
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree new compounds from the flower branch of Gastrodia elata Blume and anti-microbial activity†
Liu Yang‡
 a,
Rong Jiang‡c,
Hui-Hui Li‡b,
Ya-Ping Panc,
Jing-Jin Luc,
Hong Zhangb,
Shou-Jin Liu*b,
Ji-Lu Shen*d and
Jiang-Miao Hu*a
a,
Rong Jiang‡c,
Hui-Hui Li‡b,
Ya-Ping Panc,
Jing-Jin Luc,
Hong Zhangb,
Shou-Jin Liu*b,
Ji-Lu Shen*d and
Jiang-Miao Hu*a
aState Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming 650204, Yunnan, China
bCollege of Pharmacy, Anhui University of Traditional Chinese Medicine, Hefei 230038, China
cDepartment of Pharmacy, First Affiliated Hospital of Anhui Medical University, Anhui Medical University, Hefei, 230022, Anhui, China
dDepartment of Laboratory Medicine, Forth Affiliated Hospital of Anhui Medical University, Anhui Medical University, Hefei, 230022, Anhui, China
First published on 13th April 2020
Abstract
Three new compounds (1–3), including novel tetra-p-cresol substituted cyclopenta[a]naphthalene derivatives, named gastrodinol (1), 2-(4′-hydroxybenzoyl)-3-hydroxyethyl indole (2), 2-(4′-hydroxybenzoyl)-3-(4′′-hydroxybenzyl)indole (3) were isolated from the flower branch of G. elata, along with five known compounds (4–8). Among them, compound 1 exhibited the most anti-microbial activity against Streptococcus agalactiae, with the minimum inhibitory concentration of 1 μg ml−1. This study demonstrated that the novel gastrodinol 1 found in the flower branch of G. elata may be responsible for the anti-microbial effect. It will lead to the development of new antibiotics, and how to utilize the TCM ′′Tianma′′ better.
1. Introduction
Due to increased microbial resistance, anti-microbial resistance to antibiotics used today has become a serious health problem. The increasing resistance of microorganisms to anti-microbial agents is one of the major concerns for scientists and clinicians around the world, and has become a global problem in recent years.1,2 Hence, there is an urgent need to discover new anti-microbial agents with diverse chemical structures and novel mechanisms of action.Most of the clinically used antibiotics are isolated from microorganisms or obtained by synthesis.3 Many traditional herbal medicines contain a wide variety of compounds that prevent or ameliorate many diseases, including cancer, cardiovascular diseases, diabetes, and etc. It should be noted that fewer side effects are known to result from traditional herbal medicines. In the last few decades, interest in the search for anti-microbial natural agents has risen.4
Gastrodia elata Blume (G. elata), commonly called Tian ma ( ) in Chinese, is a perennial parasitic herb also called Chi Jian (
) in Chinese, is a perennial parasitic herb also called Chi Jian ( ), Ding feng cao (
), Ding feng cao ( ), or Du yao zhi (
), or Du yao zhi ( ), belongs to the genus Gastrodia, family Orchidaceas is used as medicinal material in the clinical practice of traditional Chinese medicine (TCM) and registered in the Pharmacopeia of People's Republic of China.5 G. elata is found primarily in eastern Asia, specifically in the mountainous areas of China, Korea, Japan and India,6,7 it grows in the forest at 400–3200 meters above level. In China, wild G. elata is naturally distributed in many provinces such as Sichuan and Yunnan. The folk application of G. elata could be traced back to the first Chinese dispensatory ′′Shennong's Herbal Classic of Materia Medica′′ (Shennong Bencao Jing), which recorded the TCM as ′′Chi Jian′′. May be shape of the flower branch of the plant looks like as an arrow in crimson. Medicinal record for the TCM in the ancient book described as killing Gui Jing Wu (ghost essence), cure Gu Du (poison produced by venomous insects) and evil Qi, then strengthening the body and enhancing health.8 Afterwards, the dried rhizome (tuber) of G. elata as the useful part in the Newly Revised Materia Medica (Xin xiu ben cao) written in the period of the Song Dynasties. There are also changes in its function of G. elata in compendium of Materia Medica (Ben cao gang mu), G. elata is a medicine that has been commonly used to treat dizziness, paralysis, convulsion and epilepsy, and is called ′′Ding feng cao′′.9,10
), belongs to the genus Gastrodia, family Orchidaceas is used as medicinal material in the clinical practice of traditional Chinese medicine (TCM) and registered in the Pharmacopeia of People's Republic of China.5 G. elata is found primarily in eastern Asia, specifically in the mountainous areas of China, Korea, Japan and India,6,7 it grows in the forest at 400–3200 meters above level. In China, wild G. elata is naturally distributed in many provinces such as Sichuan and Yunnan. The folk application of G. elata could be traced back to the first Chinese dispensatory ′′Shennong's Herbal Classic of Materia Medica′′ (Shennong Bencao Jing), which recorded the TCM as ′′Chi Jian′′. May be shape of the flower branch of the plant looks like as an arrow in crimson. Medicinal record for the TCM in the ancient book described as killing Gui Jing Wu (ghost essence), cure Gu Du (poison produced by venomous insects) and evil Qi, then strengthening the body and enhancing health.8 Afterwards, the dried rhizome (tuber) of G. elata as the useful part in the Newly Revised Materia Medica (Xin xiu ben cao) written in the period of the Song Dynasties. There are also changes in its function of G. elata in compendium of Materia Medica (Ben cao gang mu), G. elata is a medicine that has been commonly used to treat dizziness, paralysis, convulsion and epilepsy, and is called ′′Ding feng cao′′.9,10
It can be seen from the search of traditional Chinese medicine classics that the major function of G. elata has changed with the changes of medicinal part. G. elata has been recorded to kill ghost essence and louisvuitt that means anti-inflammatory, anti-microbial, and antiviral activities in pharmacology ′′Shennong Bencao Jing′′. However, ′′Xin xiu ben cao′′ and ′′Ben cao gang mu′′ record its effectiveness in dizziness and convulsion. Hence, we could argue that the tuber of G. elata is charactered as a medicine in enhancing health, strengthening the body, anticonvulsant and sedative product, while flower branch of the plant was used as medicine for anti-inflammatory, anti-microbial and antiviral activities. Therefore, we carried out pharmaceutical investigation herein on the flower branch of G. elata and led to the isolation of eight natural compounds (Fig. 1) with anti-microbial activity against clinical isolated strains. Most of all, a novel natural tetra-p-cresol substituted cyclopenta[a]naphthalene derivatives, named gastrodinol was get from the material and verified the traditional usage of ′′Tianma′′ by further bioassay. This manuscript will describe the isolation, elucidation and anti-microbial activities of isolates of the flower branch of G. elata.
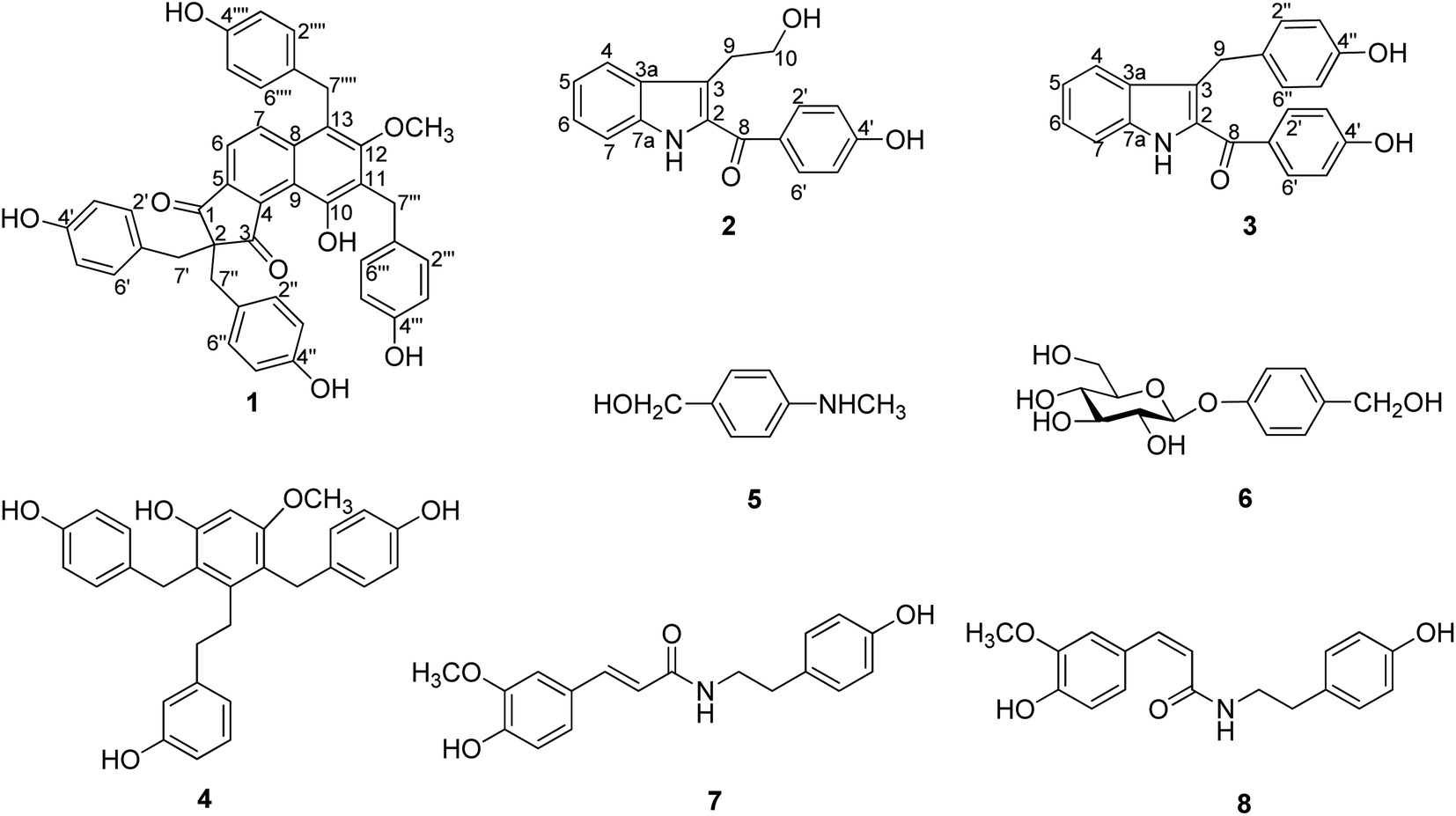 | ||
| Fig. 1 The structures of compounds 1–8. | ||
2. Results and discussion
2.1. Characterization of the isolates
The flower branch of G. elata was extracted with 70% EtOH to give the total extraction, which was partitioned with ethyl acetate and n-butanol. The ethyl acetate extract was separated by repeated silica gel CC, MCI gel CHP 20P CC, Sephadex LH-20, MPLC and HPLC to afford three new compounds (1–3), along with five known compounds (Fig. 1).Compound 1 was obtained as white amorphous powder and had molecular formula of C42H34O8 on the basis of HRESI-MS at m/z: 711.2247 [M + COOH]− (calcd for 711.2236) with 26 degrees of unsaturation. The absorption maxima at 306 (3.6), 277 (3.8), 203 (0.51) in the UV spectrum, and the IR spectrum showed hydroxyl (3439 cm−1), carbonyl (1630 cm−1), phenyl (2920 cm−1) absorptions of compound 1 strongly indicated the presence of a phenolic structure. The 1H spectrum (Table 1) of 1 indicated signals for four p-hydroxybenzyl [δH 6.78 (4H, d, J = 8.6 Hz), 6.38 (4H, d, J = 8.6 Hz), 3.20 (4H, s); 7.06 (2H, d, J = 8.6 Hz), 6.59 (2H, d, J = 8.6 Hz), 4.15 (2H, s); 6.70 (2H, d, J = 8.6 Hz), 6.68 (2H, d, J = 8.6 Hz) and 4.26 (2H, s)], one tetra-substituted benzene moiety [δH 8.07 (1H, d, J = 8.8 Hz), 7.39 (1H, d, J = 8.8 Hz)] and a methoxyl group δH 3.51 (3H, s). The 13C- and DEPT NMR date (Table 1) of 1 showed 42 carbon signals including one oxygenated methyl, four methylenes, eighteen methines and nineteen quaternary carbons. Comparison the NMR data of 1 with the bis-ketone11 suggested that it shared the similar basic framework of a five-membered diphenyl ring, C-1, C-3 of the five-membered ring are replaced by carbonyl groups, and C-2 are all replaced by quaternary carbon. This was further confirmed by the HMBC (Fig. 2) correlations of H-6/C-1, C-4 and C-8; H-7/C-5, C-8, C-9 and C-13; H-7′/C-1, C-2, C-1′, C-2′ and C-6′; H-7′′/C-2, C-3, C-1′′, C-2′′, and C-6′′. However, the 13C-NMR resonances were different with those of the known bis-ketone because 1 had two additional aromatic rings. The structures of diphenyl bis-ketone and the additional aromatic rings were further determined by the HMBCs from H-7′′′ to C-10, C-11, C-12, C-1′′′, C-2′′′ and C-6′′′ and thus supported that an bi-substituted benzene ring was linked to C-11. In addition, the HMBC correlations of H-7′′′′/C-7, C-12, C-13, C-1′′′′, C-2′′′′ and C-6′′′′ supported that the other bi-substituted benzene ring was linked to C-13. The HMBC cross-peak from OCH3 (δH 3.51, 3H, s) to C-12 (δC 161.7) was also observed favorably supported this methoxy group was linked to C-12. On the basis of above evidences, the structure of compound 1 was elucidated as 10-hydroxy-12-methoxy-2,2,11,13-tetra(p-hydroxybenzyl)-8,9(1H)-indibenzo-1,3(2H)-dione, named gastrodinol.
| No. | δH | δC | No. | δH | δC |
|---|---|---|---|---|---|
| a 1H-NMR recorded in 600 MHz, 13C-NMR recorded in 150 MHz. | |||||
| 1 | — | 203.3, s | 4′ | — | 157.3, s |
| 2 | — | 64.0, s | 7′ | 3.20 (2H, s) | 41.8, t |
| 3 | — | 211.8, s | 1′′ | — | 127.4, s |
| 4 | — | 141.9, s | 2′′, 6′′ | 6.78 (2H, d, J = 8.6 Hz) | 131.8, d |
| 5 | — | 145.5, s | 3′′, 5′′ | 6.38 (2H, d, J = 8.6 Hz) | 116.3, d |
| 6 | 7.39 (1H, d, J = 8.8 Hz) | 117.6, d | 4′′ | — | 157.3, s |
| 7 | 8.07 (1H, d, J = 8.8 Hz) | 137.3, d | 7′′ | 3.20 (2H, s) | 41.8, t |
| 8 | — | 137.8, s | 1′′′ | — | 129.9, s |
| 9 | — | 117.9, s | 2′′′, 6′′′ | 7.06 (2H, d, J = 8.6 Hz) | 130.4, d |
| 10 | — | 154.6, s | 3′′′, 5′′′ | 6.59 (2H, d, J = 8.6 Hz) | 116.3, d |
| 11 | — | 124.4, s | 4′′′ | — | 156.5, s |
| 12 | — | 161.7, s | 7′′′ | 4.15 (2H, s) | 30.5, t |
| 12-OCH3 | 3.51 (3H, s) | 62.6, q | 1′′′′ | — | 133.0, s |
| 13 | — | 121.7, s | 2′′′′, 6′′′′ | 6.70 (2H, d, J = 8.6 Hz) | 130.4, d |
| 1′ | — | 127.4, s | 3′′′′, 5′′′′ | 6.68 (2H, d, J = 8.6 Hz) | 116.3, d |
| 2′, 6′ | 6.78 (2H, d, J = 8.6 Hz) | 131.8, d | 4′′′′ | — | 156.3, s |
| 3′, 5′ | 6.38 (2H, d, J = 8.6 Hz) | 116.3, d | 7′′′′ | 4.26 (2H, s) | 31.4, t |
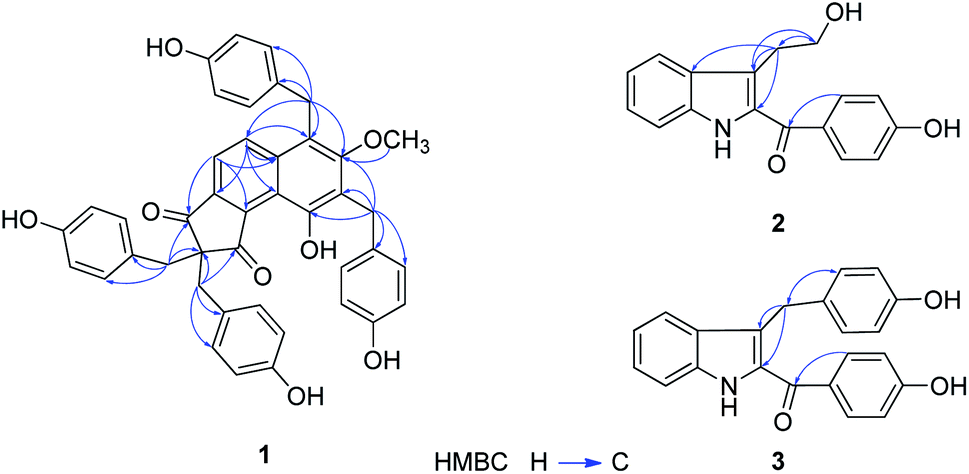 | ||
| Fig. 2 Key HMBC correlations for compounds 1–3. | ||
Compound 2 was obtained as yellow amorphous powder and had molecular formula of C17H15NO3 which was proposed from the positive HRESI-MS at m/z: 304.0943 [M + Na]+ (calcd for 304.0950), indicating 11 indices of hydrogen deficiency. The UV spectrum showed absorption maxima at λmax (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) 328 (4.1), 218 (4.3), 202 (4.3) suggested an typical of indole chromophores,12 while the IR absorption bands at 3443, 1625 cm−1 resulted from the –NH and carbonyl group. In 1H-NMR spectrum (Table 2) displayed an unsubstituted indole moiety with signals at δH 7.71 (1H, d, J = 8.1 Hz), 7.10 (1H, t, J = 7.3 Hz), 7.27 (1H, t, J = 7.7 Hz), and 7.41 (1H, d, J = 8.3 Hz); an disubstituted benzene ring moiety with signals at δH 7.75 (2H, d, J = 8.6 Hz) and 6.90 (2H, d, J = 8.6 Hz) together with two methylene ([3.19 (2H, t, J = 7.2 Hz)], [3.75 (2H, t, J = 7.2 Hz)]) in the structure of 2. The 13C-NMR and DEPT spectra of 2 (Table 2), in association with the MS spectrum, suggested that 2 possessed 17 carbonds, including two methylene, eight methane and seven quaternary carbones. In addition, as evidenced by the HMBC (Fig. 2) correlations from H-9 (δH 3.19, 2H, t, J = 7.2 Hz) to C-2 (δC 133.6), C-3 (δC 121.3), C-3a (δC 129.3), C-10 (δC 63.8), and from H-10 (δH 3.75, 2H, t, J = 7.2 Hz) to C-3 (δC 121.3), C-9 (δC 29.5), as confirmed by the upfield shift of the carbon resonance at δC 63.8 (C-10) and corresponding proton H-10 (δH 3.75, 2H, t, J = 7.2 Hz) was supported that an hydroxyethyl was linked to C-3. The linkage of carbonyl group and the disubstituted benzene ring by the HMBC correlations from H-2′, 6′ to C-8 (δC 190.3). Thus, the structure of 2 was established as 2-(4′-hydroxybenzoyl)-3-hydroxyethyl indole.
ε) 328 (4.1), 218 (4.3), 202 (4.3) suggested an typical of indole chromophores,12 while the IR absorption bands at 3443, 1625 cm−1 resulted from the –NH and carbonyl group. In 1H-NMR spectrum (Table 2) displayed an unsubstituted indole moiety with signals at δH 7.71 (1H, d, J = 8.1 Hz), 7.10 (1H, t, J = 7.3 Hz), 7.27 (1H, t, J = 7.7 Hz), and 7.41 (1H, d, J = 8.3 Hz); an disubstituted benzene ring moiety with signals at δH 7.75 (2H, d, J = 8.6 Hz) and 6.90 (2H, d, J = 8.6 Hz) together with two methylene ([3.19 (2H, t, J = 7.2 Hz)], [3.75 (2H, t, J = 7.2 Hz)]) in the structure of 2. The 13C-NMR and DEPT spectra of 2 (Table 2), in association with the MS spectrum, suggested that 2 possessed 17 carbonds, including two methylene, eight methane and seven quaternary carbones. In addition, as evidenced by the HMBC (Fig. 2) correlations from H-9 (δH 3.19, 2H, t, J = 7.2 Hz) to C-2 (δC 133.6), C-3 (δC 121.3), C-3a (δC 129.3), C-10 (δC 63.8), and from H-10 (δH 3.75, 2H, t, J = 7.2 Hz) to C-3 (δC 121.3), C-9 (δC 29.5), as confirmed by the upfield shift of the carbon resonance at δC 63.8 (C-10) and corresponding proton H-10 (δH 3.75, 2H, t, J = 7.2 Hz) was supported that an hydroxyethyl was linked to C-3. The linkage of carbonyl group and the disubstituted benzene ring by the HMBC correlations from H-2′, 6′ to C-8 (δC 190.3). Thus, the structure of 2 was established as 2-(4′-hydroxybenzoyl)-3-hydroxyethyl indole.
| No. | 2 | 3 | ||
|---|---|---|---|---|
| 1H-NMR | 13C-NMR | 1H-NMR | 13C-NMR | |
| a 1H-NMR recorded in 600 MHz, 13C-NMR recorded in 150 MHz. | ||||
| 1 | — | — | — | — |
| 2 | — | 133.6, s | — | 133.6, s |
| 3 | — | 121.3, s | — | 123.5, s |
| 3a | — | 129.3, s | — | 129.3, s |
| 4 | 7.71 (1H, d, J = 8.1 Hz) | 121.0, d | 7.51 (1H, d, J = 8.1 Hz) | 120.8, d |
| 5 | 7.10 (1H, t, J = 7.3 Hz) | 126.3, d | 7.00 (1H, t, J = 7.3 Hz) | 126.3, d |
| 6 | 7.27 (1H, t, J = 7.7 Hz) | 121.5, d | 7.24 (1H, t, J = 7.6 Hz) | 122.3, d |
| 7 | 7.41 (1H, d, J = 8.3 Hz) | 113.4, d | 7.40 (1H, d, J = 8.3 Hz) | 113.2, d |
| 7a | 138.3, s | 138.5, s | ||
| 8 | 190.3, s | 190.6, s | ||
| 9 | 3.19 (2H, t, J = 7.2 Hz) | 29.5, t | 4.15 (2H, s) | 40.0, t |
| 10 | 3.75 (2H, t, J = 7.2 Hz) | 63.8, t | — | — |
| 1′ | — | 131.4, s | — | 131.5, s |
| 2′, 6′ | 7.75 (2H, d, J = 8.6 Hz) | 133.3, d | 7.70 (2H, d, J = 8.6 Hz) | 133.2, d |
| 3′, 5′ | 6.90 (2H, d, J = 8.6 Hz) | 116.4, d | 6.83 (2H, d, J = 8.6 Hz) | 116.4, d |
| 4′ | — | 163.9, s | — | 164.0, s |
| 1′′ | — | — | — | 133.3, s |
| 2′′, 6′′ | — | — | 6.91 (2H, d, J = 8.6 Hz) | 130.3, d |
| 3′′, 5′′ | — | — | 6.58 (2H, d, J = 8.6 Hz) | 115.9, d |
| 4′′ | — | — | — | 156.3, s |
Compound 3 was obtained as yellow amorphous powder and determined to have the molecular formula C22H17NO3 based on the positive HRESI-MS at m/z: 366.1101 [M + Na]+ (calcd for 366.1106) with 15 degrees of unsaturation. The UV spectrum showed absorption maxima at λmax (logε) 326 (4.4), 219 (4.6), 201 (4.6), and the IR absorptions at 3442, 2924, 1620, 1529 cm−1 indicated the presence of NH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and aromatic functionalities, suggested compound 3 was also a structure of a indole. A detailed comparison of the 1H-NMR and 13C-NMR data of 3 (Table 2) with those of 2 shown that 3 and 2 were similar in structure except that the C-10 (δC 63.8) was reduced, one disubstituted benzene ring signals were increased. In the HMBC spectrum (Fig. 2) interaction of H-2′′, 6′′ (δH 6.91, 2H, d, J = 8.6 Hz)/C-9 (δC 40.0) was observed, which indicated that the disubstituted benzene ring was located at C-9. Hence, the structure of 3 was determined as 2-(4′-hydroxybenzoyl)-3-(4′′-hydroxybenzyl)indole.
O and aromatic functionalities, suggested compound 3 was also a structure of a indole. A detailed comparison of the 1H-NMR and 13C-NMR data of 3 (Table 2) with those of 2 shown that 3 and 2 were similar in structure except that the C-10 (δC 63.8) was reduced, one disubstituted benzene ring signals were increased. In the HMBC spectrum (Fig. 2) interaction of H-2′′, 6′′ (δH 6.91, 2H, d, J = 8.6 Hz)/C-9 (δC 40.0) was observed, which indicated that the disubstituted benzene ring was located at C-9. Hence, the structure of 3 was determined as 2-(4′-hydroxybenzoyl)-3-(4′′-hydroxybenzyl)indole.
The known compounds were identified as 3,3′-dihydroxy-2,6-bis(4-hydroxybenzyl)-5-methoxybibenzyl (4),13 4-(methylamino)-benzyl alcohol (5),14 gastrodin (6),15 N-cis-feruloyltyra-mine (7) (ref. 16) and N-trans-feruloyltyra-mine (8) (ref. 16) by comparing their spectra data with those reported in literatures.
2.2. Anti-microbial activity of the isolates
The anti-microbial activity of compounds 1–8 was evaluated on pathogenic microorganisms (Table 3), including Enterococcus faecium, E. faecium (VRE), E. faecalis, Staphylococcus aureus (MSSA), S. aureus (MRSA), S. epidermidis (MRCNS), S. epidermidis, S. (MRCNS), Staphylococcus (MSCNS), Streptococcus agalactiae, Viridans group Streptococci, and S. pyogenes. Penicillin was taken as control drug and anti-microbial activity data are presented in Table 3. The MIC was determined using the microdilution broth method according to the CLSI M100-S27. Only compound 1 showed significant anti-microbial activity except Staphylococcus (MIC > 64 μg ml−1). Our first observation is that compound 1 show good anti-microbial activity compared with the control drug (penicillin). Among them, compound 1 exhibited the most anti-microbial activity against Streptococcus agalactiae, with the minimum inhibitory concentration of 1 μg ml−1. Meanwhile we have noticed that compound 1 showed significant anti-microbial activity against E. faecium (MIC = 8 μg ml−1), E. faecium (VRE) (MIC = 16 μg ml−1), S. aureus (MSSA) (MIC = 4 μg ml−1), S. aureus (MRSA) (MIC = 16 μg ml−1), and S. epidermidis (MIC = 16 μg ml−1), which is more active than penicillin (MIC ≥ 64 μg ml−1).| Target microorganism | Anti-microbial activity (MIC, μg ml−1) | |
|---|---|---|
| 1 | Penicillina | |
| a Penicillin as positive control.b VRE: vancomycin resistant Enterococcus.c MSSA: methicillin sensitive Staphylococcus aureus.d MRSA: methicillin resistant Staphylococcus aureus.e MRCNS: methicillin resistant coagulase negative Staphylococcus.f MSCNS: methicillin sensitive coagulase negative Staphylococcus. | ||
| Enterococcus faecium | 8 | >64 |
| Enterococcus faeciumb (VRE) | 16 | >64 |
| Enterococcus faecalis | 8 | 32 |
| Staphylococcus aureusc (MSSA) | 4 | >64 |
| Staphylococcus aureusd (MRSA) | 16 | >64 |
| Staphylococcus epidermidise (MRCNS) | 64 | 32 |
| Staphylococcus epidermidis | 16 | 64 |
| Staphylococcuse (MRCNS) | >64 | 0.5 |
| Staphylococcusf (MSCNS) | 32 | 64 |
| Streptococcus agalactiae | 1 | 0.5 |
| Viridans group Streptococci | 4 | 0.5 |
| Streptococcus pyogenes | 2 | 0.5 |
Among these pathogenic microorganisms, compound 1 shows the most anti-microbial activity against all twelve pathogenic microorganisms tested, with MIC values in the range of 1–64 μg ml−1, which is significantly more active than the positive drug penicillin against E. faecium, E. faecium (VRE), E. faecalis, S. aureus (MSSA), S. aureus (MRSA), S. epidermidis, and Staphylococcus. In addition, compound 1 exhibited the most anti-microbial activity against Streptococcus agalactiae, with the minimum inhibitory concentration of 1 μg ml−1.
3. Experimental
3.1. General
UV spectra were taken on a Shimadzu UV-2401PC (Shimadzu, Kyoto, Japan) spectrometer with KBr pellets. 1D- and 2D-NMR spectra were acquired on Bruker AVANCE III-600 (Bruker BioSpin GmbH, Rheinstetten, Germany) instruments, using tetramethylsilane (TMS) as an internal standard: chemical shifts (δ) are given in ppm, coupling constants (J) in Hz, the solvent signals were used as references (CD3OD: δC = 49.0 ppm; residual CH3OH in CD3OD: δH = 4.78 ppm). Electrospray ionization (ESI) and High-resolution electrospray ionization (HRESI) mass spectra were carried out using an Agilent 6540 Q-TOF mass spectrometer (Agilent Technologies, Santa Clara, CA, USA). MCI gel CHP 20P (75–150 μm, Mitsubishi Chemical Corp., Tokyo, Japan), Sephadex LH-20 (25–100 μm, Pharmacia Fine Chemical Co. Ltd.), and LiChroprep RP-18 gel (40–63 μm, Merck, Darmstadt, Germany), and silica gel (200–300 mesh, Qingdao Haiyang Chemical Co.) were used for normal pressure column chromatography (CC). Thin-layer chromatography (TLC) was carried on silica gel G precoated plates (Qingdao Haiyang Chemical Co.) and spots were visualized by ultraviolet light (254 nm) or 9% sulfuric acid/EtOH.3.2. Plant material
The scapes of G. elata was collected from Dafang county (27°04′49′′N, 105°33′44′′E), Guizhou Province, China, in August 2013, and identified by Professor Shou-Jin Liu (College of Pharmacy, Anhui University of Traditional Chinese Medicine). A voucher specimen (No. Zsh-20) was deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences.3.3. Extraction and isolation
The dried scapes of G. elata (15 kg) were powdered and extracted three times with 70% EtOH under reflux. After removing the solvent under reduced pressure using a rotary evaporator at 50 °C, the resulting gum was dried to yield a solid residue (2.5 kg). A portion of crude extract was suspended in distilled water and partitioned with ethyl acetate (yield: 191 g) and n-butanol (yield: 133 g). The ethyl acetate extract (191 g) was subjected to chromatography using silica gel (10 × 120 cm, 2.0 kg), and eluting with CHCl3–CH3OH (200:1–1:1, v/v) gradient, six fractions were collected on the basis of their TLC characteristics as follows: A (33.4 g, white solid), B (21.7 g, brown solid), C (13.2 g, brown solid), D (20.4 g, white solid), E (10.2 g, black solid), F (19.6 g, black solid). Fraction D (18 g) was further submitted to MCI gel CHP 20P CC (6 × 40 cm, 500 mL) eluted with gradient CH3OH–H2O (10:90–90:10, v/v) to obtain ten fractions (D1–D10). Fraction D1 (2.1 g) was gel filtrated by Sephadex LH-20 (3.0 × 120 cm, 140 g) (CH3OH) to obtain D1-1–D1-5. Compound 5 (5 mg) was obtained from fraction D1-4 (700 mg) by silica gel CC using petroleum ether/ethyl acetate (20:1, v/v) as the eluent and then Sephadex LH-20 (1.5 × 120 cm, 60 g) (CH3OH) and RP-C18 gel (CH3OH/H2O gradient, 20% to 100% CH3OH). Fraction D6 (1.5 g) was purified by repeated chromatography over silica gel (petroleum ether/ethyl acetate, 20:1–0:1, v/v) to give fractions D6-1–D6-5. Fraction D6-2 (200 mg) was separated on RP-C18 gel (CH3OH/H2O gradient, 30% to 100% CH3OH) to give fractions D6-2-1–D6-2-5. Fraction D6-2-3 was submitted on a Sephadex LH-20 (1.0 × 120 cm, 40 g) (CHCl3–CH3OH, 1:1, v/v) and further purified by preparative HPLC with CH3OH–H2O (55%) to afford compounds 7 (3 mg, tR = 18 min) and compounds 8 (6 mg, tR = 23 min). Fraction D8 (900 mg) was separated by silica gel (CHCl3–CH3OH, 80:1, v/v) to obtain two factions (D8-1–D8-2). Fraction D8-1 (120 mg) was subjected to Sephadex LH-20 (1.0 × 120 cm, 40 g) (CH3OH) and further separated by silica gel (1.5 × 40 cm, 40 g) using petroleum ether–ethyl acetate (15:1, v/v) to yield compound 3 (5 mg) and compound 4 (7 mg). Fraction E (9.5 g) was purified by repeated chromatography over silica gel (petroleum ether/acetone, 8:2, v/v) to give fractions E1–E4. Fraction E3 (1.4 g) was separated by silica gel (petroleum ether/acetone, 10:1, v/v) and subjected to Sephadex LH-20 (3.0 × 120 cm, 100 g) (CH3OH) to give fractions E3-1–E3-2. Fraction E3-2 (300 mg) was subjected to Sephadex LH-20 (1.5 × 120 cm, 90 g) (CH3OH) and further purified by preparative HPLC (40%, CH3OH–H2O, tR = 12 min) to give compounds 1 (4 mg). Fraction E4 (1.0 g) was submitted to MCI gel CHP 20P CC (3 × 40 cm, 300 mL) eluted with gradient CH3OH–H2O (10:90, 30:70, 50:50, 70:30, MeOH, v/v) to obtain four fractions (E4-1–E4-4). Fraction E4-2 (250 mg) was separated on RP-C18 gel (CH3OH/H2O gradient, 20% to 100% CH3OH) to give fractions E4-2-1–E4-2-5. Fraction E4-2-2 (90 mg) was separated by silica gel (CHCl3/CH3OH, 10:1, v/v) and subjected to Sephadex LH-20 (1.0 × 120 cm, 40 g) (CH3OH) and further purified by preparative HPLC with CH3OH–H2O (25%) to afford compound 2 (1.5 mg, tR = 10 min). Fraction F (18.0 g) was submitted to MCI gel CHP 20P CC (6 × 40 cm, 500 mL) eluted with gradient CH3OH–H2O (10:90, 30:70, 50:50, CH3OH, v/v) to obtain four fractions (F1–F4). Compound 6 (23 mg) was obtained from Fraction F2 (1.2 g) by silica gel CC using CHCl3/CH3OH (9:1, v/v) and further subjected to Sephadex LH-20 (2.0 × 120 cm, 120 g) (CH3OH). All the compounds had a degree of purity > 95%.
3.4. Spectroscopic data of the isolates
Gastrodinol (1): white amorphous powder (MeOH); UV (MeOH): λmax (logε) 306 (3.6), 277 (3.8), 203 (4.6) nm; IR (KBr) νmax 3439, 2920, 2851, 1630, 1384 cm−1; 1H-NMR (CD3OD, 600 MHz) and 13C-NMR (CD3OD, 150 MHz) data see Table 1; ESIMS (negative ion): m/z = 711 [M + COOH]−; HRESIMS (negative ion): m/z = 711.2247 [M + COOH]− (calcd for C42H34O8, 711.2236).
2-(4′-Hydroxybenzoyl)-3-hydroxyethyl indole (2): yellow amorphous powder (MeOH); UV (MeOH): λmax (logε) 328 (4.1), 218 (4.3), 202 (4.3) nm; IR (KBr) νmax 3443, 2924, 1625, 1530 cm−1; 1H-NMR (CD3OD, 600 MHz) and 13C-NMR (CD3OD, 150 MHz) data see Table 2; ESIMS (positive ion): m/z = 304 [M + Na]+; HRESIMS (positive ion): m/z = 304.0943 [M + Na]+ (calcd for C17H15NNaO3, 304.0950).
2-(4′-Hydroxybenzoyl)-3-(4′′-hydroxybenzyl)indole (3): yellow amorphous powder (MeOH); UV (MeOH): λmax (logε) 326 (4.4), 319 (4.6), 201 (4.6) nm; IR (KBr) νmax 3442, 2924, 1620, 1529 cm−1; 1H-NMR (CD3OD, 600 MHz) and 13C-NMR (CD3OD, 150 MHz) data see Table 2; ESIMS (positive ion): m/z = 366 [M + Na]+; HRESIMS (positive ion): m/z = 366.1101 [M + Na]+ (calcd for C22H17NNaO3, 366.1106).
3.5. Anti-microbial activity assay
The anti-microbial activities were evaluated against 12 pathogenic microorganisms belong to Enterococcus, Staphylococcus, Streptococcus and Viridans which were obtained from clinical at the first affiliated hospital of Anhui Medical University. The anti-microbial assay was performed as described previously.17,18 The minimum inhibitory concentration (MIC) was determined using the microdilution broth method according to the Clinical Laboratory Standards Institute (CLSI M100-S27). The broth dilution method using 96-well microtiter plates was applied, and the final concentration of test compounds ranged from 0.5 to 64 μg ml−1. The lowest concentration of sample that prevented microbial growth was used to determine the MIC. Penicillin was used as positive control.4. Conclusions
Gastrodia elata Blume (Tian ma) as a famous TCM documented in Chinese Pharmacopoeia. Although the isolation and various biological activities of the tube of G. elata has been revealed in the previous investigation. This study on the stems of G. elata led to the isolation and identification of three new compounds (1–3): gastrodinol (1), 2-(4′-hydroxybenzoyl)-3-hydroxyethyl indole (2), 2-(4′-hydroxybenzoyl)-3-(4′′-hydroxybenzyl)indole (3), along with five known compounds (4–8). The anti-microbial activity of compounds 1–8 was evaluated on pathogenic microorganisms shown that 1 shows the most anti-microbial activity against all twelve pathogenic microorganisms tested, with MIC values in the range of 1–64 μg ml−1. It is noted that compound 1 exhibited the most anti-microbial activity against Streptococcus agalactiae (MIC, 1 μg ml−1) which suggested that the stems of G. elata kill ghost essence and louisvuitt “Shennong Bencao Jing”. Our results suggest that gastrodinol 1 has potential for further development anti-microbial agent. In addition, the investigations on the part of stems of G. elata are need, it will lead to the development of new antibiotics, and how to utilize it better should be paid more attention to.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors would like to thank the staff of analytical group of the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, for measurements of all spectra. This work was financial supported by the Yunnan Provincial Science and Technology Department (No. 2017ZF003-04, 2015HB093 and 2019HA001).Notes and references
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Nat. Rev. Microbiol., 2015, 13, 42–51 CrossRef CAS PubMed
.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089–1093 CrossRef CAS PubMed
- P. M. Wright, I. B. Seiple and A. G. Myers, Angew. Chem. Int. Ed., 2014, 53, 8840–8869 CrossRef CAS PubMed
- I. Ahmad, F. Aqil and M. Owais, Modern Phytomedicine: Turning Medicinal Plants into Drugs, John Wiley & Sons, Germany, 2006 Search PubMed
- Chinese Pharmacopoeia Commission, Pharmacopoeia of the People's Republic of China I, China Medical Science Press, Beijing, 2015, p. 58 Search PubMed
- C. Shuan and S. Chen, Acta Bot. Yunnanica, 1983, 5, 361–368 Search PubMed
- D. L. Jones, Austral. Orch. Res., 1991, 2, 62–65 Search PubMed
- G. G. Gu and P. J. Yang, Shennong’s Classic of Materia Medica, Academy Press, Beijing, 2013 Search PubMed
- Y. Li, Y. Ke, J. Y. Jiang, X. J. Li and Y. B. Jiang, Chin. J. Integr. Tradit. West. Med., 2015, 35, 481–487 Search PubMed
- Y. E. Lin, S. T. Chou, S. H. Lin, K. H. Lu, S. Panyod, Y. S. Lai, C. T. Ho and L. Y. Sheen, J. Ethnopharmacol., 2017, 215, 132–139 CrossRef PubMed
- J. Vile, M. Carta, C. G. Bezzu, B. M. Kariuki and N. B. Mckeown, Polymer, 2014, 55, 326–329 CrossRef CAS
- Y. Sheludko, I. Gerasimenko, H. Kolshorn and J. Stockigt, J. Nat. Prod., 2002, 65, 1006–1010 CrossRef CAS PubMed
- S. Takagi, M. Yamaki and K. Inoue, Phytochemistry, 1983, 22, 1011–1015 CrossRef CAS
- L. Wang, H. B. Xiao and X. M. Liang, Chin. Tradit. Herb. Drugs, 2003, 34, 584–585 CAS
- J. Zhou, X. Y. Pu and Y. B. Yang, Chin. Sci. Bull., 1981, 26, 1118–1120 CrossRef
- T. Yoshihara, K. Yamaguchi, S. Takamatsu and S. Sakamura, Agric. Biol. Chem., 1981, 45, 2593–2598 CAS
- O. Taglialatela-Scafati, F. Pollastro, G. Chianese, A. Minassi, S. Gibbons, W. Arunotayanun, B. Mabebie, M. Ballero and G. Appendino, J. Nat. Prod., 2013, 76, 346–353 CrossRef CAS PubMed
- S. Chakthong, P. Weaaryee, P. Puangphet, W. Mahabusarakam, P. Plodpai, S. P. Voravuthikunchai and A. Kanjana-Opas, Phytochemistry, 2012, 75, 108–113 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00965b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |