
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust hydrophobic gold, glass and polypropylene surfaces obtained through a nanometric covalently bound organic layer†
Alice Mattiuzzi*a,
Ludovic Troian-Gautier b,
Jérémy Mertensc,
François Reniersc,
Jean-François Bergaminid,
Quentin Lenned,
Corinne Lagrost*d and
Ivan Jabin*b
b,
Jérémy Mertensc,
François Reniersc,
Jean-François Bergaminid,
Quentin Lenned,
Corinne Lagrost*d and
Ivan Jabin*b
aX4C, 128 Rue du Chêne Bonnet, 6110 Montigny-le-Tilleul, Belgium. E-mail: amattiuzzi@x4c.eu
bLaboratoire de Chimie Organique, Université libre de Bruxelles (ULB), CP 160/06, 50 Avenue F. D. Roosevelt, 1050 Brussels, Belgium. E-mail: ijabin@ulb.be
cChemistry of Surfaces, Interfaces and Nanomaterials – ChemSIN, Université libre de Bruxelles (ULB), CP 255, Campus de la Plaine, Boulevard du Triomphe, 1050 Brussels, Belgium
dUniv Rennes, CNRS, ISCR-UMR 6226, F-35000 Rennes, France. E-mail: Corinne.lagrost@univ-rennes1.fr
First published on 3rd April 2020
Abstract
The (electro)chemical grafting of a polyfluorinated calix[4]arene on gold, polypropylene and glass is reported. The modified surfaces were characterized by ellipsometry, atomic force microscopy (AFM), and by X-ray photoelectron spectroscopy (XPS). A nanometric, robust and uniform monolayer of covalently surface-bound calix[4]arenes was obtained on the three different materials. For all surfaces, contact angles higher than 110° were recorded, highlighting the hydrophobic character given by this ∼2 nm thin organic monolayer. Remarkably, the contact angle values remained unchanged after 18 months under a laboratory atmosphere. The results presented herein thus present an attractive and sustainable strategy for bringing hydrophobic properties to the interface of a wide range of materials.
Introduction
The development of surfaces with controlled wettability1–3 is of great importance for applications such as anti-biofouling,4,5 anti-frosting and anti-fogging,6,7 self-cleaning,8 anti-corrosion,9,10 or water-proofing.11 Wettability is usually assessed by contact angle measurements, with regimes that span from superhydrophilic, characterized by contact angles smaller than 10°, to superhydrophobic, with contact angles greater than 150° and negligible adhesion forces.1,3,12 Hydrophilicity and hydrophobicity are intermediate behaviours that are usually characterized by a contact angle boundary of 65°.13,14An efficient approach for the development of hydrophobic surfaces consists in the grafting of polyfluorinated organic compounds bearing either a terminal thiol,15 phosphonic acid,16 silane17 or diazonium group,18 possibly combined with a roughening or a nanostructuring of the surface. In this regard, Kim et al. recently reported the modification of nanostructured aluminium with CF3(CF2)9(CH2)2–PO3H2 for anti-frosting applications.6 In this case, contact angles that ranged between 166.0° and 170.8° could be obtained thanks to the nanostructuring of the surface. However, derivatives with more than six CF2 substituents are micropollutants with deleterious environmental effects, as well as potential for bioaccumulation.19 Alternatives have focused on decreasing the number of CF2 repetitions to reduce bioaccumulation through decreased lipophilicity.20 Dichiarante et al. recently described the use of multibranched perfluoroalkyl thiol compounds and 1H,1H,2H,2H-perfluorodecanethiol to develop hydrophobic gold surfaces.21 Static contact angles of ca. 110° were measured, which marginally decreased (<10%) over two weeks. Pinson et al. grafted perfluoroalkyl-substituted aryl-diazonium derivatives on polymethylmethacrylate (PMMA) using hypophosphorous acid as chemical reductant.22 Multilayers of aryl derivatives with thickness in the range 4–10 nm were formed on PMMA, which led to static contact angles of 108°. Alternatively, stable hydrophobic surfaces with contact angles between 110° and 140° were obtained by atmospheric plasma deposition of polyfluorinated precursors.23
Some approaches for preparing hydrophobic surfaces require costly equipment and specially trained operator (plasma deposition, e-beam lithography, etc). More importantly, chemical strategies for surface modification exhibit key drawbacks. Notably, most of them are specific to a given type of materials. For example, thiols can be used only for the modification of coinage metals. Moreover, the resulting self-assembled monolayers (SAMs) show thermal and long-term stability issues, as well as non-uniform distribution across the metallic surface in the case of mixed-thiol compositions.24,25 Methods using silane derivatives have low reproducibility together with side reactions like polymerization.26 Aryldiazonium salts yield robust and stable surface coatings but with a poor control of layer thickness.27,28 Unless particular conditions or specifically designed diazonium derivatives are used, the formation of monolayers remains extremely challenging.29–39 These monolayers are nonetheless highly desired as they do not modify the bulk properties of the material but adduces specific properties.
Additionally, the formation of an organic monolayer implies the use and the presence at the surface of a very small amount of organic compounds, which is a beneficial aspect in the case of compounds with deleterious environmental effects.
Recently, calix[4]arene-tetradiazonium derivatives have been developed and used for (electro)chemical modifications of a wide variety of surfaces (Fig. 1).4,40–47 Calix[4]arenes are macrocycles composed of four phenolic units that are para-substituted and linked in the ortho position through methylene bridges. These methylene bridges prevent polymerization of the diazonium derivatives, and hence lead solely to formation of monolayers. This efficient surface modification strategy was already used on nanoparticles,46,47 conductive,41–44 semi-conductive4 and insulating surfaces.45 Importantly, the use of calix[4]arenes allows to pre-organize four reactive functions in the same direction that could yield up to four covalent bonds with the surface, and hence potentially increase the monolayer's stability. Additionally, the calix[4]arene motif can be functionalized with up to four different substituents, which yields a molecular platform that can easily be post-functionalized through standard chemical transformations such as peptide coupling or click chemistry. In the course of developing a general methodology for the preparation of thin and robust hydrophobic layers, we wanted to evaluate the grafting of a polyfluorinated calix[4]arene-tetradiazonium derivative. Note that such compounds were not described in the literature and a valuable synthetic pathway had to be developed first.
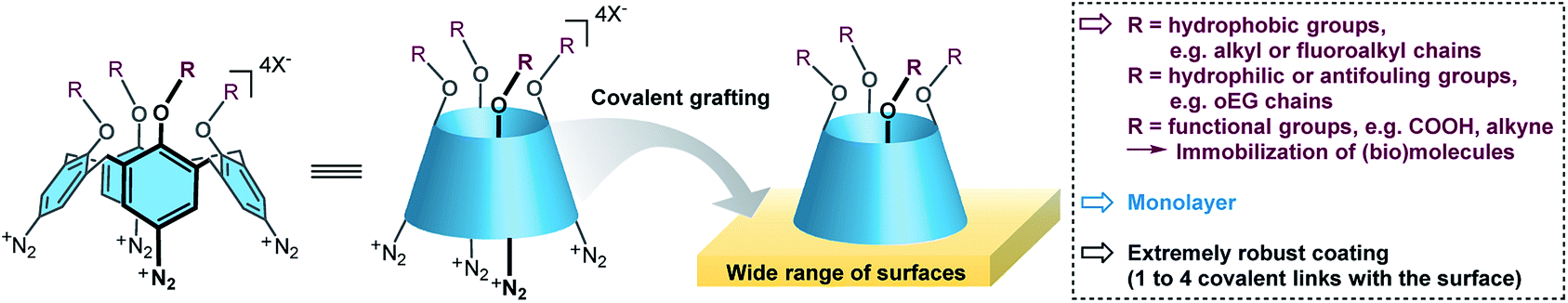 | ||
| Fig. 1 General strategy for the covalent grafting of calix[4]arene tetradiazonium derivatives. | ||
Here, we report on the synthesis of a polyfluorinated calix[4]arene-tetradiazonium derivative, 1, and its use for surface modifications of glass, gold and polypropylene (PP). This calix[4]arene derivative is decorated with four polyfluorinated chains. Due to the conductive or insulating properties of the surfaces, two grafting approaches were used, i.e. an electrochemical one or a chemical one. These grafting methodologies both led to the formation of a very robust hydrophobic monolayer that remained stable during 18 months.
Results and discussion
Synthesis of calix[4]tetra-O-(CH2)3C4F9 tetradiazonium 1
Firstly, calix[4]tris-O-(CH2)3C4F9 4 was obtained in 85% yield through the tris-O-alkylation of p-tBu-calix[4]arene 2 with TsO–(CH2)3–C4F9 3 (ref. 48) using Ba(OH)2·8H2O and BaO in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF/THF mixture (Scheme 1). The tetra-substituted product 5 was then obtained in 83% yield by reaction of calixarene 4 with 3 and sodium hydride in DMF. ipso-Nitration with fuming nitric acid and glacial acetic acid in dichloromethane afforded compound 6 in 66% yield. Reduction of 6 with hydrazine hydrate and Pd/C 10% in ethanol at 60 °C yielded compound 7 in 81% yield. Finally, the desired calix[4]tetra-O-(CH2)3C4F9 tetradiazonium 1 was obtained in 69% yield through the diazotization of 7 with NOBF4 in acetonitrile at −40 °C. The 1H NMR spectrum of 1 is characteristic of a C4v symmetrical calix[4]arene adopting a cone conformation: i.e. one doublet for the ArCH2ax and ArCH2eq protons, one broad signal for the eight CH2O protons and one singlet for the eight ArH protons. Besides, the downfield chemical shift of these ArH protons (i.e. 8.09 ppm) clearly attests of the presence of an electron-withdrawing group. Moreover, the infrared band at 2261 cm−1 is characteristic of the stretching of diazonium groups that, altogether, confirm the formation of 1.
:1 DMF/THF mixture (Scheme 1). The tetra-substituted product 5 was then obtained in 83% yield by reaction of calixarene 4 with 3 and sodium hydride in DMF. ipso-Nitration with fuming nitric acid and glacial acetic acid in dichloromethane afforded compound 6 in 66% yield. Reduction of 6 with hydrazine hydrate and Pd/C 10% in ethanol at 60 °C yielded compound 7 in 81% yield. Finally, the desired calix[4]tetra-O-(CH2)3C4F9 tetradiazonium 1 was obtained in 69% yield through the diazotization of 7 with NOBF4 in acetonitrile at −40 °C. The 1H NMR spectrum of 1 is characteristic of a C4v symmetrical calix[4]arene adopting a cone conformation: i.e. one doublet for the ArCH2ax and ArCH2eq protons, one broad signal for the eight CH2O protons and one singlet for the eight ArH protons. Besides, the downfield chemical shift of these ArH protons (i.e. 8.09 ppm) clearly attests of the presence of an electron-withdrawing group. Moreover, the infrared band at 2261 cm−1 is characteristic of the stretching of diazonium groups that, altogether, confirm the formation of 1.
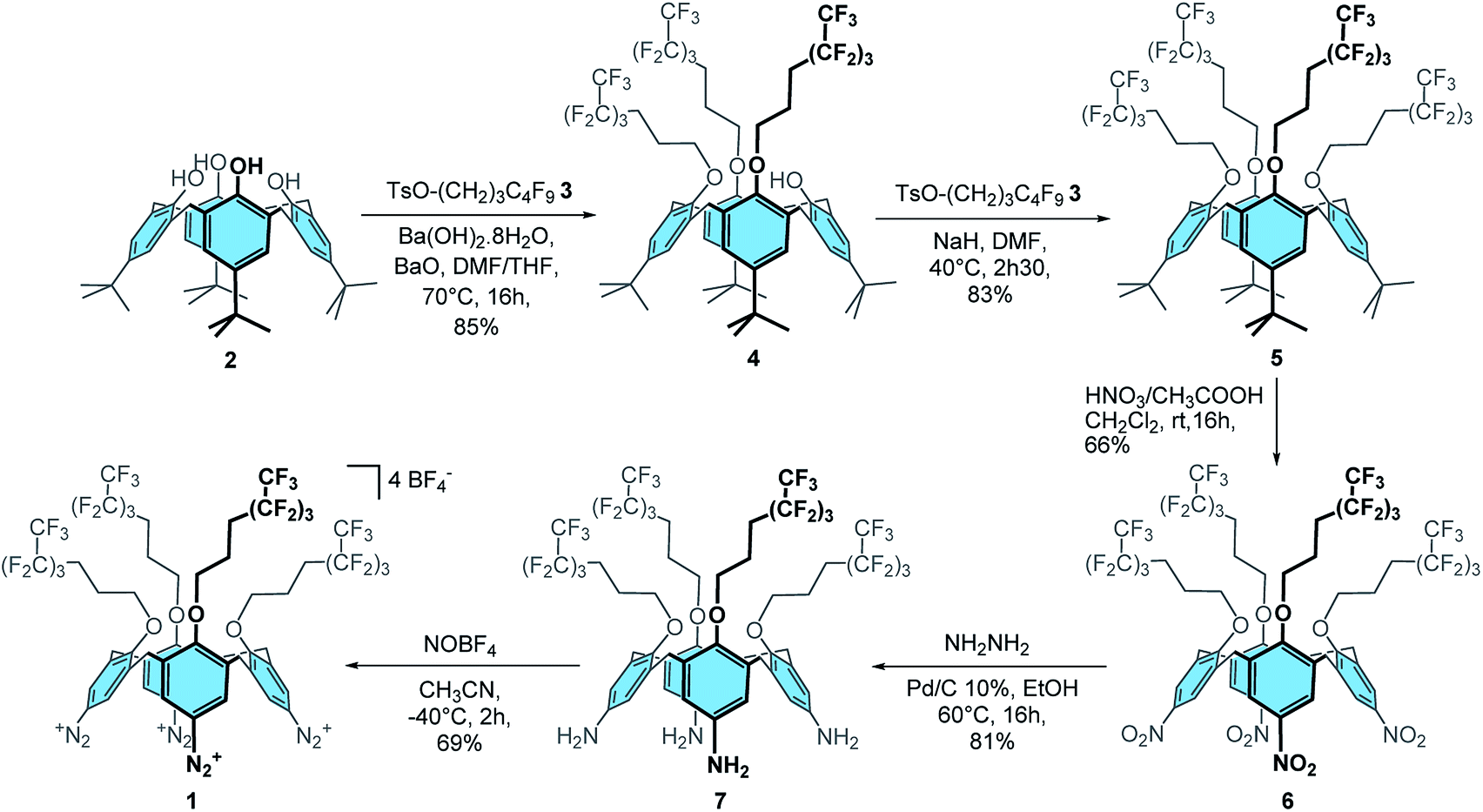 | ||
| Scheme 1 Synthesis of calix[4]tetra-O-(CH2)3C4F9 tetradiazonium 1. | ||
Grafting
Surface modification of glass, polypropylene and gold surfaces was performed through the in situ formation of diazoate salts from 1 and an aqueous NaOH solution.45,49 The procedure consists of soaking the cleaned surfaces for 2 h at room temperature in a 1:1 acetonitrile/NaOH aq (0.1 M) solution containing 1 (2 mM). Note that the presence of acetonitrile helps to solubilize the polyfluorinated calixarene species. The surfaces were then abundantly rinsed with water, acetonitrile, and exposed to ultrasonication for 10 to 30 min in dichloromethane and toluene. As a comparison, electrochemical grafting was also performed with gold substrates, by cyclic voltammetry and chronoamperometry techniques. Potential scanning between 0.5 V and −0.5 V vs. SCE at 100 mV s−1 for 5 cycles resulted in the expected progressive decrease of the current associated with the reduction of diazonium as a result of the charge-transfer blocking properties of the progressively grafted layer. Alternatively, a constant potential (−0.5 V vs. SCE) was applied for 10 minutes and the chronoamperogram also exhibited characteristic behaviour for electrografting of a gold interface with a sudden drop of current. In a previous work, we have shown that electrochemical and NaOH routes result in similar grafted layers.44
Characterization
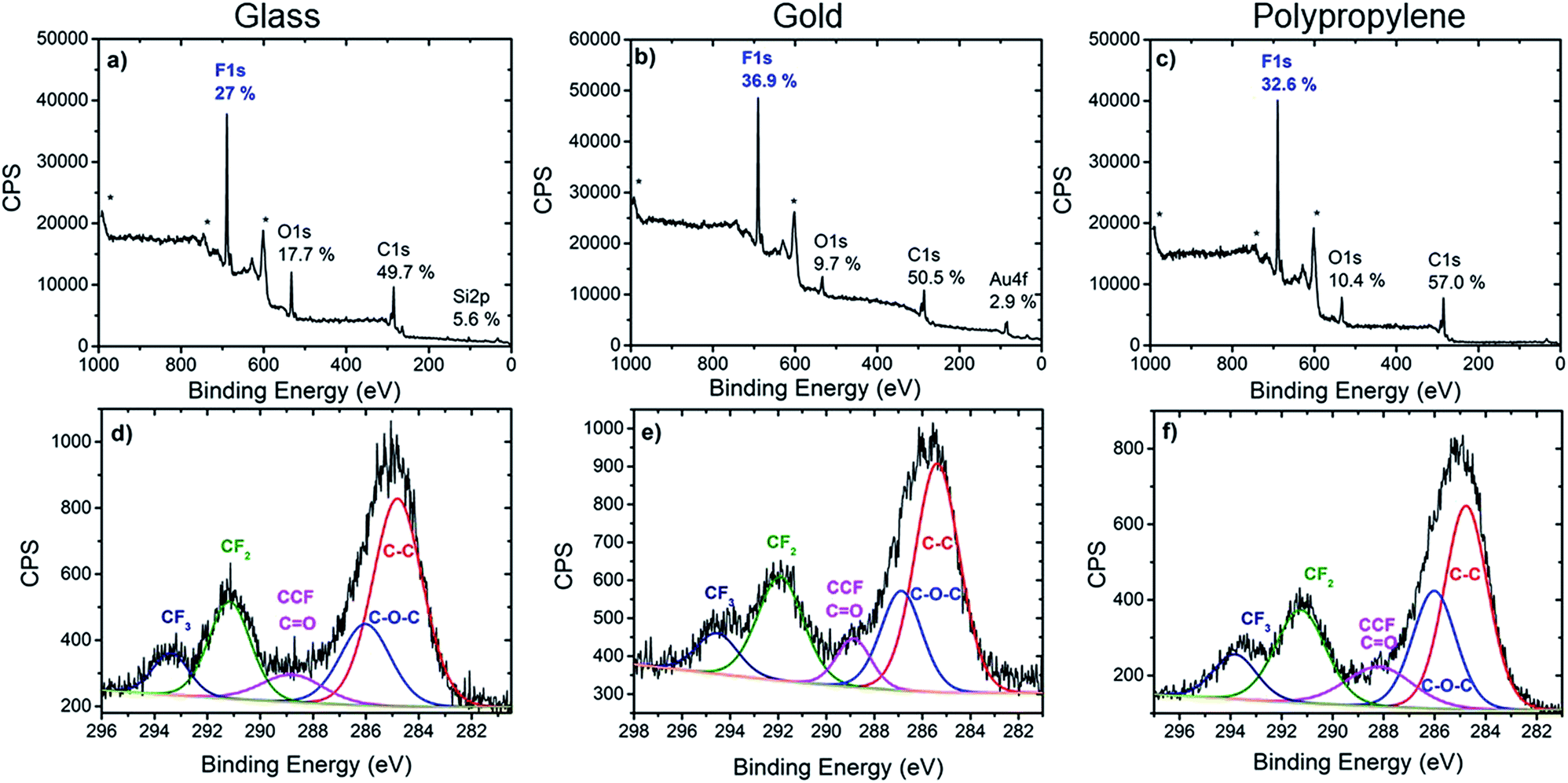 | ||
| Fig. 2 XPS survey spectra (top) and high resolution C 1s spectra (bottom) of glass (a and d), gold (b and e) and polypropylene (c and f) modified with calix[4]arene 1. * denotes Auger peaks. | ||
Further valuable information on the nature of the chemical bonds of the organic layer was extracted from the C 1s core level spectra. In all three cases, the C 1s core level spectra was decomposed into several components which reflected the chemical structure of the layer. The main component at 285 ± 0.3 eV is attributed to aryl and aliphatic C–C bonds that constitute most of the calix[4]arene backbone. Note that these C–C bonds could also arise from the surface itself in the case of polypropylene or from contamination. A component at 286.1 ± 0.3 eV is assigned to the carbon atoms involved in the C–O–C linkage present in the calix[4]arene core. The component at 288.4 ± 0.3 eV corresponds to carbon atoms in CH–CF bonds. However, these two last components could also partly contribute to oxidized contamination species.45 Importantly, components at 291.3 ± 0.4 eV and 293.5 ± 0.3 eV, that correspond to the CF2 and CF3 moieties present on the calix[4]arene small rim are clearly observed. The area ratio of CF2 and CF3 components is found in the range 0.35–0.40 for all substrates, in good agreement with 1:3 ratio expected from the molecular structure. This provides additional confirmation that the polyfluorinated calix[4]arene is indeed grafted on glass, gold and PP surfaces.
 | ||
| Fig. 3 AFM topography images of (A) bare gold surface 9 μm2, gold surfaces after grafting of calix[4]arene 1 by (B) immersing in hydroxide solution, 9 μm2, (C) electrochemical reduction using CV, 4 μm2 and (D) electrochemical reduction using chronoamperometry, ca. 4 μm2. | ||
| Samples | Bare surfaces | Coated surfaces | |
|---|---|---|---|
| Fresh | 18 months-old | ||
| Glass | 24.6 ± 2.0° | 110.0 ± 1.8° | 107.2 ± 1.7° |
| Gold | 64.7 ± 2.1° | 113.7 ± 2.2° | 111.2 ± 0.7° |
| Polypropylene | 102.9 ± 3.9° | 112.6 ± 4.0° | 108.1 ± 0.5° |
 | ||
| Fig. 4 Images of 2 μL water droplets in contact with (top) bare glass, gold and polypropylene surfaces and with (bottom) the same surfaces freshly modified with calix[4]arene 1. | ||
Preliminary ageing studies were then undertaken. The coated surfaces were left on a benchtop under laboratory atmosphere for eighteen months. The surfaces were then quickly rinsed with toluene and water, and finally thoroughly dried under a stream of argon. Static contact angles of the aged surfaces were then measured. Interestingly, static contact angle values of the 18 month-old surfaces were within 5% of the values obtained for freshly modified surfaces. A control experiment with a bare gold surface that underwent the same ageing treatment led to static contact angle value of 67 ± 3.2°. These results demonstrate the remarkable robustness of the surfaces covalently modified with polyfluorinated calix[4]arene derivatives.
Conclusions
The synthesis of a calix[4]arene tetradiazonium bearing polyfluorinated chains (1) was reported for the first time. This compound led to a covalently bound monolayer onto gold, glass and polypropylene, either through an electrochemical approach (for conductive surfaces) or through a chemical modification using sodium hydroxide (for both conductive and insulating surfaces). Both approaches gave similar results but expand this grafting methodology to a broader range of materials. The grafting was validated by ellipsometry, AFM and XPS analyses. In all cases, static contact angle values greater than 110° were determined, indicating a strong hydrophobicity despite the ultrathin layer of calixarenes that was immobilized. A remarkable aspect of this methodology was the high robustness of the polyfluorinated layer, as illustrated by the almost constant contact angle values obtained despite aggressive washing treatments and ageing studies. This grafting methodology is very simple to operate. In addition, it could be easily applied to a wide range of materials whenever a robust and thin hydrophobic coating is required.Experimental
Materials and methods
All solvents and reagents were at least of reagent grade quality and were purchased either from Alfa Aesar, Sigma-Aldrich or Acros organics. 4,4,5,5,6,6,7,7,7-Nonafluoroheptanol was purchased from Fluorochem. Anhydrous DMF and CHCl3 were obtained from Acros organics. Gold coated silicon wafer (1000 Å layer thickness) and TBAPF6 (electrochemical grade) were purchased from Sigma-Aldrich. Polypropylene (biaxially oriented, 50 μm) was purchased from Goodfellow. The typical size of the surfaces reported in this paper was 1 cm2. Ultrapure water was obtained via a Millipore Milli-Q system (18.2 MΩ.cm).Caution! Although we have not encountered any problem, it is noted that diazonium salts are potentially explosive and should be handled with appropriate precautions.
Electrochemical grafting
Gold surfaces were first immersed in a “piranha” solution (H2SO4/H2O2 3:1) and sonicated for 10 minutes. Caution! This solution is a very strong oxidant and should be handled very carefully. They were then washed with concentrated H2SO4 and with ultrapure water and dried under argon atmosphere. The electrochemical grafting was performed by cyclic voltammetry (CV) or chronoamperometry (CA) in 0.1 M TBAPF6 anhydrous acetonitrile containing 1 mM of calix[4]arene-tetradiazonium salt using an Autolab PGSTAT 30 potentiostat/galvanostat (Metrohm Autolab B.V.). For CV, the potential was scanned between +0.5 V and −0.5 V vs. SCE at 100 mV s−1 (5 cycles). For CA, a constant potential of −0.5 V vs. SCE was applied for 10 min. An extensive rinsing with solvents of decreasing polarity (acetonitrile, dichloromethane, toluene) under sonication for 20–30 min was necessary to remove the accumulation of loosely adsorbed calix[4]arenes at the top of the covalently grafted layer. This accumulation of adsorbed material was probably due to strong interactions between the polyfluorinated chains. The surfaces were finally dried using an argon flow.
Chemical grafting using sodium hydroxide
Gold and glass surfaces were first immersed in a “piranha” solution (H2SO4/H2O2 3:1) and sonicated for 10 min. Gold surfaces were then washed with concentrated H2SO4 and with ultrapure water and dried under argon atmosphere. Glass surfaces were then thoroughly rinsed with ultrapure water and cleaned under sonication for 10 min in absolute ethanol, acetone, isopropanol and ultrapure water before being dried under argon atmosphere. Polypropylene surfaces were cleaned with isopropanol under sonication for 10 min, washed with ultrapure water and dried using an argon flow. The surfaces were then immersed in a 2 mM suspension of the diazonium salt in a 1:1 mixture of acetonitrile and aqueous 0.1 M sodium hydroxide. The surfaces were left soaking for 2 h without stirring in order to avoid any mechanical friction of the surface with the magnetic stirrer. An extensive rinsing with solvents of decreasing polarity (acetonitrile, dichloromethane, toluene) under sonication for 20–30 min was necessary to remove the accumulation of loosely adsorbed calix[4]arenes at the top of the covalently grafted layer. This accumulation of adsorbed material was probably due to strong interactions between the polyfluorinated chains. The surfaces were finally dried using an argon flow.
Atomic force microscopy (AFM)
AFM experiments were performed with a NT-MDT Ntegra microscope. Topography images were acquired in semi-contact mode using silicon nitride tips with a resonance frequency of about 350 kHz.Ellipsometry measurements
The thicknesses of the layers deposited on gold were measured using a spectroscopic a-SE ellipsometer (J. A. Woollam, Co.). The polarization angles Ψ and Δ were recorded in the 380–900 nm wavelength range at different incident angles, 65°, 70° and 75°. Prior to modification, Ψ and Δ were measured to build a substrate optical model; the refractive index (n) and the extinction coefficient (k) were determined as 0.48 and 3.54, respectively. This model was used after modification to consider the substrate's contribution in the ellipsometry measurements. The thickness of the organic layer was estimated through a fitting according to the Cauchy model with n = 1.5 and k = 0. The validity of the fitting was quantified by the mean square error (MSE), with value below 4. All the values given in the manuscript are averaged values of 3–5 measurements performed on four different substrates.XPS measurements
XPS analysis was performed on a Physical Electronics PHI-5600 photoelectron spectrometer. Survey scans were used to determine the elemental chemical composition of the surface. Narrow-region photoelectron spectra were used for the chemical study of the C 1s. The spectra were acquired using the Mg anode (1253.6 eV) operating at 300 W. Wide surveys were acquired at a pass-energy of 187.5 eV with a five-scans accumulation (time per step: 50 ms, eV per step: 0.8) and high-resolution spectra of the C 1s peaks were recorded at a pass-energy of 23.5 eV with an accumulation of 5 scans (time per step: 150 ms, eV per step: 0.05). Spectral calibration was determined by setting C 1s at 285 eV. The atomic concentration for surface composition was estimated using the integrated peaks areas. The peaks areas were normalized by the manufacturer-supplied sensitivity factor (SC 1s = 0.205, SF 1s = 1, SO 1s = 0.63, SN 1s = 0.38, SSi 2p = 0.17, SAu 4f = 3.32). The core level C 1s spectra were peak-fitted using the CasaXPS software (Casa Software, Ltd., version 2.3.16).Contact angles measurements
The static contact angles of 2 μL Milli-Q water drops were measured on five different spots and two or three different samples with an easy drop goniometer (Krüss). The contact angles were determined using tangent 1 circle fitting, or Young–Laplace fitting models.Synthetic procedures
:3), affording compound 3 as a colourless liquid (29.3 g, 68 mmol, 81%). 1H NMR (300 MHz, CDCl3, 298 K): δ(ppm) = 1.91–2.00 (m, 2H, CH2CH2C4F9), 2.05–2.21 (m, 2H, CH2C4F9), 2.45 (s, 3H, CH3Ts), 4.11 (t, 3J = 6.0 Hz, 2H, TsOCH2), 7.36 (d, 3J = 8.3 Hz, 2H, ArH), 7.79 (d, 3J = 8.3 Hz, 2H, ArH). The 1H NMR spectrum of 3 is in accordance with the one reported in the literature.48:1 DMF/THF mixture. Ba(OH)2·8H2O (9.3 g, 29 mmol) and BaO (8.9 g, 65 mmol) were then added. The reaction mixture was stirred at room temperature and TsO-(CH2)3C4F9 3 (25.9 g, 60 mmol) was added. The mixture was then heated at 70 °C for 14 h. The mixture was evaporated under reduced pressure and the resulting residue was dissolved in 450 mL of dichloromethane. The organic layer was washed once with 100 mL of 2 M HCl and then with water until the aqueous layer reached a pH of 7. The organic layer was then evaporated under reduced pressure to yield compound 4 as a yellow solid (11.1 g, 7.8 mmol, 85%). Mp = 60 °C. IR (NaCl) ν (cm−1) = 2963, 1642, 1481, 1228, 1134, 1016. 1H NMR (400 MHz, CDCl3, 298 K): δ(ppm) = 0.82 (s, 18H, tBu), 1.33 (s, 9H, tBu), 1.34 (s, 9H, tBu), 2.10–2.60 (m, 12H, CH2CH2C4F9), 3.23 (d, 2J = 12.4 Hz, 2H, ArCH2eq), 3.27 (d, 2J = 13.2 Hz, 2H, ArCH2eq), 3.89 (t, 3J = 6.4 Hz, 4H, OCH2), 3.99 (t, 3J = 8.0 Hz, 2H, OCH2), 4.23 (d, 2J = 12.8 Hz, 2H, ArCH2ax), 4.27 (d, 2J = 12.4 Hz, 2H, ArCH2ax), 5.17 (s, 1H, OH), 6.52 (mult., 4H, ArH), 7.08 (s, 2H, ArH), 7.17 (s, 2H, ArH). 13C NMR (100 MHz, CDCl3, 298 K): δ(ppm) = 20.9, 21.2, 27.4 (t, 3J = 21.3 Hz), 28.1 (t, 3J = 22.4 Hz), 31.1, 31.2, 31.4, 31.7, 31.8, 33.8, 34.0, 34.3, 73.2, 74.7, 124.9, 125.2, 125.3, 126.1, 129.4, 131.6, 131.8, 135.7, 142.0, 145.8, 146.4, 150.6, 151.2, 153.3. HRMS (ESI+): m/z calcd for C65H75F27NO4 [M + NH4]+: 1446.5294; found: 1446.5259.:1), affording compound 5 as a white solid (4.9 g, 2.9 mmol, 83%). Mp = 121 °C. IR (NaCl) ν (cm−1) = 2962, 1643, 1480, 1228, 1134. 1H NMR (400 MHz, CDCl3, 298 K): δ(ppm) = 1.09 (s, 36H, tBu), 2.03–2.36 (m, 16H, CH2CH2C4F9), 2.19 (d, 2J = 12.8 Hz, 4H, ArCH2eq), 3.91 (t, 3J = 7.6 Hz, 8H, OCH2), 4.29 (d, 2J = 12.4 Hz, 4H, ArCH2ax), 6.81 (s, 8H, ArH). 13C NMR (100 MHz, CDCl3, 298 K): δ(ppm) = 21.1, 27.5 (t, 3J = 23.1 Hz), 31.2, 31.5, 34.0, 74.1, 125.4, 133.6, 145.4, 153.0. HRMS (ESI+): m/z calcd for C72H80F36NO4 [M + NH4]+: 1706.5487; found: 1706.5507.:1 mixture of fuming nitric acid and glacial acetic acid were then added. The reaction mixture was stirred at room temperature overnight. The mixture was then evaporated under reduced pressure. The residue was solubilized in 500 mL of dichloromethane and extracted with water until the aqueous phase reached a pH of 7. The organic layer was then evaporated under reduced pressure. The residue was dissolved in methanol, filtered and the filtrate was evaporated under reduced pressure to yield compound 6 as a yellow solid (3.2 g, 1.9 mmol, 66%). Mp = 160 °C (dec.). IR (NaCl) ν (cm−1) = 2965, 1642, 1527, 1349, 1227, 1132, 1007. 1H NMR (400 MHz, CDCl3, 298 K): δ(ppm) = 2.03–2.27 (m, 16H, CH2CH2C4F9), 3.51 (d, 2J = 14.0 Hz, 4H, ArCH2eq), 4.06 (t, 3J = 7.2 Hz, 8H, OCH2), 4.43 (d, 2J = 14.0 Hz, 4H, ArCH2ax), 7.62 (s, 8H, ArH). 13C NMR (100 MHz, CDCl3, 298 K): δ(ppm) = 21.3, 27.2 (t, 3J = 22.9 Hz), 31.2, 74.8, 124.5, 135.3, 143.7, 160.6. This compound could not be detected by ESI-HRMS.Conflicts of interest
L. T.-G. was a postdoctoral researcher for X4C between October 2014 and September 2015. I. J. and C. L. are shareholders of X4C. I. J. is a consultant for X4C. All other authors declare that they have no conflict of interest.Acknowledgements
L. T.-G. is a postdoctoral researcher of the Fonds de la Recherche Scientifique – FNRS. The Microgravity Research Center (ULB), and especially Jean-Charles Dupin, is acknowledged for granting access to the goniometer.Notes and references
- X.-M. Li, D. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 1350–1368 RSC.
- S. Wang, K. Liu, X. Yao and L. Jiang, Chem. Rev., 2015, 115, 8230–8293 CrossRef CAS PubMed.
- J. Yong, F. Chen, Q. Yang, J. Huo and X. Hou, Chem. Soc. Rev., 2017, 46, 4168–4217 RSC.
- P. Blond, A. Mattiuzzi, H. Valkenier, L. Troian-Gautier, J.-F. Bergamini, T. Doneux, E. Goormaghtigh, V. Raussens and I. Jabin, Langmuir, 2018, 34, 6021–6027 CrossRef CAS PubMed.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- A. Kim, C. Lee, H. Kim and J. Kim, ACS Appl. Mater. Interfaces, 2015, 7, 7206–7213 CrossRef CAS PubMed.
- J. A. Howarter and J. P. Youngblood, Adv. Mater., 2007, 19, 3838–3843 CrossRef CAS.
- P. Ragesh, V. Anand Ganesh, S. V. Nair and A. S. Nair, J. Mater. Chem. A, 2014, 2, 14773–14797 RSC.
- F. Xiao, S. Yuan, B. Liang, G. Li, S. O. Pehkonen and T. Zhang, J. Mater. Chem. A, 2015, 3, 4374–4388 RSC.
- Y. Wei, L. Hongtao and Z. Wei, RSC Adv., 2015, 5, 103000–103012 RSC.
- C.-H. Xue, X.-J. Guo, M.-M. Zhang, J.-Z. Ma and S.-T. Jia, J. Mater. Chem. A, 2015, 3, 21797–21804 RSC.
- C. W. Extrand, Langmuir, 2016, 32, 8608–8612 CrossRef CAS PubMed.
- C. Guo, S. Wang, H. Liu, L. Feng, Y. Song and L. Jiang, J. Adhes. Sci. Technol., 2008, 22, 395–402 CrossRef CAS.
- A. J. Patel, P. Varilly and D. Chandler, J. Phys. Chem. B, 2010, 114, 1632–1637 CrossRef CAS PubMed.
- X. Yu, Z. Wang, Y. Jiang, F. Shi and X. Zhang, Adv. Mater., 2005, 17, 1289–1293 CrossRef CAS.
- W. Ma, H. Wu, Y. Higaki, H. Otsuka and A. Takahara, Chem. Commun., 2012, 48, 6824–6826 RSC.
- J. A. Finlay, S. Krishnan, M. E. Callow, J. A. Callow, R. Dong, N. Asgill, K. Wong, E. J. Kramer and C. K. Ober, Langmuir, 2008, 24, 503–510 CrossRef CAS PubMed.
- G. Deniau, L. Azoulay, L. Bougerolles and S. Palacin, Chem. Mater., 2006, 18, 5421–5428 CrossRef CAS.
- M. P. Krafft and J. G. Riess, Curr. Opin. Colloid Interface Sci., 2015, 20, 192–212 CrossRef CAS.
- Z. Wang, I. T. Cousins, M. Scheringer and K. Hungerbühler, Environ. Int., 2013, 60, 242–248 CrossRef CAS PubMed.
- V. Dichiarante, M. I. Martinez Espinoza, L. Gazzera, M. Vuckovac, M. Latikka, G. Cavallo, G. Raffaini, R. Oropesa-Nuñez, C. Canale, S. Dante, S. Marras, R. Carzino, M. Prato, R. H. A. Ras and P. Metrangolo, ACS Sustain. Chem. Eng., 2018, 6, 9734–9743 CrossRef CAS.
- M. M. Chehimi, A. Lamouri, M. Picot and J. Pinson, J. Mater. Chem. C, 2014, 2, 356–363 RSC.
- J. Hubert, N. Vandencasteele, J. Mertens, P. Viville, T. Dufour, C. Barroo, T. Visart de Bocarmé, R. Lazzaroni and F. Reniers, Plasma Process. Polym., 2015, 12, 1174–1185 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS PubMed.
- S.-i. Imabayashi, D. Hobara, T. Kakiuchi and W. Knoll, Langmuir, 1997, 13, 4502–4504 CrossRef CAS.
- W. Yoshida, R. P. Castro, J.-D. Jou and Y. Cohen, Langmuir, 2001, 17, 5882–5888 CrossRef CAS.
- D. Bélanger and J. Pinson, Chem. Soc. Rev., 2011, 40, 3995–4048 RSC.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429–439 RSC.
- C. Combellas, F. Kanoufi, J. Pinson and F. I. Podvorica, J. Am. Chem. Soc., 2008, 130, 8576–8577 CrossRef CAS PubMed.
- L. T. Nielsen, K. H. Vase, M. Dong, F. Besenbacher, S. U. Pedersen and K. Daasbjerg, J. Am. Chem. Soc., 2007, 129, 1888–1889 CrossRef CAS PubMed.
- K. Malmos, M. Dong, S. Pillai, P. Kingshott, F. Besenbacher, S. U. Pedersen and K. Daasbjerg, J. Am. Chem. Soc., 2009, 131, 4928–4936 CrossRef CAS PubMed.
- T. Menanteau, E. Levillain, A. J. Downard and T. Breton, Phys. Chem. Chem. Phys., 2015, 17, 13137–13142 RSC.
- O. Fontaine, J. Ghilane, P. Martin, J.-C. Lacroix and H. Randriamahazaka, Langmuir, 2010, 26, 18542–18549 CrossRef CAS PubMed.
- G. Shul, C. A. C. Ruiz, D. Rochefort, P. A. Brooksby and D. Bélanger, Electrochim. Acta, 2013, 106, 378–385 CrossRef CAS.
- J. Carvalho Padilha, J.-M. Noël, J.-F. Bergamini, J. Rault-Berthelot and C. Lagrost, ChemElectroChem, 2016, 3, 572–580 CrossRef CAS.
- Y. R. Leroux, H. Fei, J.-M. Noël, C. Roux and P. Hapiot, J. Am. Chem. Soc., 2010, 132, 14039–14041 CrossRef CAS PubMed.
- Y. R. Leroux and P. Hapiot, Chem. Mater., 2013, 25, 489–495 CrossRef CAS.
- L. Lee, Y. R. Leroux, P. Hapiot and A. J. Downard, Langmuir, 2015, 31, 5071–5077 CrossRef CAS PubMed.
- Y. R. Leroux, F. Hui, J.-M. Noël, C. Roux, A. J. Downard and P. Hapiot, Langmuir, 2011, 27, 11222–11228 CrossRef CAS PubMed.
- J. P. Buttress, D. P. Day, J. M. Courtney, E. J. Lawrence, D. L. Hughes, R. J. Blagg, A. Crossley, S. E. Matthews, C. Redshaw, P. C. Bulman Page and G. G. Wildgoose, Langmuir, 2016, 32, 7806–7813 CrossRef CAS PubMed.
- G. De Leener, F. Evoung-Evoung, A. Lascaux, J. Mertens, A. G. Porras-Gutierrez, N. Le Poul, C. Lagrost, D. Over, Y. R. Leroux, F. Reniers, P. Hapiot, Y. Le Mest, I. Jabin and O. Reinaud, J. Am. Chem. Soc., 2016, 138, 12841–12853 CrossRef CAS PubMed.
- V. Malytskyi, L. Troian-Gautier, A. Mattiuzzi, S. Lambotte, B. Cornelio, C. Lagrost and I. Jabin, Eur. J. Org. Chem., 2018, 2018, 6590–6595 CrossRef CAS.
- A. Mattiuzzi, I. Jabin, C. Mangeney, C. Roux, O. Reinaud, L. Santos, J.-F. Bergamini, P. Hapiot and C. Lagrost, Nat. Commun., 2012, 3, 1130 CrossRef PubMed.
- L. Santos, A. Mattiuzzi, I. Jabin, N. Vandencasteele, F. Reniers, O. Reinaud, P. Hapiot, S. Lhenry, Y. Leroux and C. Lagrost, J. Phys. Chem. C, 2014, 118, 15919–15928 CrossRef CAS.
- L. Troian-Gautier, D. E. Martínez-Tong, J. Hubert, F. Reniers, M. Sferrazza, A. Mattiuzzi, C. Lagrost and I. Jabin, J. Phys. Chem. C, 2016, 120, 22936–22945 CrossRef CAS.
- L. Troian-Gautier, H. Valkenier, A. Mattiuzzi, I. Jabin, N. V. den Brande, B. V. Mele, J. Hubert, F. Reniers, G. Bruylants, C. Lagrost and Y. Leroux, Chem. Commun., 2016, 52, 10493–10496 RSC.
- H. Valkenier, V. Malytskyi, P. Blond, M. Retout, A. Mattiuzzi, J. Goole, V. Raussens, I. Jabin and G. Bruylants, Langmuir, 2017, 33, 8253–8259 CrossRef CAS PubMed.
- S. Ishihara, Y. Furuki, J. P. Hill, K. Ariga and S. Takeoka, J. Nanosci. Nanotechnol., 2014, 14, 5130–5137 CrossRef CAS PubMed.
- F. I. Podvorica, F. Kanoufi, J. Pinson and C. Combellas, Electrochim. Acta, 2009, 54, 2164–2170 CrossRef CAS.
- Molecular modeling was performed with Chem3D software.
- H.-T. Liu, X.-G. Xiong, P. Diem Dau, Y.-L. Wang, D.-L. Huang, J. Li and L.-S. Wang, Nat. Commun., 2013, 4, 2223 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR of the newly synthesized compounds. See DOI: 10.1039/d0ra01011a |
| This journal is © The Royal Society of Chemistry 2020 |