. Abbasov, A. Tursunbayeva, V. Portnov, H. Ibrahimov, G. Mukhtarova and Y. Lvov, ACS Appl. Mater. Interfaces, 2013, 5, 4464–4471 CrossRef CAS PubMed.
- A. L. Chong, M. Forsyth and D. R. Macfarlane, Electrochim. Acta, 2015, 159, 219–226 CrossRef CAS.
- Y. Guo, B. Xu, Y. Liu, W. Yang, X. Yin, Y. Chen, J. Le and Z. Chen, J. Ind. Eng. Chem., 2017, 56, 234–247 CrossRef CAS.
- Q. B. Zhang and Y. X. Hua, Electrochim. Acta, 2009, 54, 1881–1887 CrossRef CAS.
- S. Velusamy, S. Sakthivel, L. Neelakantan and J. S. Sangwai, J. Earth Sci., 2017, 28, 949–961 CrossRef CAS.
- A. Yousefi, S. Javadian, N. Dalir, J. Kakemam and J. Akbari, RSC Adv., 2015, 5, 11697–11713 RSC.
- C. Verma, E. E. Ebenso and M. A. Quraishi, J. Mol. Liq., 2017, 233, 403–414 CrossRef CAS.
- N. Saurín, J. Sanes and M. Bermúdez, Tribol. Lett., 2015, 58, 4 CrossRef.
- H. Mąka, T. Spychaj and R. Pilawka, eXPRESS Polym. Lett., 2014, 8(10), 723–732 CrossRef.
- E. Maza, J. S. Tuninetti, N. Politakos, W. Knoll, S. Moya and O. Azzaroni, Phys. Chem. Chem. Phys., 2015, 17, 29935–29948 RSC.
- T. Alonso, J. Irigoyen, J. J. Iturri, I. L. Larena and S. E. Moya, Soft Matter, 2013, 9, 1920–1928 RSC.
- S. T. Dubas and J. B. Schlenoff, Macromolecules, 2001, 34, 3736–3740 CrossRef CAS.
- Y. Ma, J. Dong, S. Bhattacharjee, S. Wijeratne, M. L. Bruening and G. L. Baker, Langmuir, 2013, 29, 2946–2954 CrossRef CAS PubMed.
- R. A. Ghostine, M. Z. Markarian and J. B. Schlenoff, J. Am. Chem. Soc., 2013, 135, 7636–7646 CrossRef CAS PubMed.
- P. Bieker and M. Schönhoff, Macromolecules, 2010, 43, 5052–5059 CrossRef CAS.
- S. M. Notley, M. Eriksson and L. Wågberg, J. Colloid Interface Sci., 2005, 292, 29–37 CrossRef CAS PubMed.
- N. Y. Abu-Thabit and A. S. Hamdy, Surf. Coat. Technol., 2016, 303, 406–424 CrossRef CAS.
- Q. Zhang and Y. Hua, Electrochim. Acta, 2009, 54, 1881–1887 CrossRef CAS.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- H. Kim and R. Fassihi, J. Pharm. Sci., 1997, 86, 323–328 CrossRef PubMed.
- S. A. Dharaskar, M. N. Varma, D. Z. Shende, C. K. Yoo and K. L. Wasewar, Sci. World J., 2013, 2013, 9 Search PubMed.
- H. Yu, Y. Zhang, X. Sun, J. Liu and H. Zhang, Chem. Eng. J., 2014, 237, 322–328 CrossRef CAS.
- S. E. Burke and C. J. Barrett, Langmuir, 2003, 19, 3297–3303 CrossRef CAS.
- M. Michel, V. Toniazzo, D. Ruch and V. Ball, ISRN Mater. Sci., 2012, 2012, 701695 Search PubMed.
- X. Xing, J. Wang, Q. Li, W. Hu and J. Yuan, Colloids Surf., A, 2018, 553, 295–304 CrossRef CAS.
- K. A. Curtis, D. Miller, P. Millard, S. Basu, F. Horkay and P. L. Chandran, PLoS One, 2016, 11, e0158147 CrossRef PubMed.
- Y.-C. Li, J. Schulz and J. C. Grunlan, ACS Appl. Mater. Interfaces, 2009, 1, 2338–2347 CrossRef CAS PubMed.
- S. W. Cranford, C. Ortiz and M. J. Buehler, Soft Matter, 2010, 6, 4175–4188 RSC.
- H. Pulikkalparambil, S. Siengchin and J. Parameswaranpillai, Nano-Struct. Nano-Objects, 2018, 16, 381–395 CrossRef CAS.
- L. C. Murulana, A. K. Singh, S. K. Shukla, M. M. Kabanda and E. E. Ebenso, Ind. Eng. Chem. Res., 2012, 51, 13282–13299 CrossRef CAS.
- R. Farahmand, B. Sohrabi, A. Ghaffarinejad and M. R. Z. Meymian, Corros. Sci., 2018, 136, 393–401 CrossRef CAS.
- N. Mat Nor, L. Ismail, S. Jamari, K. Ramesh, B. Vengadaesvaran and A. Arof, Pigm. Resin Technol., 2014, 43, 371–378 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01215g |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Muhammad Rashid
Shamsuddin
*b,
Muhammad Rashid
Shamsuddin