
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of lixisenatide analogues as long-acting hypoglycemic agents using novel peptide half-life extension technology based on mycophenolic acid†
Chunli Tang‡
ac,
Qing Li‡d,
Xiaoyan Dengd,
Weiwei Wua,
Liufeng Liaoa,
Kai Lianga,
Rongrui Huoc,
Chenglin Lie,
Jing Han *f,
Weizhong Tang*b and
Neng Jiang*a
*f,
Weizhong Tang*b and
Neng Jiang*a
aDepartment of Pharmacy, Affiliated Tumor Hospital of Guangxi Medical University, Nanning, PR China. E-mail: cpujnh@163.com
bDepartment of Gastrointestinal Surgery, Affiliated Tumor Hospital of Guangxi Medical University, Nanning, PR China. E-mail: tangweizhong@gxmu.edu.cn
cEditorial Department, Affiliated Tumor Hospital of Guangxi Medical University, Nanning, PR China
dPharmaceutical College, Guangxi Medical University, Nanning 530021, China
eJiangsu Key Laboratory of New Drug Research and Clinical Pharmacy, Xuzhou Medical University, Xuzhou, China
fSchool of Chemistry & Materials Science, Jiangsu Normal University, Xuzhou 221116, PR China. E-mail: hj1986424@jsnu.edu.cn
First published on 25th March 2020
Abstract
Noncovalent binding of peptides to human serum albumin protects against renal clearance and enzymatic degradation. Herein, we investigated the effect of mycophenolic acid (MPA) albumin binders for improving the stability of peptides. For proof-of-principle, the short acting glucagon-like peptide-1 (GLP-1) receptor agonist lixisenatide was selected and functionalized with different MPA albumin binders. In vitro, all lixisenatide analogues showed well preserved GLP-1 receptor activation potency. High performance affinity chromatography (HPAC) and ultrafiltration analyses indicated that DiMPA was able to confer high albumin affinity to lixisenatide and revealed that affinity is increased for DiMPA modified lixisenatide analogues containing OEG spacers. In db/db mice, the selected peptide 2c showed comparable efficacies to lixisenatide with respect to glucose-lowering and insulinotropic activities. Furthermore, the duration of action of glucose homeostasis of 2c was comparable to semaglutide in db/db mice. Importantly, DiMPA albumin binder did not bring significant toxicity of lixisenatide, as reflected by the comparable toxicity indexes in 2c and semaglutide groups after 2 weeks dosing in normal Kunming mice. Short-term study (21 days) conducted on db/db mice showed the better therapeutic efficacies of 2c than semaglutide on pancreas islets protection. Importantly, in chronic studies (84 days) on db/db mice, 2c exhibited a sustained improvement in glycaemic control, to a greater extent than that of semaglutide. Thus, we propose DiMPA modification as a novel and general method for development of long-acting GLP-1 receptor agonists for type 2 diabetes treatments, and 2c as a promising antidiabetic candidate.
1. Introduction
Over 400 million people are living with type 2 diabetes mellitus (T2DM) which account for around 90% of diabetes cases all over the world.1–3 Worryingly, over the past 30 years, the global prevalence of type 2 diabetes is estimated to be doubled.4–6 T2DM is a complex and costly disease which can cause cardiovascular disorder, blindness, kidney failure, lower limb amputation, and some other long-term complications to decline substantially the quality of life with disability.7–10 The eventual need for multiple medications due to T2DM and its complications in most patients promotes the development of new drugs. Glucagon-like peptide-1 (GLP-1) is a promising pharmacological target in T2DM due to its multiple biological activities including activation of GLP-1 receptor, stimulating the insulin secretion from pancreatic cells, increasing the appetite suppression and weight loss.11,12 However, the half-life is short due to the quick degradation the two N-terminal amino acids of native GLP-1 by dipeptidyl peptidase IV (DPP-IV).13,14 Researchers are committed themselves to utilize numerous approaches to prolong the half-life of GLP-1.15–18Facilitating the physical interaction of the target peptides with human serum albumin (HSA) has been proved an effective way to prolong half-life.19,20 Fatty acids are albumin bound molecules possessed high albumin affinities.21 Therefore, peptide lipidation can be exploited to improve the pharmacokinetic profiles of peptide pharmaceuticals by promoting albumin interactions. This approach has been successfully used in the discovery of liraglutide and semaglutide with reduced proteolytic degradation and renal clearance due to the affinity to HSA.22,23 However, lipidation can bring to a drastic reduction in solubility and is not generally applicable for peptides. Thus, alternative strategies for mediating serum albumin interactions with enhanced solubility, HSA affinity or additional therapeutic property continues to be attractive.24,25 Herein, we explored an alternative methodology to conjugate of novel small molecule albumin binders to peptides.
Some anionic aromatic small molecules have high albumin binding affinities and can be used as an alternative to lipidation. Mycophenolic acid (MPA) has high albumin binding affinity and could extensively bound to HSA (97–98%). In our previous research, we have demonstrated that MPA albumin binders modified Xenopus GLP-1 analogues has longer hypoglycemic activities in db/db mice, as compared to liraglutide.26 In nature, multivalent binding strategies are frequently used to increase the weak ligand–receptor interactions, and the affinity of a dimer ligand can be significantly higher than the sum of avidities for its inherent components.27 Therefore, we hypothesized those small molecule albumin binders with two MPA moieties (DiMPA) could serve as an effective method to further improve the half-life of peptide pharmaceuticals.
Lixisenatide is a potent GLP-1 receptor agonist based on exendin-4 with 3–4 h in vivo half-life, and is clinical approved as once-daily administration.28 To investigate our hypothesis, we selected lixisenatide as a model peptide and conducted a series of proof-of-principle studies on DiMPA modified lixisenatide analogues. First, to avoid serious biological activities lost and identify the suitable modification site on lixisenatide, three MPA albumin binders with different length of alkyl chain were site-specific linked to three lysine residues of lixisenatide (Lys12, Lys27 and Lys39). Using this strategy, we prepared nine different MPA modified lixisenatide analogues at Lys12, Lys27 and Lys39 (1a–1i, Fig. 1), and then explored their in vitro GLP-1 receptor activation potency, stabilities and in vivo glucose lowering activities. Compound 1f with high in vitro receptor activation potency, plasma stability and in vivo hypoglycemic activity was selected for the next step DiMPA modification.
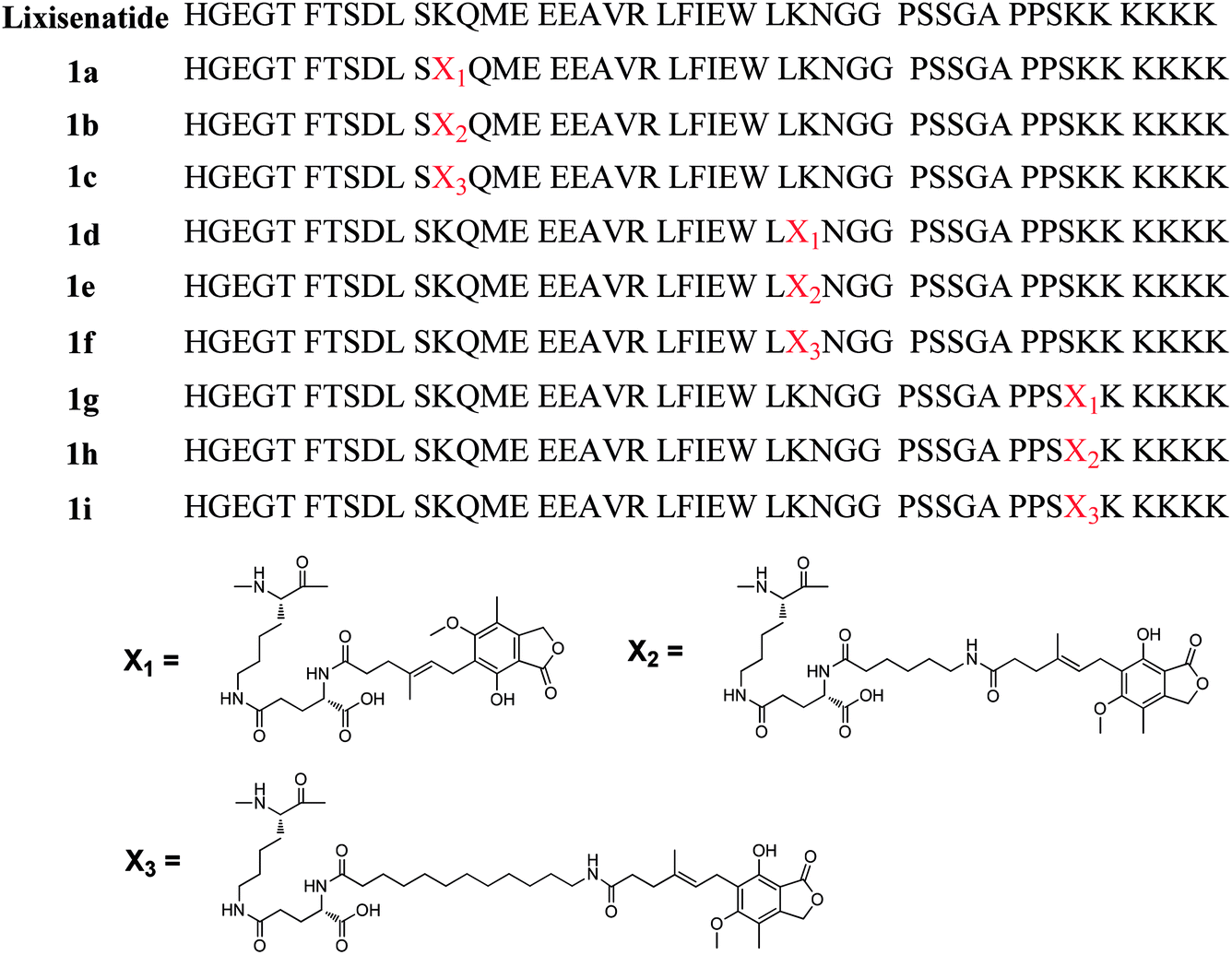 | ||
| Fig. 1 Structures of mono-MPA lixisenatide conjugates 1a–1i. | ||
Next, by using the two amino groups on lysine, we designed a DiMPA albumin ligand with C11 alkyl chain and linked to Lys27 of lixisenatide, affording 2a (Fig. 3). Considering the DiMPA modification could bring solubility issues and also investigate of the importance of a spacer, different length of 8-amino-3,6-dioxa-octanoic acid (OEG) spacers were introduced between the DiMPA albumin binding ligand and lixisenatide, giving 2b–2d (Fig. 3). MPA is an inhibitor of inosine monophosphate dehydrogenase (IMPDH) and has been used as an immunosuppressive agent.29 We hypothesize that MPA conjugated to peptide hormones like lixisenatide will not generate immunosuppressive effects. Mycophenolate mofetil is a prodrug of MPA, which is normally administered in thousands of mg per day and with ∼90% bioavailability,30 it pharmacologically relevant dose far exceed that of lixisenatide (recommended dose of lixisenatide is 10–20 μg per day).31
In the present study, we investigated the DiMPA albumin binders as a novel approach to improve peptide pharmacokinetics. For this purpose, we conducted proof-of-principle studies using short-acting GLP-1 receptor agonist lixisenatide and site-specific modified by DiMPA albumin ligands. Moreover, the physicochemical, stabilities, antidiabetic and insulinotropic characteristics, toxicities, and long-term treatment effects of these lixisenatide analogues were extensively investigated.
2. Materials and methods
2.1. Materials and animals
Rink Amide MBHA resin, DIC, HOBT, N-Fmoc-amino acids, Boc-His(Boc)-OH, Fmoc-Lys(Dde)-OH, Fmoc-Glu-OtBu, Fmoc-Lys(Fmoc)-OH, lixisenatide, semaglutide were obtained from the GL Biochem (Shanghai, China) Ltd. Fmoc protected 6-aminocaproic acid (Fmoc-6-Ahx-OH) and 12-aminolauric acid (Fmoc-12-Ado-OH) were purchased from Sigma-Aldrich Co. (St. Louis, MO). Fmoc-8-amino-3,6-dioxa-octanoic acid (Fmoc-AEEAc-OH) was purchased from Bachem (Bachem AG, Switzerland). The cAMP kit was purchased from the CIS Bio International (Bedford, MA, USA) and the insulin kit was obtained from Nanjing Jiancheng Bioengineering Institute (Jiangsu, China). All other reagents were obtained from Sigma-Aldrich Co. (St. Louis, MO) unless otherwise indicated.Male Kunming mice (20–25 g) were purchased from Qinglongshan Animal Breeding Farm (Nanjing, China). Male db/db mice (BKS-Leprdb/Nju, 8 weeks, 30–40 g) were purchased from Model Animal Research Center of Nanjing University (Nanjing, China). All animals were housed six in a cage with a 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 h light–dark cycle and the temperature was set at 25 ± 2 °C. All mice had free access to food (standard chow) and water and all experiments were approved by the institutional committee at Jiangsu Normal University and were in compliance with the Laboratory Animal Management Regulations in China, the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (revised 2011).
:12 h light–dark cycle and the temperature was set at 25 ± 2 °C. All mice had free access to food (standard chow) and water and all experiments were approved by the institutional committee at Jiangsu Normal University and were in compliance with the Laboratory Animal Management Regulations in China, the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (revised 2011).
2.2. General procedure for peptide synthesis
The peptide backbones of lixisenatide analogues (1a–1i, 2a–2d) were carried out on Rink amide MBHA resin with a loading of 0.382 mmol g−1 using Fmoc synthesis strategy on a semi-automated peptide synthesizer (mode PSI-200, Peptide Scientific Inc., USA).32 The N-terminal His was replaced with Boc-His(Trt)-OH and the Lys on the conjugation site was replaced with Fmoc-Lys(Dde)-OH to prepare peptides with MPA side chain. The Dde protection group was removed selectively by washing 5 times (10 min) with 2% (v/v) hydrazine hydrate in DMF. For 1a–1i, Fmoc-Glu-OtBu, Fmoc-6-Ahx-OH or Fmoc-12-Ado-OH, and MPA (4 equiv.) were coupling in order by using DIC/HOBT according to their structure. For 2a–2d, Fmoc-Lys(Fmoc)-OH was used to construct DiMPA ligand. Specifically, the Dde protection group on the modification site was removed using the same method described above. Then Fmoc-AEEAc-OH, Fmoc-Glu-OtBu, Fmoc-12-Ado-OH, and Fmoc-Lys(Fmoc)-OH (4 equiv.) were added in order according to their structure and coupled using the same condition described above. The two Fmoc protected groups of Fmoc-Lys(Fmoc)-OH were removed, and MPA (8 equiv.) was dissolved with DIC (8 equiv.) and HOBt (8 equiv.) in DMF (5 mL), and coupled with the two amine groups on Lys for 3 h. The crude products were precipitated in cold ether and washed with cold ether for 4 times after cleaved from the resin with the usage of EDT/phenol/water/thioanisole/TFA (2.5:5:5:5:82.5). The crude analogues were further purified by RP-HPLC (LC-20AP, Shimadzu) with C18 column (Hypersil GOLD, 250 × 20 mm, 12 μM). For 1a–1i, the isolated yields were between 45–55% due to unavoidable loss during purification. For 2a–2d, the isolated yields decreased to 30–40% due to the more complicated synthetic process. All products were confirmed characterized by Agilent 6538 Q-TOF mass or Bruker MicroTOF Q2 with the application of ESI ionization method and the purity was confirmed by analytical HPLC (Agilent 1260 infinity).
2.3. In vitro GLP-1 receptor activation assay
HEK293 cells expressing the hGLP-1 receptor were used for functional assay as previously described.33 Cells were suspended in DMEM growth medium containing 0.5% FBS, 20 mM HEPES, 1% penicillin–streptomycin and 2 mM L-glutamine. Cells were plated into 384-well microplates. The tested compounds were dissolved in DMSO, then diluted in assay buffer and transferred to the microplate to make the assay concentrations at 1 × 10−13 to 1 × 10−6 M. Then, the plate was incubated at 5% CO2 for 30 min at 37 °C. The cAMP concentrations was measured by Cisbio cAMP dynamic 2 kit with the use of Envision 2104 multilabel reader (PerkinElmer, U.K.). The cAMP data was exported and the EC50 values were calculated by sigmoidal curve fitting by GraphPad Prism 5.0.2.4. In vitro stability assay
The in vitro stability of lixisenatide and 1a–1i were determined using a previously described method with small modification.34 In brief, plasma were obtained from SD rats (male, 200–250 g) and stored at −20 °C until use. Lixisenatide and 1a–1i were dissolved in PBS (pH 7.4) and 0.5 mL of the compound were added to the 1 mL plasma to achieve concentration of 1000 ng mL−1. The mixture was vortex mixed for 60 s and then incubated at 37 °C for 6, 12, 24, and 48 h. At each time point, 100 μL of sample was removed and mixed with acetonitrile (200 μL) containing 2% formic acid for the precipitation of plasma protein. Then the mixture was centrifuged at 14000 rpm for 15 min, and the supernatant (50 μL) was analyzed via liquid chromatography-mass spectrometry (LC-MS/MS) system (AB SCIEX TripleTOF 4600, USA). The degradation curves of each tested compounds were determined in triplicate.
2.5. Intraperitoneal glucose tolerance test (IPGTT) in normal Kunming mice
IPGTT were carried out in normal male Kunming mice (n = 6) using a previously described method with small modification.35 Mice were fasted overnight (12 h) prior to a 2 g kg−1 i.p. injected of glucose. The mice were administered a single i.p. injection of saline (control), lixisenatide, 1d, 1e, and 1f (25 nmol kg−1) 30 min prior to the i.p. glucose injection. Tail vein blood was drawn for glucose measurements 30 min before glucose load and at t = 0, 15, 30, 45, 60, and 120 min. One-touch glucometer (GA-3, Sinocare, Changsha, China) was used to measure the blood glucose levels.2.6. Relative and direct albumin binding assay
The relative serum albumin binding abilities of lixisenatide, 1f, and 2a–2d were measured by high-performance affinity chromatography (HPAC) analysis using CHIRALPAK-HSA column (Chiral Technologies Europe) according to previous described method with small modifications.36 In brief, an Agilent 1260 HPLC equipped with an immobilized HSA column (2 mm × 50 mm, 5 μm) was used for chromatographic separation. Considering the possible high albumin binding rate of 2a–2d, 20 mM phosphate buffer containing 25% 2-propanol was used as the isocratic mobile phase. The tested peptides were injected into the column at 25 °C and the detection wavelength was 214 nm. The retention times (tr) of the tested compounds were the average of three independent measurements. The column void time (to) was determined by injecting a non-binding compound (acyclovir).The direct serum albumin binding abilities of lixisenatide, semaglutide, 1f, and 2a–2d were measured using a modified ultra filtration method according to previous described method with small modifications.26 In brief, test peptides were dissolved in 1 mL of PBS phosphate buffer (pH 7.4, 100 μg mL−1), and 3 mL of 10 mg mL−1 of HSA in PBS (pH 7.4) was added. The solution was vortex mixed for 2 min. The mixture was incubated at 37 °C for 1 h, and 1 mL of the mixture was removed and loaded into Centricon centrifugal filter device (30 kDa molecular weight cutoff, Millipore, Bedford, USA). Then, the filter device was subjected to centrifuge for 40 min at 3500 rpm. The filtrate was determined using HPLC to analysis the content of tested peptides. The adsorption of each peptide to the filter membrane was also determined using the similar method. The albumin binding rates of tested peptides were determined in triplicate.
2.7. IPGTT in diabetic db/db mice
Male db/db mice were used to evaluate acute actions of peptide on blood glucose.37 Briefly, db/db mice (n = 6) were subjected to 16 h of fasting and injected i.p. with glucose (1 g kg−1). Saline (control), lixisenatide, 2a, 2b, 2c, and 2d (25 nmol kg−1) were i.p. injected 30 min prior to the glucose load. Tail blood glucose levels were determined with a one-touch glucometer before (−30 min) and at 0, 15, 30, 60, and 120 min after the injection of glucose.2.8. Glucoregulatory and insulinotropic assay
The effects of lixisenatide and 2c on glucoregulatory and insulinotropic were tested by IPGTT on db/db mice as previous reported.38 Mice (n = 6) were fasted overnight (16 h) and then i.p. injected with saline (control), lixisenatide and 2c (25 nmol kg−1) 30 min prior to the glucose load. The glucose (1 g kg−1) was i.p. loaded at 0 min. Tail vein blood was drawn for blood glucose levels measurement at −30 and 0, 10, 15, 30, 45, 60, and 120 min by using one-touch glucometer, and for plasma insulin measurement at 0, 15, 30, and 60 min by using mouse insulin ELISA kit (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China).2.9. Glucose-stabilizing activity in db/db mice
The antidiabetic duration of lixisenatide, semaglutide, and 2c were determined using two methods on db/db mice. In the first pre-oral glucose tolerance test (OGTT) experiment, glucose was administered orally to mimic the postprandial glycemic condition.39 Male db/db mice (n = 6) were fasted overnight (16 h) and saline (control), lixisenatide, semaglutide, and 2c (25 nmol kg−1) were pre-injected (i.p.) at 4 and 8 hours before oral glucose administered (t = 0 h, 1.5 g kg−1). Blood samples were obtained at −4 or −8, 0, 0.25, 0.5, 1, 1.5 and 2 h from the cut of the tip tail, and blood glucose concentrations were determined by one-touch glucometer. In the second experiment, the hypoglycemic activities of lixisenatide, semaglutide and 2c were measured in db/db mice (n = 6) under non-fasted condition.40 The mice were allowed to free access to food. On the experiment day, mice were s.c. injected with saline (control), lixisenatide, semaglutide, and 2c (25 nmol kg−1) at t = 0 h. The blood was obtained and the blood glucose levels were measured using the same above described method at 0, 2, 4, 6, 12, 24 and 48 h.2.10. Acute toxicity assay of 2c
The toxicity test of 2c was conducted on normal Kunming mice for two weeks using previously described method with small modifications.41 Mice were randomly divided into three groups, and saline (control), semaglutide (100 nmol kg−1) and 2c (1000 nmol kg−1) were once daily s.c. injected for 14 days. Water and food were provided ad libitum during the experiment. At day 15, each group of mice were sacrificed and the whole blood were collected, and the blood samples were centrifuged to obtain serum. Blood urea nitrogen (BUN), alanine aminotransferase (ALT), serum creatinine (SCr), and aspartate aminotransferase (AST) were measured using biochemical analyzer (Beckman Coulter AU5811, Japan).2.11. Immunogenicity testing of 2c
The immunogenicity test of 2c was conducted by evaluated the ability of 2c to induce T cell proliferation in peripheral blood mononuclear cells (PBMC) from 50 Chinese individuals (obtained from LDEBiO, Guangzhou, China), using previous described method.42 Briefly, PBMC were cultured in AIMV medium (Life Technologies, Carlsbad, CA) and added to the 24-well plates (2 mL) to reach a final concentration of ∼3 ×106 cells per mL, and then stimulated by addition of lixisenatide, semaglutide and 2c in AIMV medium with a final concentration of 30 μg mL−1 of each tested peptides. The 24-well plates were incubated in CO2 incubator (5%) at 37 °C for 8 days. The cells in each well of the plate were transferred to 96-well plate on days 5, 6, 7 and 8. The cultures were pulsed with [3H]-Thymidine (PerkinElmer) and incubated for further 18 h and counts per minute (cpm) for each well were determined. The stimulation index (SI) was calculated by dividing the proliferative response of the test well (cpm) by the proliferative response of the medium-only treated well (cpm) for each donor, and SI greater than 2.0 was considered positive. The % donors responding was calculated by taking the number of donors that had a positive response over the entire time course (5–8 days) as a percentage of the total number of donors that were tested.2.12. Short-term effects of 2c on pancreas development by once daily administration
Seven days before the first dosing, tail vein blood was obtained from non-fasted animals and DCA 2000 chemistry analyzer (Siemens/Bayer, USA) was used to determine of baseline HbA1c levels using previous described method.43 Based on the HbA1c levels, male db/db mice (n = 6) were stratified into the following groups: control (saline); semaglutide (25 nmol kg−1); 2c (25 nmol kg−1). The mice were once daily s.c. treated with saline, semaglutide and 2c. Food intake and body weight were determined daily in the morning. The treatment study was lasting 21 days and tail vein blood was collected again from non-fasted mice for determination of HbA1c levels. Finally, mice were sacrificed by cervical dislocation, and the pancreases were removed and stored in 4% paraformaldehyde. The insulin immunohistochemistry studies were performed using previously described method. The integrated optical density (IOD) values of insulin in each group were analyzed by software Image-pro plus 6.0 (Media Cybernetics Inc., USA).2.13. Short-term effects of 2c on pancreas development by once-weekly administration
Male db/db mice (n = 6) were stratified into the following groups: control (saline); lixisenatide (25 nmol kg−1); semaglutide (25 nmol kg−1); 2c (25 nmol kg−1) based on the HbA1c levels, using the same method described above. The mice were once weekly s.c. treated with saline, lixisenatide, semaglutide and 2c. Body weight were determined daily in the morning. Fasting blood glucose concentrations were measured before and at the end of the study from mice fasted for 6 h. The experiment was lasted for three weeks and HbA1c levels were determined again using the same method described above. Finally, each group of mice were sacrificed and the pancreases were removed to perform insulin immunohistochemistry studies using the same method described above.2.14. Chronic treatment of 2c on glycaemic control
Male db/db mice were subjected to an OGTT performed as previously described. In brief, mice were fasted 16 h and glucose (1.5 g kg−1) were loaded orally, and glucose tolerance was measured in relation to area under the curve (AUC) of blood glucose levels. Based on the OGTT results, mice exhibiting similar glucose tolerances were divided into 3 groups (n = 6). Three groups of mice were once daily s.c. injected with saline, semaglutide, and 2c (25 nmol kg−1) based on their antidiabetic duration profiles for 42 days, and then followed by 42 days of vehicle (saline) treatment. Glucose tolerance was assessed on days 42, 56, 70 and 84 during the treatment period using the same method described above. Furthermore, fasting glucose levels were determined from 10 h fasted mice on days 0, 35, 49, 63 and 77 of the study. HbA1c levels were determined on days −1, 43 and 85 using the same method described above.442.15. Data analysis
Results are expressed as means ± SD unless otherwise stated. Statistical significances of the data were performed by one-way ANOVA followed by appropriate post hoc tests. All statistical calculations were conducted with GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA) and P < 0.05 values were considered statistically significant.3. Results
3.1. Design and synthesis of lixisenatide analogues 1a–1i
To investigate whether DiMPA small molecule albumin binders could be an effective methodology to improve peptide pharmacokinetics, DiMPA was linked to a short acting GLP-1 agonist, lixisenatide. However, to avoid the serious biological activities lost caused by DiMPA modification and identify the suitable modification site on lixisenatide, the three lysine residues on lixisenatide (Lys12, Lys27 and Lys39) were selected and site-specific modified by three MPA albumin binders with one MPA moiety and different length of alkyl chain, affording 1a–1i (Fig. 1). Rink Amide MBHA resin was used as a polymer support to obtain an amidated C-terminus.Site-specific MPA modification at the side chain of Lys12, Lys27 and Lys39 was achieved by replacing the corresponding Lys to Fmoc-Lys(Dde)-OH, and the N-terminal His was replaced with Boc-His(Boc)-OH. The Dde group was specially removed by 2% hydrazine hydrate/DMF (v/v), and Fmoc-Glu-OtBu was coupled with the side amino group of lysine. Then MPA was directly attached to the amine of a glutamyl spacer, affording 1a, 1d, 1g. For other compounds with fatty chain spacer, N-Fmoc-protected ω-amino acids (6-aminocaproic acid and 12-aminolauric acid) were attached to the amine of a glutamyl spacer. Finally, after removal of Fmoc group, the ω-amino groups were attached to the carboxyl group of MPA. Using this method, the whole synthetic procedure of the crude products was conducted on solid support (Scheme S1, see ESI†). Crude products were characterized by HPLC and MS and further purified using semi-preparative RP-HPLC (see ESI†).
3.2. Biological activities and stabilities of 1a–1i
The in vitro GLP-1 receptor activation potency of 1a–1i was studied by a GLP-1 receptor cAMP activity assay to evaluate whether attachment of the MPA had changed the GLP-1 receptor potency of lixisenatide. The EC50 values of 1a–1i are summarized in Table 1 and the concentration response curves of lixisenatide and 1a–1i were shown in Fig. 2A and B. In general, MPAs modification was well tolerated at Lys27 (1d–1f), and the length of alkyl chain has no significant impact on potency. Meanwhile, the analogues with MPAs modification on Lys12 (1a–1c) exhibited a more profound reduction in potency compared to lixisenatide and 1d–1f. The greatest impact on potency was found by introducing MPAs at Lys39, as the serious reductions in receptor activation potency observed for 1g–1i. The in vitro stabilities of 1a–1i were examined in rat blood plasma at 37 °C for 48 h in order to examine their stability against peptidases and proteases in a life-near system. The degradation profiles of 1a–1i were shown in Fig. 2C and D. The stabilities of 1a–1i were exhibited positive correlation with length of the alkyl chain and the modification site of MPA has little impact on stabilities (Table 1). Considering the high potencies and stabilities of 1d–1f, IPGTT was performed on Kunming mice to validate their in vivo antidiabetic activities. As shown in Fig. 2E and F, the administration of lixisenatide significantly blunted IPGTT-evoked blood glucose increases compared to the vehicle. The same applied to 1d–1f, which reduced blood glucose excursion levels to similar level as lixisenatide. The hypoglycemic activity of 1f was similar with 1d and 1e, and 1f only showed slightly decreased activity toward glucose-lowering as compared with 1d and 1e. Moreover, the in vitro stability of 1f (33.4 h) was significant longer than that of 1d (13.1 h) and 1e (21.2 h). Considering the high in vitro stability is essential for the in vivo long-acting glucose-lowering activity, together with the similar in vivo glucose-lowering activities of 1d–1f, 1f was finally selected for the next step DiMPA modification.| Peptide | EC50a (nM) | Plasma half-lifeb (h) | Peptide | EC50a (nM) | Plasma half-lifeb (h) |
|---|---|---|---|---|---|
| a Data are represented as EC50. Values are the means ± SD from three independent experiments.b The plasma half-lives of lixisenatide and 1a–1i were calculated after incubation of peptides in rat plasma over 48 h. | |||||
| Lixisenatide | 0.076 ± 0.013 | 7.1 | 1e | 0.13 ± 0.03 | 21.2 |
| 1a | 0.19 ± 0.02 | 13.2 | 1f | 0.32 ± 0.13 | 33.4 |
| 1b | 0.49 ± 0.11 | 19.1 | 1g | 0.59 ± 0.16 | 11.9 |
| 1c | 0.98 ± 0.24 | 30.0 | 1h | 1.97 ± 0.53 | 17.8 |
| 1d | 0.085 ± 0.021 | 13.1 | 1i | 4.68 ± 0.85 | 31.6 |
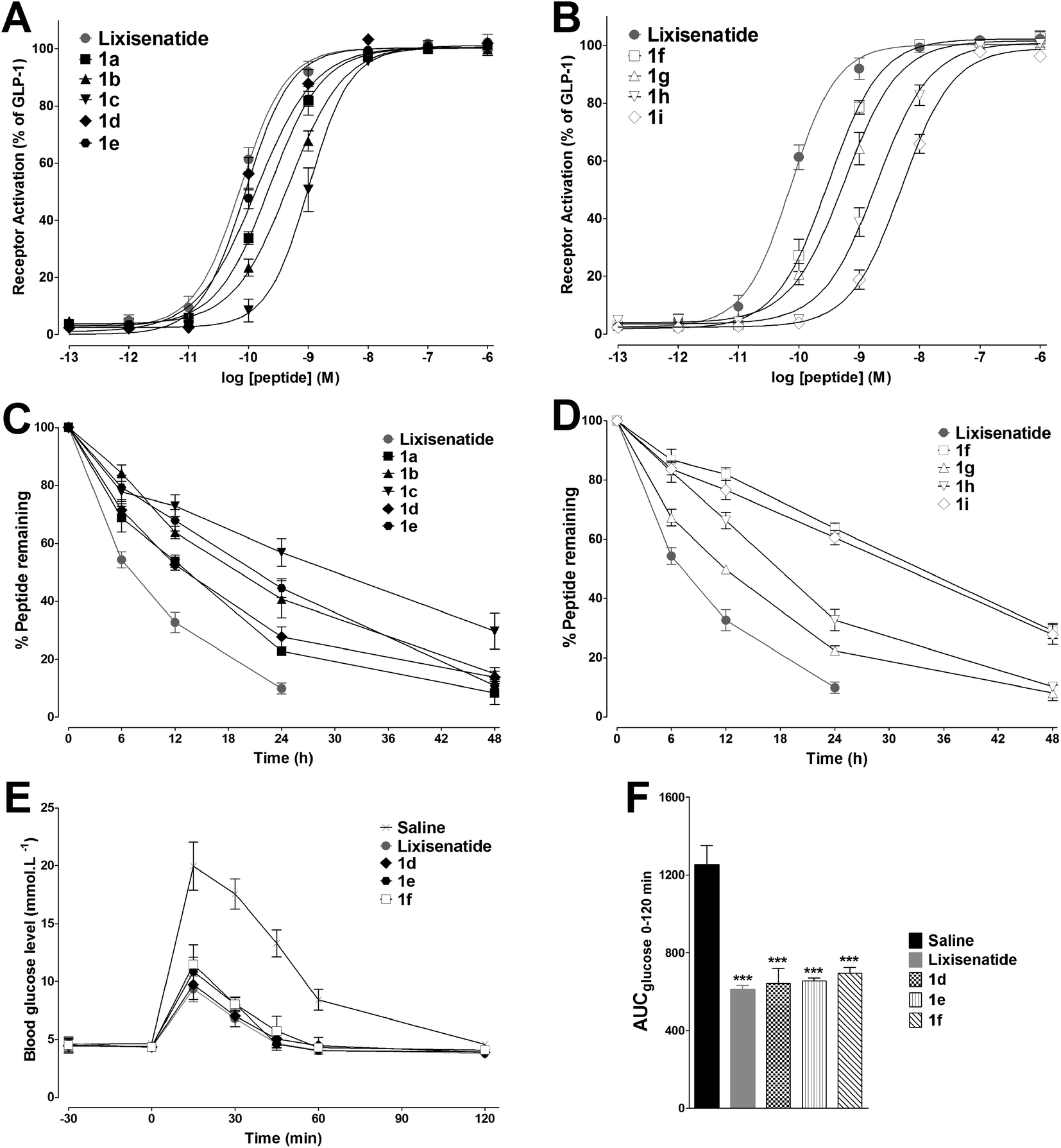 | ||
| Fig. 2 In vitro bioactivity and stability tests of 1a–1i and in vivo biological activities of 1d–1f. (A and B) Dose–response curves of 1a–1i for activation at the GLP-1 receptor. Means ± SD. Tests were performed in triplicate and conducted in three independent experiments. (C and D) Degradation profiles of 1a–1i in rat plasma. Means ± SD, n = 3. (E) Acute effects on IPGTT in normal Kunming mice treated with saline, lixisenatide or 1d–1f. (F) Hypoglycemic effects of lixisenatide and 1d–1f expressed as AUCglucose 0–120 min values. Means ± SD, n = 6. ***P < 0.001 vs. saline. | ||
3.3. Design and synthesis of DiMPA modified lixisenatide analogues 2a–2d
The above results showed that MPA modification significant enhanced the in vitro half-life of 1f to ∼33 h. However, this duration is far shorter than the serum half-life of HSA, which is up to 3 weeks in humans.15 Therefore, we envisioned that an increased affinity to HSA could increase the in vitro half-life beyond ∼33 h seen for 1f. We hypothesized that the HSA binding affinity for MPA albumin ligands with two MPA moieties (DiMPA) could be much higher than those with one MPA moiety. Based on the MPA side chain of 1f, we conducted two steps structure optimization. First, to achieve DiMPA modification, an additional Lys was introduced to the ω-amino group of 12-aminolauric acid, and the carboxyl group of MPA was attached to the two amino groups of Lys to obtain DiMPA structure. Next, different length of 8-amino-3,6-dioxaoctanoic acid (O2Oc) spacers were introduced between Lys27 and DiMPA side chain to investigate the importance of a spacer and also minimize the high hydrophobicity brings by DiMPA modification. A total of four DiMPA modified lixisenatide analogues (2a–2d, Fig. 3) were designed and synthesized. The synthesis process of 2a–2d was similar to 1a–1i, and Fmoc-Lys(Fmoc)-OH was used to achieve DiMPA modification (Schemes S2 and S3, see ESI†). Crude products were characterized and purified using the same method described above (see ESI†).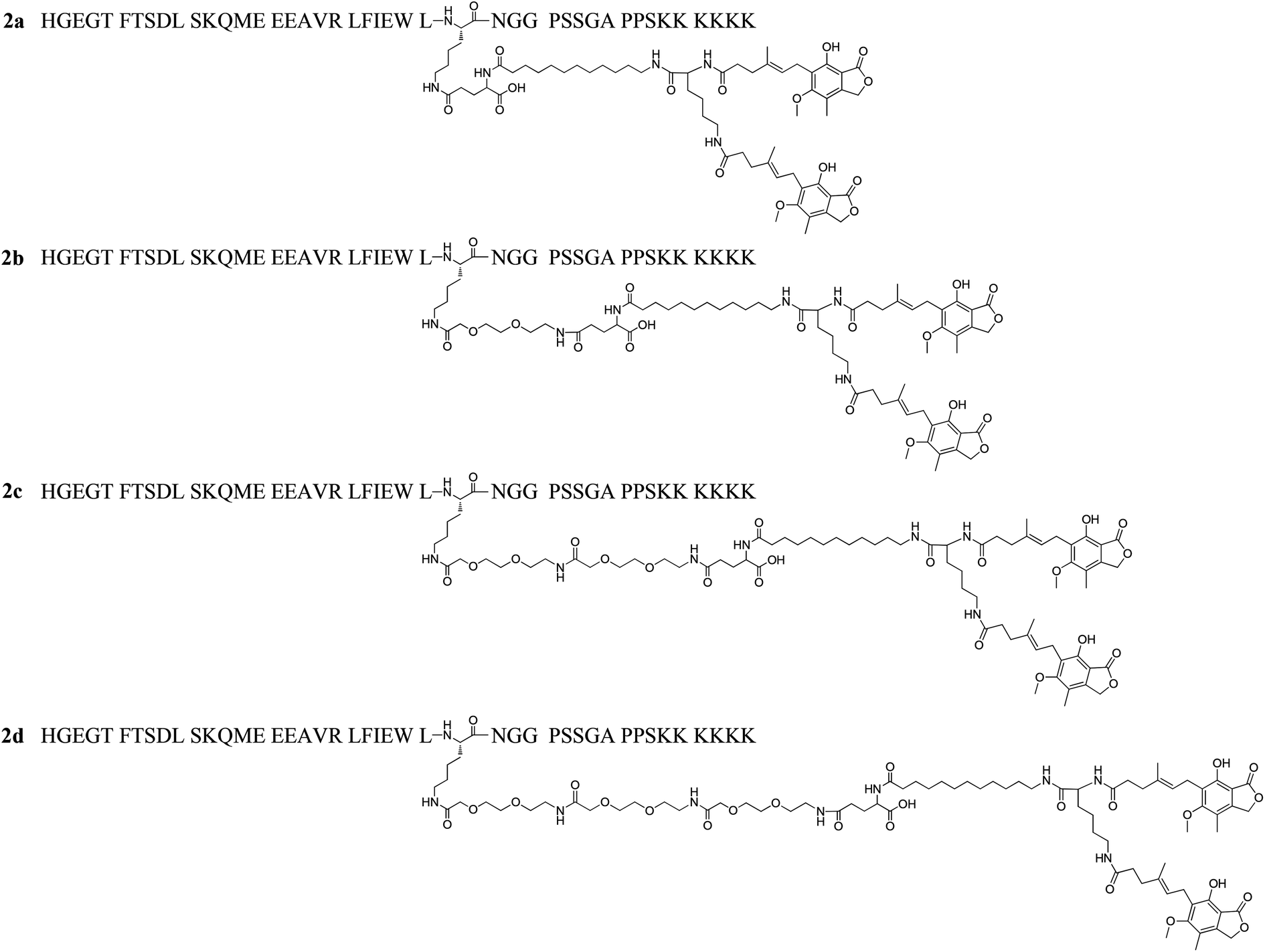 | ||
| Fig. 3 Structures of dimeric-MPA lixisenatide conjugates 2a–2d. | ||
3.4. MPA → DiMPA substitution to further improve the albumin binding affinity
Multivalent binding strategies such as dimeric interactions are frequently occurred in nature and used to increase the weak ligand–receptor interactions,45 we predicted that DiMPA substitution could further increased the albumin binding affinities of 2a–2d compared with 1f. The albumin binding abilities of lixisenatide, 1f, and 2a–2d was firstly evaluated by HPAC analysis, using the retention factor (k′) as the indicator. The value of k′ was obtained according to the equation below: k′ = (tr − to)/to.36 As shown in Fig. 4A, the k′ values of lixisenatide and 1f were ∼1.1 and ∼6.7, respectively. As expected, the albumin affinity of 2a (k′ = ∼13.3) was higher than that of lixisenatide and 1f, for 2b–2d with OEG spacers, there is a clear trend that with increasing length of the OEG spacer, the compounds display an increased affinity to albumin binding relative to 2a (2b, k′ = ∼14.1; 2c, k′ = ∼15.2; 2d, k′ = ∼17.2). Next, the absolute albumin binding rates of lixisenatide, 1f, and 2a–2d were measured using an ultrafiltration method. As illustrated in Fig. 4B, the albumin binding rates of lixisenatide and 1f were 21.3 ± 2.1% and 81.7 ± 3.5%, respectively, which were significant lower than those of 2a (92.7 ± 2.5%), 2b (95.0 ± 2.0%), 2c (96.3 ± 1.5%) and 2d (97.7 ± 0.6%). Similar with the results in relative albumin binding study, 2b–2d with OEG spacers showed higher albumin binding rates than that of 2a, indicated the favorable effect of OEG spacer on albumin binding.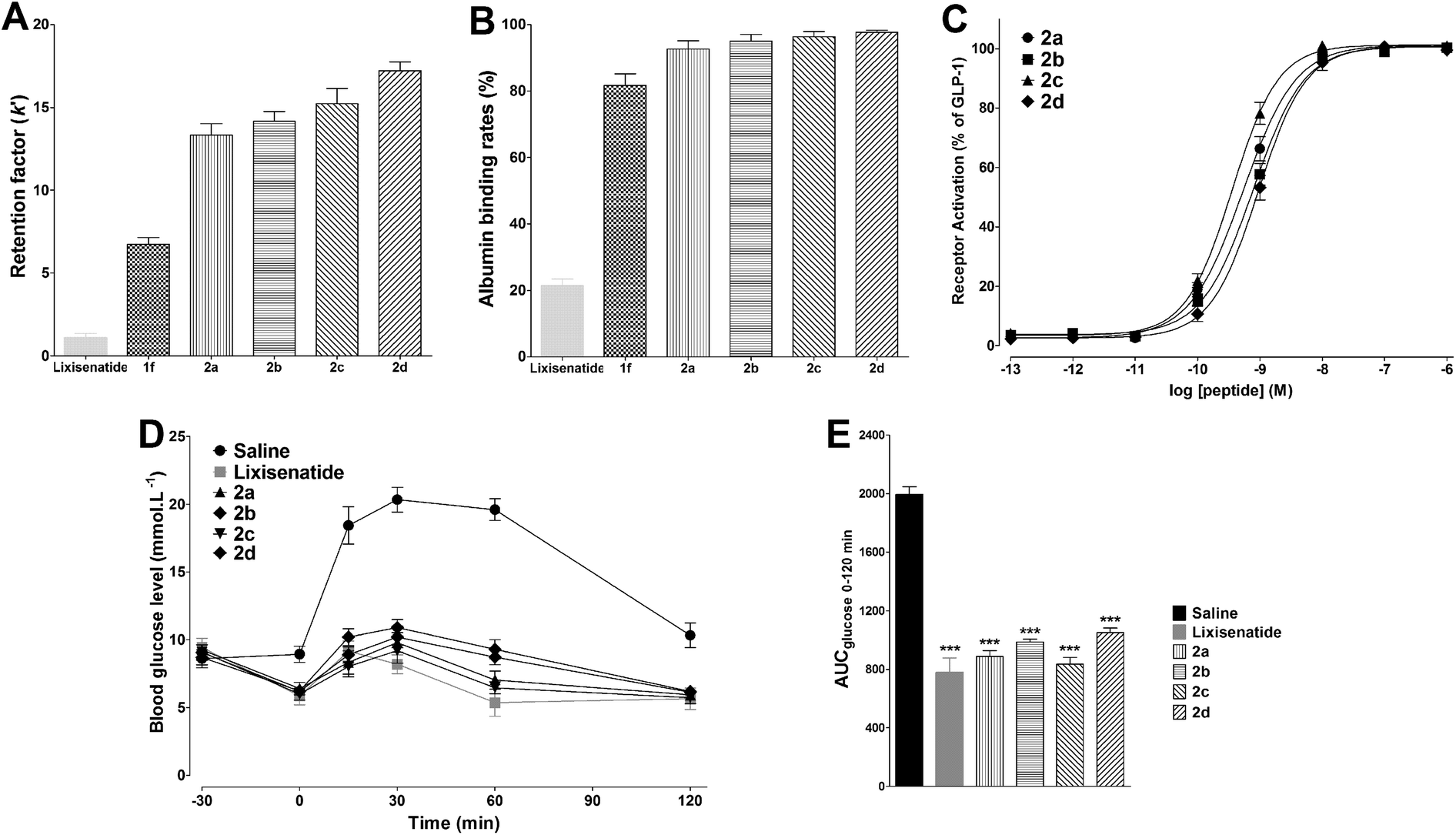 | ||
| Fig. 4 Physicochemical and bioactivity profiles of 2a–2d. (A) The relative albumin binding affinities of lixisenatide, 1f and 2a–2d were determined on an immobilized HSA column using retention factor (k′) as the indicator. Means ± SD, n = 3. (B) The direct albumin binding rates of lixisenatide, 1f and 2a–2d were determined by ultrafiltration method. Means ± SD, n = 3. (C) Dose–response curves of 2a–2d for activation at the GLP-1 receptor. Means ± SD. Tests were performed in triplicate and conducted in three independent experiments. (D) Acute effects on IPGTT in diabetic db/db mice treated with saline, lixisenatide or 2a–2d. (E) Hypoglycemic effects of lixisenatide and 2a–2d expressed as AUCglucose 0–120 min values. Means ± SD, n = 6. ***P < 0.001 vs. saline. | ||
3.5. Selection of a lead compound: 2c exhibited comparable efficacious with lixisenatide in vivo
The in vitro GLP-1 receptor activation potency of 2a–2d was determined using the same method described above. As shown in Fig. 4C, compared with 1f, 2a (EC50 = 0.53 ± 0.11 nM) showed a slight reduction in potency as a consequence of DiMPA modification. For 2b–2d, the OEG spacer containing two OEG moieties was more favorable for the potency, as reflected by the higher potency of 2c (EC50 = 0.36 ± 0.10 nM) compared to 2a, 2b (EC50 = 0.77 ± 0.22 nM) and 2d (EC50 = 0.93 ± 0.25 nM). Next, the in vivo glucose-lowering activities of 2a–2d were studied in the IPGTT in type 2 diabetic db/db mice. Fig. 4D shows the time course for blood glucose changes during IPGTT. Consistent with the in vitro potency results, 2c significantly reduced glucose excursion after glucose challenge, comparable with that observed in lixisenatide group, and glucose AUC values further proved that the antidiabetic effects of 2c and lixisenatide were similar (Fig. 4E). The hypoglycemic activity of 2a was less effective compared with lixisenatide and 2c, while the hypoglycemic activities of 2b and 2d were much worse than 2c. Based on the above in vitro and in vivo studies, we identified 2c as a novel and potent GLP-1 receptor agonist with high albumin binding affinity and hypoglycemic activity, and 2c was selected as lead compound and more biological evaluations were conducted to explore its therapeutic utilities.3.6. 2c Exhibited potent glucose-lowering and insulinotropic activities in db/db mice
In the IPGTT, lixisenatide and 2c significantly improved glucose tolerance after i.p. administration at doses of 25 nmol kg−1 relative to the control (saline). As shown in Fig. 5A and C, accompanied by the enhanced plasma insulin levels, the blood glucose levels were decreased. Both lixisenatide and 2c resulted in increased plasma insulin concentrations at 15 min after the glucose load compared with control. The time courses and AUC values for blood glucose and plasma insulin concentrations were very similar following treatment with either of lixisenatide and 2c (Fig. 5).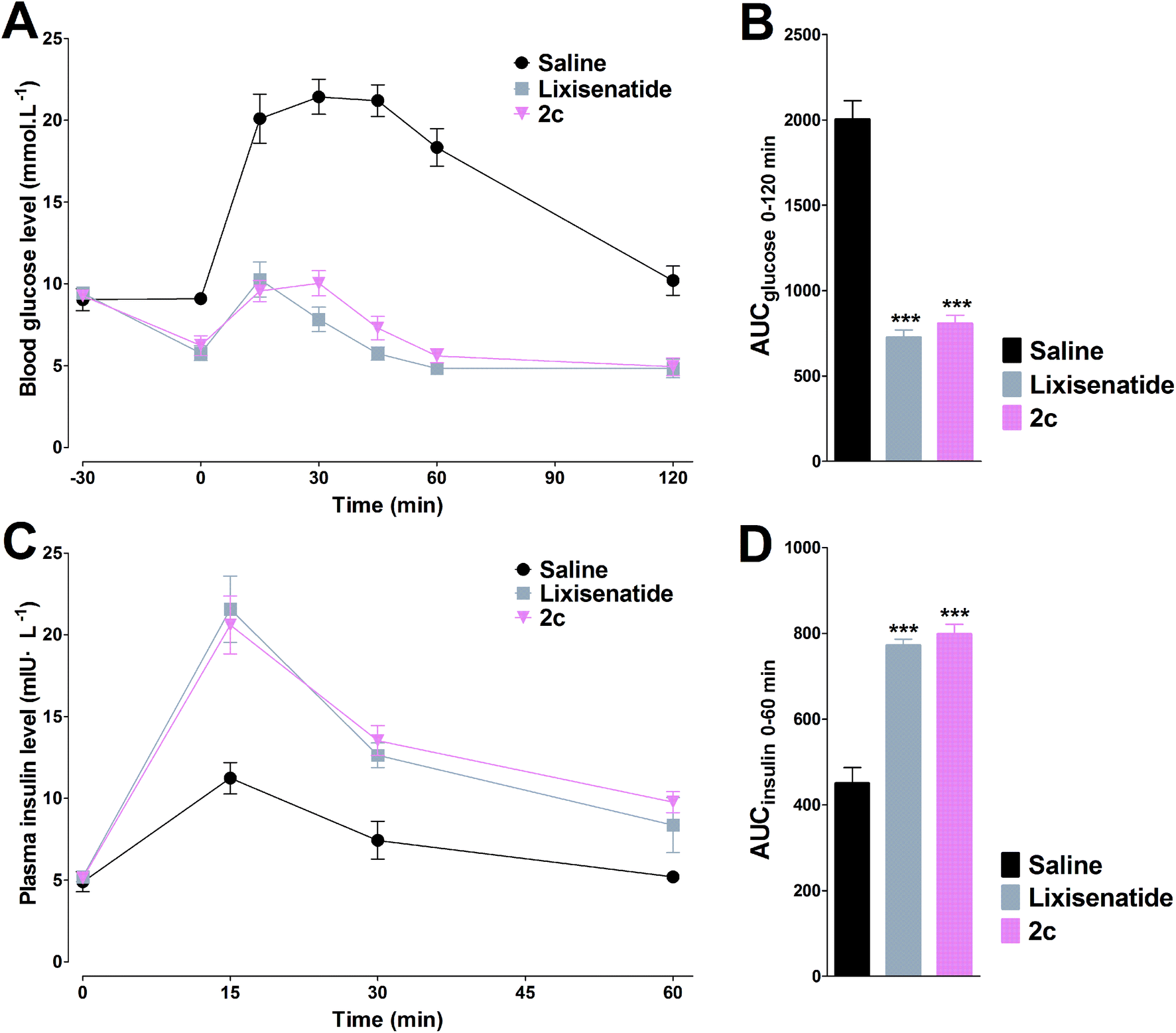 | ||
| Fig. 5 Glucose-lowering and insulinotropic activities of lixisenatide and 2c were determined by IPGTT in db/db mice. The time–response curves of blood glucose (A) and plasma insulin concentrations (C). The calculated AUC values of blood glucose (B) and plasma insulin (D). Means ± SD, n = 6. ***P < 0.001 vs. saline. | ||
3.7. 2c displayed potent long-acting anti-hyperglycemic effect
Two different experiments were conducted on db/db mice to verify whether the high albumin binding affinity of 2c could transfer to the long-acting hypoglycemic activity in vivo. In the pre-OGTT experiments, compared with control, both semaglutide and 2c could potently reduce blood glucose when mice were pre-injected of tested compounds at 4 or 8 h pre-OGTT. However, the glucose-stabilizing effects of lixisenatide were significantly compromised when administered at 4 h pre-OGTT, and completely lost at 8 h pre-OGTT (Fig. 6A and B). Normally, the blood glucose levels in db/db mice were higher than 15 mmol L−1 under non-fasting condition. By using the hyperglycemic properties of db/db mice, we conducted the second experiment on non-fasting db/db mice. As shown in Fig. 6C, semaglutide and 2c could maintain normoglycemia state up to 48 h, and lixisenatide clearly showed a significant shorter duration of action compared to semaglutide and 2c. Importantly, semaglutide and 2c exhibited similar glucose-lowering activities during 0–48 h. Blood glucose AUC0–48 h values also verified that 2c had similar antidiabetic activity compared with semaglutide.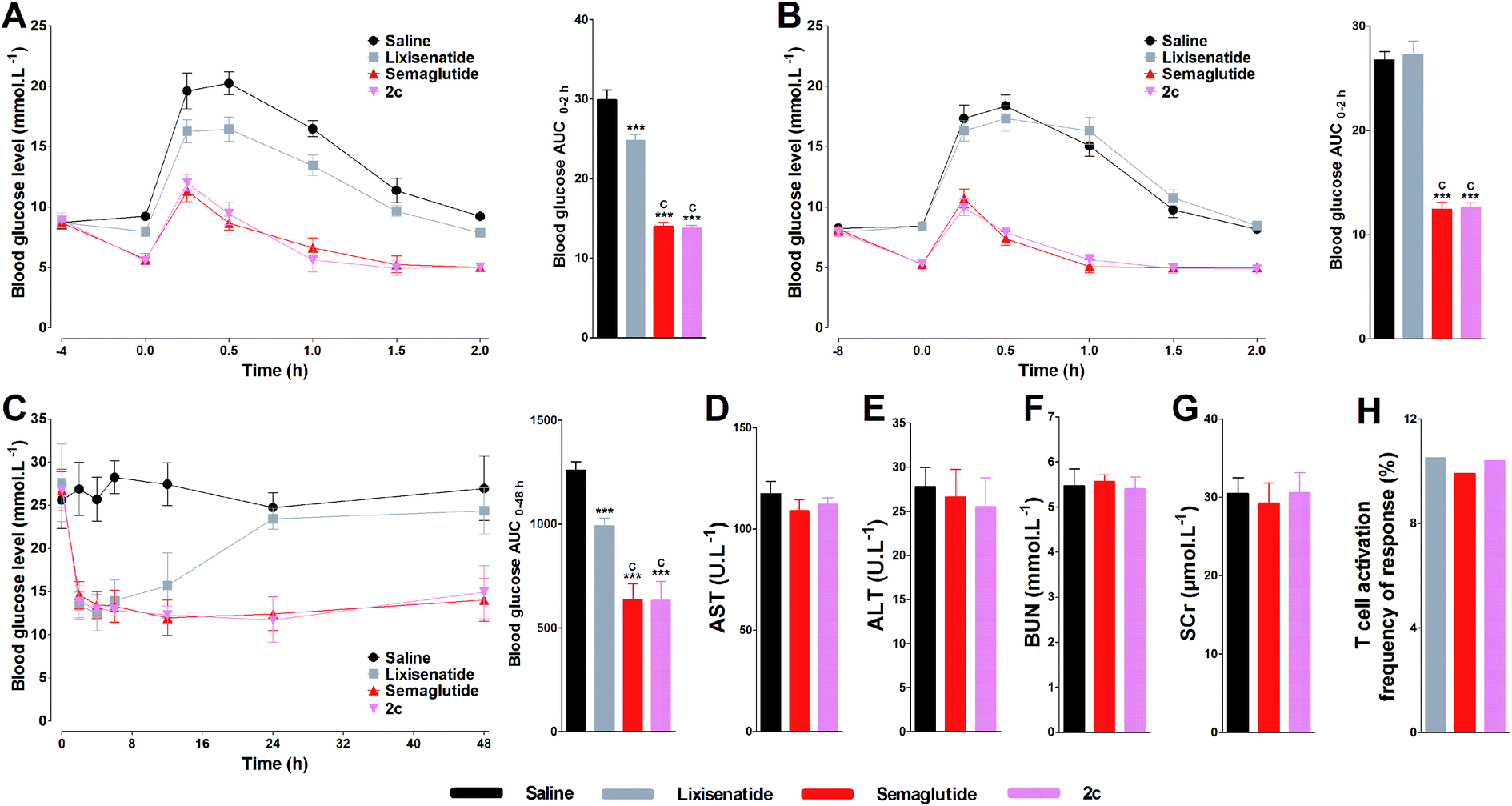 | ||
| Fig. 6 Glucose-stabilizing activity of 2c on db/db mice and acute toxicity assay of 2c on normal Kunming mice. Glucose-stabilizing profiles of lixisenatide, semaglutide and 2c in db/db mice fasted for 16 h, after the administrations (i.p.) of these agents at 4 h (A) and 8 h (B) before OGTT. (C) Hypoglycemic activities of lixisenatide, semaglutide and 2c on non-fasted db/db mice. Biochemical analysis results of 2c on AST (D), ALT (E), BUN (F) and SCr (G) after two weeks dosing on normal Kunming mice. Means ± SD, n = 6. ***P < 0.001 vs. saline, cP < 0.001 vs. lixisenatide. The immunogenicity testing of 2c was evaluated by in vitro T cell proliferation assay and compared with lixisenatide and semaglutide (H). The frequency of response (%) that showed a positive T-cell proliferative response over the course of the entire study (n = 50). | ||
3.8. Acute in vivo toxicity study and immunogenetic testing of 2c
The acute in vivo toxicity study of 2c was studied by loading extreme high dose of 2c (1000 nmol kg−1) on Kunming mice for two weeks. As shown Fig. 6D–G, compared with saline and low dose of semaglutide (100 nmol kg−1), no significant difference was observed for the key indexes of renal toxicity (SCr and BUN) and liver toxicity (AST and ALT) between saline, semaglutide and 2c treated groups, preliminary indicating the safety of 2c. The immunogenicity test of 2c was conducted by evaluating the ability of 2c to induce T cell proliferation in peripheral blood mononuclear cells (PBMC). As shown in Fig. 6H, the percentage of responding donors was 10.4% for 2c, similar to that of lixisenatide (10.5%) and semaglutide (9.9%). Considering that lixisenatide and semaglutide were both approved pharmaceutics, the above results preliminarily suggest the low immunogenicity of 2c.3.9. Short-term effects of 2c on body weight, food intake, and pancreas by once daily administration
Twenty-one days of once daily treatment with semaglutide or 2c potently reduced cumulated food intake and body weight gain (Fig. 7A–C). In addition, treatment with semaglutide or 2c prohibited worsening of the HbA1c in db/db mice (Fig. 7D). The immunohistochemistry analysis of pancreata revealed that the staining of insulin in islets of semaglutide or 2c treated mice were significantly improved compared with saline treated mice, as reflected by the significant enhanced IOD values of insulin in semaglutide or 2c treatment groups (Fig. 7E). Representative images of insulin staining histologic samples are shown in Fig. 7F–H. Importantly, we did not observed significant difference of IOD values of insulin between semaglutide and 2c treatment groups, indicating that DiMPA modification did not change the beneficial treatment effects of lixisenatide. The results of the immunohistochemistry also revealed the beneficial treatment effects of semaglutide and 2c on amelioration of pancreas deficiencies.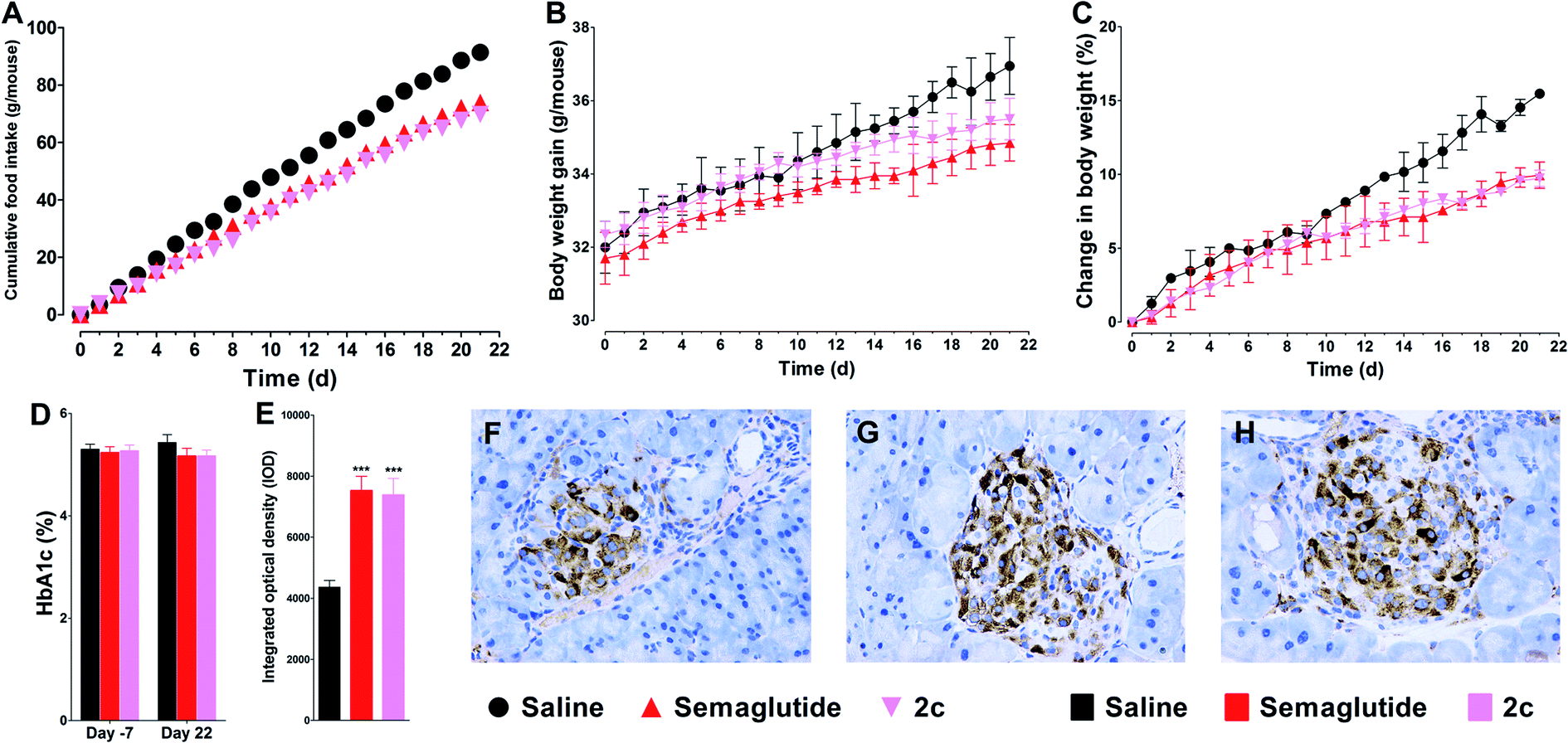 | ||
| Fig. 7 Beneficial treatment effects of semaglutide and 2c in db/db mice on food intake (A), body weight (B and C) and HbA1c (D). (E) Effects on pancreas development after 21 days treatment with saline, semaglutide and 2c. Representative images of insulin staining in saline (F), semaglutide (G) and 2c (H) groups. Means ± SD, n = 6. ***P < 0.001 vs. saline. | ||
3.10. Short-term effects of 2c on body weight, food intake, and pancreas by once weekly administration
Due to 2c exhibited similar long-acting anti-hyperglycemic effect compared with semaglutide, and semaglutide was approved as once weekly administration GLP-1 agonist, we further tested the effect of weekly s.c. injections of lixisenatide, semaglutide or 2c in db/db mice. As shown in Fig. 8, once weekly treatment with lixisenatide did not achieve prominent beneficial effects on body weight, HbA1c and pancreas function. However, both semaglutide and 2c potently reduced body weight gain (Fig. 8A and B). In addition, once weekly treatment with semaglutide and 2c prohibited worsening of the fasted blood glucose and HbA1c levels in db/db mice (Fig. 8C–F). The IOD values of insulin in semaglutide or 2c treatment groups were significantly higher than that of in saline group (Fig. 8G). Unlike the similar IOD values of insulin between semaglutide and 2c treatment groups in once daily administration experiment, the IOD value of insulin in 2c group was significant higher than that of in semaglutide group (P < 0.05). Representative images of insulin staining histologic samples are shown in Fig. 8H–K.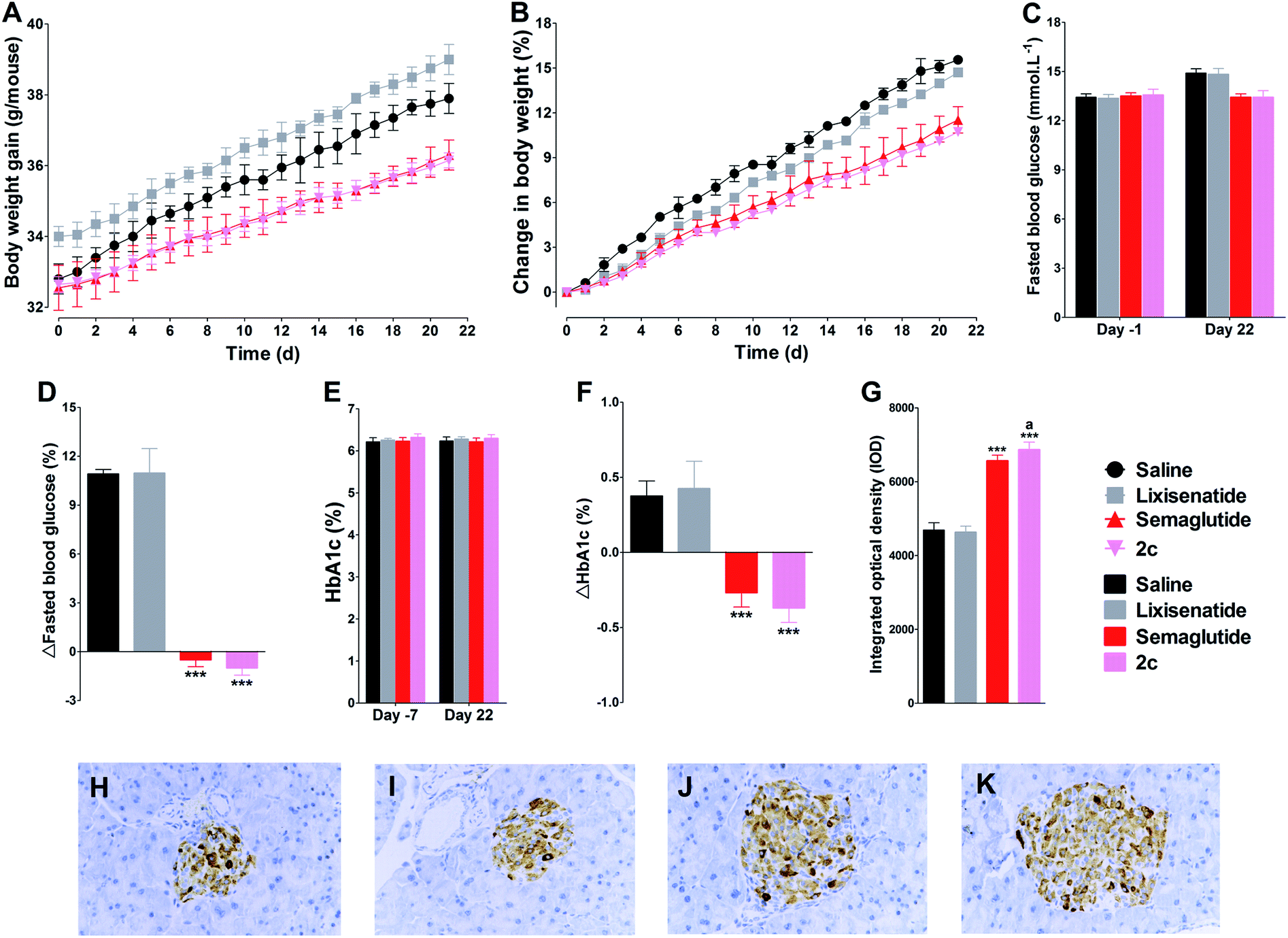 | ||
| Fig. 8 Beneficial treatment effects of lixisenatide, semaglutide and 2c by once weekly administration in db/db mice on body weight (A and B), fasted blood glucose (C and D) and HbA1c (E and F). Effects on immunohistochemistry for insulin after three weeks treatment with saline, lixisenatide, semaglutide and 2c (G). Representative images of insulin staining in saline (H), lixisenatide (I), semaglutide (J) and 2c (K) groups. Means ± SD, n = 6. ***P < 0.001 vs. saline, aP < 0.05 vs. semaglutide. | ||
3.11. Long-term effects of 2c on glycaemic control
The time course of blood glucose and AUCglucose of repeated OGTTs in db/db mice were shown in Fig. 9A–F. During the 84 days of the study, there was progress impairment in glucose tolerance in saline treatment group. This phenomenon was expected, as the diabetic state in db/db mice is known to progress over time. During the 42 days treatment period in all groups of mice treated with semaglutide or 2c, we observed significant improvement in glucose tolerance compared with control group. In the following 42 days saline treatment period, both semaglutide and 2c caused long lasting improvement in glucose tolerance (Fig. 9F), and an effect that was most pronounced in mice treated with 2c, as reflected by the significant lower glucose AUC values in 2c group compared with that observed in semaglutide group at days 56 (P < 0.001), 70 (P < 0.01) and 84 (P < 0.001). Fasting blood glucose concentrations were also determined throughout the study on different weeks from the OGTT. The results of OGTT and fasting blood glucose levels showed a similar trend (Fig. 9G). HbA1c values were reduced in semaglutide and 2c groups relative to control after 6 weeks treatment. Furthermore, HbA1c values remained lower in semaglutide and 2c groups than control for a further 6 weeks after cessation of semaglutide and 2c treatment (Fig. 9H).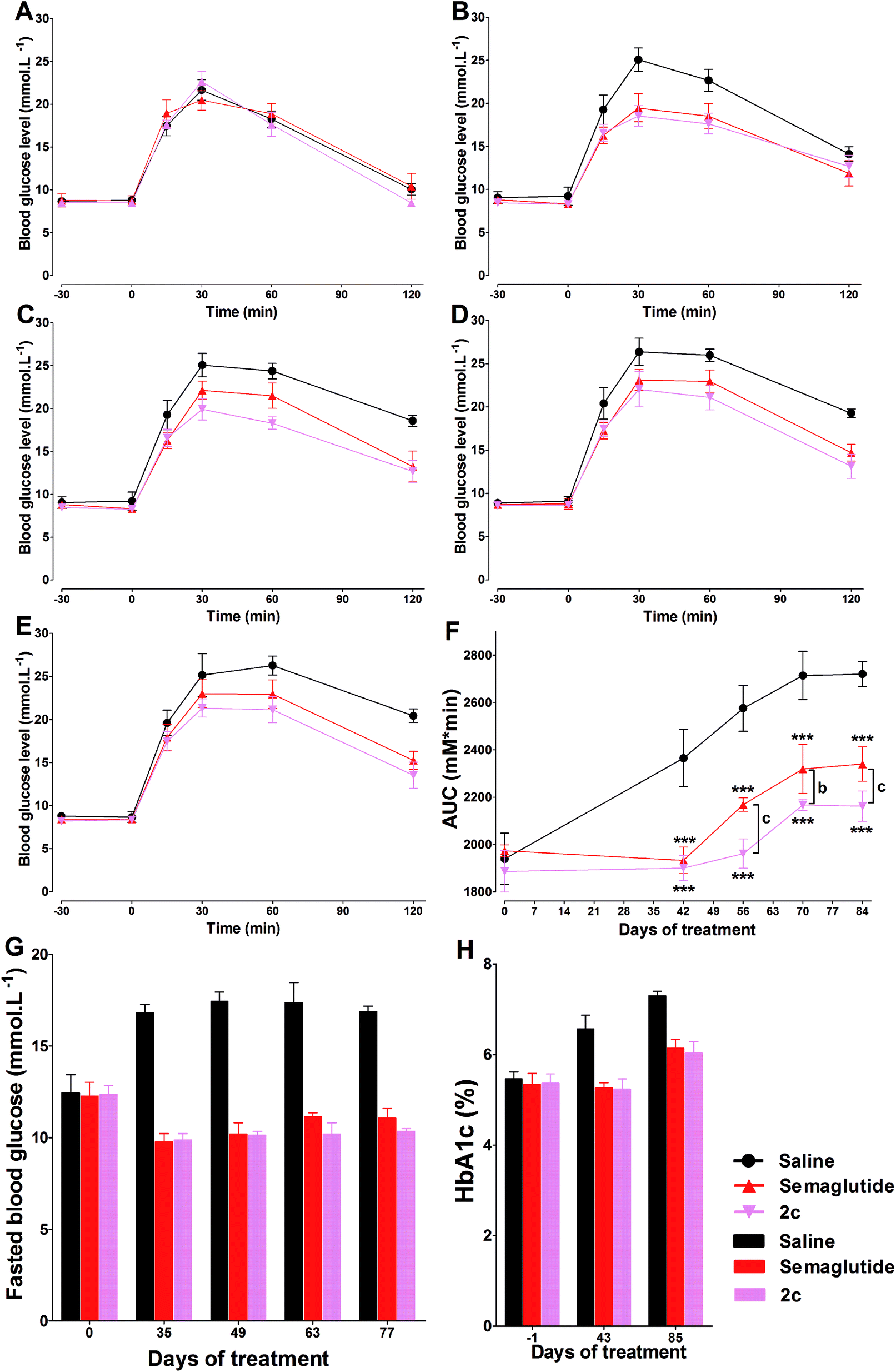 | ||
| Fig. 9 Chronic treatment effects of semaglutide and 2c in db/db mice. Effects of saline, semaglutide and 2c on glucose tolerance at days 0 (A), 42 (B), 56 (C), 70 (D) and 84 (E) during the treatment period. (F) Effects of s.c. administration of saline, semaglutide and 2c on glucose tolerance as measured by the glucose AUC in db/db mice. Effects of saline, semaglutide and 2c on fasted blood glucose (G) and HbA1c (H) in db/db mice, see Methods for details. Means ± SD, n = 6. ***P < 0.001 vs. saline, bP < 0.01 vs. semaglutide, cP < 0.001 vs. semaglutide. | ||
4. Discussion
Peptides are subject to rapid renal clearance and extensive enzymatic degradation. Thus, half-life prolonging is a prerequisite for the development of peptide pharmaceuticals. In the present study, a novel method for prolonging peptide half-life using DiMPA small-molecule albumin ligands was explored and demonstrated for short acting GLP-1 receptor agonist lixisenatide. In the past decade, many research efforts were focused on chemical modification on exenatide to develop of long-acting incretin mimetics, however, little research has been done on lixisenatide.12,46 Thus, the present research was using lixisenatide instead of exenatide as a model peptide. Although promote peptide to bind with albumin is a well-established method for peptide half-life extension, while the way to achieve it is variable and directly affect the potential therapeutic utility of peptide. The suitable albumin binders should possess two important characteristics, which are low toxicity and high albumin binding affinity, and to discover the proper albumin binders from the approved therapeutic drugs should be a practical way due to the high safety of the approved therapeutic drugs.24 As mentioned above, MPA is an effective immunosuppressive agent, and is extensively (∼98%) binds to HSA. The recommended dosage of mycophenolate mofetil (an ester prodrug of MPA) is 2 g per day, and this drug can be safely used as anti-rejection therapy in diabetic animal models, and in transplant recipients.47,48 Considering the high safety and albumin binding affinity of MPA, we explored the possibility of using albumin binders based on MPA for half-life extension on lixisenatide analogues.Lixisenatide had the same Lys12 and Lys27 residues compared with exenatide, and had additional six lysine residues on the C-terminal region. Previous researches have confirmed that both Lys12 and Lys27 on exenatide are suitable sites for chemical modification, and consequentially, Lys12 and Lys27 on lixisenatide were firstly selected for MPA modification.49 Moreover, to further explore whether the C-terminal region of lixisenatide was suitable for chemical modification, Lys39 was also selected and site-specific connected with MPA albumin binders, and a total of twelve MPA modified lixisenatide analogues (1a–1i) were firstly synthesized to identify the suitable modification site on lixisenatide.
The following in vitro biological studies showed direct evidence of the relevant structure–activity relationships. The MPAs modification on Lys27 has little impact on potency, while the modification on Lys12 exhibited a more profound reduction in potency and Lys39 was clearly not suitable for chemical modification.
By using structure-inducing probe (SIP) technology, Zealand Pharma successfully discovered lixisenatide. The principle of the SIP technology is that a short sequence (5–10 amino acids) of certain charged amino acids (e.g., asparagine or lysine) attached to the C-terminus of another peptide may constrain it into a more ordered conformation based on intermolecular hydrogen bonds.50 The C-terminal hexalysine extension of lixisenatide is the main reason that lixisenatide could bind to GLP-1 receptor more tightly than exendin-4 (4-fold increase).51 The serious potency lost observed for MPA modified lixisenatide analogues 1g–1i (derivatized at Lys39) might be attributed to two reasons. First, the chemical modification on Lys39 may interrupt the interaction between lixisenatide and GLP-1 receptor, thus causing serious biological activity lost. Second, the phenolic hydroxyl group on MPA may compete with other amino acid residues to form intermolecular hydrogen bonds with the C-terminus hexalysine, interfering the hexalysine-assisted formation of a more stable α-helix of the whole peptide. The N terminal portion of GLP-1 receptor agonists is crucial for mediating the cellular response to GLP-1 signaling. This explains the moderate potency lost observed for 1a–1c, as the MPA was modified close to the N terminal of lixisenatide at Lys12.
The protraction of 1a–1i in vitro, however, was exhibited positive correlation with length of the alkyl chain between MPA and peptide backbone. These results provide direct evidence on the important role of the long alkyl chain linker on half-life extension. Next, the in vivo IPGTT study further proved that the higher GLP-1 receptor activation potencies of 1d–1f successfully transferred to the potent in vivo hypoglycemic activities, and 1f with balanced stability and biological activity was selected for the DiMPA modification. As was mentioned above, multivalent interaction commonly occurred in nature as a means to enhance the weak ligand–receptor interactions. Proper structure optimization on MPA to design DiMPA might further improve the HSA binding affinity. In the present study, DiMPA greatly improved the HSA binding affinity of lixisenatide and could further enhance when DiMPA combined with OEG spacer. We found that 2c with proper length of OEG spacer (two OEG moieties) showed the best in vitro potency and in vivo hypoglycemic activity, this interesting finding was similar to the previous research of semaglutide, which OEG spacer played an important role in albumin binding and potency. The hypoglycemic and insulinotropic activities of 2c were further evaluated by IPGTT on db/db mice, and lixisenatide and 2c administration potent reduced blood glucose levels and the insulin levels also significantly increased at 15 min. The decreasing in blood glucose levels in 2c treated group was accompanied by the increasing plasma insulin levels which were in accordance with GLP-1R-dependent mechanism.
Subsequent long-acting hypoglycemic activity studies revealed that the duration of action of 2c on glucose-lowering was similar to semaglutide, indicating that 2c might has the potential to develop as a once-weekly administration antidiabetic agent. The acute toxicity test of 2c demonstrated its low in vivo renal toxicity and hepatoxicity. Normally, MPA exhibited profound toxicity in mice when the administered dose over 1000 mg per kg per day,52 which is thousands times higher than the dose of 2c in acute toxicity test (1000 mg kg−1 of MPA corresponds to ∼19386.1 mg kg−1 of 2c). Moreover, in clinical, the adverse effect profile of MPA is comparatively benign, and gastrointestinal adverse effects are a major concern.53 These adverse effects are partially explained by the enhanced immune suppression. Considering the administration dose of 2c is far lower (thousands times) than that of MPA, we envision that introduce of MPA to lixisenatide will not generate immunosuppressive effects, thus avoid potential toxicity brings by MPA modification. However, the systematic chronic toxicity tests of 2c still need to be conducted to more carefully evaluate the toxicity of 2c. The in vitro T cell proliferation test of 2c revealed the similar immunogenic potential of 2c with lixisenatide and semaglutide, preliminary indicated the low immunogenicity of 2c.
The short-term and long-term anti-diabetic effects of 2c were evaluated on db/db mice, in which the development of diabetes status was characterized by increased apoptosis and reduced proliferation in pancreatic β-cells.54 A once daily injection of 2c on db/db mice for 22 days achieved beneficial effects on HbA1c lowering and effectively suppressed food intake and body-weight gain. Thorough histological investigation of pancreas revealed a significant improved β-cell function following 2c treatment. Importantly, the IOD value of insulin in 2c group by once weekly administration was significant higher than that of in semaglutide group, indicated the better effect of 2c on amelioration of pancreas deficiencies than semaglutide. The different results observed in once daily and once weekly treatment experiments might attribute to the different biological activities of 2c and semaglutide, and further studies should be conducted to more carefully evaluate the stability, PK profiles and bioactivity of 2c, and compared with semaglutide or other once weekly GLP-1 receptor agonists, such as albiglutide and dulaglutide. When we treated db/db mice for 42 days followed by 42 days of no treatment, compared with the benchmark of control, 2c exhibited a sustained improvement in glycaemic control, and the beneficial treatment effects of 2c on glucose tolerance clearly should be attributed to the restoration of pancreatic function of 2c, and were significantly better than that of semaglutide.55 Taken together, our studies indicated that a better sustainable effect on glycaemic control through improved pancreatic function is exerted by the DiMPA modified lixisenatide in comparison with the once-weekly GLP-1 agonist semaglutide.
5. Conclusion
Novel lixisenatide analogues with covalently attached MPA or DiMPA albumin binders were synthesized. In vitro studies proved the ability of MPA albumin binders to confer HSA affinity. Moreover, the unprecedented DiMPA albumin binders had significant increased affinity for HSA, which confirmed our original design. DiMPA modified lixisenatide analogues exhibited superior long-acting hypoglycemic activities in vivo, as a consequence of enhanced HSA affinity. We show that selected compound 2c has a prominent preventive effect on the progressive pancreatic function deterioration seen in the db/db mice. In addition, we report that treatment of db/db mice with 2c, improves glycaemic control greater than that of commercially available once-weekly GLP-1 agonist semaglutide. We expect that 2c could be used for the treatment of diabetes with once-weekly administration. The present study also represents a novel method for half-life extension of peptide pharmaceuticals, which is how DiMPA albumin binders can promote longer-acting in vivo activity of GLP-1 analogs. We can envision that further customization of the DiMPA albumin binder in applications for specific proteins and peptides. This could include replacing the ligand moieties themselves, replacing functional groups of the ligands, changing the linker, and so forth. It can be anticipated that DiMPA albumin binder technology has potential as an alternative to traditional lipidation and PEGylation.Abbreviations
| GLP-1 | Glucagon-like peptide-1 |
| HPAC | High performance affinity chromatography |
| T2DM | Type 2 diabetes mellitus |
| DPP-IV | Dipeptidyl peptidase IV |
| HSA | Human serum albumin |
| MPA | Mycophenolic acid |
| IMPDH | Inosine monophosphate dehydrogenase |
| LC-MS/MS | Liquid chromatography-mass spectrometry |
| IPGTT | Intraperitoneal glucose tolerance test |
| OGTT | Oral glucose tolerance tests |
| BUN | Blood urea nitrogen |
| ALT | Alanine aminotransferase |
| SCr | Serum creatinine |
| AST | Aspartate aminotransferase |
| IOD | Integrated optical density |
| AUC | Area under the curve |
| SIP | Structure-inducing probe |
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (Grants No. 31970371, 81973533, 21762009), Guangxi Science and Technology Base and Special Talents Program (Grant No. AD18281032), the Natural Science Foundation of Guangxi Zhuang Autonomous Region of China (Grant No. 2017GXNSFAA198077), Guangxi First-class Discipline Project for Pharmaceutical Sciences (Grant No. GXFCDP-PS-2018), Guangxi Key R&D Project (Grant No. AB17195009), and the Natural Science Foundation of Xuzhou (Grant No. KC19154).References
- Q. Lu, W.-Z. Zuo, X.-J. Ji, Y.-X. Zhou, Y.-Q. Liu, X.-Q. Yao, X.-Y. Zhou, Y.-W. Liu, F. Zhang and X.-X. Yin, Phytomedicine, 2015, 22, 1071–1078 CrossRef CAS PubMed.
- Q. Lu, X.-J. Ji, Y.-X. Zhou, X.-Q. Yao, Y.-Q. Liu, F. Zhang and X.-X. Yin, Pharmacol. Res., 2015, 99, 237–247 CrossRef CAS PubMed.
- X. Zhu, Y.-Q. Cheng, L. Du, Y. Li, F. Zhang, H. Guo, Y.-W. Liu and X.-X. Yin, Phytother. Res., 2015, 29, 295–302 CrossRef CAS PubMed.
- X. Zhu, Y.-q. Cheng, Q. Lu, L. Du, X.-x. Yin and Y.-w. Liu, Naunyn-Schmiedeberg's Arch. Pharmacol., 2018, 391, 1237–1245 CrossRef CAS PubMed.
- Z. Shang, F. Han, X. Zhou, Z. Bao, J. Zhu, T. Wang, Q. Lu, L. Du, W. Li, D. Lv and X. Yin, Drug Dev. Res., 2018, 79, 129–135 CrossRef CAS PubMed.
- Y.-W. Liu, Y.-C. Hao, Y.-J. Chen, S.-Y. Yin, M.-Y. Zhang, L. Kong and T.-Y. Wang, Phytother. Res., 2018, 32, 1574–1582 CrossRef CAS PubMed.
- T. Wang, Y. Wang, D.-M. Lv, J.-F. Song, Q. Lu, X. Gao, F. Zhang, H. Guo, W. Li and X.-X. Yin, Pharmacotherapy, 2014, 34, 131–139 CrossRef CAS PubMed.
- Q. Yang, W.-W. Wang, P. Ma, Z.-X. Ma, M. Hao, T. I. Adelusi, D. Lei, X.-X. Yin and Q. Lu, Iran. J. Basic Med. Sci., 2017, 20, 1220–1226 Search PubMed.
- M. Guo, D. Liu, Q. Sha, H. Geng, J. Liang and D. Tang, J. Pharm. Biomed. Anal., 2019, 164, 309–316 CrossRef CAS PubMed.
- D. Tang, Y. Yu, X. Zheng, J. Wu, Y. Li, X. Wu, Q. Du and X. Yin, J. Pharm. Biomed. Anal., 2014, 100, 1–10 CrossRef CAS PubMed.
- J. J. Meier, Nat. Rev. Endocrinol., 2012, 8, 728–742 CrossRef CAS PubMed.
- B. Manandhar and J.-M. Ahn, J. Med. Chem., 2014, 58, 1020–1037 CrossRef PubMed.
- S. E. Alavi, P. J. Cabot and P. M. Moyle, Mol. Pharmaceutics, 2019, 16, 2278–2295 CrossRef CAS PubMed.
- E. M. Bech, S. L. Pedersen and K. J. Jensen, ACS Med. Chem. Lett., 2018, 9, 577–580 CrossRef CAS PubMed.
- J. Lau, P. Bloch, L. Schäffer, I. Pettersson, J. Spetzler, J. Kofoed, K. Madsen, L. B. Knudsen, J. McGuire and D. B. Steensgaard, J. Med. Chem., 2015, 58, 7370–7380 CrossRef CAS PubMed.
- J. G. Lee, J. H. Ryu, S.-M. Kim, M.-Y. Park, S.-H. Kim, Y. G. Shin, J.-W. Sohn, H. H. Kim, Z.-Y. Park, J. Y. Seong and J. I. Kim, Biochem. Pharmacol., 2018, 151, 59–68 CrossRef CAS PubMed.
- J. Fremaux, C. Venin, L. Mauran, R. H. Zimmer, G. Guichard and S. R. Goudreau, Nat. Commun., 2019, 10, 924 CrossRef PubMed.
- I. Tom, V. Lee, M. Dumas, M. Madanat, J. Ouyang, J. Severs, J. Andersen, J. M. Buxton, J. P. Whelan and C. Q. Pan, AAPS J., 2007, 9, E227–E234 CrossRef PubMed.
- S. B. van Witteloostuijn, K. Mannerstedt, P. Wismann, E. M. Bech, M. B. Thygesen, N. Vrang, J. Jelsing, K. J. Jensen and S. L. Pedersen, Mol. Pharmaceutics, 2016, 14, 193–205 CrossRef PubMed.
- W. Zhang, C. Chen, C. Zhang, J. Duan, Y. Li, H. Ma, H. Yao, A. Meng and J. Shi, Lat. Am. J. Pharm., 2016, 35, 1399–1406 CAS.
- S. Curry, H. Mandelkow, P. Brick and N. Franks, Nat. Struct. Biol., 1998, 5, 827–835 CrossRef CAS PubMed.
- K. Madsen, L. B. Knudsen, H. Agersoe, P. F. Nielsen, H. Thøgersen, M. Wilken and N. L. Johansen, J. Med. Chem., 2007, 50, 6126–6132 CrossRef CAS PubMed.
- R. E. Pratley, V. R. Aroda, I. Lingvay, J. Lüdemann, C. Andreassen, A. Navarria and A. Viljoen, Lancet Diabetes Endocrinol., 2018, 6, 275–286 CrossRef CAS PubMed.
- E. M. Bech, M. C. Martos-Maldonado, P. Wismann, K. K. Sørensen, S. B. van Witteloostuijn, M. B. Thygesen, N. Vrang, J. Jelsing, S. L. Pedersen and K. J. Jensen, J. Med. Chem., 2017, 60, 7434–7446 CrossRef CAS PubMed.
- J. Han, L. Sun, Y. Chu, Z. Li, D. Huang, X. Zhu, H. Qian and W. Huang, J. Med. Chem., 2013, 56, 9955–9968 CrossRef CAS PubMed.
- J. Han, J. Fu, L. Sun, Y. Han, Q. Mao, F. Liao, X. Zheng and K. Zhu, Med. Chem. Commun., 2018, 9, 67–80 RSC.
- G. Wei, A. I. Jewett and J.-E. Shea, Phys. Chem. Chem. Phys., 2010, 12, 3622–3629 RSC.
- U. Werner, G. Haschke, A. W. Herling and W. Kramer, Regul. Pept., 2010, 164, 58–64 CrossRef CAS PubMed.
- C. R. Chong, D. Z. Qian, F. Pan, Y. Wei, R. Pili, D. J. Sullivan and J. O. Liu, J. Med. Chem., 2006, 49, 2677–2680 CrossRef CAS PubMed.
- R. Bullingham, S. Monroe, A. Nicholls and M. Hale, J. Clin. Pharmacol., 1996, 36, 315–324 CrossRef CAS PubMed.
- M. Lorenz, C. Pfeiffer, A. Steinsträßer, R. H. A. Becker, H. Rütten, P. Ruus and M. Horowitz, Regul. Pept., 2013, 185, 1–8 CrossRef CAS PubMed.
- J. Han, Y. Huang, X. Chen, F. Zhou, Y. Fei and J. Fu, Eur. J. Med. Chem., 2018, 157, 177–187 CrossRef CAS PubMed.
- J. Han, Y. Wang, Q. Meng, G. Li, F. Huang, S. Wu, Y. Fei, F. Zhou and J. Fu, Eur. J. Med. Chem., 2017, 132, 81–89 CrossRef CAS PubMed.
- J. Han, X. Chen, L. Zhao, J. Fu, L. Sun, Y. Zhang, F. Zhou and Y. Fei, Mol. Pharmaceutics, 2018, 15, 2840–2856 CrossRef CAS PubMed.
- J. Han, T. Meng, X. Chen, Y. Han, J. Fu, F. Zhou, Y. Fei and C. Li, FASEB J., 2019, 33, 7113–7125 CrossRef CAS PubMed.
- D. Ashton, C. Beddell, G. Cockerill, K. Gohil, C. Gowrie, J. Robinson, M. Slater and K. Valko, J. Chromatogr. B: Biomed. Sci. Appl., 1996, 677, 194–198 CrossRef PubMed.
- J. Han, Y. Fei, F. Zhou, X. Chen, Y. Zhang, L. Liu and J. Fu, Br. J. Pharmacol., 2018, 175, 544–557 CrossRef CAS PubMed.
- J. Han, Y. Huang, X. Chen, F. Zhou, Y. Fei and J. Fu, Eur. J. Pharm. Sci., 2018, 123, 111–123 CrossRef CAS PubMed.
- S. Y. Chae, Y. G. Chun, S. Lee, C.-H. Jin, E. S. Lee, K. C. Lee and Y. S. Youn, J. Pharm. Sci., 2009, 98, 1556–1567 CrossRef CAS PubMed.
- J. Han, F. Zhou, Y. Fei, X. Chen, J. Fu and H. Qian, Bioconjugate Chem., 2018, 29, 390–402 CrossRef CAS PubMed.
- J. Han, Y. Fei, F. Zhou, X. Chen, W. Zheng and J. Fu, Mol. Pharmaceutics, 2017, 14, 3954–3967 CrossRef CAS PubMed.
- M. K. Joubert, M. Deshpande, J. Yang, H. Reynolds, C. Bryson, M. Fogg, M. P. Baker, J. Herskovitz, T. J. Goletz and L. Zhou, PLoS One, 2016, 11, e0159328 CrossRef PubMed.
- J. Han, X. Chen, Y. Wang, Y. Fei, F. Zhou, Y. Zhang, L. Liu, P. Si and J. Fu, Biochem. Pharmacol., 2017, 142, 155–167 CrossRef CAS PubMed.
- K. Fosgerau, L. Jessen, J. Lind Tolborg, T. Østerlund, K. Schæffer Larsen, K. Rolsted, M. Brorson, J. Jelsing and T. Skovlund Ryge Neerup, Diabetes, Obes. Metab., 2013, 15, 62–71 CrossRef CAS PubMed.
- T. H. Kim, H. H. Jiang, S. Lee, Y. S. Youn, C. W. Park, Y. Byun, X. Chen and K. C. Lee, Bioconjugate Chem., 2011, 22, 625–632 CrossRef CAS PubMed.
- S. Y. Chae, Y. G. Choi, S. Son, S. Y. Jung, D. S. Lee and K. C. Lee, J. Controlled Release, 2010, 144, 10–16 CrossRef CAS PubMed.
- J. J. Lipsky, Lancet, 1996, 348, 1357–1359 CrossRef CAS.
- P. A. Gottlieb, S. Quinlan, H. Krause-Steinrauf, C. J. Greenbaum, D. M. Wilson, H. Rodriguez, D. A. Schatz, A. M. Moran, J. M. Lachin and J. S. Skyler, Diabetes Care, 2010, 33, 826–832 CrossRef CAS PubMed.
- S. Son, S. Y. Chae, C. W. Kim, Y. G. Choi, S. Y. Jung, S. Lee and K. C. Lee, J. Med. Chem., 2009, 52, 6889–6896 CrossRef CAS PubMed.
- D. R. Kapusta, C. Thorkildsen, V. A. Kenigs, E. Meier, M. M. Vinge, C. Quist and J. S. Petersen, J. Pharmacol. Exp. Ther., 2005, 314, 652–660 CrossRef CAS PubMed.
- M. Pinget, R. Goldenberg, E. Niemoeller, I. Muehlen-Bartmer, H. Guo and R. Aronson, Diabetes, Obes. Metab., 2013, 15, 1000–1007 CrossRef CAS PubMed.
- L. Sebastian, S. N. Madhusudana, V. Ravi and A. Desai, Chemotherapy, 2011, 57, 56–61 CrossRef CAS PubMed.
- M. Behrend, Drug Saf., 2001, 24, 645–663 CrossRef CAS.
- C. Clemmensen, B. Finan, T. D. Müller, R. D. DiMarchi, M. H. Tschöp and S. M. Hofmann, Nat. Rev. Endocrinol., 2019, 15, 90–104 CrossRef PubMed.
- M. Davies, T. R. Pieber, M.-L. Hartoft-Nielsen, O. K. Hansen, S. Jabbour and J. Rosenstock, JAMA, J. Am. Med. Assoc., 2017, 318, 1460–1470 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic route of compounds 1a–1i and 2a–2d. Characterization profiles of compounds 1a–1i and 2a–2d. The studies of the impurity profile of 2c and the impact of the impurity in 2c on the biological activity of 2c. In vivo toxicity of the impurity in 2c. See DOI: 10.1039/d0ra01002b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |