
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMannosylated hemagglutinin peptides bind cyanovirin-N independent of disulfide-bonds in complementary binding sites†
Philipp E. Schillinga,
Georg Kontaxisb,
Martin Dragositsc,
Robert H. Schiestlde,
Christian F. W. Becker a and
Irene Maier*ae
a and
Irene Maier*ae
aFaculty of Chemistry, Institute of Biological Chemistry, University of Vienna, Währinger Straße 38, A-1090 Vienna, Austria
bDepartment of Structural and Computational Biology, Max Perutz Laboratories, University of Vienna, Campus Vienna Bohrgasse 5, A-1030 Vienna, Austria
cDepartment of Chemistry, Division of Biochemistry, University of Natural Resources and Life Sciences, Muthgasse 18, A-1190 Vienna, Austria
dDepartment of Pathology and Laboratory Medicine, Geffen School of Medicine, University of California, Los Angeles, CA-90095, USA
eDepartment of Environmental Health Sciences, Fielding School of Public Health, University of California, Los Angeles, 650 Charles E. Young Dr. South, Los Angeles, CA-90095, USA. E-mail: irene.maier@meduniwien.ac.at; Fax: +1-310-267-2578; Tel: +1-310-267-2087
First published on 17th March 2020
Abstract
Cyanovirin-N (CV-N) has been shown to reveal broad neutralizing activity against human immunodeficiency virus (HIV) and to specifically bind Manα(1→2)Manα units exposed on various glycoproteins of enveloped viruses, such as influenza hemagglutinin (HA) and Ebola glycoprotein. Chemically synthesized dimannosylated HA peptides bound domain-swapped and dimeric CV-N with either four disulfide-bonds (Cys–Cys), or three Cys–Cys bonds and an intact fold of the high-affinity binding site at an equilibrium dissociation constant KD of 10 μM. Cys–Cys mutagenesis with ion-pairing amino-acids glutamic acid and arginine was calculated by in silico structure-based protein design and allowed for recognizing dimannose and dimannosylated peptide binding to low-affinity binding sites (KD ≈ 11 μM for one C58–C73 bond, and binding to dimannosylated peptide). In comparison, binding to HA was achieved based on one ion-pairing C58E–C73R substitution at KD = 275 nM, and KD = 5 μM for two C58E–C73R substitutions. We were utilizing a triazole bioisostere linkage to form the respective mannosylated-derivative on the HA peptide sequence of residues glutamine, glycine, and glutamic acid. Thus, mono- and dimannosylated peptides with N-terminal cysteine facilitated site-specific interactions with HA peptides, mimicking a naturally found N-linked glycosylation site on the HA head domain.
Introduction
CV-N, an 11 kDa lectin originating from Nostoc ellipsosporum shows antiviral activity against HIV and other enveloped viruses.1–3 CV-N variants are more potent anti-HIV agents than plant lectins4 and similar in avidity and nature to broadly neutralizing antiviral antibodies.5 The multivalent binding character of cyanobacterial lectins allows for direct binding to trimeric envelope glycoproteins at low surface density6 and reversible binding with glycoproteins linked to the cell membrane.7–9 High-mannose oligosaccharides are known to bind CV-N based on Man-9-GlcNAc2, with N-acetyl-glucosamine attached to viral surface proteins.10,11 Here, we focused on the preparation and incorporation of chemically linked mono- and dimannose units to propargylglycine to form a side chain derivative on a 12mer peptide, mimicking part of the antiviral active HA head domain H1.12,13 Neutralizing glycan-targeting antibodies and lectins has been studied in terms of stoichiometry of bound molecules7,14 and their interactions with the rapidly modifying glycan shield of influenza A subtypes,15 such as H1N1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 14 and H3N2.12,16
14 and H3N2.12,16
We report kinetic and thermodynamic binding studies on dimeric head-to-tail linked tandem CV-N (CVN2L0) with at least one native high-affinity binding site for oligosaccharides with high-mannose content, that showed enhanced HIV neutralization compared to wild-type CV-N.17 This lectin consists of 101 amino-acids with two repeat units (residues 1–50 and 51–101), bearing four Cys residues that allow two intramolecular disulfide bonds forming a monomer, or an intertwined dimer.18 We recombinantly expressed CVN2L0 and variants with less than four Cys–Cys bridges. Cys58 and Cys73 were mutated to charged residues Glu and Arg in analogy to the glycan-contacting Arg76–Glu41 amino-acid pair, or by amphipathic Trp–Met pairs. Recognition through the Manα(1→2)Manα unit11,19,20 was studied using chemically prepared (di-) mannosylated peptides derived from the HA head domain for binding CV-N and disulfide bridge variants.13,21 We emphasized symmetric replacement of native Cys residues by multi-specific mutations as established for protein–protein interactions before.22 The commonly used docking procedure treats the proteins as rigid bodies and uses the Fourier correlation theorem and the fast Fourier transform to efficiently search for dimers with the highest interfacial surface complementarities.23–25 In the present study, a pair of hydrophilic or amphipathic amino-acids were newly introduced in vicinity to either one, or two carbohydrate binding sites of the domain-swapped dimer CVN2L0. Interactions between polar residues and the resulting helix dipole have been demonstrated to impact the stability of both model peptides and proteins, which cannot be predicted by simple electrostatics in protein design on its own.26,27 Therefore, peptide backbone and the distance (of about the size of asparagine) to a mannosylated glycine-derivative, or the number of free rotamers for computational protein design on CV-N molecule were not modelled together.
However, short peptides were chemically linked with mannose or dimannose and measured for binding affinity to CVN2L0 and variants in surface-bound and in solution interaction studies, applying SPR, ITC and NMR.
Results and discussion
Chemical synthesis of HA-derived mannosylated peptides
We chemically prepared dimannosylated peptide (DM), bearing the Manα(1→2)Manα unit, for which CVN2L0 shows high affinity. The model peptide (P1) was synthesized via SPPS, with a propargylglycine building block for site-specific modification, and by using regioselective copper(I)-catalyzed azide–alkyne cycloaddition with mannosyl-azides (Fig. 1).28 The peptide sequence corresponds to a 12mer peptide of the HA sequence,12 representing a model for the linkage and recognition of the carbohydrate units without the larger protein context. Introducing an azide at C1 of the carbohydrate initiated copper(I)-catalyzed formation of a triazole-linkage between the peptide and the carbohydrate moiety.29 While N-linked glycosylation on asparagine is the naturally occurring modification, triazoles are excellent amide bioisosteres30,31 that have been established as directly accessible bridging moiety in N-linked glycopeptides and N-linked glycoproteins.32 Acetylated methyl-dimannose formation of the disaccharide and introduction of the azide-functionality in the C1-α position made this building block available for CuAAC on the solid support with a yield of 20% over 7 steps57,58 (Fig. S1–S6, ESI†). The exclusive 1,2-trans selectivity, as reported by Salunke et al.,33 for other carbohydrates, was confirmed for the introduction of the azide into dimannose, as NMR analysis indicated the characteristic coupling constant for both α-anomers. Dimannosylated peptide P2 (DM) and monomannosylated peptide P3 (MM) were obtained by the reaction of azido-dimannose and azido-mannose with fully-protected peptide on resin (Fig. S10 and S12, ESI†).34 Subsequent cleavage and global deprotection of these glycans released fully unprotected DM and MM. Purified mannosylated peptides were obtained in overall yields of 10–20% based on the peptide synthesis scale. | ||
| Fig. 1 Synthesis of di- and monomannosylated peptides DM and MM. 1,2-Mannobiose azide 8 or azidomannose 9 were used to perform CuAAC for introducing the carbohydrate moiety to propargylglycine-substituted peptide P1 on resin. Conditions: CuSO4·5H2O, TBTA, sodium ascorbate, DMF/H2O, r.t., 30–40 h, side chains of resin-bound P1 were still protected during CuAAC; DMF/DCM/MeOH wash; resin cleavage, global deprotection (TFA/phenol/water/triisopropylsilyl ether/EDT (82.5/5/5/5/2.5, v/m/v/v/v)), r.t., 3 h, purification, 10% (P2), 20% (P3); DM = dimannosylated peptide P2; MM = monomannosylated peptide P3. | ||
High-affinity carbohydrate binding sites reveal protein folds without disulfide bonds
The concatenated tandem dimer of CV-N (CVN2L0) is a domain-swapped dimer (Fig. 2A) that shows high stability and binding characteristics comparable to the wild-type protein. Contrarily, other variants of CVN2 had to be modified by mutagenesis in the domain-linker region in order to increase monomeric protein formation or to stabilize a dimer in solution.35,36 Pseudo-binding domain A (residues 1–39 of one monomer and 90–101 of the other half-dimer with a disulfide bond between Cys8–Cys22) assigns for low-affinity binding to trimannose or hexamannose. Binding domain B (residues 40–51 of one monomer and 52–89 of the other chain with a Cys58–Cys73 disulfide bond) forms the primary binding site to mannobiose independent of structural differences (Fig. 2B).18,37 Computational protein design using ORBIT (Optimization of Rotamers By Iterative Techniques) software was applied to evaluate CV-N structures for experimental intermolecular protein–peptide binding studies, where 20 possible mutations may employ domain-swapped and folded intermediates of protein conformations.27,38 Calculations guided to possible mutation sites at C58W–C73M and C58E–C73R. One or two disulfide bonds (Cys58-Cys73), in one or both high-affinity domain B structures, were replaced by incorporating Glu58–Arg73 pairs additionally to the existing intermolecular glycan-contacts Glu41–Arg76. On the other hand amphipathic, nonpolar Trp58–Met73 amino-acid residues were inserted to replace the same disulfide bonds; or a combination of both mutation pairs was created (Fig. 2C and D). Thus, the numbers of disulfide bonds were decreased from four (CVN2L0) to three (variant 2) or two (variants 3, 4 and 5), respectively (Fig. S14, ESI†). All variants were expressed in Escherichia coli. Thermal stability of CVN2L0, as well as corresponding melting temperatures (Tm), of its carbohydrate binding site variants (CVN2L0-V2, and V4-5) were assessed by circular dichroism (CD, Table S1†).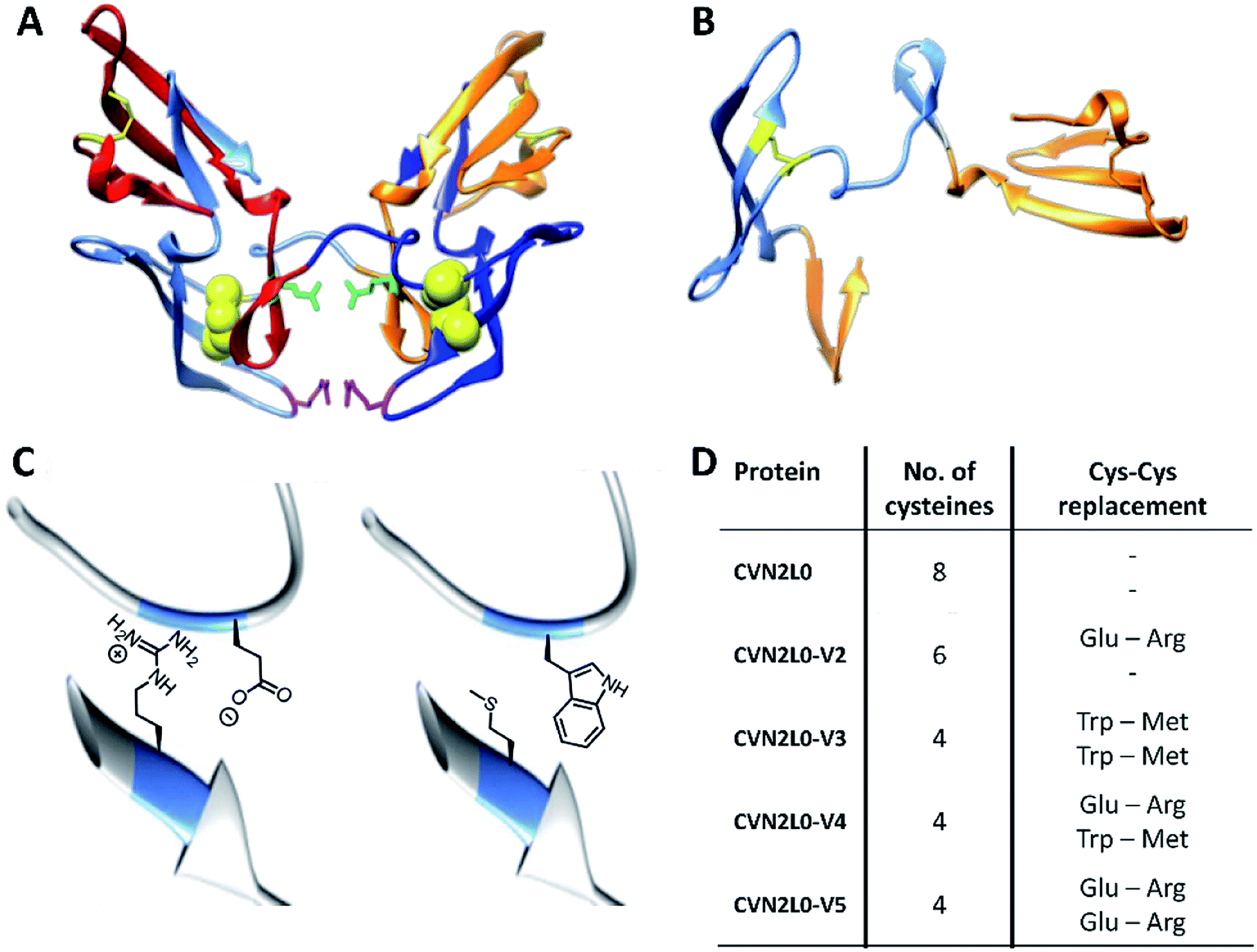 | ||
| Fig. 2 (A) X-ray structures of dimeric CVN2L0 (reproduced from PDB ID: 3EZM). Disulfide bridges highlighted in yellow, mutation sites in disulfide bridges presented as yellow spheres. Glu41 and Arg76 of each domain highlighted in green and magenta, respectively. (B) Structure of domain-swapped monomeric CVN; low-affinity binding domains (residues 1–39, 90–101) and high-affinity binding domain B (residues 40–89). (C) Enlarged view of C58E and C73R mutations; enlarged view of C58W and C73M mutations using chimera protein software. (D) Table of recombinantly expressed CVN2L0 variants V2-5, numbers of Cys in disulfide bridges and replacement of Cys–Cys bonds. | ||
Two functional low-affinity domains and a disulfide bond facilitate CVN2L0 binding to DM
Given conformational selectivity, we investigated whether reduced numbers of disulfide bridges affected CVN2L0's ability to bind immobilized HA full length protein from A/Wisconsin/67/05 strain by SPR. The secondary binding site with 700 ± 50 μM affinity to mannobiose and 3.5 μM affinity to trimannose37,39,40 is located far from the stabilized linker region and formed a semicircular cleft that surrounds the carbohydrate moiety.18 We observed binding of CVN2L0 to HA with nanomolar affinity (KD = 255 nM), and compared binding of CVN2L0 and its variants V2-5 with a total of three, or two disulfide bridges around the glycan pockets, to HA, glycopeptides DM and MM. HA and glycopeptides were immobilized onto different SPR chips; CVN2L0 and CVN2L0 variants V2-5 were injected as analytes. Concentration-dependent association to ligand HA, followed by equilibrium binding and dissociation after terminated injection was measured for binding of CVN2L0, CVN2L0-V2 (-V4 and -V5). The obtained sensorgrams and kinetics data are shown in Fig. 3 and Table S2.† CVN2L0 and CVN2L0-V2 showed nanomolar affinity in binding to the glycoprotein (HA), which compares with CV-N's HIV neutralization activity.17 CVN2L0-V4 and -V5, bearing two Cys–Cys bond mutations, however, showed low micromolar affinity to HA (Fig. 3A–D).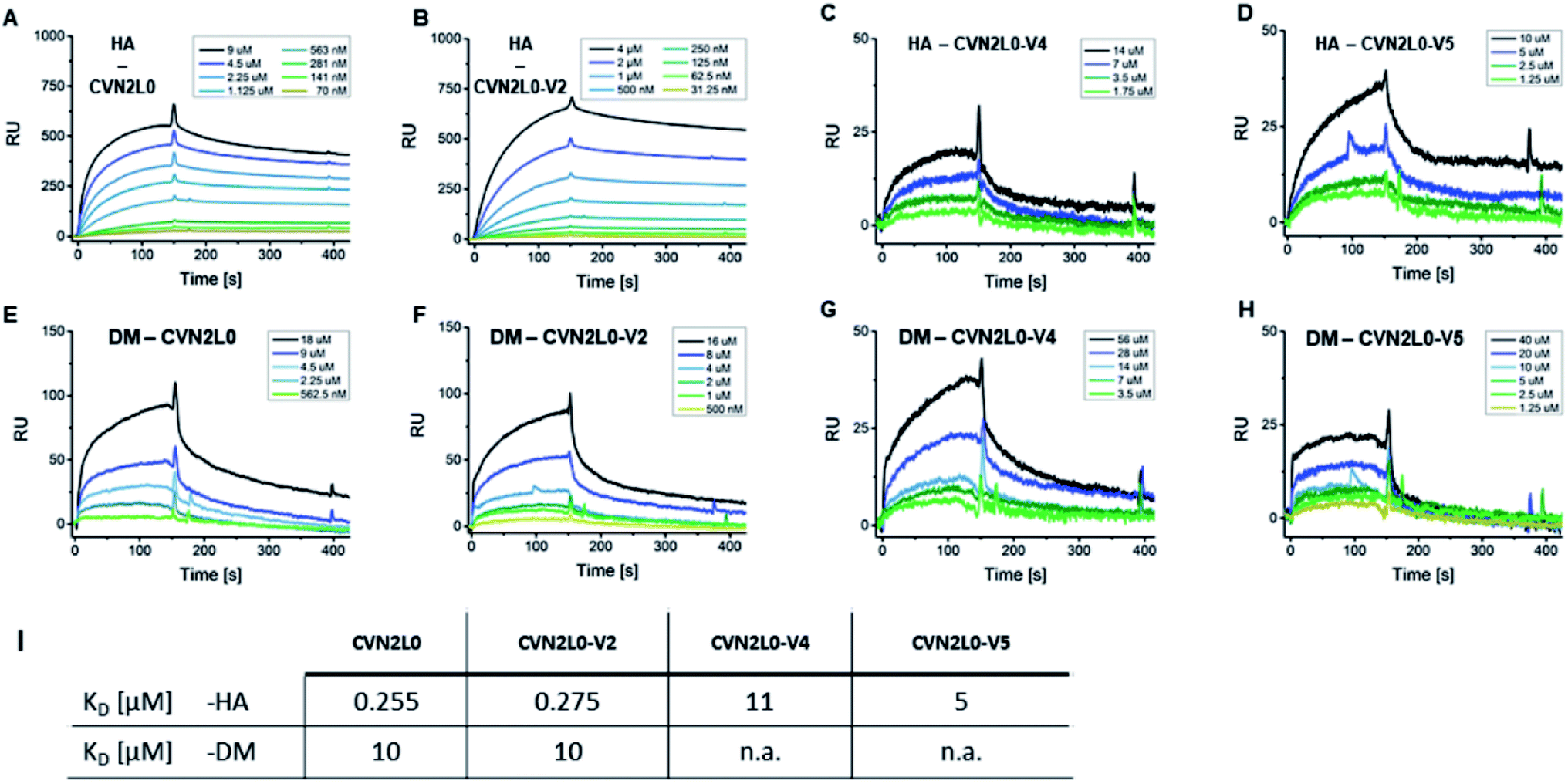 | ||
| Fig. 3 CVN2L0 and variants binding to HA and DM. (A–D) SPR sensorgrams showing CVN2L0 (and variants V2, V4 and V5) binding to HA which is covalently immobilized onto a Xantec CMD500D sensorchip at 20 μg mL. (E–H) SPR binding assays of CVN2L0 and variants V2, V4 and V5 with DM under the same conditions. Scrubber (Biological Software) was used to fit real-time SPR association and dissociation curves (kon = 0.9–5 × 10−3 M−1s−1; koff = 1.3 × 10−3 s−1 for CVN2L0; koff = 1.1 × 10−3 s−1 for V2; koff = 1.3 × 10−2 s−1 for V4; koff = 5.8 × 10−2 s−1 for V5) and to calculate equilibrium dissociation constants from both kinetic and equilibrium data. The SPR dissociation rate constant koff for CVN2 binding to DM (koff = 9.2 × 10−3 s−1 for CVN2L0 and 1.2 × 10−2 s−1 for CVN2L0-V2) was to the maximum 10-fold higher concerning CVN2L0-V2, if compared to the dissociation off-rates in binding to HA (Table S2†). The residual response for dissociation of V4 (Cys–Cys mutagenesis to Trp–Met and Glu–Arg) from DM was still higher than 25% of the response maximum. V5 (2× Glu–Arg) failed in dissociation from surface-bound DM. (I) Values of KD = equilibrium dissociation constant. RU = Response Units. (A) Rmax = 520 RU, (B) 723 RU, (C) 34 RU, (D) 52 RU, (E) 161 RU, (F) 132 RU. | ||
Next, the interaction of CVN2L0 with immobilized DM was measured and showed binding of CVN2L0 and -V2 with also micromolar affinity (both 10 μM; Fig. 3E and F). V4 and V5 displayed even lower responses to DM (Fig. 3G and H). Kinetic dissociation constants indicated that low-affinity binding sites were involved in peptide binding to CVN2L0-V4 with a single Trp58–Met73 substitution in the high-affinity binding site (Fig. 3I and Table S2, ESI†). We observed that two Cys–Cys substitutions resulting in additionally two positively and two negatively charged side chains formed the actively binding CVN2L0-V5, thereby supported in affinity by the involvement of electrostatic interactions to steady-state binding with DM. Furthermore, we confirmed that a disaccharide unit was required for recognition of CVN2L0 and its binding to HA,39,40 but no essential binding to monomannosylated peptide P3 (MM) was detected by SPR. The interaction with DM involved at least one pseudo-domain with the favorable high-affinity to dimannose, independent of CVN2L0's number of intramolecular disulfide bonds. Binding of CVN2L0-V3 containing two Trp58–Met73 substitutions instead of the four buried Cys in high-affinity domains B was largely abrogated though.
A mannose residue interacts with Trp-mutated glycan-pocket of CVN2L0
To investigate the influence of the various mannose units in a defined binding pose, binding to MM was studied. None of the CVN2L0 variants achieved significant response to bind monomannose in SPR binding studies, revealing the importance of a dimannose moiety that has been shown to be bound to the high-affinity binding site, for the recognition by CV-N.41 According to CD spectra, the secondary structure of CVN2L0 variants has remained to be unchanged, if compared with CVN2L0: largely β-stranded structures were recorded and minor contribution by α-helices (calculated with the BeStSel algorithm).42 The minima found in the CD spectra were shifted to lower wavelength for all variants (212 nm → 203–205 nm). Mannosylated peptide binding was measured in the μM range with a corresponding association constant and relaxation time for the binding mode in NMR spectroscopy. The interactions of DM and MM with CVN2L0 and symmetrically incorporated C58W–C73M mutations in CVN2L0-V3 were studied by NMR-based STD spectroscopy.43 DM and MM were chosen to mimic part of HA top with a spatially exposed glycosylation site on Asp81 (Fig. 4A).13 STD-NMR spectra of DM and MM were different in the 3–4 ppm range which corresponded to the typical shift region of saccharides (Fig. 4B and S18A†). Higher numbers of scans were required to display complex formation between glycan DM and CVN2L0 (ns = 4000 scans) than between MM and CVN2L0 (ns = 1000 scans). Assuming conformational selectivity to specific high-mannose peptides, the turn-over i.e., the exchange between free and bound peptide with protein, was tremendously enhanced for a single monomannose unit. Chemical exchange in monomannose-linked peptide at the recognition site Q80-G*81-E82-T83, when in contact with the primary high-affinity carbohydrate binding site, was found in agreement with its lower affinity to CVN2L0. Nonpolar mutated residues in V3 can donate and accept hydrogen bonds through the indole ring of Trp for its basal binding affinity to both DM and MM (Fig. 4C and D). The typical STD signals were seen when MM was measured against CVN2L0 (δ1H = 3.5 ppm), but not for the interaction with V2 (Fig. S18A and B†). The STD effect was particularly attributed to the alkyl side chain (–CH3, δ1H = 1 ppm) of residue Thr83, that was closest to the glycosylation site in the peptide.44 Together, the high-affinity glycan-binding pocket, which equals primary binding sites, consists of the same composition of seven proton-accepting and hydrophilic amino-acids: Gln78, Thr57, Glu56, Glu41, Arg76, Asp44 and Ser52, as like those used in the influenza HA peptide to complement an isoelectric protein–peptide complex. Ser52 in the CV-N-linker region corresponded with triazole-linked Gly81 mutation in the peptide sequence CD--QGET-D--. Another five hydrophobic or nonpolar amino-acids (V, F, W, L, F) of the peptide were spacing the glycine-attached mannose-units to N-terminal Cys and C-terminal Phe. A network of hydrogen bonds can be formed with Thr83 and hydroxyl-groups in order to make the mannose sugar interact with the mutated Trp58–Met73 pair in both binding pockets.45,46 By contrast, V2 incorporated polar contacts with Glu58-Arg73 into a single high-affinity binding pocket, thereby replacing an intramolecular Cys–Cys bond for the attachment of DM. The peptide itself constitutes of another accessible Cys, when bound to the protein with a second functional low-affinity binding site on the same molecule or other domain A.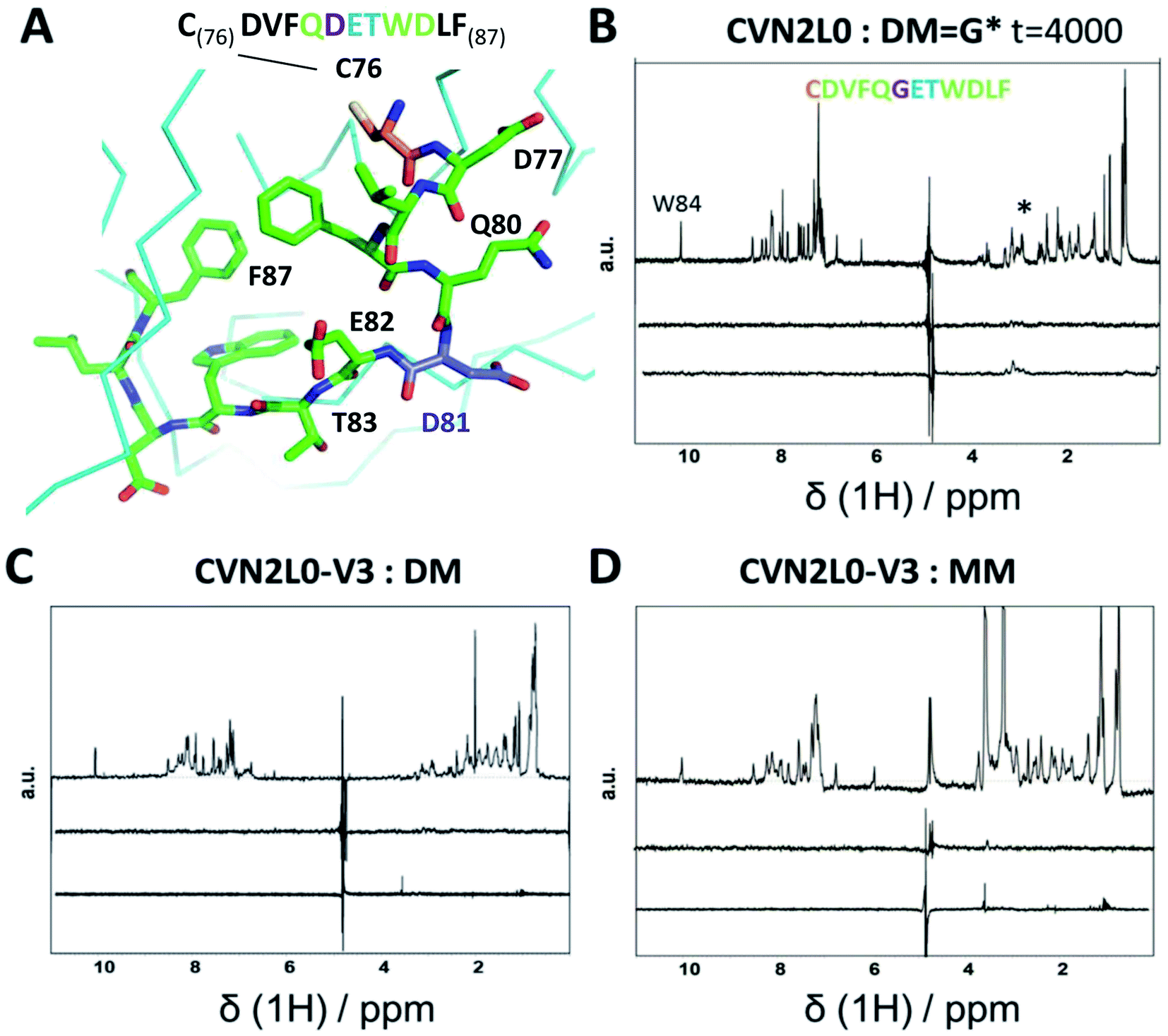 | ||
| Fig. 4 (A) Part of 3D X-ray crystal structure of HA top (PDB ID: 2VIR; Fleury et al. 1998)13 with the residues of the peptide (C76–F87) highlighted with sticks. A disulfide bond in HA top (C64–C76) provides C76 (highlighted in orange) as the N-terminal amino-acid of the model peptide. The residue bearing the mannosylation (D/N81, or mannosylated residue in the model peptide) is shown and labelled in purple. (B) STD-NMR of CVN2L0-peptide complex: upper trace: 1H NMR spectrum of the native peptide P2 (DM), *(asterisk) marked the appr. shift range of saccharides. Middle trace: STD-NMR spectrum of the free peptide (negative control experiment). Lower trace: STD-NMR spectrum of DM in the presence of CVN2L0 indicating complex formation at 3 ppm. a.u. = arbitrary units. (C) STD-NMR of CVN2L0-V3 and DM. (D) STD-NMR of CVN2L0-V3 and peptide P3 (MM). STD in the difference spectrum related to peptide alkyl-resides (δ1H = 1 ppm) and its mannosylation site (δ1H = 3–4 ppm). Chemical exchange effects arise due to lower affinity with MM upon site-specific recognition of D81X-E82-T83 (X = mannosylated glycine-derivative). CVN2L0-V3 (C58W and C73M in both high-affinity carbohydrate binding sites). 100 μM peptide was measured over 10 μM protein. Saturation was performed with a Gaussian pulse train lasting for 4 s at −1.0 ppm (1000–4000 scans were needed). Water suppression was achieved with a double Watergate echo (excitation sculpting). | ||
DM binds domain-swapped CVN2L0 with functional low-affinity binding sites
Binding of CVN2L0 to DM was confirmed by ITC. In solution binding of DM to CVN2L0 was determined with dissociation constants KD1 (306 nM at high-affinity domain B) and KD2 (4 μM at low-affinity domain A; Fig. 5A and B). Domain B binding site knock-outs have been reported to bind in the nM range (KD = 65 nM) compared to wild-type CV-N (KD = 5.7 nM).11 Calculated from kinetic binding curves measured by SPR (KD = 10 μM), CVN2L0 binding affinity to DM correlated with the numbers of disulfide bonds and low-affinity binding sites. Thermal denaturation curves were recorded from CD spectra at invariant wavelength (235 nm). All variants with substitutions of one disulfide bridge by ion-pairing Glu58-Arg73 mutagenesis retained stability (V2 and V4). Ionic interaction of oppositely charged amino-acids in domain B changed helical components when located in one of the two in opposite to each other placed and also mutated high-affinity binding sites (Fig. S17A and B, ESI†). CVN2L0-V4 and -V5 without any disulfide bridges in the high-affinity binding sites were melting approximately three degrees lower for each substituted disulfide bridge compared to CVN2L0 (Fig. S17B, ESI†). Variant V4 added Glu–Arg and Trp–Met mutation pairs to binding domain B unsymmetrically, but showed binding to DM via SPR, while the protein was bearing two functional low-affinity binding sites. Cross-contacts between either type of low- and high-affinity binding sites in the same domain-swapped monomer were found diminished because of insufficient DM binding in the double Trp–Met mutated variant (V3). | ||
| Fig. 5 Bivalent binding of DM to folded CVN2L0. Isothermal calorimetric titration of DM by CVN2L0 at 298 K. (A) Aliquots (2 μL) of ligand (>600 μM) were injected into CVN2L0 (50 μM) in 100 mM PBS, pH 7.0, in excess of ligand (DM) for the saturation of binding sites. Two sets of binding sites fitting model was used to calculate KD values for the 2 low-affinity and 2 high-affinity carbohydrate binding sites on each CVN2L0 molecule. (B) The ratio between CVN2L0 = 50 μM and DM was chosen to be below 1:12 for CVN2L0, and 1:6 for other variants. Heats of binding were measured as delta (Δ)H, and one set of sites fitting model used to determine KD for the binding affinity of DM to soluble CVN2L0. | ||
Together, CV-N shows inhibitory properties against several viruses47 and is considered a potential glycan-targeting agent for preventing seasonal and pandemic influenza diseases48,49 because of its lack of cytotoxicity. Its simultaneous broad activity against human immunodeficiency17,18 and enveloped Ebola virus50 is an additional benefit. Various studies on binding of CV-N to HIV were performed, as well as CV-N mutants and oligomers designed.17,36,47 The stabilizing character of disulfide bridges for folding and maintaining secondary structure elements is in particular interesting to domain-swapped CVN2L0 dimer due to their proximity to CVN2L0 binding sites for mannose units. Recently, STD-NMR has been used to probe conformation of carbohydrate-mimetic peptides associated with O-polysaccharide of Shigella flexneri Y,51 but not N-linked glycosylation sites, that are the most ubiquitous modification to Asn in a consensus sequence of Asn-Xaa-Ser/Thr.52
In this study, homogeneously mannosylated peptides were synthesized, partially covering a sequence of the influenza surface glycoprotein HA to serve as mimicry for a direct complementary binding sequence to CV-N. HA top domain constitutes of three possible Asn-Xaa-Thr glycosylation sites, of which the single polar Asn–Glu–Thr sequence was used for the model peptide, in contrast to the more hydrophobic Asn–Ala–Thr and Asn–Val–Thr sites.13 Although CVN2L0 binding affinity was lower to dimannosylated peptide (DM) compared to HA, CVN2L0 and disulfide bond variants V2 and V4 showed both kinetic and in solution thermodynamically controlled binding to DM. The replacement of one disulfide bridge in the high-affinity binding region (C58E–C73R) compensated for binding to the second primary binding site in opposite to the modified one (Fig. 2), and the nearby disulfide bridge in the bound HA protein. Binding of this destabilized variant CVN2L0-V2 to DM (with three instead of four disulfide bridges) has been determined in the range of 10 micromolar concentrations. Similarly, intermolecular domain-swapped CVN2L0-V4 dimer, where another Cys–Cys bridge was replaced by nonpolar hydrogen-bond forming Trp–Met side chains, achieved binding to DM with native two functional low-affinity binding sites. Hydrophobic residues formed an interface for binding studies with the monomannose-linked peptide itself (Fig. 4D). By contrast, dimeric CVN2L0-V2 showed significant affinity to HA (KD = 275 nM). When replacing the Cys–Cys bond in the high-affinity binding site in both monomers, as performed for V3, V4, and V5, binding was mainly impaired, and in the range of μM affinity for variants with introduced ion-pairing Glu–Arg residues (V4, KD = 11 μM; and V5, KD = 5 μM).
In summary, our studies specifically tested carbohydrate units linked to a complementary peptide sequence of the high-affinity binding site on CVN2L0. We found that the substitution of disulfide bonds with charged amino-acids allowed for low-affinity binding of dimannose on the peptide sequence Gln–X–Glu–Thr (DM, X= dimannosylated residue), whereas another N-terminal Cys next to ionizing Asp formed polar interactions to the primary high-affinity binding site in CVN2L0. In addition, binding of CVN2L0 and variants to MM was investigated. Trp-mutagenesis in both high-affinity glycan-binding sites of CVN2L0-V3 was applied for binding studies on MM, showing STD effects related to Thr83, not necessarily Met, and nuclear Overhauser effect. The hydrophilic environment next to mannosylated peptide position Gly81 may have interacted with possibly interface-forming planar Trp from the peptide and mutated Trp58 in the protein that needed to be closest to position Met73 in the folded protein (Fig. 4D).46 Therefore, the viral neutralization process that is mediated by recognizing and binding N-linked oligo-mannose moieties, demonstrated selective binding of pseudoantibody-like lectins to heterogeneously cross-linked oligosaccharide binding sites. Other heterocyclic peptide backbone modifications have been shown to functionalize dipeptide surrogates in α-helical coiled coils.53
Experimental
Peptide P1 was synthesized using Fmoc-based, automated and microwave-assisted SPPS (Liberty Blue™, CEM, Kamp-Lintfort, Germany). Acetylated methylether-dimannose was synthesized using the trimethylsilyl trifluoromethanesulfonate (TMSOTf)-catalyzed method. Compound 2,3,4,6-tetra-O-acetyl-α-D-mannopyranosyl-(1→2)-3,4,6-tri-O-acetyl-α-D-mannopyranoside methylether (5) was first converted to the fully acetylated dimannose (6). The final dimannose azide (8) was obtained by deacetylation. The presented route allowed for isolation of this building block with a yield of 20% over 7 steps (Fig. S1–S6, ESI†). For the introduction of the azide, TMSN3 and catalytic amounts of anhydrous iron(III) chloride were used (Fig. S6 and S7, ESI†). Incorporation of the di- and monomannose building blocks (8) and (9) to the model peptide P1 (Fig. S8, ESI†) was performed using CuAAC, where all protecting groups and the linkage of the peptide to the solid support were found intact. Cleavage of the synthesized glycopeptides from the solid support using high concentrations of trifluoroacetic acid (TFA) showed good stability of the O-glycoside. Purity and homogeneity of these peptides were verified by HRMS and LC-MS (Fig. S9, S11 and S13, ESI†).CVN2L0 was expressed in Escherichia coli and purified from extracellular non-reducing periplasm. CVN2L0 binding variants were designed on C58–C73 disulfide bond of PDB ID: 3EZM using ORBIT force field as described in.27 For each of the six CVN2L0 variants (Fig. S14, ESI†) and CVN2L0 wild-type protein with an N-terminal pelB leader sequence and His-tag, lyophilized plasmid DNA was obtained from GenScript® (pET-27b(+) vector; Piscataway, USA) and dissolved in sterile ddH2O to a final concentration of 100 ng μL−1. Transformation into competent E. coli BL21 (DE3) was performed for each variant (V1–V6) by chemical transformation (ESI†). Proteins were purified using Ni-NTA column chromatography (ÄKTA prime plus). Binding of CVN2L0 to HA H3 was verified applying ELISA in microtitration plate format. The sequence for the same protein was cloned into pET27b(+) and re-analyzed by ESI-LC-MS (Fig. S15 and S16, ESI†).
SPR
Affinity binding studies were performed on Reichert's SR7500DC (Reichert, Buffalo, USA), a two channel SPR instrument. CMD500D SPR sensorchips were purchased from Xantec biosensors (Düsseldorf, Germany). Measurements were performed at 25 °C and at a flow rate of 30 μL min−1. Sensorchips were activated using single-channel amine coupling, where 0.4 M EDC/HCl and 0.1 M NHS were used to activate the carboxymethyldextran hydrogel chip surface. The ligand (HA or glycopeptides P2, P3) was diluted to 20 μg mL−1 in 10 mM sodium acetate buffer (pH 5) and injected to the activated chip surface, followed by ethanolamine coupling to block excess reactive carboxyl groups on sensor chips. Recombinant influenza A virus hemagglutinin H3 protein (H3N2 A/Wisconsin/67/05) was obtained from abcam (Cambridge, UK). Primary kinetics data was analyzed according to the integrated rate of association and rate of dissociation equations which describe the interaction of soluble CVN2L0, variants V2, V4 and V5 with surface-immobilized ligand and the dissociation of the complex between protein and ligand (see ESI†).STD-NMR
1H-NMR experiments were acquired on Bruker NEO 500 or 600 spectrometers equipped with either a 5 mm BBFO (broadband observe probe at 500 MHz) or a 5 mm TXI (H/N/C triple resonance probe at 600 MHz) each equipped with shielded z-gradients. An NMR sample of CVN2L0 and each variant was prepared in phosphate-buffered saline (100 mM, pH 7.0) added to a peptide solution in phosphate buffer for a final peptide concentration of 100 μM and a ratio of peptide/CVN2L0 = 10:1. STD-NMR experiments were recorded at 298 K with 500–1000 scans for MM (and up to 4000 scans for DM). Selective saturation of protein resonances was performed at −1 ppm (40 ppm for off resonance spectra) using a series of Gaussian-shaped pulses (1.4 ms, γB1/2π = 200 Hz), for total saturation times ranging from 2 s to 4 s.
ITC
Calorimetric titrations were performed using a PEAQ-ITC isothermal titration calorimeter (Malvern Pananalytical, UK). The protein solution (35–50 μM) was placed in the calorimeter cell (1.5 mL active volume), and 2 μL aliquots of the DM and MM solutions (300–600 μM) were added at 2 min intervals from a stirring syringe (operated at 750 rpm).Conclusions
We used peptide synthesis and recombinant protein expression methods for the preparation of glycopeptides and CVN2L0 variants and studied the binding between these interacting molecules using SPR and ITC. CVN2L0 (four disulfide bonds) and V2 (three disulfide bonds) showed similar binding affinity in the low μM range, while V4 and V5 (only two disulfide bonds) responded above measurable concentration (Fig. 3). This could be explained by the loss of the main binding motifs and alternate binding with low-affinity sites in CVN2L0 as measured by ITC,2,11 and by STD-NMR, a method to study transient carbohydrate protein interactions. Binding of monomannose-conjugates to CV-N has not been shown before. The number of disulfide bonds in the bound and unbound CVN2 molecule, however, was found relevant for the chemical exchange between free and bound state of the monomannose bearing peptide (Fig. 4). According to our results, mannosylated peptides constitute and interrogate new protein scaffolds for screening binding characteristics of antiviral agents by SPR and NMR. Binding of lectin CVN2L0 and variants with fully stabilized carbohydrate binding sites to mannosylated peptides were investigated in this study to search for glycan-interacting homo-dimeric CVN2L0 scaffolds. Here presented variants may further allow studies on improved binding capacities at CV-N low-affinity binding sites by computational protein design,54,55 while a comparison of binding with HA H3N2 full length protein was achieved. Overall, the ability of lectins and pseudo-antibodies to bind highly conserved epitopes on the influenza HA protein is of key importance to the rational design of next-generation vaccines for prophylactic and therapeutic use.56Abbreviations
| CuAAC | Copper(I)-catalyzed azide alkyne cycloaddition |
| CV-N | Cyanovirin-N |
| Fmoc | Fluorenylmethyloxycarbonyl |
| ITC | Isothermal titration calorimetry |
| KD | Equilibrium dissociation constant |
| HA | Hemagglutinin |
| HIV | Human immunodeficiency virus |
| Man | Mannose |
| GlcNAc | N-Acetyl glucosamine |
| NMR | Nuclear magnetic resonance |
| ORBIT | Optimization of rotamers by iterative techniques |
| SPPS | Solid phase peptide synthesis |
| SPR | Surface plasmon resonance |
| STD | Saturation transfer difference |
| TMSOTf | Trimethylsilyl Trifluoromethanesulfonate |
| TFA | Trifluoroacetic acid |
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the Mayo Lab and the Protein Expression Center at Caltech, Dr Oz Sharabi from the Hebrew University of Jerusalem and Gerhard Niederacher from the University of Vienna for scientific discussions and technical support, as well as the NMR Facility in the Department of Structural and Computational Biology and the MS Facility at the University of Vienna for technical support.References
- M. T. Esser, T. Mori, I. Mondor, Q. J. Sattentau, B. Dey, E. A. Berger, M. R. Boyd and J. D. Lifson, J. Virol., 1999, 73, 4360–4371 CrossRef CAS PubMed.
- L. G. Barrientos, B. R. O'Keefe, M. Bray, A. Sanchez, A. M. Gronenborn and M. R. Boyd, Antiviral Res., 2003, 58, 47–56 CrossRef CAS PubMed.
- B. R. O'Keefe, D. F. Smee, J. A. Turpin, C. J. Saucedo, K. R. Gustafson, T. Mori, D. Blakeslee, R. Buckheit and M. R. Boyd, Antimicrob. Agents Chemother., 2003, 47, 2518–2525 CrossRef PubMed.
- R. C. Cheung, J. H. Wong, W. Pan, Y. S. Chan, C. Yin, X. Dan and T. B. Ng, Appl. Microbiol. Biotechnol., 2015, 99, 3755–3773 CrossRef CAS PubMed.
- J. Wang, H. Li, G. Zou and L. X. Wang, Design, synthesis, and antibody binding study, Org. Biomol. Chem., 2007, 5, 1529–1540 RSC.
- Y. Liu, J. R. Carroll, L. A. Holt, J. McMahon, B. Giomarelli and G. Ghirlanda, Biopolymers, 2009, 92, 194–200 CrossRef CAS PubMed.
- B. Brandenburg, W. Koudstaal, J. Goudsmit, V. Klaren, C. Tang, M. V. Bujny, H. J. Korse, T. Kwaks, J. J. Otterstrom, J. Juraszek, A. M. van Oijen, R. Vogels and R. H. Friesen, PLoS One, 2013, 8, e80034 CrossRef PubMed.
- A. F. S. Santos, M. D. C. da Silva, T. H. Napoleao, P. M. G. Paiva, M. T. S. Correia and L. C. B. B. Coelho, Curr. Top. Pept. Protein Res., 2014, 15, 41–62 Search PubMed.
- S. R. Shenoy, B. R. O'Keefe, A. J. Bolmstedt, L. K. Cartner and M. R. Boyd, J. Pharmacol. Exp. Ther., 2001, 297, 704–710 CAS.
- E. Matei, S. Andre, A. Glinschert, A. S. Infantino, S. Oscarson, H. J. Gabius and A. M. Gronenborn, Chemistry, 2003, 19, 5364–5374 CrossRef PubMed.
- C. A. Bewley, S. Kiyonaka and I. Hamachi, J. Mol. Biol., 2002, 322, 881–889 CrossRef CAS PubMed.
- I. A. Wilson, J. J. Skehel and D. C. Wiley, Nature, 1981, 289, 366–373 CrossRef CAS PubMed.
- D. Fleury, S. A. Wharton, J. J. Skehel, M. Knossow and T. Bizebard, Nat. Struct. Biol., 1998, 5, 119–123 CrossRef CAS PubMed.
- J. J. Otterstrom, B. Brandenburg, M. H. Koldijk, J. Juraszek, C. Tang, S. Mashaghi, T. Kwaks, J. Goudsmit, R. Vogels, R. H. Friesen and A. M. van Oijen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E5143–E5148 CrossRef CAS PubMed.
- J. Balzarini, Nat. Rev. Microbiol., 2007, 5, 583–597 CrossRef CAS PubMed.
- J. P. Julien, P. S. Lee and I. A. Wilson, Immunol. Rev., 2012, 250, 180–198 CrossRef PubMed.
- J. R. Keeffe, P. N. Gnanapragasam, S. K. Gillespie, J. Yong, P. J. Bjorkman and S. L. Mayo, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14079–14084 CrossRef CAS PubMed.
- F. Yang, C. A. Bewley, J. M. Louis, K. R. Gustafson, M. R. Boyd, A. M. Gronenborn, G. M. Clore and A. Wlodawer, J. Mol. Biol., 1999, 288, 403–412 CrossRef CAS PubMed.
- C. A. Bewley, K. R. Gustafson, M. R. Boyd, D. G. Covell, A. Bax, G. M. Clore and A. M. Gronenborn, Nat. Struct. Biol., 1998, 5, 571–578 CrossRef CAS PubMed.
- C. A. Bewley, Structure, 2001, 9, 931–940 CrossRef CAS PubMed.
- T. Bizebard, B. Gigant, P. Rigolet, B. Rasmussen, O. Diat, P. Bosecke, S. A. Wharton, J. J. Skehel and M. Knossow, Nature, 1995, 376, 92–94 CrossRef CAS PubMed.
- O. Sharabi, J. Shirian, M. Grossman, M. Lebendiker, I. Sagi and J. Shifman, PLoS One, 2014, 9, e93712 CrossRef PubMed.
- R. Chen and Z. P. Weng, Proteins, 2002, 47, 281–294 CrossRef CAS PubMed.
- P. S. Huang, J. J. Love and S. L. Mayo, J. Comput. Chem., 2005, 26, 1222–1232 CrossRef CAS PubMed.
- C. Yin and S. S. Yau, PLoS One, 2017, 12, e0174862 CrossRef PubMed.
- B. M. P. Huyghues-Despointes, J. M. Scholtz and R. L. Baldwin, Protein Sci., 1993, 2, 1604–1611 CrossRef CAS PubMed.
- S. A. Marshall, C. S. Morgan and S. L. Mayo, J. Mol. Biol., 2002, 316, 189–199 CrossRef CAS PubMed.
- E. Oueis, M. Jaspars, N. J. Westwood and J. H. Naismith, Angew. Chem., 2016, 55, 5842–5845 CrossRef CAS PubMed.
- A. C. Conibear, K. Farbiarz, R. L. Mayer, M. Matveenko, H. Kahlig and C. F. Becker, Org. Biomol. Chem., 2016, 14, 6205–6211 RSC.
- S. R. Tala, A. Singh, C. J. Lensing, S. M. Schnell, K. T. Freeman, J. R. Rocca and C. Haskell-Luevano, ACS Chem. Neurosci., 2018, 9, 1001–1013 CrossRef CAS PubMed.
- D. C. Schröder, O. Kracker, T. Fröhr, J. Góra, M. Jewginski, A. Nieß, I. Antes, R. Latajka, A. Marion and N. Sewald, Front. Chem., 2019, 7, 155, DOI:10.3389/fchem.2019.00155.
- W. Huang, S. Groothuys, A. Heredia, B. H. Kuijpers, F. P. Rutjes, F. L. van Delft and L. X. Wang, Chembiochem, 2009, 10, 1234–1242 CrossRef CAS PubMed.
- S. B. Salunke, N. S. Babu and C. T. Chen, Chem. Commun., 2011, 47, 10440–10442 RSC.
- Z. Szurmai, L. Balatoni and A. Liptak, Carbohydr. Res., 1994, 254, 301–309 CrossRef CAS PubMed.
- L. G. Barrientos, J. M. Louis, I. Botos, T. Mori, Z. Han, B. R. O'Keefe, M. R. Boyd, A. Wlodawer and A. M. Gronenborn, Structure, 2002, 10, 673–686 CrossRef CAS PubMed.
- V. Patsalo, D. P. Raleigh and D. F. Green, Biochemistry, 2011, 50, 10698–10712 CrossRef CAS PubMed.
- L. C. Chang and C. A. Bewley, J. Mol. Biol., 2002, 318, 1–8 CrossRef CAS PubMed.
- D. B. Gordon, S. A. Marshall and S. L. Mayo, Curr. Opin. Struct. Biol., 1999, 9, 509–513 CrossRef CAS PubMed.
- L. G. Barrientos, E. Matei, F. Lasala, R. Delgado and A. M. Gronenborn, Protein Eng., Des. Sel., 2006, 19, 525–535 CrossRef CAS PubMed.
- X. Wang, E. Matei, A. M. Gronenborn, O. Ramstrom and M. Yan, Anal. Chem., 2012, 84, 4248–4252 CrossRef CAS PubMed.
- Z. Li, A. Bolia, J. D. Maxwell, A. A. Bobkov, G. Ghirlanda, S. B. Ozkan and C. J. Margulis, Biochemistry, 2015, 54, 6951–6960 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y. H. Lee, Y. Goto, M. Refregiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- M. Mayer and B. Meyer, Angew. Chem., 1999, 38, 1784–1788 CrossRef CAS.
- Y. Jamin, T. R. Eykyn, E. Poon, C. J. Springer and S. P. Robinson, Mol. Imaging Biol., 2014, 16, 152–157 CrossRef PubMed.
- G. Nestor, T. Anderson, S. Oscarson and A. M. Gronenborn, J. Am. Chem. Soc., 2018, 140, 339–345 CrossRef CAS PubMed.
- J. Anglister, R. Levy and T. Scherf, Biochemistry, 1989, 28, 3360–3365 CrossRef CAS PubMed.
- R. S. Singh, A. K. Walia, J. S. Khattar, D. P. Singh and J. F. Kennedy, Int. J. Biol. Macromol., 2017, 102, 475–496 CrossRef CAS PubMed.
- J. R. Chen, Y. H. Yu, Y. C. Tseng, W. L. Chiang, M. F. Chiang, Y. A. Ko, Y. K. Chiu, H. H. Ma, C. Y. Wu, J. T. Jan, K. I. Lin, C. Ma and C. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2476–2481 CrossRef CAS PubMed.
- D. F. Smee, K. W. Bailey, M. H. Wong, B. R. O'Keefe, K. R. Gustafson, V. P. Mishin and L. V. Gubareva, Antiviral Res., 2008, 80, 266–271 CrossRef CAS PubMed.
- A. R. Garrison, B. G. Giomarelli, C. M. Lear-Rooney, C. J. Saucedo, S. Yellayi, L. R. Krumpe, M. Rose, J. Paragas, M. Bray, G. G. Olinger Jr, J. B. McMahon, J. Huggins and B. R. O'Keefe, Antiviral Res., 2014, 112, 1–7 CrossRef CAS PubMed.
- M. G. Szczepina, D. W. Bleile and B. M. Pinto, Chemistry, 2011, 17, 11446–11455 CrossRef CAS PubMed.
- D. F. Zielinska, F. Gnad, J. R. Wisniewski and M. Mann, Cell, 2010, 141, 897–907 CrossRef CAS PubMed.
- W. S. Horne, M. K. Yadav, C. D. Stout and M. R. Ghadiri, J. Am. Chem. Soc., 2004, 126, 15366–15367 CrossRef CAS PubMed.
- S. Rämisch, U. Weininger, J. Martinsson, M. Akke and I. André, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17875–17880 CrossRef PubMed.
- J. A. Fallas, G. Ueda, W. Sheffler, V. Nguyen, D. E. McNamara, B. Sankaran, J. H. Pereira, F. Parmeggiani, T. J. Brunette, D. Cascio, T. R. Yeates, P. Zwart and D. Baker, Nat. Chem., 2017, 9, 353–360 CrossRef CAS PubMed.
- D. Lingwood, P. M. McTamney, H. M. Yassine, J. R. Whittle, X. Guo, J. C. Boyington, C. J. Wei and G. J. Nabel, Nature, 2012, 489, 566–570 CrossRef CAS PubMed.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem. Int. Ed. Engl., 2002, 41(14), 2596–2599 CrossRef CAS PubMed.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67(9), 3057–3064 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra01128b |
| This journal is © The Royal Society of Chemistry 2020 |