
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering a protease-based and site-specific PEGylation-based strategy for the controlled release of exenatide
Aihong Zhangab,
Yin Lina,
Shirly Nongc,
Wei Zhaoc and
Mei Dong *a
*a
aDepartment of Pharmaceutical, The Third Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China. E-mail: dmmd10101@outlook.com
bDepartment of Clinical Medicine,School of Basic Medical Sciences, Harbin Medical University, Harbin, Heilongjiang 150040, P. R. China
cCollege of Life Science and Technology, Sichuan University, Chengdu, Sichuan 610041, P. R. China
First published on 30th June 2020
Abstract
Using the commercially available antidiabetic drug exenatide (exendin-4) as a model peptide, we designed a novel exenatide derivative, termed LEX-1, comprising a 12-mer albumin-binding peptide, a protease-sensitive linker and a native exenatide. In addition, site-specific PEGylation was performed using LEX-1 as a lead sequence to generate four exenatide derivatives (LEX-2 to LEX-5). Moreover, we determined the optimal molecular weight of maleimide-derivatized PEG for the site-specific PEGylation of LEX-1 by an in vitro stability assay and an in vivo hypoglycemic efficacy test. As a result, LEX-3 (PEG10 kDa) exerted enhanced proteolytic stability, rational release rate of free exenatide and the best glucose-stabilizing capability compared with others. In addition, the prolonged hypoglycemic effects of LEX-1 and LEX-3 were demonstrated in type 2 diabetic mice by multiple OGTTs and a hypoglycemic duration test. Furthermore, a pharmacokinetic test was conducted using Sprague Dawley (SD) rats; LEX-3 (PEG10 kDa) showed the best circulating t1/2 of ∼119.7 h for exenatide release from LEX-3, suggesting that LEX-3 has the potential to be developed into a once-weekly antidiabetic agent. The consecutive 8 week treatment of both LEX-1 and LEX-3 exhibited enhanced beneficial efficacies on body weight gain, cumulative food intake, % fat and hemoglobin A1c (HbA1c) reduction compared with exenatide treatment. Meanwhile, the chronic administration of LEX-1 and LEX-3 also effectively improved the blood biochemical indexes. Our results indicate the enhanced antidiabetic effects of LEX-1 and LEX-3, and our strategy of PEGylation and albumin conjugation can be applied to other bioactive agents.
Introduction
Numerous therapeutic peptides have already been investigated for medical applications.1 It is well-known that the efficacy of a therapeutic agent is mainly dependent on its in vivo lifespan.2 However, the efficacies of pharmaceutical peptides are limited due to their rapid clearance in circulation, resulting in poor druggability.3,4 To date, mechanisms including glomerular filtration in the kidney and proteolysis by proteases are the reasons for the short t1/2 of peptide drugs.4,5 The increase in the molecular size of protein drugs reduces the renal clearance and further extends the in vivo exposure of peptide therapies.6 As a result, extensive research has been carried out to prolong the in vivo lifespan of peptide drugs.7,8 Among these methods, a certain domain of polypeptides, which can bind to a plasma protein, such as human serum albumin (HSA), or suitable site-specific PEGylation has shown potential to prolong the short in vivo t1/2 of the therapeutic agents.9,10HSA, as one of the most useful in vivo molecule transporters, exerts a maximum t1/2 of ∼19 days.11,12 Peptides or proteins that bind tightly with HSA exert longer lifespans compared with the free ones. Similarly, a site-specific PEGylation technology, covalent attachment of PEG to targeted peptides or has been applied for drug modification to make the existing biopharmaceuticals clinically more effective than before.13–15 The active sites of modified peptides or proteins for enzymes or antibodies are protected by PEG conjugation,16 but the size of the PEG chain and the modified sites are closely related to the biological activity of the peptides or proteins. For example, the application of random PEGylation may result in an exponential decrease in biological activity because the attachment of PEGs may reject the binding of the target molecules.17
According to a previously reported protease-based strategy,18 we used exendin-4, a clinically approved therapeutic drug for type 2 diabetes with a short elimination t1/2 of 3–4 h, as a domain of bioactive peptides to design new agents. We selected the albumin-binding domain (LPHSHRAHSLPP), which possesses high binding affinity (Kd = 1.67 μM) for HSA, to construct an albumin-binding fusion peptide. Previous research has proved that thrombin is specific for the sequence Phe (F)-Asn (N)-Pro (P)-Arg (R) or Lys (K)-Xxx (X)-Zzz (Z), while dipeptidyl peptidase-4 (DPP4) is specific for an N-terminal dipeptide with Pro (P) or Ala (A).19 The optimized protease-cleavable linker sequence was determined as FNPKCP, which is scissile to protease hydrolysis by human thrombin and DPP4, and generated a novel fusion exendin-4 derivative, termed LEX-1. Furthermore, we used LEX-1 as an active intermediary agent for site-specific PEGylation (5, 10, 20, and 30 kDa) at the 17th position of cysteine to generate LEX-2 to LEX-5 for the delayed release rate of exendin-4. After entering into the blood circulation, the newly designed exendin-4 derivatives can be protected from elimination by glomerulus and from proteinase due to the dual protection activities of albumin binding and PEGylation. When the derivative was dissociated from HSA, the linker was consequentially digested by proteinase including thrombin and DPP4, and thereby the native exendin-4 was slowly released into the circulation at a proper rate controlled by PEGylation.
In the present study, we determined the suitable molecular weight of PEG by evaluating the hypoglycemic effects of LEX-2 to LEX-5 in fasted C57BL/6J mice. The antidiabetic efficacies and pharmacokinetic characteristics of LEX-1 and its PEGylated analog were further assessed using db/db mice and SD rats, respectively.
Experimental
Materials and animals
Exendin-4 and LEX-1 were prepared by a previously reported method of solid-phase synthesis.20,21 PEGylated derivatives were prepared by site-specific conjugation of LEX-1 with a free thiol group with maleimide-derivatized PEG. Peptides were evaluated by mass spectrometry and reversed-phase high-performance liquid chromatography (RP-HPLC) with a purity of more than 95%. Six to eight-week-old male C57BL/6J mice, db/db mice and SD rats were allowed to access water freely and fed separately in accordance with their different sources and different experimental purposes. All animal experimental procedures were approved by the Harbin Medical University Animal Ethics Committee (approval code: HMAE192013) and performed according to the experimental regulations.Preparation and characterizations of LEX peptides
Maleimide-derivatized PEG (1 mM) was mixed with LEX-1 (1 mM) in 1 mL of 0.1% DMSO at 4 °C for 24 h. The final mixture was purified by RP-HPLC using a SHIMADZU C18 column (10 × 250 mm, 5 μm), and LEX-2 to LEX-5 were collected. Separately, LEX peptides were detected by high-performance liquid chromatography. The accurate molecular weights of LEX peptides were identified using an Autoflex III smartbeam-MALDI-TOF/MS. The characterization of the fusion peptides bound to HSA were performed by the surface plasmon resonance (SPR) using a BIAcore 8K system (G&E Electronics, USA) according to the manufacturer's instructions.Plasma stability tests
Compared with the native exendin-4, in vitro stabilities of five exendin-4 analogues in a rat kidney homogenate and plasma were researched.22 At 37 °C, six kinds of samples (native exendin-4, LEX-1 and LEX-2 to -5) were incubated in the plasma and supernatant of the homogenized rat kidney at a final concentration of 1 mM. Sex replicates were carried out and the collected samples were frozen in liquid nitrogen. Subsequently, the collected samples were measured using the commercially available exendin-4 ELISA kits brought from Millipore (Merck, Germany).Protease cleavage test
The controlled release of exendin-4 from LEX-1 to LEX-5 was analysed in a PBS buffer (pH 7.4) containing thrombin at a concentration of 0.4 U mL−1.23,24 The reaction mixture was incubated at 37 °C for 104 h shielded from light and was taken out at 0, 8, 16, 24, 32, 40, 48, 56, 64, 72, 80, 88, 96 and 104 hours, respectively. Finally, the ELISA and LC-MS/MS methods were used to analyze the released exendin-4, respectively.Multiple OGTTs in fasted db/db mice
In this study, modified OGTTs were conducted in male C57BL/6J mice and db/db mice under overnight (18 h) fasting conditions. The animals were allocated based on body weight and initial BGLs. A single subcutaneous injection of exendin-4 and LEX-1 to LEX-5 (25 nmol kg−1) was given to the male C57BL/6J mice 0.5 h before the glucose administration at a dose of 2.0 g kg−1 bodyweight. Meanwhile, the glucose values of blood samples were detected pre-dose and at 0, 0.25, 0.5, 0.75, 1, 1.5, 2, 3 h after each administration of D-glucose using a hand-held glucometer. The AUC was calculated from 0 to 3 h, 72 to 75 h and 144 to 147 h, respectively, to assess the in vivo hypoglycemic efficacies.Hypoglycemic efficacies in non-fasted db/db mice
The animals received a single s.c. administration of saline, exendin-4, LEX-1 and LEX-3 (25 and 250 nmol kg−1 body wt). The samples were measured at various time-points (0, 0.5, 1, 2, 4, 8, 12, 16, 24, 32, 48, 72, 96, 120 and 144 h). The glucose-lowering effects within the treatment period (0–144 h) were also assessed by calculating the area under the curve (AUC) of different groups.Pharmacokinetics tests
The samples were collected after a single subcutaneous injection of exendin-4 (25 nmol kg−1), LEX-1 (25 and 250 nmol kg−1) and LEX-3 (25 and 250 nmol kg−1) at various time-points (0, 0.5, 1, 2, 3, 4, 6, 12, 24, 48, 72, 96, 120 and 144 h). The free exendin-4 concentration of the collected samples was detected to be within the range of 0.01–3.20 μg mL−1 using commercial exendin-4 ELISA kits as mentioned above. The intact LEX-1 or LEX-3 concentration was measured by the established LC-MS/MS method according to the previous reports.25Chronic studies
Male type 2 diabetic mice were grouped according to the average body weight and were treated with exendin-4 (250 nmol kg−1), LEX-1 (25 and 250 nmol kg−1) and LEX-3 (25 and 250 nmol kg−1) for 8 consecutive weeks. The cumulative food intake and body weight were measured once every four days, and % fat of body mass was detected after the treatment cycle. After 8 week administration, OGTT was performed after a week wash-out period. The measurement of % HbA1c values and blood biochemical parameter levels was performed using an automatic biochemical analyzer.Statistical analysis
All data are expressed as means ± SD. The differences in all parameters were tested using one-way ANOVA. The P values lower than 0.05 were considered significant.Results
Characterization of exendin-4 derivatives
LEX-1 was prepared by a previously reported method of solid-phase synthesis, and optimized site-specific PEGylation was employed to obtain LEX-2 (5 kDa), LEX-3 (10 kDa), LEX-4 (20 kDa) and LEX-5 (30 kDa).20,21 The elution and purity profiles of LEX peptides were identified by RP-HPLC, and the results indicated that the elution time of exendin-4 was 15.4 min, LEX-1 was 16.4 min, LEX-2 was 16 min, LEX-3 was 15.7 min, LEX-4 was 14.2 min and LEX-5 was 13.2 min. In addition, the purity of all the peptides was over 99% (Fig. 2A). The MALDI-TOF/MS was further performed to identify exendin-4 and its derivatives. By further peptide mapping, the calculated molecular weights of the five molecules well match the theoretical values.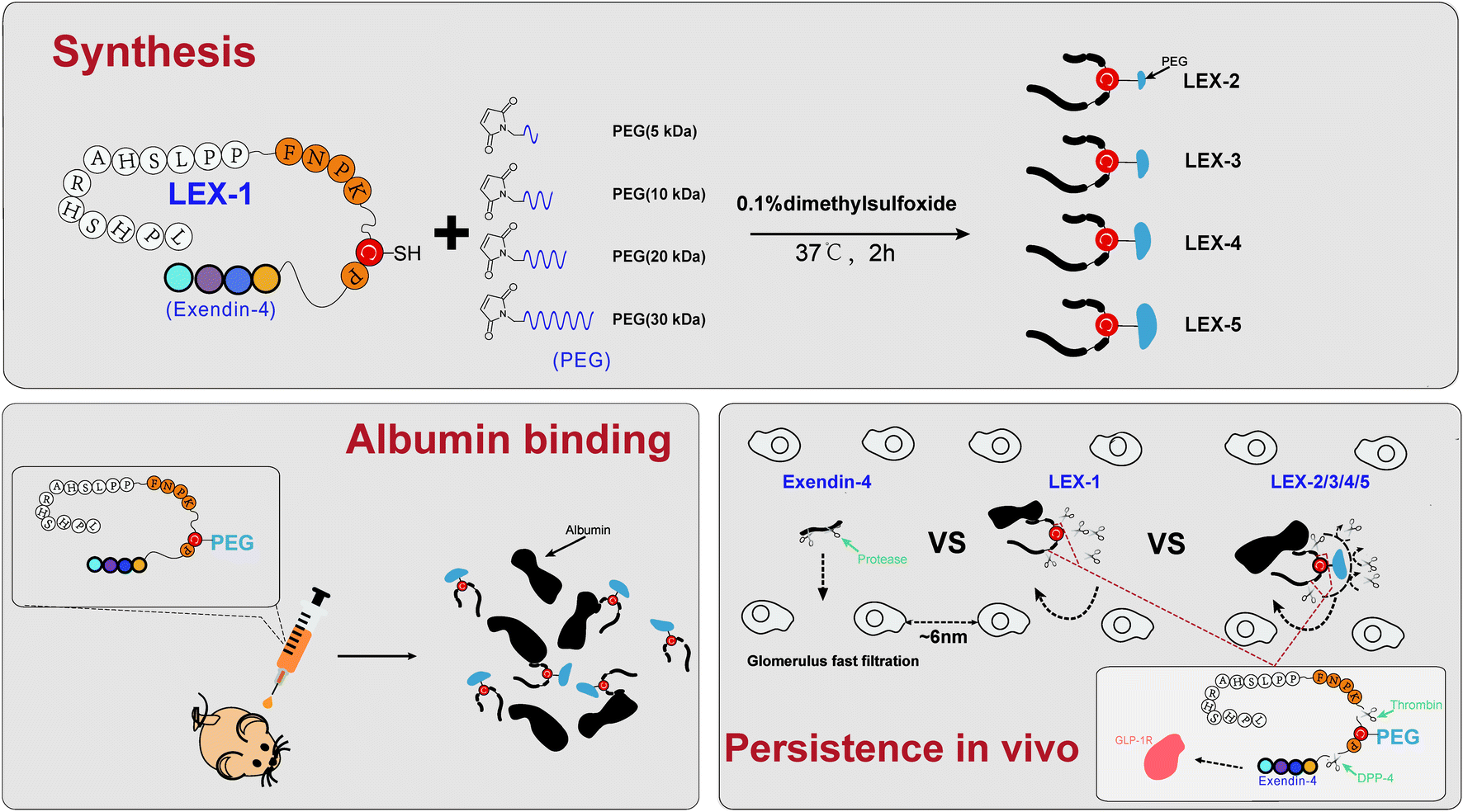 | ||
| Fig. 1 Strategy for multistage sustained release of bioactive peptides via protease cleavage and site-specific PEGylation. | ||
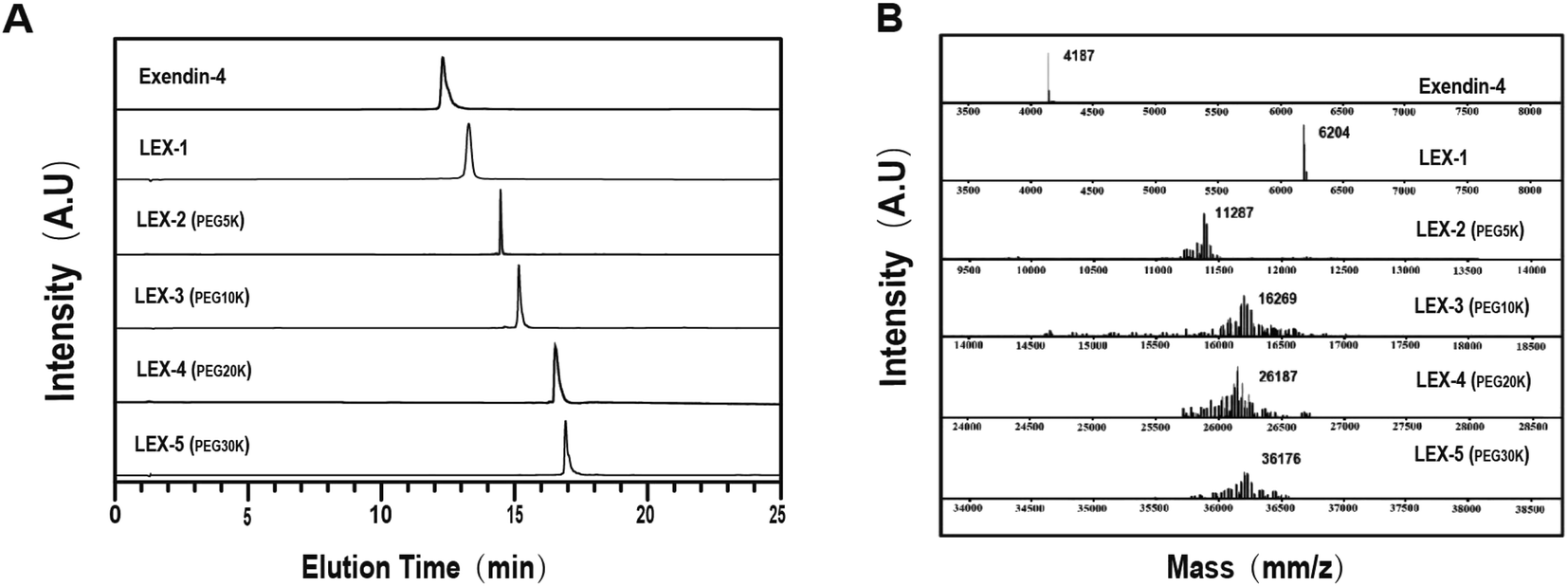 | ||
| Fig. 2 Characterizations of exenatide derivatives. (A) HPLC chromatograms and (B) mass spectra of exendin-4, and LEX peptides. | ||
Binding constants of LEX peptides for HSA including the association rate constant (ka, M−1 s−1), dissociation rate constant (kd, s−1) and overall affinity constant (KD, M) are listed in Table 1. The results indicated that LEX-3 (0.22 × 10−6 M) associated with HSA with higher affinity than LEX-1 (1.33 × 10−6 M), LEX-2 (3.16 × 10−6 M), LEX-4 (4.31 × 10−6 M) or LEX-5 (3.12 × 10−6 M).
| Molecule | HSA | ||
|---|---|---|---|
| ka (M−1 s−1) | kd (s−1) | KD (M) | |
| LEX-1 | 1.38 × 104 | 1.83 × 10−2 | 1.33 × 10−6 |
| LEX-2 | 1.14 × 104 | 3.60 × 10−2 | 3.16 × 10−6 |
| LEX-3 | 1.28 × 104 | 0.28 × 10−2 | 0.22 × 10−6 |
| LEX-4 | 1.24 × 104 | 5.33 × 10−2 | 4.31 × 10−6 |
| LEX-5 | 2.01 × 104 | 6.27 × 10−2 | 3.12 × 10−6 |
Proteolytic stabilities of LEX peptides
The susceptibility of peptides or proteins to enzymatic digestion is one of the barriers to obtain prolonged bioactivities.26 The stability tests were performed with the rat kidney homogenate and plasma to evaluate the proteolytic stabilities of LEX-1 to LEX-5. As demonstrated in Fig. 3, a rapid decrease in the concentration of exendin-4 was shown with a t1/2 of 31.2 ± 5.9 min in the rat kidney homogenate. In contrast, LEX-1 to LEX-5 exert significantly prolonged half-lives compared with the native exendin-4 (56.5 ± 12.3 min, 92.2 ± 13.4 min, 123.1 ± 21.5 min, 152.9 ± 21.3 min and 169.4 ± 22.0 min vs. 31.2 ± 5.9 min, respectively). Similarly, the t1/2 of LEX-1 to LEX-5 was also notably prolonged compared with exendin-4 in rat plasma (6.8 ± 0.8 h, 7.5 ± 1.7 h, 17.0 ± 2.3 h, 17.1 ± 2.0 h and 34.8 ± 4.2 h vs. 3.2 ± 0.5 h), suggesting that the strategy of albumin binding and PEGylation in LEX-1 strikingly improved the in vitro proteolytic stability of the native exendin-4.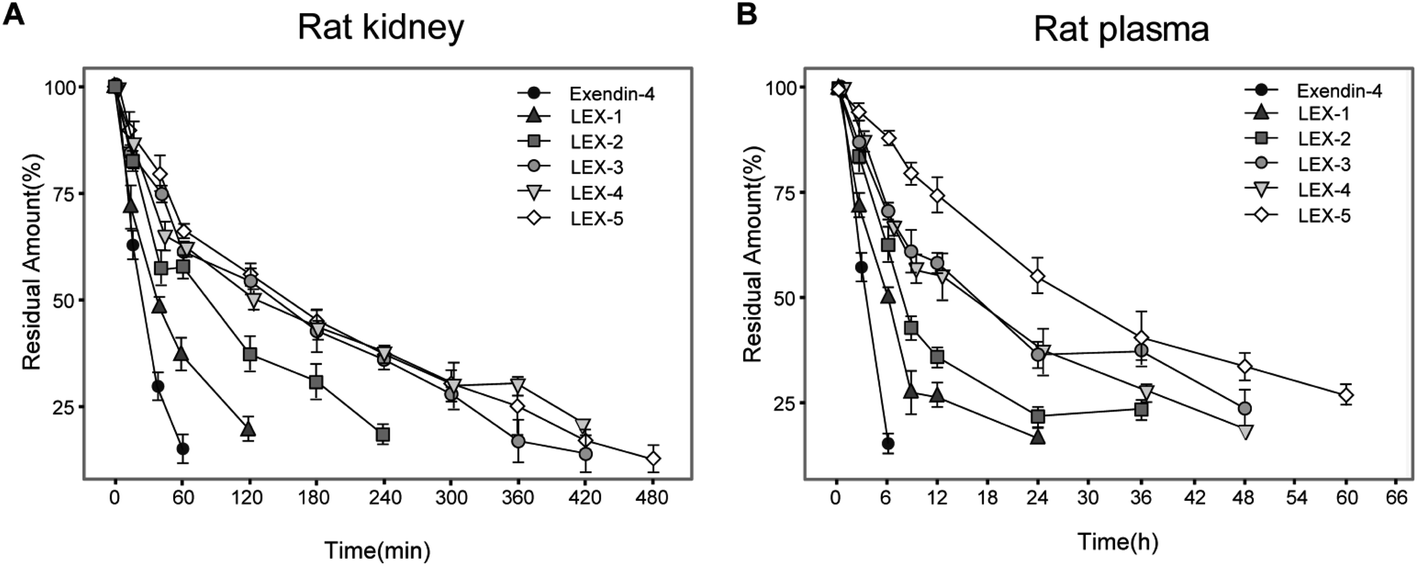 | ||
| Fig. 3 The degradation profiles of exendin-4 and its derivatives when incubated in the rat kidney homogenate (A) and rat plasma (B). Residues were quantified by ELISA. Data are expressed as means ± SD (n = 6). | ||
Evaluation of the controlled release of exendin-4
As shown in Fig. 4, the concentration of exendin-4 transiently released from LEX peptides via the plasma TBN was detected. The concentration of released exendin-4 varies as a function of time during protease digestion and was monitored by an ELISA method. As a result, LEX-1 exerts a very fast release of exendin-4 due to thrombin digestion when they enter the blood circulation. However, the concentration of the intact LEX-1 was also rapidly decreased and became nearly exhausted after 48 h. PEGylation of LEX-1 effectively slows digestion effects of blood proteases and the peak concentration of free exendin-4 in LEX-2 to LEX-5 groups, especially for LEX-4 and LEX-5, all lower than that of LEX-1. Moreover, the duration of controlled release of exendin-4 in PEGylated LEX-1 was significantly prolonged. However, the concentration of transient exendin-4 in both the LEX-4 and LEX-5 groups was obviously lower than those of LEX-1 to LEX-3 probably due to the larger molecular weight of PEG. Furthermore, a very low concentration of free exendin-4 may not exert effective hypoglycemic actions. Therefore, we need to perform in vivo experiments to screen the suitable molecule for further preclinical studies.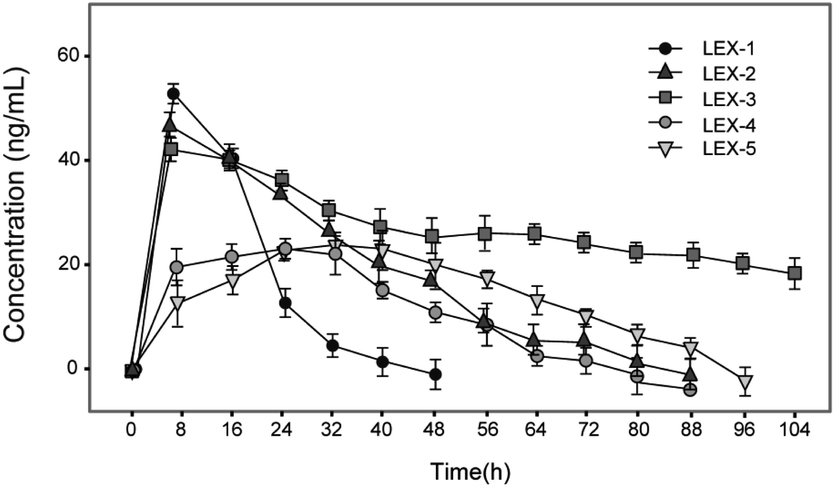 | ||
| Fig. 4 Controlled release of exendin-4 from LEX peptides. Data are expressed as means ± SD (n = 6). | ||
Glucose-stabilizing capabilities of LEX peptides
In order to confirm the suitable molecule weight of PEG, we further identified the glucose-stabilizing capabilities of LEX-1 to LEX-5 by modified multiple oral glucose tolerance tests (OGTTs) in normal male C57BL/6J mice. As evidenced in Fig. 5, the blood glucose levels (BGLs) of the placebo-treated group promptly reached the peak at 15 min and then slowly dropped to baseline (approximately 5 mmol L−1) after 2 h, while the native exendin-4, LEX-1 and LEX-2 to LEX-5 resulted in considerable hypoglycemic effects in the first OGTT. Remarkably, the glucose-lowering effects of LEX-4 and LEX-5 were worse than those of other conjugates probably due to the increased steric hindrance caused by the larger molecular weight of PEG, which affected the proper release of exendin-4. Interestingly, better glucose-stabilizing capability of LEX-3 was observed during the third OGTT, suggesting longer hypoglycemic duration and effects than other conjugates. Therefore, LEX-3 was selected to evaluate further in vivo bioactivities.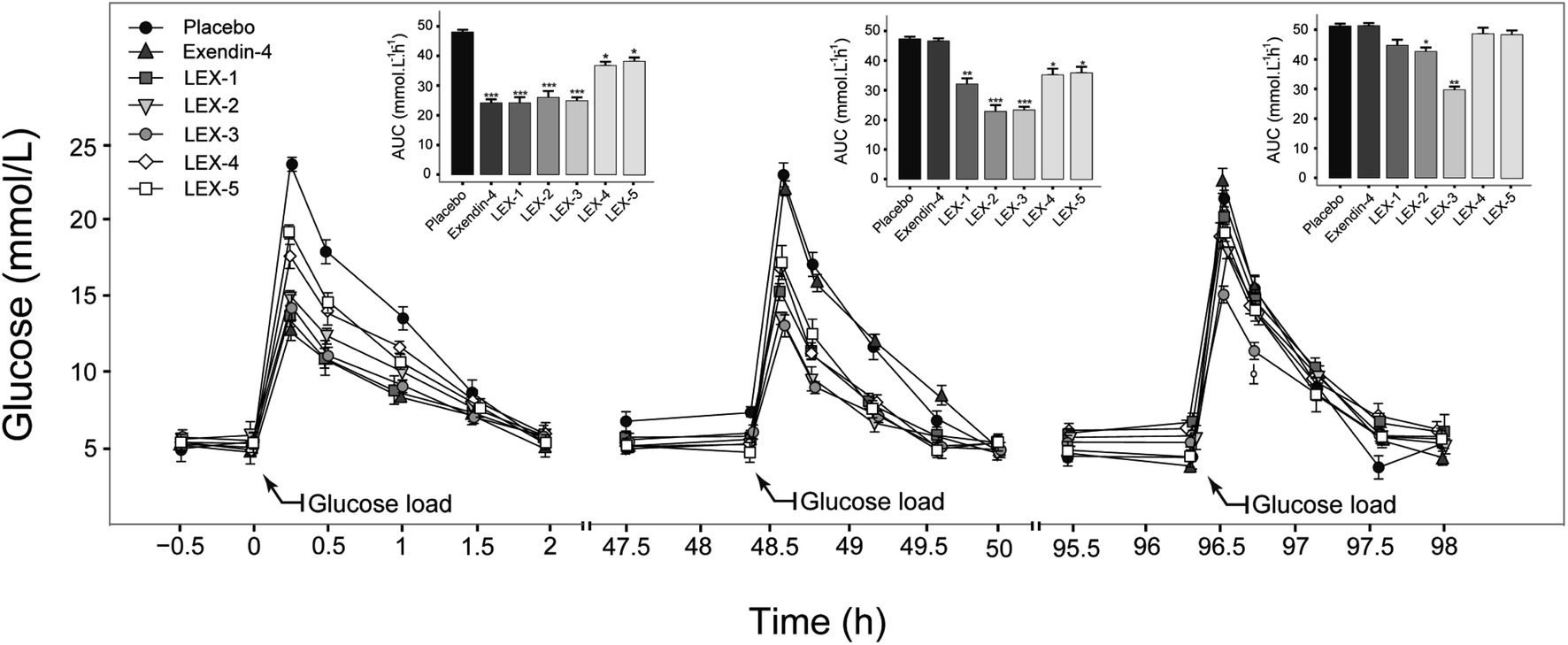 | ||
| Fig. 5 Multiple OGTTs in normal C57BL/6J mice. Data are expressed as means ± SD (n = 6). ***P < 0.001, **P < 0.02, *P < 0.05. | ||
Hypoglycemic duration tests of LEX-1 and LEX-3
To further evaluate the antihyperglycemic efficacies of LEX-1 and LEX-3, hypoglycemic duration tests were investigated in non-fasting diabetic mice. The BGLs of exendin-4- (250 nmol kg−1), LEX-1- (25 and 250 nmol kg−1) and LEX-3- (25 and 250 nmol kg−1) treated groups all rapidly decreased to the hypoglycemic state (< 8.35 mmol L−1) in 4 h and the time required to rebound to initial level were nearly 48, 72, 120, 120 and 144 h, respectively, while the placebo-treated group exhibited a hyperglycemic state (>20 mmol L−1) during the entire experiment (0–144 h) (Fig. 5A). Furthermore, LEX-3 at 250 nmol L−1 remarkably decreased the area under the curve (AUC) to 72.5% for a treatment period of 0–144 h compared with the placebo-treated group, while for exendin-4- and LEX-1-treated groups it was only 18.5% and 58.0% at the same dose, indicating that the sustained release effects from protease cleavage and site-specific PEGylation could notably prolong the hypoglycemic duration of LEX-3 (Fig. 5B). In short, LEX-3 showed enhanced hypoglycemic efficacy compared with LEX-1 or exendin-4.Glucose-stabilizing capability tests of LEX-1 and LEX-3
We further assessed in vivo glucoregulatory activities of LEX-1 and LEX-3 using multiple OGTTs in overnight fasted diabetic mice. These mice were subcutaneously injected with the doses of exenatide (250 nmol kg−1), LEX-1 (25 and 250 nmol kg−1), and LEX-3 (25 and 250 nmol kg−1) at 0.5 h before first treatment of D-glucose. As illustrated in Fig. 7A, the glucose level of placebo-treated groups rapidly showed a peak at half an hour after each round of administration of glucose and then slowly returned to the baseline level within 3 h. As shown in Fig. 7A, the injection of exendin-4, LEX-1 and LEX-3 all obviously reduced the BGL compared with the control during the first period of multiple OGTTs (P < 0.01). Nevertheless, exendin-4 was completely ineffective after 10 h, while the treatment of LEX-1 and LEX-3 resulted in significant glucose-lowering effects during the experimental period (0–144 h) at the same dose. In particular, a single s.c. administration of LEX-3 exerts higher AUC-lowering percentage than LEX-1 in the third period of multiple OGTTs (24.0% vs. 18.3%, versus placebo-treated groups) (Fig. 7B–D). The above-mentioned results suggested that glucose-stabilizing capabilities of LEX-1 and LEX-3 at 250 nmol kg−1 could sustain over 74 h and 144 h, respectively.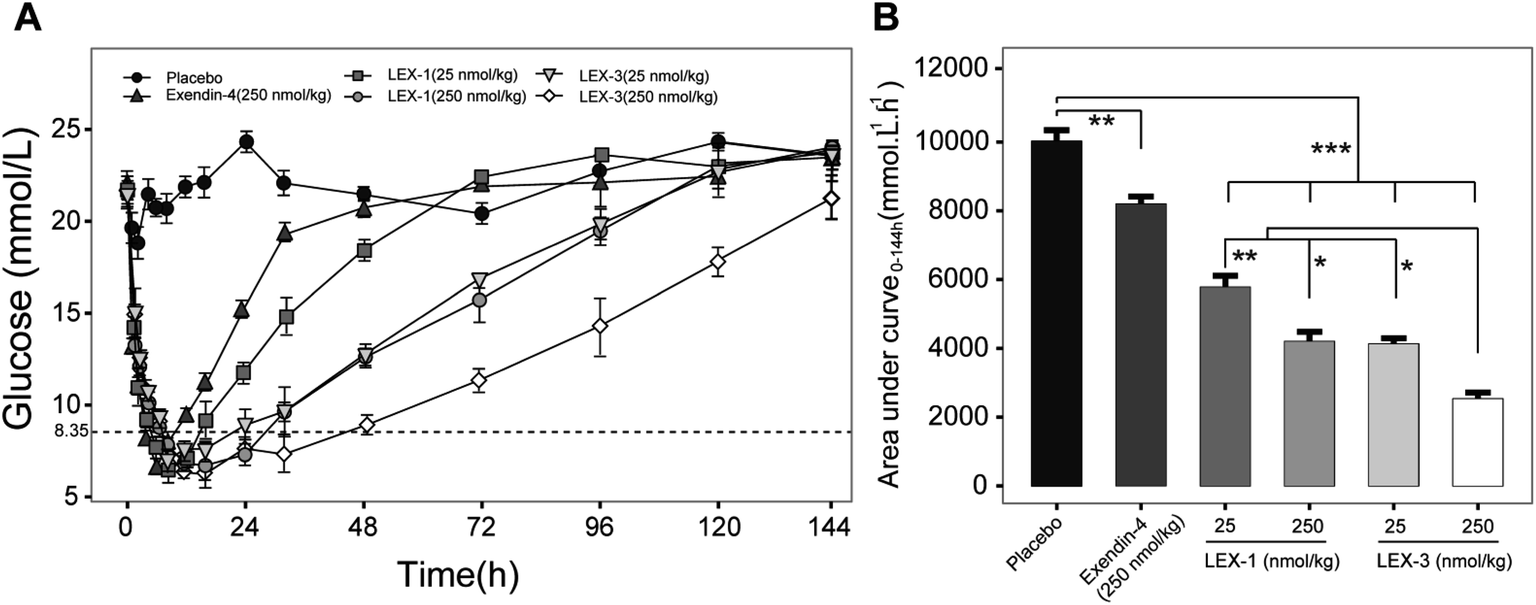 | ||
| Fig. 6 Anti-hyperglycemic efficacy of exendin-4, LEX-1 and LEX-3 in type 2 diabetic mice. (A) Time–BGLs curve. (B) Calculated AUC0–120 h values of exendin-4, LEX-1 and LEX-3. Data are expressed as means ± SD (n = 6). ***P < 0.001, **P < 0.02, *P < 0.05. | ||
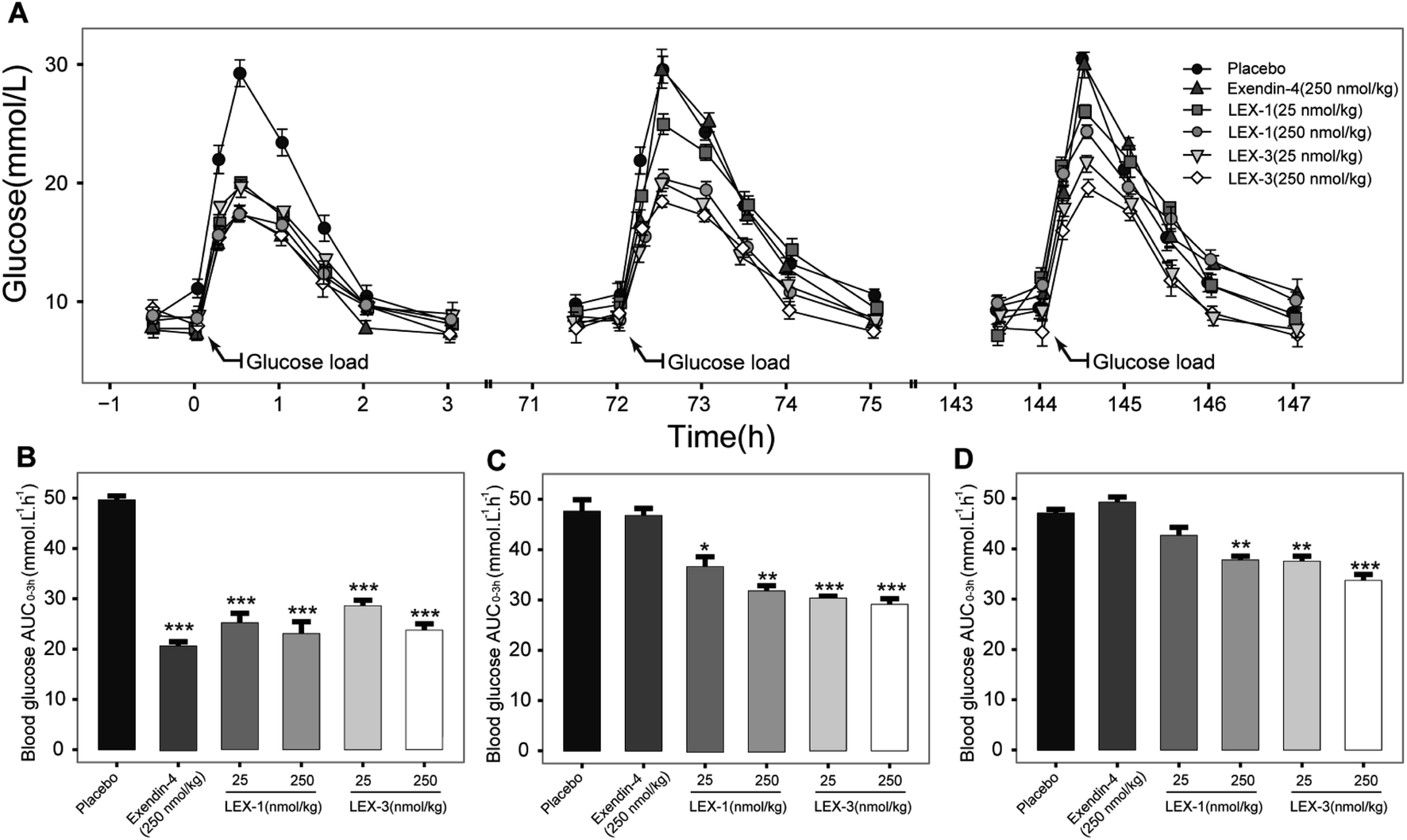 | ||
| Fig. 7 Glucose-stabilizing capabilities of exendin-4 and its derivatives was investigated by using multiple OGTTs in male diabetic mice. (A) Time–BGLs curve; calculated (B) AUC0–3 h, (C) AUC72–75 h, (D) AUC144–147 h of exenatide, LEX-1 and LEX-3 in diabetic mice. All results are shown as the means ± SD (n = 6). ***P < 0.001, **P < 0.02, *P < 0.05. | ||
In vivo pharmacokinetics of LEX-1 and LEX-3
Albumin-binding and PEGylation significantly improved the pharmacokinetic properties of modified exendin-4 derivatives in Sprague Dawley (SD) rats. As we expected, the plasma concentration of exendin-4 peaked within an hour, and then quickly returned to the original blood glucose level at 12 h post-injection with a calculated t1/2 of 4.4 ± 0.9 h, whereas the t1/2 of intact LEX-1 or LEX-3 were 78.4 ± 11.5 h or 119.7 ± 15.0 h, and that of released exendin-4 were 67.4 ± 9.8 h or 109.6 ± 10.4 h, respectively, at a dose of 250 nmol kg−1 (Fig. 8). From the combined results of the hypoglycemic duration test and multiple OGTTs, it was found that LEX-3 has the potential to be developed as a once-weekly antidiabetic agent.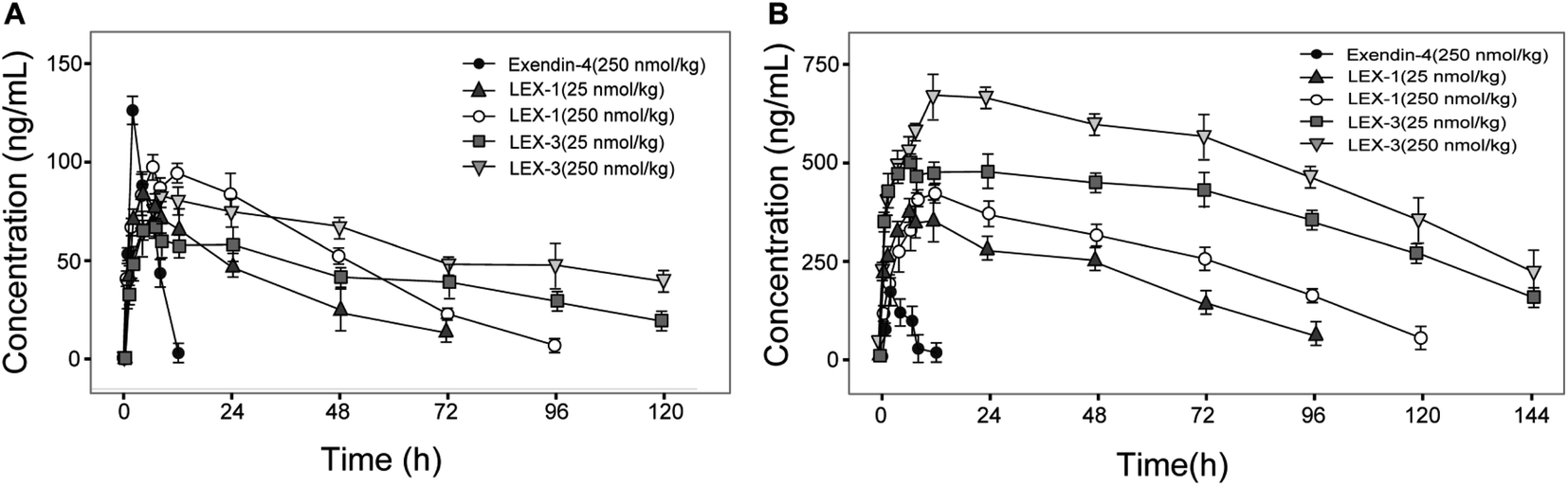 | ||
| Fig. 8 The concentration–time curve of (A) released exendin-4 and (B) intact LEX-1, LEX-3 in SD rats after a single subcutaneous administration of LEX-1 and LEX-3 at the doses of 25 and 250 nmol kg−1. All results are shown as the means ± SD (n = 6). | ||
Chronic studies
As LEX-1 and LEX-3 exert remarkable in vivo efficacies and metabolic characters in acute pharmacodynamic and pharmacokinetic studies, we further performed chronic studies to evaluate their ulteriorly therapeutic potential. The diabetic mice subcutaneously received LEX-1 and LEX-3 at doses of 25 and 250 nmol kg−1 twice-weekly for 8 consecutive weeks using exendin-4 as the positive control. As shown in Fig. 9A and B, cumulative food intake and body weight gain significantly decreased in both LEX-1- and LEX-3 (25 and 250 nmol kg−1 body wt)-treated mice but continuously increased for the placebo-treated ones. Moreover, LEX-3 treatment exerts more significant suppression effects than exendin-4 and LEX-1 at the same dose (P < 0.05). Similarly, long-term treatment of LEX-1 and LEX-3 at 250 nmol kg−1 resulted in a reduction in fat (% of body mass) for 41.1% and 56.0%, respectively, while it was only 10.6% after exendin-4 treatment compared with the placebo-treated ones (Fig. 9C).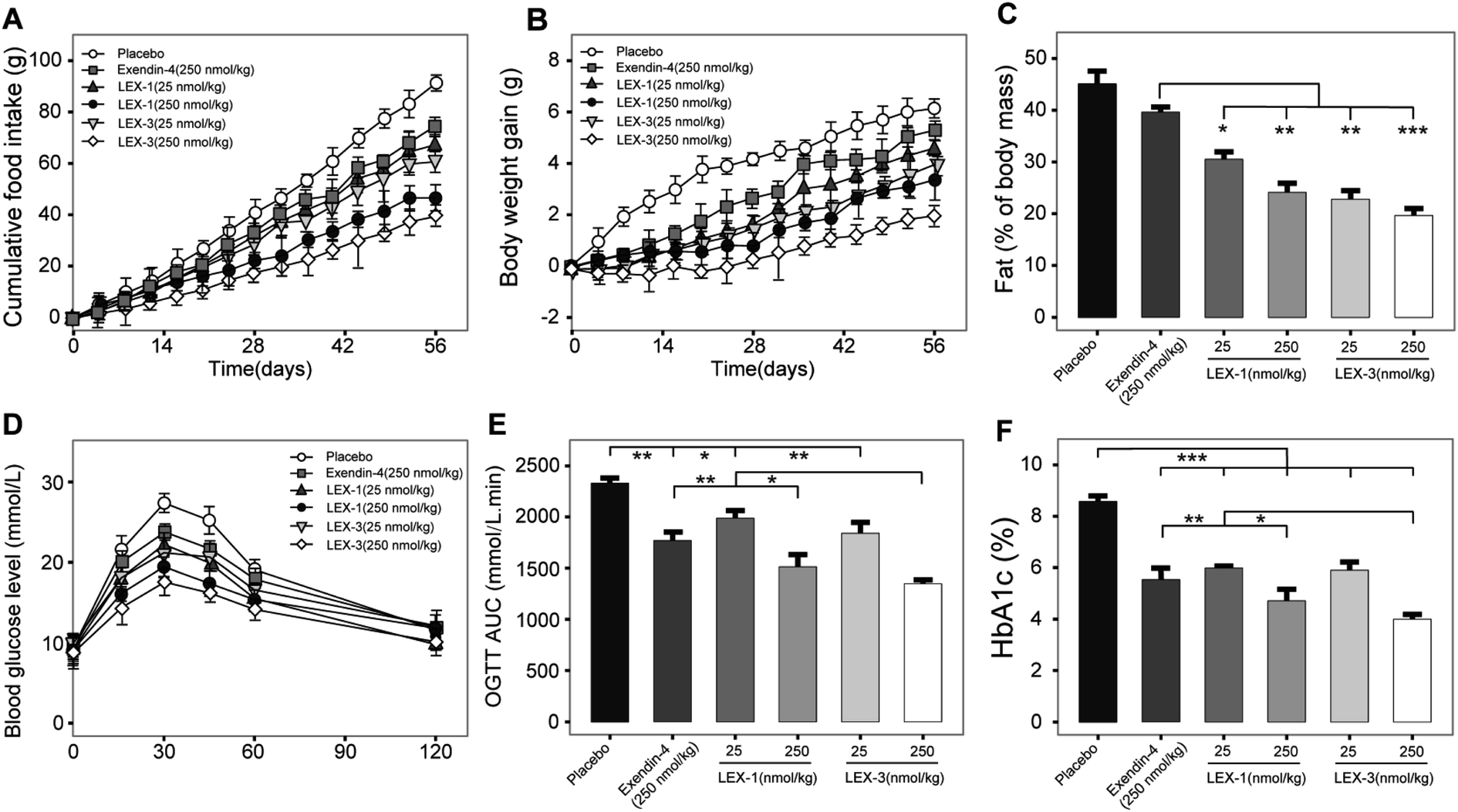 | ||
| Fig. 9 Chronic in vivo studies of LEX-1 and LEX-3 in diabetic mice. Efficacies of chronic treatment of LEX peptides on (A) food intake, (B) body weight gain, (C) % fat, (D) time–BGLs curve, (E) OGTT AUC and (F) % HbA1c of db/db mice. All results are shown as the means ± SD (n = 8). ***P < 0.001, **P < 0.02, *P < 0.05. | ||
To determine whether long-term administration of LEX peptides effectively improved glucose metabolism in diabetic mice, we conducted an OGTT at weeks 8 and 9 (after a week wash-out period). It was found that 8 week treatment of LEX-3 exerted significantly improved glucose-stabilizing effects in db/db mice compared with LEX-1- or exendin-4-treated ones (Fig. 9D). Interestingly, the results graphed in Fig. 9E revealed that the LEX-3-treated group exhibited a significantly higher AUC-lowering effect than the exendin-4- and LEX-1-treated group (44.3% vs. 26.7% and 37.3%, P < 0.02 and P < 0.05, respectively, compared with the placebo-treated group). The following % HbA1c values, which represent the cumulative blood glucose concentrations for more than 3 months,27 were further detected, and the results are shown in Fig. 9F. LEX-3 treatment significantly improved the reduction of % HbA1c values compared with exendin-4 or LEX-1 treatment, indicating that PEG conjugation notably enhanced the in vivo stability and induced better potential therapeutic utilities.
After the 8 week experimental period, the mice were killed and the blood biochemical indexes were measured (Fig. 10). A significant decline in values of either total cholesterol (TC) or triglyceride (TG) was observed in LEX-1- and LEX-3- (250 nmol kg−1) treated groups. Herein, the activities of ALT and AST were determined to assess the hepatocyte damage. As we expected, treatment with both LEX-1 and LEX-3 at a dose of 250 nmol kg−1 resulted in a remarkable reduction compared with placebo treatment (P < 0.05). Furthermore, long-term treatment with LEX-3 at a dose of 250 nmol kg−1 decreased the LDL/HDL ratio up to 30.9%, while exendin-4 and LEX-1 exert a reduction of only 20.9% and 25.4% at the same doses, respectively.
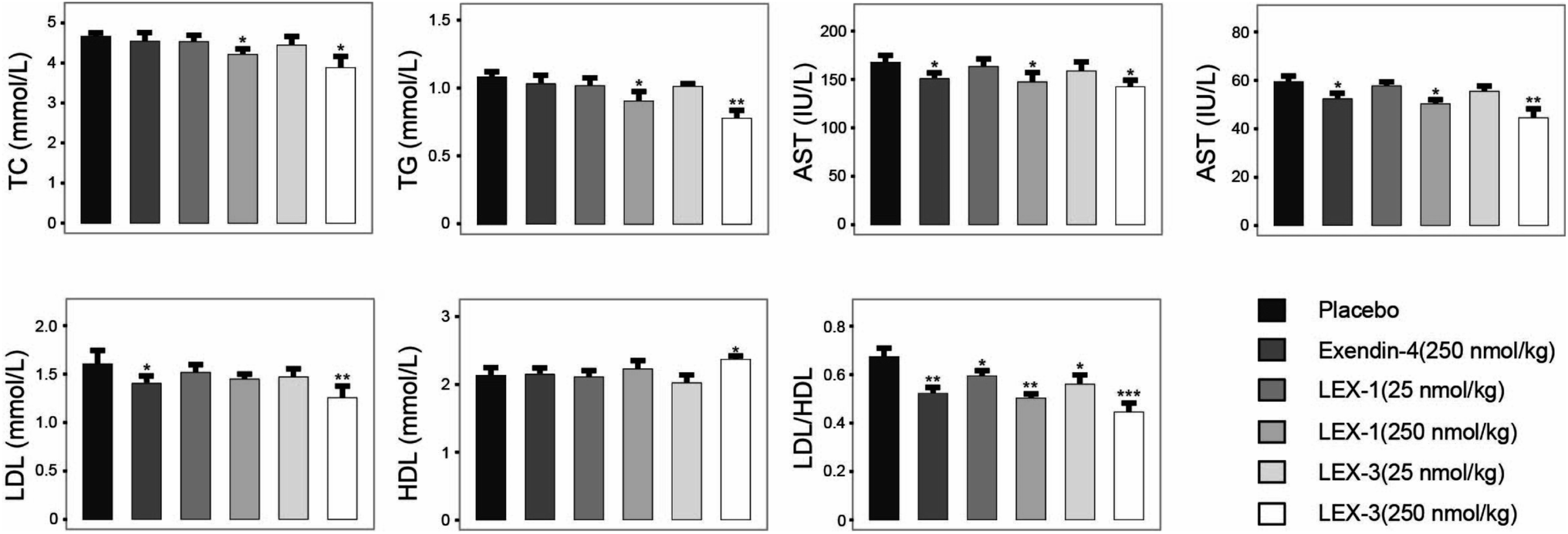 | ||
| Fig. 10 Efficacies of 8 consecutive weeks of administration of LEX peptides on the blood biochemical indexes in diabetic mice. All results are expressed as means ± SD (n = 8). ***P < 0.001, **P < 0.02, *P < 0.05. | ||
Discussion
Therapeutic effects of polypeptide drugs need frequent injection to sustain the effective duration due to rapid renal filtration or proteolysis. Hence, many strategies have been applied to achieve administration efficacies of peptide drugs in vivo.7,28,29 it is well known that albumin binding and site-specific PEGylation are two effective choices of prolonging short half-lives of therapeutic peptides in the systemic circulation by increasing the molecular size and reducing the renal clearance.2,30,31As reported previously,32,33 the extremely short in vivo lifespan of exendin-4 necessitates twice-daily injections. To address this, both HSA binding and PEGylation were utilized to reduce the renal clearance and further extend the exposure of exendin-4 in vivo. Based on our strategy reported before,18 we designed a novel exendin-4 derivative, termed LEX-1, which was performed by fusing exendin-4 with a protease-cleavable linker (FNPKCP) and an albumin-binding 12-mer peptide, which exerts high affinity for HSA. Additionally, different lengths of Mal-PEG-NHS (5, 10, 20 and 30 kDa) were conjugated to LEX-1 using the cysteine- and amine residue-specific coupling reactions to generate four modified peptides (called LEX-2 to LEX-5) (Fig. 1). As illustrated in Fig. 2, the purities of exendin-4 derivatives were all above 99.0% and the molecular weights were calculated, which were consistent with the theoretical values. It is worth discussing that the impurity peaks of all exenatide derivatives were captured in the mass spectra, as those PEG molecules themselves are mixtures. Generally, it is of extreme importance in the characterisation of impurity profiles in biological candidates. Several reports indicated that the smaller molecular weight of PEG exerts stronger toxicity for experimental animals.35 In the present study, the selected PEG molecule in the exendin-4 derivative held a larger molecular weight (>10 kDa), which means that it has less toxicity theoretically. Moreover, we have previously determined the suitable doses of exendin-4 derivatives by toxicological pre-experiments. As a result, all animals remained healthy for the entire period of the study, showing no evident sign of toxicity or other adverse effects. Furthermore, the following in vitro and in vivo efficacy evaluation tests all showed that the exenatide derivatives still exert favorable pharmacodynamic and pharmacokinetic properties without obvious side effects. Nevertheless, we still will separate out the impurities in the subsequent tests and carefully investigate their toxicity to healthy and model animals.
The binding constant measurement revealed that LEX-3 exerts the highest affinity with HSA compared with other conjugates (Table 1). As shown in Fig. 3 and 4, we further evaluated the proteolytic stabilities and the controlled release of exendin-4 of LEX peptides. As expected, LEX-1 to LEX-5 exert improved stabilities in both the plasma and kidney homogenate compared with the native exendin-4. In addition, PEGylation of LEX-1 effectively retarded the digestive effects of blood proteases and reduced the peak concentration of the free exendin-4 in LEX-2 to LEX-5 groups. However, a very low concentration of free exendin-4 was observed in LEX-4 and LEX-5 groups probably due to excessively large PEG hindering the enzyme digestion, and hence, less free exendin-4 molecules were released. To determine the suitable length of PEG, multiple OGTTs were further performed to assess the glucose-lowering capabilities of LEX-1 to LEX-5. As a result, LEX-3 exerts better glucose-stabilizing capability and duration compared with other derivatives in the normal C57BL/6J mice, indicating that the suitable molecular weight of PEG chain was 10 kDa (Fig. 5). Therefore, LEX-3 was selected to perform further in vivo efficacy evaluation tests.
The results shown in Fig. 5 revealed that C57BL/6J mice can be used for the initial screening and pharmacodynamic evaluation of LEX peptides. Moreover, C57BL/6J mice and db/db mice were derived from the same strain. Hence, the glucose-stabilizing effects of LEX-1 and LEX-3 were further evaluated in db/db mice by the hypoglycemic duration test and multiple OGTTs. As illuminated in Fig. 6 and 7, both LEX-1 and LEX-3 exert greater sustained hypoglycemic effects and glucose-lowering abilities compared with the native exendin-4. It is worth to mention that LEX-3 exerts the longest duration (∼39.9 h) for the BGL to rebound to 8.35 mmol L−1, which was considered as the hypoglycemic duration. Moreover, LEX-3 exhibited significantly higher AUC-lowering percentage than LEX-1 in third period of multiple OGTTs at the same dose, indicating the enhanced glucose-stabilizing ability probably due to site-specific PEGylation. In addition, the results of multiple OGTTs in C57BL/6J mice and diabetic mice revealed that both C57BL/6J mice and diabetic mice are suitable for the pharmacodynamic evaluation of LEX peptides. To rationalize in vivo efficacy results of LEX peptides, pharmacokinetic tests were then performed in rhesus monkeys. As shown in Fig. 8, the elimination half-lives of released exendin-4 from LEX-3 (250 nmol kg−1) were approximately 119.7 h, suggesting that it holds the potential to be developed as a once-weekly antidiabetic agent.
In order to comprehensively assess the efficacies of LEX peptides, we investigated the effects of chronic treatment in the diabetic mice. As shown in Fig. 9A–C, twice-weekly injection of LEX-3 achieved the best long-term beneficial effects on the appetite suppression, body weight and % fat in diabetic mice compared with the exendin-4- and LEX-1-treated groups after the 8 week treatment. Moreover, OGTT was conducted to confirm whether chronic treatment with LEX peptides effectively improved the glucose metabolism in db/db mice. It was found that LEX-3 exerts a significantly higher AUC-lowering percentage than exendin-4 or LEX-1 at the same dose (P < 0.02 and 0.05) (Fig. 9D–E). % HbA1c, a well-known indirect indicator of cumulative blood glucose concentrations, was a more sensitive index of glycemic control than glucose concentration.34 As presented in Fig. 9F, an obvious reduction in the % HbA1c value was observed in LEX-3-treated groups, which was significantly lower than those of exendin-4- and LEX-1-treated ones (both P < 0.05). Furthermore, chronic treatment with LEX peptides significantly improved the blood biochemical indexes in diabetic mice (Fig. 10).
Conclusion
A novel anti-diabetic drug, LEX-1, has been designed by the fusion of an albumin-binding domain and a thrombin-cleavable linker to a native exendin-4. Furthermore, the site-specific PEGylation (10 kDa) of LEX-1 generated a long-acting exendin-4 derivative, termed LEX-3. Considering that exenatide (exendin-4) is a FDA-approved anti-diabetic drug, LEX-1 and LEX-3, which could stably release the native exendin-4, hold potential to be developed as promising once-weekly anti-diabetic agents. This multistage sustained-release strategy could also be applied for other peptides with short lifespans.Author contributions
Conceptualization, Mei Dong; Data curation, Aihong Zhang; Methodology, Aihong Zhang and Yin Li; Supervision, Aihong Zhang, and Yin Li; Validation, Mei Dong; Writing-original draft, Aihong Zhang; Writing-review & editing, Yin Li.Conflicts of interest
Authors declare no conflict of interest.Acknowledgements
We indeed appreciate the linguistic assistance from Mogoedit during the writing and revising this manuscript. We thank Prof Tanner for insightful and helpful discussion and excellent technical assistance.Notes and references
- J. S. Ryu, A. Y. Cho, W. S. Sang and H. Min, Methods Mol. Biol., 2014, 1088, 35–50 CrossRef CAS PubMed.
- Y. Shechter, M. Mironchik, S. Rubinraut, A. Saul, H. Tsubery and M. Fridkin, Bioconjugate Chem., 2005, 16, 913–920 CrossRef CAS PubMed.
- D. P. Mcgregor, Curr. Opin. Pharmacol., 2008, 8, 616–619 CrossRef CAS PubMed.
- L. Benzi, P. Cecchetti, A. Ciccarone, A. Pilo, G. Di Cianni and R. Navalesi, Diabetes, 1994, 43, 297–304 CrossRef CAS PubMed.
- D. B. M. Fcp and D. H. Z. Fcp, J. Clin. Pharmacol., 2013, 52, 54S–62S Search PubMed.
- J. Lin and A. Lu, Pharmacol. Rev., 1997, 49, 403–449 CAS.
- D. Kumar Malik, S. Baboota, A. Ahuja, S. Hasan and J. Ali, Curr. Drug Delivery, 2007, 4, 141–151 CrossRef PubMed.
- R. Pawar, A. Ben-Ari and A. J. Domb, Expert Opin. Biol. Ther., 2004, 4, 1203–1212 CrossRef CAS PubMed.
- H. Chen, W. Guohao, L. Lixin, J. Orit, D. Kiesewetter, L. Yi, M. Ying, Z. Xianzhong, W. Hua and Z. Lei, Theranostics, 2016, 6, 243–253 CrossRef CAS PubMed.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- S. Mitragotri, P. A. Burke and R. Langer, Nat. Rev. Drug Discovery, 2014, 13, 655–672 CrossRef CAS PubMed.
- D. Sleep, J. Cameron and L. R. Evans, Biochim. Biophys. Acta, 2013, 1830, 5526–5534 CrossRef CAS PubMed.
- N. Gong, A.-N. Ma, L.-J. Zhang, X.-S. Luo, Y.-H. Zhang, M. Xu and Y.-X. Wang, Br. J. Pharmacol., 2011, 163, 399–412 CrossRef CAS PubMed.
- V. Gabercporekar, I. Zore, B. Podobnik and V. Menart, Curr. Opin. Drug Discovery Dev., 2008, 11, 242–250 CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- G. Pasut, A. Guiotto and F. Veronese, Expert Opin. Ther. Pat., 2004, 14, 859–894 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS PubMed.
- H. Li, Y. Ma, Y. Chen, Y. Sang and Z. Su, Angew. Chem., 2010, 49, 4930–4933 CrossRef CAS PubMed.
- L. L. Nielsen, J. Am. Board Fam. Med., 2005, 10, 703–710 CAS.
- Y. Chi, H. Zhang, J. Zhou, W. Huang and S. Ni, Lett. Org. Chem., 2008, 5, 399–402 CrossRef CAS.
- C. Yushi, Z. Huibin, H. Wenlong and Z. Jinpei, Bioorg. Med. Chem., 2008, 16, 7607–7614 CrossRef PubMed.
- J. Han, L. Sun, X. Huang, Z. Li and W. Huang, Br. J. Pharmacol., 2014, 171, 5252–5264 CrossRef CAS PubMed.
- M. C. Guillin, A. Bezeaud, M. C. Bouton and M. Jandrot-Perrus, Thromb. Haemostasis, 1995, 74, 129–133 CrossRef CAS.
- K. Borensztajn, M. P. Peppelenbosch and C. A. Spek, Trends Mol. Med., 2008, 14, 429–440 CrossRef CAS PubMed.
- T. J. Wang, J. Li, L. J. Liang, M. Zhong, X. H. Chen and B. I. Kai shun, Chin. J. Pharm. Anal., 2007 Search PubMed.
- A. Zorzi, S. J. Middendorp, J. Wilbs, K. Deyle and C. Heinis, Nat. Commun., 2017, 8, 16092 CrossRef CAS PubMed.
- R. Malka, D. M. Nathan and J. M. Higgins, Sci. Transl. Med., 2016, 8, 359ra130 CrossRef PubMed.
- J. Lee, C. Lee, I. Kim, H. R. Moon, T. H. Kim, K. T. Oh, E. S. Lee, K. C. Lee and Y. S. Youn, Int. J. Pharm., 2012, 424, 50–57 CrossRef CAS PubMed.
- L. Diao and B. Meibohm, Clin. Pharmacokinet., 2013, 52, 855–868 CrossRef CAS PubMed.
- A. P. Chapman, Adv. Drug Delivery Rev., 2002, 54, 531–545 CrossRef CAS PubMed.
- S. Choi, M. Baudys and S. Kim, Pharm. Res., 2004, 21, 827–831 CrossRef CAS PubMed.
- J. Buse, R. Henry, J. Han, D. Kim, M. Fineman and A. Baron, Diabetes Care, 2004, 27, 2628–2635 CrossRef CAS PubMed.
- T. H. Kim, H. H. Jiang, S. Lee, Y. S. Youn, C. W. Park, Y. Byun, X. Chen and K. C. Lee, Bioconjugate Chem., 2011, 22, 625–632 CrossRef CAS PubMed.
- A. K. Saleh and M. A. A. Moussa, Clin. Chem., 1985, 31, 1872–1876 CrossRef CAS.
- R. Webster, V. Elliott, B. Park, D. Walker, M. Hankin and P. Taupin, PEGylated Protein Drugs: Basic Science and Clinical Applications, 2009, pp. 127–146, DOI:10.1007/978-3-7643-8679-5_8.
| This journal is © The Royal Society of Chemistry 2020 |