
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect cyanidation of silver sulfide by heterolytic C–CN bond cleavage of acetonitrile†
Biraj Das‡
a,
Pinku Saikia‡a,
Mukesh Sharmaa,
Manash J. Baruaha,
Subhasish Royb and
Kusum K. Bania *a
*a
aDepartment of Chemical Sciences, Tezpur University, Assam-784028, India. E-mail: kusum@tezu.ernet.in
bDepartment of Chemistry, School of Applied Sciences, University of Science and Technology, Meghalaya, Techno City, Kling Road, Baridua 9th Mile, Ri-Bhoi, Meghalaya, India 793101
First published on 27th February 2020
Abstract
Extraction of silver as silver cyanide from silver sulfide was made possible using acetonitrile as the source of cyanide. The process of cyanidation took place through the oxidation of sulfide to sulfur oxides and cleavage of the C–CN bond of acetonitrile. The reaction was found to be catalyzed by vanadium pentoxide and hydrogen peroxide. The different species involved in the cyanidation process were duly characterized using FTIR, ESI-MS, HRMS, XPS and UV-vis spectroscopic analysis. The mechanism of the cyanidation process was confirmed through in situ FTIR analysis.
Silver (Ag) is a precious noble metal that has found applications in photography, nanocatalysis, antibacterial agents and jewelry.1–3 Most importantly, it contributes to the economic growth of numerous countries. Silver metal extracted from its main ores contributes to a high percentage (∼30%) of the total world production of Ag. Silver sulfide (Ag2S) is the most common ore from which it is extracted.4,5 However, the traditional way of extracting Ag from Ag2S ore has many disadvantages, as it involves the use of poisonous sodium cyanide (NaCN) or potassium cyanide (KCN), which are also used in gold (Au) extraction.4,5 The use of cyanide to achieve Ag leaching has led to great public concern, due to the damage it can cause to both human health and the environment. Because of the high toxicity of cyanide salts, strict regulation has been imposed to regulate or to ban the leaching of cyanide into the environment.6 It has been reported that the use of such cyanide salts in Au mining industries has led to the death of people working in those industries.7 Because of the devastating impacts of cyanide poisoning, researchers are constantly searching for alternative methods and new lixiviants to replace cyanide salt in silver extraction from Ag2S. So far, researchers have adopted various alternative methods for silver extraction, such as leaching of Ag2S using ferricyanide-cyanide solution.8,9 Some have also used the precipitation method using high pressure and so on.10 Recently, the use of thiosulfate solution with buffer solution, most commonly ammonium acetate buffer at pH = 4, has drawn great attention, and has been found to be a potential candidate for Ag extraction from its ores.11 Although the method is cheaper, it consumes a high concentration of the reagent and also depends on different conditions such as pH and the kinetics of oxygen reduction.11,12 The reaction of silver ions (Ag+) with thiosulfate solution in the presence of a base (NH3) is thermodynamically favorable, but the slow oxygen reduction process leads to deliberate leaching of Ag as silver thiosulfate complex, Ag(S2O3)35−.12 Hence, the search for an alternative method is still an ongoing research process.
Apart from alkali cyanides, alkyl nitriles such as acetonitrile (CH3CN) are much less toxic and can be used as a source of cyanide ions (CN−).13 However, the thermodynamic stability and high bond energy associated with the C–CN bond restrict the application of this green solvent as a source of cyanide (CN−) in the silver extraction process.14 Literature reports suggest that organometallic catalysts can scissor the C–CN bond very effectively.15–17 However, to the best of our knowledge, there is no such report that applies CH3CN as a lixiviant in silver extraction as AgCN from Ag2S. Recently, we reported that CH3CN can be a suitable solvent to act as a cyanide source for the direct cyanidation of silver nitrates (AgNO3) in the presence of vanadium pentoxide (V2O5) and hydrogen peroxide (H2O2).18 However, Ag is mostly extracted from Ag2S in mining industries. Therefore, with our growing interest in finding a solution to replace cyanide salt, herein we demonstrate a highly effective method that allows for the direct precipitation of AgCN from Ag2S within a very short period of time.
Initially, to achieve the cyanidation of Ag2S and obtain silver (Ag) as silver cyanide (AgCN), silver sulfide (Ag2S) and vanadium pentoxide (V2O5) were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio using a mortar and pestle. The mixture was ground for few minutes and then dried in oven at 100 °C. The process of grinding and heating was continued for 2 h by making a paste using an ethanol/water mixture as a solvent. The whole solid mixture was transferred to a 25 mL round bottom flask, followed by dropwise addition of 2 mL of hydrogen peroxide (30% w/v) in 10 mL CH3CN as a solvent. The reaction mixture was then stirred for 90 min at room temperature. The solid material became completely dissolved and formed a homogeneous mixture, and the color of the solution was found to transform from black to yellow and finally to red, as shown in Scheme 1. Along with the change in color, white particles also began to precipitate from the solution, and this precipitation process was completed upon allowing the solution to stand overnight. The white precipitate was obtained by filtration using Whatmann filter paper, and was washed several times with double deionized water, and collected for further characterization. The obtained red filtrate was also kept for further analysis.
:1 ratio using a mortar and pestle. The mixture was ground for few minutes and then dried in oven at 100 °C. The process of grinding and heating was continued for 2 h by making a paste using an ethanol/water mixture as a solvent. The whole solid mixture was transferred to a 25 mL round bottom flask, followed by dropwise addition of 2 mL of hydrogen peroxide (30% w/v) in 10 mL CH3CN as a solvent. The reaction mixture was then stirred for 90 min at room temperature. The solid material became completely dissolved and formed a homogeneous mixture, and the color of the solution was found to transform from black to yellow and finally to red, as shown in Scheme 1. Along with the change in color, white particles also began to precipitate from the solution, and this precipitation process was completed upon allowing the solution to stand overnight. The white precipitate was obtained by filtration using Whatmann filter paper, and was washed several times with double deionized water, and collected for further characterization. The obtained red filtrate was also kept for further analysis.
 | ||
| Scheme 1 Synthesis of AgCN by cyanidation of Ag2S. | ||
The formation of AgCN was found to be dependent on the amounts of V2O5 and the oxidant H2O2. The optimization values for these are depicted in Tables 1 and 2. From Table 1, it was found that when 1 mmol of V2O5 reacted with 1 mmol of Ag2S in presence of 2 mL of H2O2 in 10 mL CH3CN, the amount of AgCN was found to be at its maximum value. On decreasing the amount of V2O5, keeping all other values constant, the % yield of AgCN was found to decrease. Therefore, a 1:1 ratio of Ag2S:V2O5 was considered as the optimum amount (Table 1). Similarly, from Table 2, it was ascertained that 2 mL of H2O2 was sufficient to give 96% of AgCN. 10 mL of CH3CN was found to be the most appropriate amount for complete precipitation of AgCN.
| Ag2S mg (mmol) | H2O2 (mL) | V2O5 (mg) | AgCN mg (mmol) | % Yield |
|---|---|---|---|---|
| a The values in the parentheses represent the amounts in mmol. | ||||
| 247.8 (1) | 2 | 181.8 (1) | 258.6 (1.9) | 96.6 |
| 2 | 90.9 (0.5) | 240.6 (1.8) | 89.9 | |
| 2 | 60.6 (0.3) | 230.8 (1.7) | 86.2 | |
| 2 | 45.4 (0.25) | 218.0 (1.6) | 81.6 | |
:1 ratio of V2O5:Ag2S
| V2O5 (mmol) | Ag2S (mmol) | CH3CN (mL) | H2O2 (mL) | % Yield AgCN |
|---|---|---|---|---|
| 1 | 1 | 10 | 0.5 | — |
| 1 | 1 | 10 | 1 | 58 |
| 1 | 1 | 10 | 1.5 | 79 |
| 1 | 1 | 10 | 2 | 96 |
The white precipitate obtained from the reaction was characterized by XRD (Fig. 1a) and FTIR (Fig. 1b) and was found to be exactly the same as that of our previous reports that confirmed the white precipitate to be silver cyanide (AgCN).18,19 The presence of peaks at 2θ values of 23.9, 29.8, 38.4, 49.3, 52.8, 58.5 and 61.8 confirmed the formation AgCN (Fig. 1a). The FTIR spectra also revealed the formation of AgCN with the presence of a weak vibrational band at 2166 and sharp, intense bands at 2139 cm−1 for –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N and 476 cm−1 for Ag–C (Fig. 1b). The AgCN material was found to be highly pure without any contamination.
N and 476 cm−1 for Ag–C (Fig. 1b). The AgCN material was found to be highly pure without any contamination.
 | ||
| Fig. 1 (a) XRD pattern and (b) FTIR spectra of AgCN. | ||
In order to investigate the role of vanadium oxide and H2O2, different analyses were performed. From the XRD analysis it was evident that the 1:1 mixture of V2O5 and Ag2S formed a composite containing the crystalline planes of both Ag2S and V2O5 (Fig. 2).20–22 For example, the planes at 2θ values of 15.4 (200) and 20.1 (001) were present in V2O5 and the composite but were absent in Ag2S. Similarly, the planes at 2θ values of 28.9 (110), 36.7 (121) and 43.4 (202) were absent in V2O5 but were found in Ag2S and the composite.20–22 This alternation of XRD planes clearly confirmed the good mixing of V2O5 with Ag2S. The Raman spectra of Ag2S, V2O5 and V2O5–Ag2S composite were also compared. However, the Raman signals in the V2O5–Ag2S composites were mostly dominated by the signals of the strong vibrational bands of V2O5 (Fig. S1†), as the Raman signals for Ag2S were much weaker than those of V2O5. The scanning electron microscopy (SEM) images of the composite (Fig. S2†) were also found to differ from those of Ag2S (Fig. S3†). The elemental compositions of the composite (Fig. S4†) and Ag2S (Fig. S5†) were assessed through energy dispersive X-ray (EDX) analysis. The presence of all the elements, i.e., Ag, V, S and O, clearly revealed the formation of the composite and also the purity of the Ag2S ore which was used as the source of silver.
 | ||
| Fig. 2 XRD pattern of V2O5 (black), Ag2S (brown) and V2O5–Ag2S composite (blue). | ||
FTIR analyses were performed before and after the addition of H2O2 (Fig. 3). When the ground mixture of Ag2S and V2O5 (1:1) was kept at 100 °C overnight, a new band was found at 1109 cm−1 in the FTIR spectrum, characteristic of the S–O bond (Fig. 3a).23 The appearance of this new band in the region of ∼1100 cm−1 implies the oxidation of sulfide (S2−) to sulfur oxides (SxOy, x = 1–2, y = 2–4).24 The appearance of this peak was dependent on temperature, as it was not clearly visible when the sample was treated below 100 °C (Fig. 3a). The peak was found to become more intense upon treatment with different amounts of H2O2 (0.5 and 1 mL), keeping the amount of CH3CN fixed (1 mL), as shown in Fig. 3b. The conversion of S2− to SxOy was also confirmed from the XPS analysis. The presence of S (2p3/2) at 168.7 eV and 166.0 eV, corresponding to S2O3 and SO32− ions, confirmed the oxidation of S2− to SxOy (Fig. S6†).25,26 Usually, the binding energy values of sulfur (S) in sulfides are observed in the range of 162–164 eV.27,28 It is pertinent to mention that the oxidation of S2− to SxOy did not happen in the absence of V2O5. This clearly suggests that the oxidation of S2− to SxOy was catalyzed by V2O5 in the presence of H2O2. Various other reports are also available in the literature supporting the oxidation of S2− to SxOy by vanadium-oxide catalysts.29,30 Thus, from these analyses, it was confirmed that Ag2S in the presence of V2O5 and H2O2 became oxidized and formed ionic silver sulfur oxide (AgnSxOy) compounds that easily dissociated in solution to form nAg+ and SxOyn−.
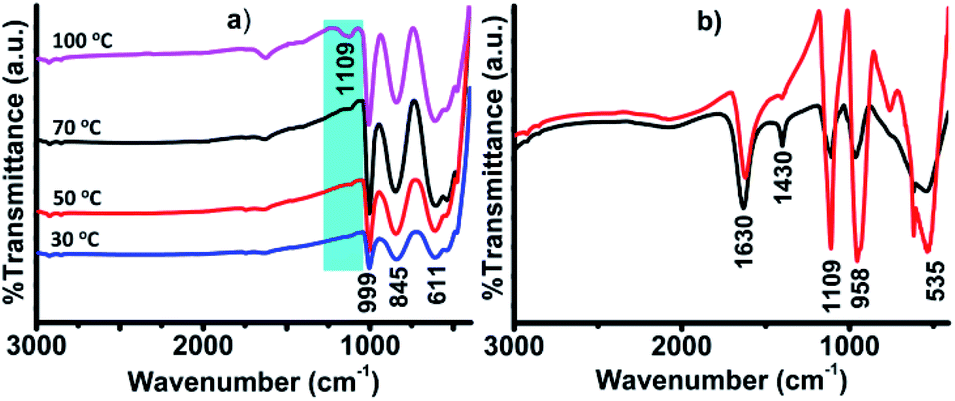 | ||
| Fig. 3 (a) FTIR spectra of the composites recorded at different temperatures and (b) FTIR spectra of the composite after treating with H2O2 and CH3CN; (1 mL H2O2 + 1 mL CH3CN, red line) and (0.5 mL H2O2 + 1 mL CH3CN, black line). | ||
Electron Spray Ionization-Mass Spectrometry (+ESI-MS) analysis was performed to detect the formation of silver ions (Ag+) in solution. The mass spectrum of the solution just after it became reddish in color showed a sharp intense peak with m/z values of 106.9, 109 and 148 corresponding to molecular ion peaks of Ag+ (m/z = 107), its isotope (m/z = 109) and [Ag(CH3CN)]+ species (m/z = 148), as shown in Fig. S7.† The presence of an isotopic mass signal for [Ag(CH3CN)]+ was also confirmed from the High Resolution Mass Spectrometry (HRMS) analysis of the same sample showing isotopic m/z values at 148 and 150, as shown in Fig. S8.† HRMS analysis of the solution recorded after separating the AgCN precipitate gave sharp signals at m/z values of 171, 153, 150 and 148, corresponding to [OV(O2)(H2O)4]+, [OV(O2)(H2O)3]+, [109Ag(CH3CN)]+ and [107Ag(CH3CN)]+, as shown in Fig. S9.† These m/z values for the peroxo-vanadium species were completely matched with those previously reported by Conte and his co-workers.31 This implied the formation of peroxo-vanadate species in solution during AgCN precipitation.
The presence of peroxovanadate species was also confirmed from the UV-vis analysis of the red solution. The UV-vis spectra presented a weak band at 378 nm due to ligand to metal charge transfer (LMCT) originating from the peroxo to vanadium (O22− → V) transition.32 As the absorption behavior of peroxo-vanadate species is pH dependent,32 the UV-vis spectra were recorded with the addition of 10 μL of 0.1 M HCl solution. After titrating with dilute HCl, two sharp new bands were observed at 219 and 286 nm. The band at 219 nm is due to the LMCT transition of πh* → dσ*.33 The band at 286 nm was attributed to the formation of [VO(OH)] species resulting from the protonation of peroxo-vanadate species (Fig. 4a).34
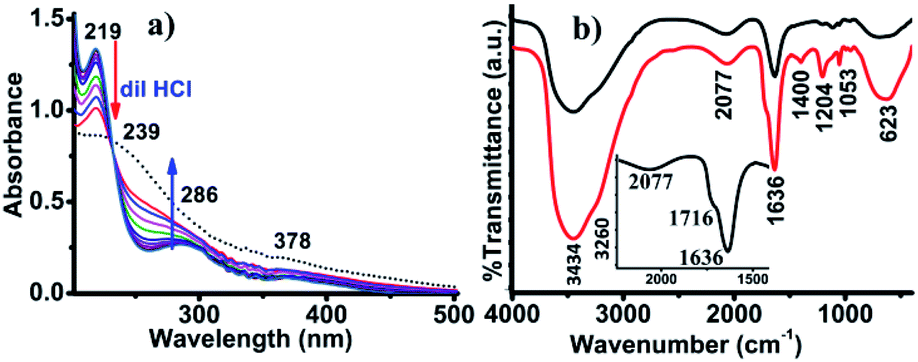 | ||
| Fig. 4 (a) UV-vis spectra of the solution titrated against of 0.1 M HCl by addition of 10 μL at a time, and (b) in situ FTIR-analysis performed at 273 K (red) and 300 K (black). | ||
Based on the above analysis, a plausible mechanism for the formation of AgCN has been proposed in Scheme 2. In the presence of V2O5 and H2O2, the Ag2S transforms into silver sulfur oxides, as is evident from the FTIR spectroscopy. This ionic compound then becomes dissociated to release the Ag+. In the meantime, the CH3CN becomes activated by the anionic yellow diperoxo species [V(O)(O2)2]− generated during part of the reaction (Scheme 2). The formation of diperoxo species was assumed based on the change in the color of the reaction mixture from black to yellow, and then finally to red. Further, the formation of such species was evident from UV-vis analysis (Fig. 4a). Li et al. also proposed the generation of such diperoxo-vanadium species upon treatment of H2O2 with V2O5.35 This anionic diperoxo species abstracts a proton from CH3CN to form −CH2CN, which then binds to the vanadium center (Scheme 2).36 A similar mechanism was also proposed by Brazdil et al. on the gas phase oxidation of CH3CN at high temperature.36 In the subsequent step, this −CH2CN species gets transformed into hydroxyacetonitrile (OHCH2CN) and finally produces the CN− ion, and HCHO or HCOOH. The released CN− ion then combines with Ag+ to form AgCN. The formation of this species was confirmed from in situ IR operation at 273 K where a band for surface-absorbed free CN− ions was observed at 2077 cm−1 (Fig. 4b).37 In addition to this, a few other peaks were also observed at 1716, 1400 and 1204 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration (inset) and the C–H bending mode vibration,23 as shown in Fig. 4b (red line). The O–H vibrations were observed at 3434 and 3260 cm−1. These results match well with those reported by Danger et al.38 The appearance of these vibrational bands truly implies the oxidation of CH3CN to formic acid via the formation of methanol with the liberation of CN−. The vibrational frequency band for CN− was clearly visible when the in situ operation was performed at 300 K, as the C–H vibration disappeared and the C–N vibration became more prominent along with the presence of the S–O vibration at 1113 cm−1, as shown in Fig. 4b (black line).
O vibration (inset) and the C–H bending mode vibration,23 as shown in Fig. 4b (red line). The O–H vibrations were observed at 3434 and 3260 cm−1. These results match well with those reported by Danger et al.38 The appearance of these vibrational bands truly implies the oxidation of CH3CN to formic acid via the formation of methanol with the liberation of CN−. The vibrational frequency band for CN− was clearly visible when the in situ operation was performed at 300 K, as the C–H vibration disappeared and the C–N vibration became more prominent along with the presence of the S–O vibration at 1113 cm−1, as shown in Fig. 4b (black line).
 | ||
| Scheme 2 Plausible mechanism for CH3CN oxidation and AgCN formation. | ||
In summary, the long-term challenge in finding an alternative approach for the direct cyanidation of silver sulfide ore in the absence of alkali metal cyanide has been realized by using acetonitrile as the cyanide source or as a green lixiviant for the direct precipitation of silver as silver cyanide. We are currently optimizing the process towards the batch-scale production of silver cyanide from silver sulfide. We believe that the current approach offers an alternative green pathway for silver extraction.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
KKB thanks DST-SERB for financial support (SB-EMEQ-2014/463, CRG/2019/00096). BD thanks UGC-MHRD, Govt. of India, for the National Fellowship (RGNF-2017-18-SC-ASS-43132). MS also acknowledges CSIR-HRDG, New Delhi, for the SRF fellowship (No. 09/796(0097)/19-EMR-I). MJB thanks the Department of Science and Technology (DST), Govt. of India, for the DST-INSPIRE Fellowship (No. DST/INSPIRE Fellowship/2018/IF180217).Notes and references
- S. C. Abeyweera, K. D. Rasamani and Y. Sun, Acc. Chem. Res., 2017, 50, 1754–1761 CrossRef CAS.
- H. L. Cao, H. B. Huang, Z. Chen, B. Karadeniz, J. Lü and R. Cao, ACS Appl. Mater. Interfaces, 2017, 9, 5231–5236 CrossRef CAS.
- R. S. Diggikar, S. P. Deshmukh, T. S. Thopate and S. R. Kshirsagar, ACS Omega, 2019, 4, 5741–5749 CrossRef CAS.
- G. Deschênes, J. Rajala, A. R. Pratt, H. Guo, M. Fulton and S. Mortazavi, Miner. Metall. Process., 2011, 28, 37–42 Search PubMed.
- H. Jiang, F. Xie and D. B. Dreisinger, Hydrometallurgy, 2015, 158, 149–156 CrossRef CAS.
- M. S. Reisch, Chem. Eng. News, 2017, 95, 18–19 Search PubMed.
- G. Hilson and A. J. Monhemius, J. Cleaner Prod., 2006, 14, 1158–1167 CrossRef.
- F. Xie, D. Dreisinger and J. Lu, Miner. Eng., 2008, 21, 1109–1114 CrossRef CAS.
- F. Xie and B. D. Dreisinger, Hydrometallurgy, 2007, 88, 98 CrossRef CAS.
- P. C. Holloway, K. P. Merriam and T. H. Etsell, Hydrometallurgy, 2004, 74, 213–220 CrossRef CAS.
- S. Ayata and H. Yildiran, Miner. Eng., 2005, 18, 898–900 CrossRef CAS.
- Y. Cui, X. Tong and A. Lopez-Valdivieso, Rare Met., 2011, 30, 105–109 CrossRef CAS.
- Y. Zhu, M. Zhao, W. Lu, L. Li and Z. Shen, Org. Lett., 2015, 17, 2602–2605 CrossRef CAS PubMed.
- K. Zhou, C. Qin, X. L. Wang, L. K. Shao, K. Z. Yan and Z. M. Su, CrystEngComm, 2014, 16, 10376–10379 RSC.
- F. L. Taw, A. H. Mueller, R. G. Bergman and M. Brookhart, J. Am. Chem. Soc., 2003, 125, 9808–9813 CrossRef CAS PubMed.
- L. R. Guo, S. S. Bao, Y. Z. Li and L. M. Zheng, Chem. Commun., 2009, 2893–2895 RSC.
- F. L. Taw, P. S. White, R. G. Bergman and M. Brookhart, J. Am. Chem. Soc., 2002, 124, 4192–4193 CrossRef CAS.
- B. Das, M. Sharma, M. J. Baruah, K. K. Borah and K. K. Bania, Inorg. Chim. Acta, 2019, 498, 119160 CrossRef CAS.
- B. Das, M. Sharma, C. Kashyap, A. K. Guha, A. Hazarika and K. K. Bania, Appl. Catal., A, 2018, 568, 191–201 CrossRef CAS.
- S. Martha, D. P. Das, N. Biswal and K. M. Parida, J. Mater. Chem., 2012, 22, 10695–10703 RSC.
- J. Wang, H. Feng, K. Chen, W. Fan and Q. Yang, Dalton Trans., 2014, 43, 3990–3998 RSC.
- H. Li, F. Xie, W. Li, H. Yang, R. Snyders, M. Chen and W. Li, Catal. Surv. Asia, 2018, 22, 156–165 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, John Wiley & Sons, NY, 5th edn, 1997 Search PubMed.
- L. Li, Z. Xu, A. Wimmer, Q. Tian and X. Wang, Environ. Sci. Technol., 2017, 51, 7920–7927 CrossRef CAS PubMed.
- R. S. C. Smart, W. M. Skinner and A. R. Gerson, Surf. Interface Anal., 1999, 28, 101–105 CrossRef CAS.
- H. W. Nesbitt, M. Scaini, H. Hochst, G. M. Bancroft, A. G. Schaufuss and R. Szargan, Am. Mineral., 2000, 85, 850–857 CrossRef CAS.
- B. Das, M. Sharma, A. Hazarika and K. K. Bania, ChemistrySelect, 2019, 4, 7042–7050 CrossRef CAS.
- B. Das, M. J. Baruah, M. Sharma, B. Sarma, G. V. Karunakar, L. Satyanarayana, S. Roy, P. K. Bhattacharyya, K. K. Borah and K. K. Bania, Appl. Catal., A, 2020, 589, 117292 CrossRef CAS.
- E. Sahle-Demessie and V. G. Devulapelli, Appl. Catal., B, 2008, 84, 408–419 CrossRef CAS.
- M. D. Soriano, A. Vidal-Moya, E. N. R. I. Q. U. E. Rodríguez-Castellón, F. V. Melo, M. T. Blasco and J. L. Nieto, Catal. Today, 2016, 259, 237–244 CrossRef CAS.
- (a) V. Conte, O. Bortolini, M. Carraro and S. Moro, J. Inorg. Biochem., 2000, 80, 41–49 CrossRef CAS; (b) O. Bortolini, V. Conte, F. D. Furia and S. Moro, Eur. J. Inorg. Chem., 1998, 1193–1197 CrossRef CAS.
- M. Kaliva, C. P. Raptopoulou, A. Terzis and A. Salifoglou, Inorg. Chem., 2004, 43, 2895–2905 CrossRef CAS.
- M. Kaliva, T. Giannadaki, A. Salifoglou, C. P. Raptopoulou, A. Terzis and V. Tangoulis, Inorg. Chem., 2001, 40, 3711–3718 CrossRef CAS.
- M. R. Maurya, S. Khurana, W. Zhang and D. Rehder, J. Chem. Soc., Dalton Trans., 2002, 15, 3015–3023 RSC.
- C. Li, P. Zheng, J. Li, H. Zhang, Y. Cui, Q. Shao, X. Ji, J. Zhang, P. Zhao and Y. Xu, Angew. Chem., Int. Ed., 2003, 42, 5063–5066 CrossRef CAS PubMed.
- J. F. Brazdil, R. G. Teller, W. A. Marritt, L. C. Glaeser and A. M. Ebner, J. Catal., 1986, 100, 516–519 CrossRef CAS.
- S. Yoshikawa, D. H. O'Keeffe and W. S. Caughey, J. Biol. Chem., 1985, 260, 3518–3528 CAS.
- G. Danger, A. Rimola, N. A. Mrad, F. Duvernay, G. Roussin, P. Theule and T. Chiavassa, Phys. Chem. Chem. Phys., 2014, 16, 3360–3370 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: The details material used and physical measurements, Raman spectra of V2O5, Ag2S and V2O5–Ag2S, XPS spectra of sulfur and the +ESI-mass spectra and HRMS. See DOI: 10.1039/d0ra00940g |
| ‡ Both the authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |