
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuperior antitumor effect of self-assembly supramolecular paclitaxel nanoparticles
Na Yu†
a,
Jun Li†b,
Yuan Zhang†a,
Dan Ding b,
Xiaolin Li*c and
Huae Xu*a
b,
Xiaolin Li*c and
Huae Xu*a
aDepartment of Pharmaceutics, School of Pharmacy, Nanjing Medical University, Nanjing, 210029, China. E-mail: xuhuae@njmu.edu.cn
bState Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Bioactive Materials Ministry of Education and College of Life Sciences Nankai University, Tianjin, 300071, China
cDepartment of Geriatric Gastroenterology, Center of Neuroendocrine Tumor, The First Affiliated Hospital of Nanjing Medical University, Nanjing, 210029, China. E-mail: lxl@njmu.edu.cn
First published on 31st March 2020
Abstract
Paclitaxel (Ptx), a microtubule depolymerization inhibitor, is one of the first-line regimens in lung cancer chemotherapy. However, the poor solubility of Ptx, as well as hypersensitivity of the solvent Cremphor EL, severely limits its clinical application. Here we developed a drug-polymer conjugate of Ptx–SA–PEG, in which amphiphilic copolymers poly(ethylene glycol) (PEG) and Ptx were conjugated by succinic acid (SA). The Ptx–SA–PEG polymers self-assemble into nanoparticles (Ptx-NPs) for efficient delivery of Ptx; cell count kit-8 assay and clonogenic assay were used to analyze the antitumor effect of Ptx-NPs. Acridine orange/ethidium bromide double staining, apoptosis analysis and western blot were measured to explore the apoptotic cell death after Ptx-NPs or free Ptx treatment. Subcutaneous xenograft models were practiced to estimate its tumor cytotoxicity and nonspecific side effects in vivo. Immunohistochemistry was used to analyze the effects of apoptosis and proliferation in tumor tissue; in vitro studies demonstrated that Ptx-NPs treatment exhibited more tumor inhibitory activity compared with free Ptx, especially at the lower doses. Moreover, Ptx-NPs activated apoptotic proteins. Animal experiments showed Ptx-NPs induced less weight loss and organ damage than free Ptx. Moreover, tumor growth was slower after Ptx-NPs treatment, indicating the superior antitumor effect and slight side effect of Ptx-NPs over free Ptx. Conjugation of Ptx–SA–PEG provides a feasible way to acquire self-assembled supramolecular Ptx-loaded nanoparticles with higher drug loading efficiency, less non-specific toxicity and more stable and durable antitumor effect of Ptx, providing a potential strategy to meliorate its clinical therapeutic efficacy.
Introduction
Lung carcinoma ranks first among all cancers in China and worldwide.1 Paclitaxel (Ptx) inhibits cell proliferation by blocking the depolymerization of the microtubule. NCCN guidelines recommend Ptx as the main components of first-line chemotherapeutic regimens for lung cancer.2,3 However, the application of Ptx is severely limited by its poor water solubility and the emerging resistance in clinics.4,5 Moreover, the solvent Cremophor EL of Ptx could cause severe side effects, including nonselective toxicity and hypersensitivity reactions.6Nano-delivery systems have been developed to improve the pharmacokinetic and pharmacodynamic profiles of chemotherapeutics recently.4,7,8 Amphiphilic polymeric nanoparticles are composed of two parts. The hydrophobic part, such as poly(ε-caprolactone) (PCL), forms the core of nano-spheres with lipophilic drugs inside, while the hydrophilic part, like poly(ethylene glycol) (PEG) or poly(N-vinylpyrrolidone) (PVP), forms the outer layer to increase the solubility and avoid scavenging by the reticuloendothelial systems.9–12 Early studies of the author's group have proved that nanoparticles with amphiphilic carriers, such as PCL-b-PEG or PVP-b-PCL, could increase the solubility of Ptx.10,13–16 Moreover, amphiphilic polymeric nanoparticles with less toxicity can replace Cremophor EL as a solvent of Ptx, thus alleviating the possible toxic effect of Ptx. Besides, the Enhanced Permission and Retention (EPR) effect of these amphiphilic polymeric nanoparticles amplifies the targeting ability to tumor tissues.17,18 The preferable efficacy of Ptx-loaded nanoparticles has been confirmed in several kinds of cancers.14,15,19 However, the relatively low drug loading efficiency and the cytotoxicity of nanoparticles carrier matrix still impede the potential application of amphiphilic polymeric Ptx-loaded nanoparticles.
To solve this problem, novel delivery systems were designed through the conjugation between Ptx and polymers by ester or disulfide bonds. Compared with polymeric nanosphere systems, these compositions could self-assemble into nanoparticles with drugs themselves as carriers (also referred to as “carrier-free”), which have higher drug loading efficiency and offer more stable drug release.20,21 Previous studies have demonstrated that Ptx is a widely used model drug to achieve the “carrier-free” goal.19,22–24 As our previous study reported, Ptx loaded nanoparticles can be designed by the conjugation of Ptx and a small molecule compound, succinic acid (SA), with a very high drug loading content (90%).25 As the molecular weight and size of succinic acid are much smaller than Ptx, the sphere structure of nanoparticles was mostly conformed by Ptx themselves. Though the loading efficiency of Ptx–SA was significantly improved, the poor solubility of drug remained the bottleneck for its intravenous use.
To overcome this limitation, we hypothesized that PEG2000, as the hydrophilic part of the nano-delivery systems, could further improve the poor solubility of Ptx. The ester bond among Ptx, SA and PEG2000 slowly hydrolyzed, which contributes to the continuous drug release and longer circulation time to maintain a higher drug concentration in blood, and thereby could enhance the antitumor efficacy of Ptx.
Here, we synthesized a novel conjugation of Ptx and PEG2000 through SA which self-assembles into nanoparticles to achieve “carrier free” encapsulation of Ptx in aqueous solution. The effect against lung cancer of Ptx-NPs was investigated in both in vitro and in vivo cancer models. Lastly, we investigated the cytobiological mechanism of the anti-cancer effect of Ptx-NPs by evaluating the expression proteins related to tumor cell death.
Experimental
Human lung adenocarcinoma cell line A549 was provided by the Cell bank of Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco's Modified Eagle Medium (DMEM) medium. The medium was supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin (Thermo Fisher, USA). PEG (MW 2 kDa) and SA was obtained from GL Biochem (China). Ptx, coumarin-6, Acridine Orange (AO) and Ethidium Bromide (EB) solutions were manufactured by Sigma Aldrich (USA). Cell Counting Kit-8 (CCK8), Mito-tracker red probe and Hoechst solutions were obtained from Beyotime (China). Annexin V-FITC/PI Apoptosis Detection Kit was bought from Dojindo (Japan). Antibodies and terminal dUTP nick-end labelling (TUNEL) assay kit were all purchased from Abcam (USA).Synthesis of compound Ptx–SA
Compound Ptx–SA was synthesized according to the previous literature.22 Ptx (500 mg) and succinic anhydride (900 mg) were dissolved in 10 mL pyridine. After 3 hours reaction at room temperature, the solvent was evaporated under reduced pressure. Then water (20 mL) was added and stirred for 20 min. After filtering, precipitate was gradually dissolved in acetone and mixed water was then added. Finally, the fine crystal was collected.Synthesis of compound Ptx–SA–PEG2000
Compound Ptx–SA–PEG2000 was synthesized as mentioned before by Wei and co-worker.26 In brief, 200 mg (209.6 μmol) of Ptx–SA was dissolved in anhydrous methylene chloride (20 mL) at room temperature. To this solution N,N′-diisopropylcarbodiimide (DIPC) (98.4 μL), 4-dimethylaminopyridine (DMAP) (27.2 mg) and PEG2000 (482 mg) were added at 0 °C and stirred for 10 minutes. The mixture was further stirred for 24 hours at room temperature. The solution was washed with 0.1 N HCl, dried, and evaporated under reduced pressure. Finally, a white solid was recrystallized using 2-propanol.Cellular uptake of Ptx–SA–PEG2000
Coumarin-6, as a liposoluble green fluorescence tracer, was combined to Ptx-NPs at molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 during this test. A549 cells were first cultured with only coumarin-6 for 4 h or coumarin-6 loaded Ptx-NPs for 0.5 h, 2 h and 4 h. Then A549 cells were incubated with Mito-tracker red probe and Hoechst for other 20 min. Cells were later washed and imaged under the fluorescent microscopy.
:1 during this test. A549 cells were first cultured with only coumarin-6 for 4 h or coumarin-6 loaded Ptx-NPs for 0.5 h, 2 h and 4 h. Then A549 cells were incubated with Mito-tracker red probe and Hoechst for other 20 min. Cells were later washed and imaged under the fluorescent microscopy.
In vitro cytotoxicity
Cells were seeded at a density of 3000 cells per well (96 well plate) and allowed to adhere for 24 h. Then cells were cultured with Ptx and Ptx-NPs (25, 50, 100, 200, 400, and 800 nM, respectively). In PEG group, cells were cultured with PEG2000 at the same concentration as in corresponding Ptx or Ptx-NPs group. After 24 h treatment, 10 μL CCK8 solution was mixed with 90 μL DMEM medium. Cells were incubated in the mixture solution for 1 h. Absorbance at 450 nm was measured using a microplate reader (Bio-Rad, USA). Each concentration was reported as average value of 5 replicates.Clonogenic assay
A549 cells were seeded at a density of 300 cells per well (6 well plate). Cells were exposed to PEG2000, free Ptx and Ptx-NPs (50 nM), respectively. After 10 days, cells were fixed by 4% polyoxymethylene and stained in 0.5% crystal violet. Cell clusters with more than 30 cells were counted as colonies.AO/EB double staining
After 24 h of PEG2000, Ptx or Ptx-NPs (100 nM) treatment, the cells were washed and resuspended in 300 μL DMEM medium. Then cells were stained with dual staining of AO/EB solution (3 μL, respectively) in the dark at 37 °C for 20 min. The morphology of apoptotic tumor cells was observed within 15 min using a fluorescent microscope (OLYMPUS, Japan).Apoptosis analysis
After 24 h of PEG2000, Ptx and Ptx-NPs (100 nM) treatment, the adherent cells were collected by 0.25% trypsin without EDTA, washed, resuspended in working buffer and stained with 10% Annexin-V-FITC/PI solutions in the dark at 37 °C for 20 min. Finally, the fluorescence was analyzed using a FACScan flow cytometer (BD Biosciences, USA).Western blot
After 24 h of PEG2000, Ptx and Ptx-NPs (100 nM) treatment, cell proteins were extracted and separated by SDS-PAGE. Then total proteins were transferred to PVDF membranes. The blots were blocked in 5% BSA for 2 h and probed with primary antibodies overnight and then second antibodies for 2 h in 4 °C, followed by the activation of ECL kit.In vivo evaluation
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Nanjing Medical University and approved by the Animal Ethics Committee of Nanjing Medical University. Nude mice (Nu/Nu) were obtained from the animal center of Nanjing Medical University and hatched in specific pathogen-free (SPF) environments. One million of A549 lung cancer cells were suspended in 100 μL PBS solution and injected subcutaneously in the left shoulder of mice. When tumor grew to a size of 30–50 mm3, the mice were randomly divided into three groups (n = 7) (day 0). Ptx or Ptx-NPs (10 mg kg−1) was given intraperitoneally at day 0, day 3 and day 6, while mice in control group received 100 μL of sterile normal saline solution. Bodyweight and tumor volume were recorded every 3 day. Tumor sizes of each group were performed by a micro-CT system. Tumor volume = (widest diameter)2 × (longest diameter)/2. Relative tumor volume (RTV) = volume(day n)/volume(day 0). Tumor inhibition rate (T/C%) = RTV(experimental group)/RTV(control group). At the end of the experiment, blood was collected through orbital venous plexus and then all mice were euthanized. The samples of mixed plasma (3 mice/sample) were analyzed on fully automatic biochemical analyzer (Rayto, Chemray 240, China). Tumors were excised and fixed in 4% paraformaldehyde overnight.Histological analysis
Tumor sections were stained with TUNEL kit or antibodies according to the manufactory protocol. Sections were visualized with DAB (Dako, Denmark), and then counter-stained with hematoxylin and observed under a light microscope using Olympus Camedia Software.Statistical analysis
All experiments in this study were independently repeated at least three times. The outcomes were measured as mean ± standard deviation (SD). The difference was analyzed by independent sample T test or one-way analysis of variance (ANOVA). A probability level of p < 0.05 was considered as significant.Results and discussion
Synthesis and characterization of Ptx-NPs
The synthesis and characterization of Ptx–SA–PEG2000 were reported in a previous study.22 The synthetic route of compound Ptx–SA–PEG2000 is depicted in Scheme 1. Coumarin-6 was used to visualize cellular uptake in A549 cells. As shown in Fig. 1, two hours treatment was enough for nanoparticles to be transferred into cytoplasm. With the extension of time, the intracellular drugs further accumulated intracellularly and located in the cytoplasm. | ||
| Scheme 1 The synthetic route of Ptx–SA–PEG2000. | ||
 | ||
| Fig. 1 Cellular uptake of Ptx-NPs. Fluorescent images of coumarin 6 (green) or coumarin 6-loaded Ptx-NPs cultured A549 cells (10×) showing progressive intracellular uptake over time. Mito-tracker red probe and Hoechst were stained to locate mitochondria (red) and nuclear (blue), respectively, in cells. | ||
Subsequently, the Ptx–SA was reacted with PEG2000 through esterification reaction. From our previous work reported by Wei et al., we successfully get the compound Ptx–SA–PEG and also knew that the compound Ptx–SA–PEG form nanoparticles in the phosphate buffer saline solution at the concentration of 20 wt%. The size distributions of nanoparticles were stability maintained in the shape of sphere with diameter about 20 nm based on dynamic light scattering (DLS) measurement and transmission electron microscopy (TEM). Ptx–SA–PEG nanoparticles could release free Ptx continuously and steadily to maintain an effective plasma concentration of 50 h.26 As the length of PEG chain increases, the drug loading efficiency would decrease according to the equation of drug loading content (DLC). The releasing of drug will not be affected, as the self-hydrolysis of ester bond between Ptx and SA is determined by the pH and temperature. According to a previous study in mice, the blood circulation time will increase with the increasing molecular weight (Mw) of PEG chain within a certain range from 2000 up to 5000 g mol−1. When the molecular weight of PEG is more than 5000, no considerable increase of blood circulation time could be observed.27
As compared with other Ptx loaded nanosystems, such as functional carbon nanotubes (the grafted–CNT–PEG–PTX system) have high surface area and multiple reaction sites, which induce relatively high drug loading content (Bioconjugate Chemistry, 2013, 24(4), 626–639). Though the drug loading content of carrier free system is not particularly high, the system is simple and easy to obtain.28
Cellular uptake and cytotoxicity of Ptx-NPs in A549 cells
Anticancer efficiency of both free Ptx and Ptx-NPs were tested through CCK8 assay. Our result showed that PEG2000 was not significantly toxic to A549 within the concentration range (25–800 nM) used in our study Fig. 2. Both free Ptx and its nanoparticles suppressed cell proliferation in a dose-dependent manner. Even more, Ptx-NPs shown more serious cytotoxicity than free Ptx in vitro, especially at lower concentrations (50–200 nM) of Ptx (P < 0.05).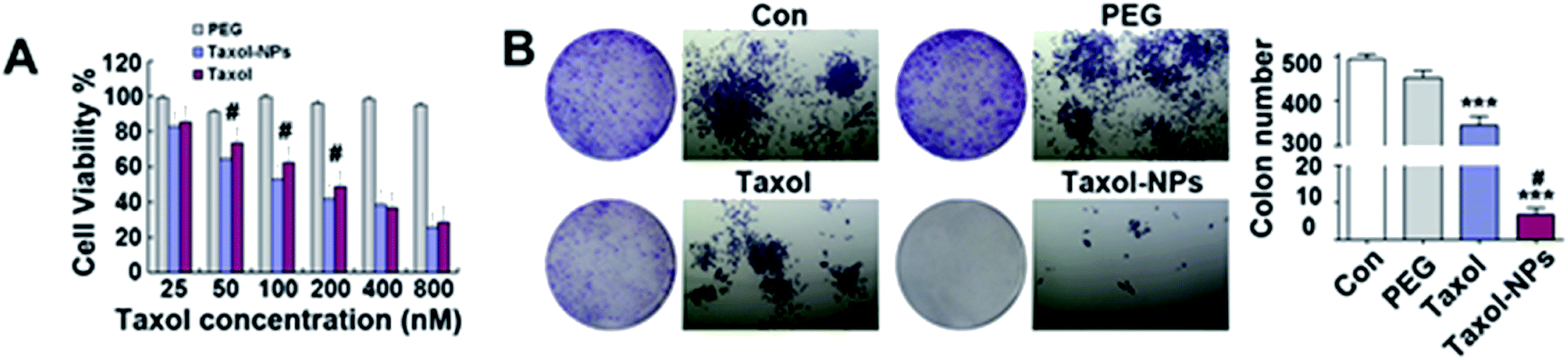 | ||
| Fig. 2 In vitro antitumor effect of Ptx-NPs. (A) Cytotoxicity of PEG, Ptx and Ptx-NPs against A549 cells assessed by the CCK8 assay. (B) Effect of PEG, Ptx and Ptx-NPs on the colony forming ability. # illustrates p < 0.05 of Ptx vs. Ptx-NPs. *** illustrates p < 0.001 vs. the control (Con) group and (or) PEG group. | ||
The IC50 of Ptx-NPs and free Ptx were calculated to be 123.9 and 204.6 nM, respectively. The lower value of IC50 (123.9 nM) of Ptx-NPs further confirmed that Ptx-NPs has stronger cytotoxicity in A549 cells as compared to free Ptx. Our results of CCK8 assay further showed enhanced dose-dependent cytotoxicity of Ptx-NPs than free Ptx, especially at the lower concentration. Higher intracellular accumulation and controlled drug release possibly explained the mechanism of greater cytotoxicity of Ptx-NPs. It has been reported earlier that the self-assembled nanostructures exhibit superior cellular uptake efficiency than small molecules, consistent with the current study.22,29,30
Plate colony formation assay of Ptx-NPs
Different from cytotoxicity, colony forming ability is the capacity of cancer cells to proliferate from one cell to a big colony, a surrogate for “reproductive integrity”.31,32 The clonogenic growth of cancer cells was evaluated through plate colony formation assay. In the current study, the number of colonies in Ptx-NPs treated group was obviously less than those in free Ptx group, while it was not obviously decreased when cells cultured with PEG2000 (Fig. 2C) (P < 0.001). Ptx-NPs group shown an enhancement of clonogenic cell death in A549 cells.Apoptotic relative protein levels were evaluated through western blot analysis. Here we measured the expression of apoptotic proteins, including the pro-apoptotic protein Bax, anti-apoptotic proteins Bcl-2 and Bcl-xl and a major apoptotic marker Caspase-3 (Fig. 3A). The ratio of Bax/Bcl-2 is positively correlated with apoptosis.33 Up-regulation of Bax/Bcl-2 ratio is regarded as the onset of cell apoptosis.34 In Fig. 3B, Ptx-NPs treatment induced a significantly rise of Bax/Bcl-2 ratio compared with free Ptx treatment, demonstrating that more apoptosis was induced by Ptx-NPs in A549 cells (P < 0.05).
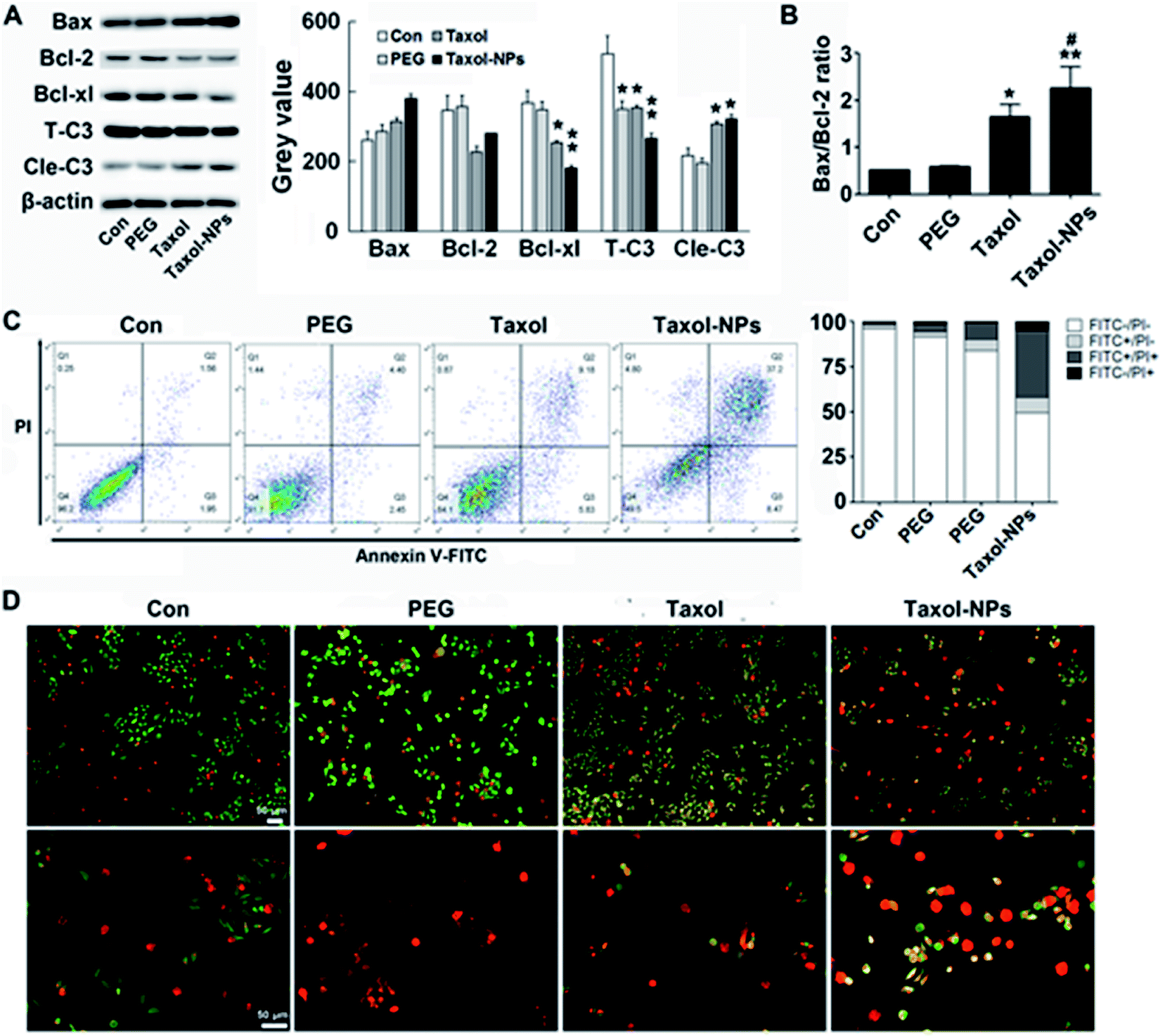 | ||
| Fig. 3 Apoptosis and related proteins expression. (A) Ptx or Ptx-NPs induce expression of apoptosis related proteins. (B) The ratio of Bax/Bcl-2 protein expression. (C) Flow cytometric assessment of cell apoptosis after treatment with Ptx or Ptx-NPs. (D) Fluorescence changes related to apoptosis following treatment with Ptx or Ptx-NPs analyzed by AO/EB double staining. * illustrates p < 0.05 vs. Con group and (or) PEG group. ** illustrates p < 0.01 vs. Con group and (or) PEG group. # illustrates p < 0.05 of Ptx vs. Ptx-NPs. | ||
After various intrinsic and extrinsic factors initiate the apoptosis process, the apoptotic signal is amplified and transmitted by progressive hydrolysis of caspase proteins. Caspase-3 is one of the final executioners of apoptosis and a key biomarker of treatment response via apoptosis. Enzymatic activity of Caspase-3 could be rapidly initiated by the caspase cascade reaction.35–37 In this study, cleaved fragments of Caspase-3 (Cle-C3) were much higher in Ptx-NPs treated group than the free Ptx treated group, again confirming enhanced induction of apoptosis by Ptx-NPs.
Double staining and FACS were performed to observe apoptosis after treatments. As shown in AO/EB double staining, more yellow (early apoptosis) and red (late apoptosis) fluorescence was observed in Ptx-NPs group, indicated the superior proapoptotic effect of Ptx-NPs. Moreover, FACS analysis with dual PI/Annexin-V FITC staining confirmed that significantly higher number of A549 cells treated with Ptx-NPs underwent apoptosis compared to the other two treatment groups (Fig. 3C and D). Therefore, both qualitative and quantitative analysis prove that Ptx-NPs exhibit enhanced cytotoxic effect through augmenting apoptosis in A549 cells (P < 0.05).
Enhanced tumor suppression of Ptx-NPs against A549 lung xenografts
We further estimated in vivo tumor suppressing effect of Ptx-NPs using A549 cell xenograft model of nude mice. First, indicators of liver function and renal function, such as aspartate aminotransferase (AST), alanine aminotransferase (ALT), urea (urea nitrogen) and Cr (creatinine), were tested in pooling plasma of mice. As shown in Table 1, both liver function and renal function were slightly impaired by xenograft planting and obviously impaired by Ptx treatment. Comparing with those groups, Ptx-NPs shown less toxicity for liver and renal in mice. Fig. 4A shown the bodyweights of mouse from the first injection (day 0) to the termination of experiment (day 15). After 3 days of free Ptx treatment, mice showed a significant reduction in bodyweights, while the weights of Ptx-NPs group keep increase as the same pattern as saline group until day 9. Bodyweights of mice represent the general physiologic status of mice. The remarkable decrease of weight after Ptx treatment indicated more serious toxicity of free Ptx than Ptx-NPs (P < 0.05). Furthermore, significantly (p < 0.01) smaller tumor sizes were observed in Ptx group or Ptx-NPs group as compared to the saline group demonstrating that both free Ptx and Ptx-NPs could delay the growth of xenograft tumors (Fig. 4B). Dynamic tumor growth was also observed in Fig. 4C. Mean tumor volume of the saline group shown sustained and rapid growth. The Ptx-NPs treated group exhibited an antitumor advantage compared with the free Ptx treated group beginning on day 6. We further estimated the therapeutic efficacy. Significantly lower values of RTV and T/C% in Ptx-NPs treatment group over free Ptx treatment group also confirmed the superior antitumor effect of Ptx-NPs, especially in day 9–12 after the third injection (Fig. 4D and E) (P < 0.05). As mentioned above, Ptx-NPs showed both the EPR effect to enhanced targeting ability and the “carrier free” effect to reduce the side effect of drug carriers, which contributed to the reduced side effects.| Groups | ALT (U L−1) | AST (U L−1) | BUN (mg dl−1) | Cr (μmol L−1) |
|---|---|---|---|---|
| Saline | 32.144 | 76.459 | 20.793 | 17.011 |
| Mod | 32.375 | 92.093 | 27.581 | 21.085 |
| Ptx | 37.704 | 96.478 | 35.154 | 28.076 |
| Ptx-NPs | 33.236 | 84.556 | 24.493 | 20.809 |
 | ||
| Fig. 4 In vivo antitumor efficacy of Ptx-NPs. (A) Dynamic bodyweights for each group. Arrows indicate timing of drugs administration. (B) CT scan of nude mice and photographs of excised tumor at the 13th day of saline, Ptx, or Ptx-NPs experiment. (C) Tumor volume of mouse xenografts following different treatments. (D) Curve of Relative Tumor Volume (RTV) of xenografts. (E) Growth curve of T/C% during therapy with different treatments. # illustrates p < 0.05 vs. the Ptx group. ** illustrates p < 0.01 vs. the saline group. | ||
Ki-67, as a nucleoprotein involving in the transcription of ribosomal RNA, is a classical marker for cell proliferation and tumor growth.38 After Ptx and Ptx-NPs treatments, Ki-67 positive cells was reduced than control group in tumor tissue. Ptx-NPs group shown the least Ki-67 positive cells percentage, reminding that Ptx-NPs could effectively inhibit tumor growth (Fig. 5). TUNEL staining visualized apoptosis, especially early apoptosis, by labelling free 3′-hydroxyl termini of fragmented DNA. In this study, tumor tissues from Ptx-NPs group shown more TUNEL positive cells and higher protein level of caspases 3, demonstrated that Ptx-NPs could induce more apoptosis, especially early apoptosis, in vivo (Fig. 5). It has been reported previously that in vivo pharmacodynamic and pharmacokinetic characteristics of nanoparticles could be affected by different shape of nanostructures.21 Comparing with the traditional amphilic polymeric nanoparticles, the new Ptx-NPs reported herer were self-assembled by Ptx–SA–PEG, in which Ptx is connected with PEG by succinic acid and the sphere structure of nanoparticles were mostly conformed by Ptx themselves, thus resulting in a relatively high drug loading content. This novel kind of nanoparticles has an advantage of a longer circulation time in vivo than traditional ones.39 The stable sustained release of Ptx-NPs caused an accumulation of Ptx in vivo, thereby leading to an enhanced tumor inhibition in vivo. The targeting ability to tumor tissues of Ptx-NPs mainly owes to the enhanced permission and retention (EPR) effect of the polymeric nanoparticles with certain molecular size.
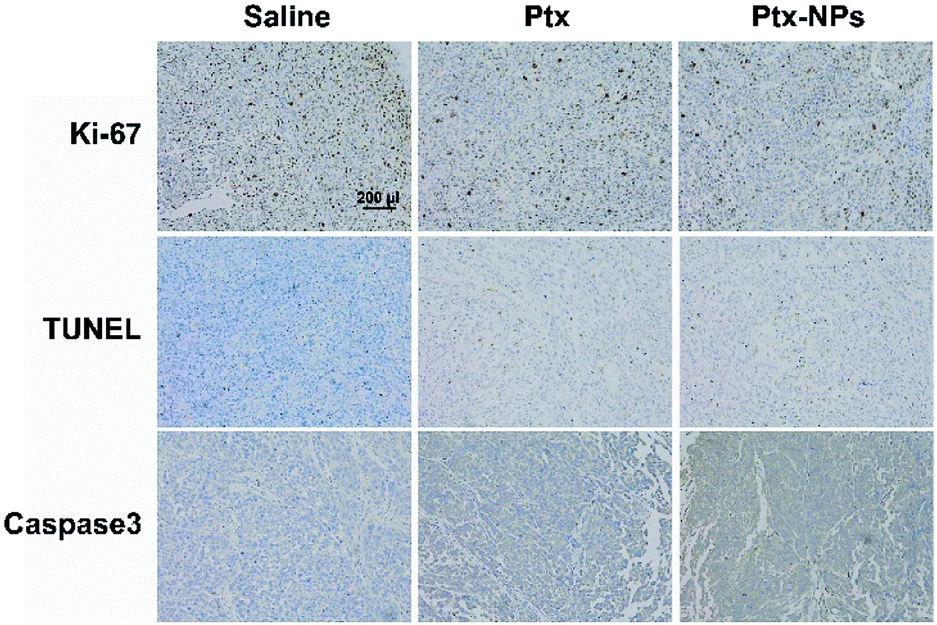 | ||
| Fig. 5 Ptx-NPs induced apoptosis and suppressed tumor proliferation. Representative histological microphotographs of tumor tissue of nude mice treated with saline, Ptx and Ptx-NPs (×100). | ||
This study reports a simplified self-assembled Ptx–SA–PEG delivery system which not only has a high drug loading efficiency but also has a seemingly non-toxic carrier. Ptx-NPs demonstrated efficient cellular uptake with a sustained release pattern in vitro. Ptx-NPs group shown lower cell proliferation and lesser number colonies compared to the equivalent concentration of Ptx. The superior tumor inhibition of Ptx-NPs was possibly induced by the stimulation of apoptotic cell death. Our in vivo data with Ptx-NPs in lung cancer xenografts model also support our observations where Ptx-NPs treatment caused more effectively suppression of tumor growth than free Ptx did. More importantly, as sustained release nanoparticles, Ptx-NPs provide a long-time anti-tumor cytotoxicity with less side effect in mice. This phenomenon possibly attributed to the removal of drug carrier in this self-assembly supramolecular nano-delivery system and the EPR effect of nano-drugs.
Conclusions
These novel self-assembly supramolecular Ptx-loaded nanoparticles lead to stronger antitumor effect and less side effect, which argues for Ptx-NPs being a potential antitumor agent to meliorate the clinical application of Ptx in lung carcinoma.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81871942, 81773211, 81572924, 81672411 and 81472781) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).References
- A. Zaman and T. G. Bivona, Ann. Transl. Med., 2018, 6, 160 CrossRef PubMed.
- T. Kubota, Y. Okano and M. Sakai, Anticancer Res., 2016, 36, 307–312 Search PubMed.
- N. A. Rizvi, M. D. Hellmann and J. R. Brahmer, J. Clin. Oncol., 2016, 34, 2969–2979 CrossRef PubMed.
- E. Bernabeu, M. Cagel and E. Lagomarsino, Int. J. Pharm., 2017, 526, 474–495 CrossRef PubMed.
- P. Baxevanos and G. Mountzios, Ann. Transl. Med., 2018, 6, 139 CrossRef PubMed.
- J. Liebmann, J. A. Cook and J. B. Mitchell, Lancet, 1993, 342, 1428 CrossRef.
- Y. Wang, H. Zhao and J. Peng, J. Biomed. Nanotechnol., 2016, 12, 2097–2111 CrossRef.
- E. Gargioni, F. Schulz and A. Raabe, Ann. Transl. Med., 2016, 4, 523 CrossRef.
- H. Xu, Z. Hou and H. Zhang, Int. J. Nanomed., 2014, 9, 231–242 Search PubMed.
- L. Zhang, Y. He and M. Yu, J. Controlled Release, 2011, 152(suppl 1), e114–e116 CrossRef.
- H. Xu, X. Li and H. Kong, J. Biomed. Nanotechnol., 2016, 12, 1699–1707 CrossRef.
- L. W. Tan, B. Y. Ma and Q. Zhao, J. Biomed. Nanotechnol., 2017, 13, 393–408 CrossRef.
- H. Xu, F. Jia and P. K. Singh, Int. J. Nanomed., 2017, 12, 29–40 CrossRef.
- X. Li, H. Xu and X. Dai, Int. J. Nanomed., 2012, 7, 5183–5190 CrossRef.
- X. Li, X. Lu and H. Xu, Mol. Pharm., 2012, 9, 222–229 CrossRef.
- H. Zhang, Y. Tian and Z. Zhu, Sci. Rep., 2016, 6, 26546 CrossRef.
- K. Greish, Methods Mol. Biol., 2010, 624, 25–37 CrossRef.
- H. Nakamura, F. Jun and H. Maeda, Expert Opin. Drug Delivery, 2015, 12, 53–64 CrossRef.
- W. Zhang, Y. Zhou and N. Yu, J. Biomed. Nanotechnol., 2018, 14, 379–388 CrossRef.
- S. Iqbal, M. H. Rashid and A. S. Arbab, J. Biomed. Nanotechnol., 2017, 13, 355–366 CrossRef.
- Y. Geng, P. Dalhaimer and S. Cai, Nat. Nanotechnol., 2007, 2, 249–255 CrossRef.
- H. Wang, J. Wei and C. Yang, Biomaterials, 2012, 33, 5848–5853 CrossRef.
- R. Tian, H. Wang and R. Niu, J. Colloid Interface Sci., 2015, 453, 15–20 CrossRef.
- M. Zhou, X. Zhang and Y. Yang, Biomaterials, 2013, 34, 8960–8967 CrossRef.
- H. Xu, X. Lu and J. Li, Int. J. Pharm., 2017, 526, 217–224 CrossRef CAS.
- J. Wei, H. Wang and M. Zhu, Nanoscale, 2013, 5, 9902–9907 RSC.
- R. Gref, M. Luck and P. Quellec, Colloids Surf., B, 2000, 18, 301–313 CrossRef.
- M. Das, R. P. Singh and S. R. Datir, Bioconjugate Chem., 2013, 24(4), 626–639 CrossRef.
- J. Liu, H. Xu and Y. Zhang, Int. J. Nanomed., 2014, 9, 197–207 Search PubMed.
- P. Xue, J. Wang and X. Han, Colloids Surf., B, 2019, 180, 202–211 CrossRef PubMed.
- J. A. Plumb, Methods Mol. Med., 1999, 28, 17–23 CAS.
- K. Kawada, T. Yonei and H. Ueoka, Acta Med. Okayama, 2002, 56, 129–134 CAS.
- X. Shu, Z. Dong and S. Shu, Ann. Transl. Med., 2019, 7, 128 CrossRef CAS.
- G. Jia, Q. Wang and R. Wang, OncoTargets Ther., 2015, 8, 303–311 CAS.
- Y. H. Choi and Y. H. Yoo, Oncol. Rep., 2012, 28, 2163–2169 CrossRef CAS PubMed.
- S. J. Lim, M. K. Choi and M. J. Kim, Exp. Mol. Med., 2009, 41, 737–745 CrossRef CAS.
- E. L. Gregoraszczuk, A. Rak-Mardyla and J. Rys, Iran. J. Pharm. Res., 2015, 14, 1153–1161 CAS.
- L. T. Li, G. Jiang and Q. Chen, Mol. Med. Rep., 2015, 11, 1566–1572 CrossRef CAS PubMed.
- Y. Geng, P. Dalhaimer and S. Cai, Nat. Nanotechnol., 2007, 2, 249 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |