
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReduced graphene oxide promoted assembly of graphene@polyimide film as a flexible cathode for high-performance lithium-ion battery†
Bin Changa,
Jian Maa,
Tiancai Jiangb,
Li Gaoa,
Yuanting Lia,
Mingan Zhoua,
Yanshan Huang *a and
Sheng Han*a
*a and
Sheng Han*a
aSchool of Chemical and Environmental Engineering, Shanghai Institute of Technology, Haiquan Road 100, Shanghai, 201418, PR China. E-mail: hysh9@126.com; hansheng654321@sina.com
bSchool of Physics and Technology, Center for Nanoscience and Nanotechnology, Wuhan University, Wuhan, 430072, Hubei, PR China
First published on 28th February 2020
Abstract
Organic carbonyl polymers have been gradually used as the cathode in lithium-ion batteries (LIB). However, there are some limits in most organic polymers, such as low reversible capacity, poor rate performance, cycle instability, etc., due to low electrochemical conductivity. To mitigate the limits, we propose a strategy based on polyimide (PI)/graphene electroactive materials coated with reduced graphene oxide to prepare a flexible film (G@PI/RGO) by solvothermal and vacuum filtration processes. As a flexible cathode for LIB, it provides a reversible capacity of 198 mA h g−1 at 30 mA g−1 and excellent rate performance of 100 mA h g−1 at high current densities of 6000 mA g−1, and even a super long cycle performance (2500 cycles, 70% capacity retention). The excellent performance results in a special layer structure in which the electroactive PI was anchored and coated by the graphene. The present synthetic method can be further applied to construct other high-performance organic electrodes in energy storage.
Introduction
Rechargeable Lithium Ion Batteries (LIBs) are often the primary choice for portable electronics due to their high power and high energy density.1–4 In the traditional LIBs, the electrode material is the most important component of LIBs, and is generally an inorganic material with a high theoretical specific capacity.3,5,6 However, inorganic electrodes cause many problems such as high energy consumption and environmental pollution during the fabrication. Therefore, the development of environmentally friendly and electrochemically stable electrode materials has become an inevitable trend.7 Recently, organic electrode materials have drawn widespread academic attention due to resource renewability, their eco-friendly nature, structural diversity, and design flexibility.8–11 In particular, polyimide (PI) as a conjugated carbonyl polymer,10,12 has received more attention than other organic electrodes13–17 because of its stable skeleton conjugated structure and high potential active carbonyl group.12 However, the poor conductivity of PI is a serious obstacle to a high energy density electrode material. To increase conductivity, one of the strategies to solve this problem is generally to design the composites electrodes with highly conductive current collectors or add a large number of conductive agents in the composites.18,19 However, the addition of such a large amount of inert additives reduces the utilization efficiency of the organic polymer common electrode, and is also disadvantageous for the assembly of the flexible electrode device. For example, Song and colleagues reported the synthesis of powdered graphene/PI nanocomposites as LIBs cathodes, but not only their active substance content was reduced, but the composites could not be folded repeatedly.20The typical two-dimensional graphene with high electrical conductivity is usually introduced into the organic polymer as a matrix. Recent studies have shown that the reinforced composite material is promising to enhance the performance of the LIB and the utilization efficiency of the cathode polymer. Graphene is introduced into PI, which has recently demonstrated not only improving the electrochemical properties of the electrodes but also directly increasing the mechanical strength of the electrode.20–25 However, due to the inconsistency between the conventional three-dimensional graphene structure and the PI particles, the electrochemical conductivity of the PI particles during the cycle is still poor. This leads to a major drawback of the poor-rate performance. One of the most promising strategies to solve the problem is to confine the particles in a densely packed graphene film structure.26–28 The main challenge in this strategy is achievement of both a high content of active particles and the flexible structure of the composites.
Herein, we developed a novel reduced graphene oxide (RGO)-promoted assembly method to fabricate a flexible G@PI with a dense layer structure (G@PI/RGO) via combination of solvothermal reaction, vacuum filtration and carbonization processing. After direct cutting, the film can serve as a binder-free electrode. Owing to the excellent conductivity of graphene and the carbonyl active site of PI, electrons can rapidly transfer during the redox reaction, giving the free-standing G@PI/RGO cathode excellent electrochemical performance for LIB: ultra-high reversible capacity of 198 mA h g−1 at current density of 30 mA g−1, excellent rate performance of 97.6 mA h g−1 at 6000 mA g−1, super long cycle performance density (2500 cycles, 70% capacity retention, 100 mA g−1). The excellent property is superior to most PI-based materials, demonstrating the enormous potential of this method in the manufacture of film mixtures with controlled morphology and energy devices.
Experiment
Material preparation
The preparation of GO was based on the improved Hummers' method, which was made by separating graphene flakes.29 The G@PI/RGO was prepared by solvothermal reaction, vacuum filtration, and carbonization processing. Weigh 40 mg of 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTCDA) into 5 mL of 2 mg mL−1 GO 1-methyl-2-pyrrolidone (NMP) solution, and mix well evenly, followed by addition of 10 mg of ethylenediamine (EDA) to the mixture. After half an hour's stirring, transfer the suspension to a 50 mL Teflon-lined stainless steel autoclave, seal, and heat at 200 °C for 20–24 hours. After cooling, wash the as-prepared suspension and vacuum filtered 6 times with deionized water. Finally, the obtained graphene@polyimide (G@PI) was dispersed in 10 mL of deionized water for further use. The G@PI/RGO film was prepared by graphene promoted assembly with G@PI suspension. First, 10 mg of GO was added to the prepared G@PI suspension and sonicated for 30 minutes, and then the flexible mixture was prepared by vacuum filtration of the above mixture. Finally, the G@PI/RGO film was subjected to 300 °C thermal annealing for 8 hours in an Ar atmosphere. As a control sample, we also prepared reduced graphene oxide (RGO) and PI samples, which was obtained by heating 5 mg mL−1 of GO in a hydrothermal environment at 200 °C for 10 hours, cooling, washing and drying to obtain RGO. The ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 NTCDA and EDA was reacted in 200 °C solvothermal for 12 h, cooled, washed and dried to obtain PI.
:1 NTCDA and EDA was reacted in 200 °C solvothermal for 12 h, cooled, washed and dried to obtain PI.
Materials characterization
Morphological characterization of the samples were done by transmission electron microscopy (TEM, JEOL 2100F) and field emission scanning electron microscopy (SEM, JEOL JSM 7500F). X-ray diffraction (XRD, Rigaku MiniFlex 600, Cu) were able to characterize the crystallinity of the samples. The samples were measured by X-ray photoelectron spectroscopy (XPS, Kratos Analytical Ltd., Axis Ultra DLD). Structural analysis of G@PI/RGO was demonstrated by Fourier transform infrared spectroscopy (FTIR, Bruker Tensor II Sample Compart RT-D LaTGS) and Raman spectroscopy were conducted with DXR Thermo-Fisher Scientific via micro-Raman system using a laser source of 532 nm. The content of PI in G@PI/RGO was measured by a thermogravimetric analyzer (TGA, NETZSCH, STA 449 F3). N2 adsorption/desorption was determined by Brunauer–Emmett–Teller (BET) measurements using a BSD-PM1 analyzer.Electrochemical measurements
G@PI/RGO was directly pressed as the working electrode of LIB, using metal lithium foil as the counter electrode. 70% active material, 20% carbon black (Super P) and 10% polytetrafluoroethylene (PVDF) were mixed and coated on aluminum foil (99.6%) as the G@PI electrode of the comparative sample, and the metal lithium foil was Electrode. A solvent of 1 M LiN(CF3SO2)2 (LiTFSI) and ethylene carbonate (EC) and dimethyl carbonate (DMC) (weight ratio of 1:1) was mutually dissolved as an electrolyte. The separator was Celgard 2400 porous polypropylene for LIB. The battery packs (CR2016) were assembled in an argon-filled glove box. Constant current charge/discharge tests were performed using a Land CT 2001A battery instrument at different battery rates from 1.5 to 3.5 V.
Result and discussion
The flexible G@PI/RGO film was synthesized by performing controlled in situ polymerization of PI particles on the GO surface to synthesize G@PI nanosheets, which were then subjected to self-assembly process by vacuum filtration with GO (Fig. 1a). First, the NTCDA and EDA monomers were prepared by in situ polymerization on the graphene surface by solvent heat treatment to prepare G@PI nanosheets, and GO was reduced to graphene in this process. The G@PI/RGO composites film were prepared by homogeneously mixing G@PI with GO by vacuum filtration followed by pyrosis treatment. After mixed with GO, the G@PI/RGO film was showed as a flexible film, while the G@PI without adding GO, the film was fragile and easily pulverized (Fig. 1c and d) in digital photography. The SEM and TEM of the G@PI/RGO and G@PI were showed in Fig. 2. For G@PI/RGO, PI particles with a size of about 150 nm were uniformly grown on the surface of graphene, and no free PI particles existed, indicating that PI needs to be nucleated and grown on the graphene surface. It also reveals that the G@PI/RGO film was with a dense layered or self-orientation structure, observed previously in graphene film prepared by vacuum filtration. The uniform growth of PI polymers on graphene is mainly due to the π–π interaction between graphene and the backbone of PI containing conjugated aromatic rings. This also revealed that the G@PI/RGO film has a dense layered or self-oriented structure, which was previously observed in graphene films prepared by vacuum filtration.30 However, the G@PI composite was only showed as an irregular structure. This result, together with the observations of the digital photographs, showed that the addition of graphene improves the mechanical flexibility of the film. Meanwhile, the G@PI nanosheets surface were covered by the RGO (Fig. 2g and h), which can improve the conductivity of film. In addition, the uniform dispersion of C, N and O elements in the G@PI/RGO element map reveals the uniform distribution of PI polymers in the composite (Fig. 2i–l), and the observations are consistent with SEM and TEM. With the excellent mechanical features, the G@PI/RGO film could be arbitrarily cut into LIB cathodes without any additional additive agents.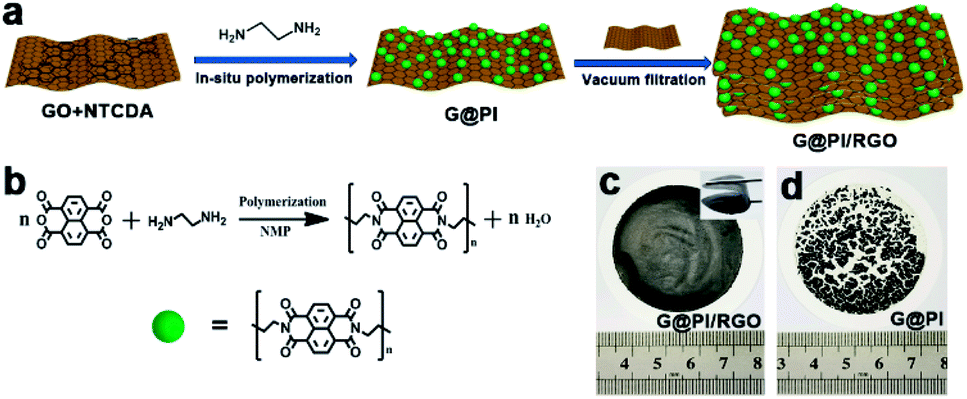 | ||
| Fig. 1 (a) Schematic of the preparation process of G@RGO; (b) synthetic route to PI; (c) photograph of G@PI/RGO; (d) photograph of G@PI. | ||
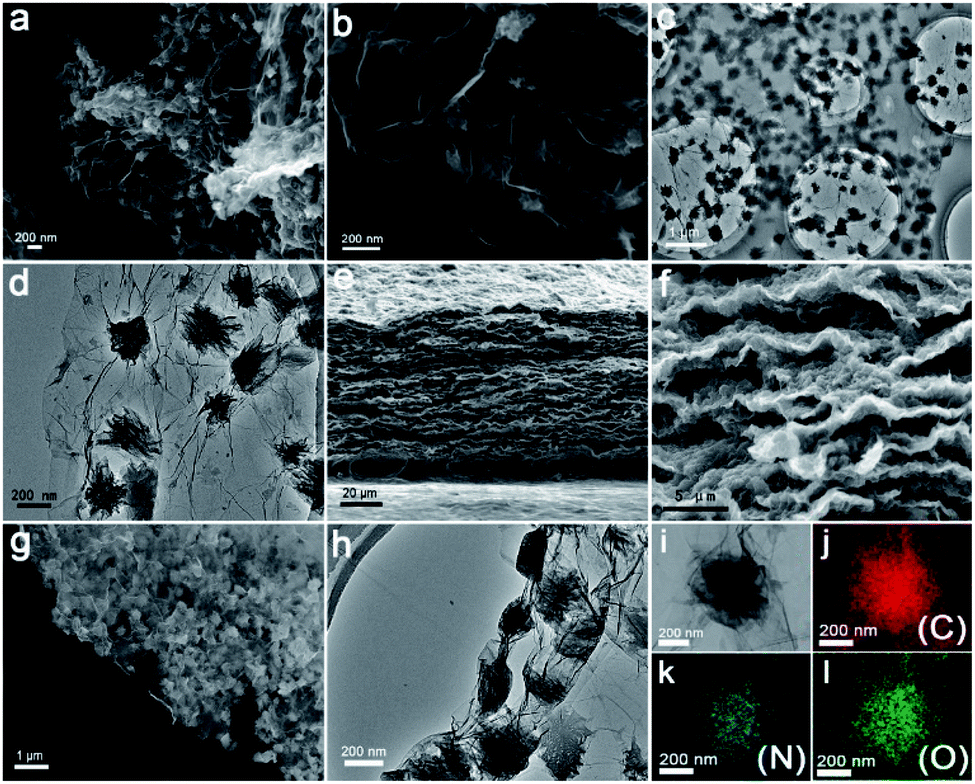 | ||
| Fig. 2 (a and b) SEM images of G@PI; (c and d) TEM images of G@PI; (e–g) SEM images of G@PI/RGO; (h) TEM images of G@PI/RGO; (i–l) elemental mapping images of G@PI/RGO (by TEM): carbon (j), nitrogen (k), and oxygen (l) distribution in the selected area. | ||
The X-ray diffraction (XRD) pattern allows for an analysis of the crystal structures of G@PI/RGO, G@PI and pure PI (Fig. 3a). Pure PI showed two peaks at 16.92° and 25.8°, which proved that it has a crystal structure. Similarly, G@PI and G@PI/RGO showed the same peaks at these positions, indicating that solvothermal and vacuum filtration did not destroy the crystal form. In addition, the peak of G@PI is shortened and broadened than PI, due to addition of graphene to prevent aggregation and overgrowth of PI particles.31 Fourier transform infrared (FT-IR) spectroscopy can further demonstrate the structural characteristics of RGO, PI, G@PI and G@PI/RGO (Fig. 3b). The peaks at 1630 and 1104 cm−1 correspond to C–C and C–O–C bond of graphene. Both PI and G@PI/RGO have strong absorption peaks at 1349 cm−1, 1703, 1670 cm−1 corresponds to the stretching vibration of the C–N bond, the asymmetric and symmetric tensile vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. It declares the successful polymerization of PI on the graphene surface in G@PI/RGO composites. X-ray photoelectron spectroscopy (XPS) can also clarify the composition of G@PI/RGO (Fig. S1a–d, ESI†). Due to the carbon atoms of the graphitic carbon, the peak CI corresponding to the C–C coordination is 284.6 eV. The peak CII at 285.3 eV corresponds to the CO double bond, and the peak CIII at 285.3 eV is associated with the C–N bond. The N1s peak shown at 399.3 eV corresponds to the N–C bond of pyridine N in PI. The O1s XPS peak corresponding to the CO bond was observed at 74.4 eV. Raman spectra of RGO, G@PI, G@PI/RGO were shown in Fig. 3c. Two peaks located near 1353 and 1591 cm−1 correspond to the disordered carbon (D-bond) and ordered graphitic carbon (G-bond), respectively. As an indicator of the degree of disorder of carbon materials, the ID/IG intensity ratio (1.11) of G@PI is slightly higher than RGO (1.07), indicating the successful attachment of PI to the graphene. Similarly, ID/IG of the G@PI/RGO was 0.97, which is lower than others, indicating that the G@PI/RGO composite combines the carbon structure in both RGO and G@PI.32,33 The G@PI and G@PI/RGO composites were investigated by thermogravimetric analysis (TGA, Fig. 3d). As could be calculated, the content of graphene and PI in G@PI is about 12% and 88%. Since the amount of GO added twice is the same, the mass content of graphene and PI in G@PI/RGO is 22% and 78%, respectively, according to the results of G@PI. The N2 adsorption/desorption isotherms and the pore size distributions of G@PI/RGO and G@PI are shown on Fig. S2 (ESI†). The pore volume and surface area of the self-assembled composite G@PI/RGO was 0.3695 cm3 g−1 and 27.68 m2 g−1. The corresponding G@PI showed a pore volume of 0.6557 cm3 g−1 and a surface area of about 151.2 m2 g−1. The pore volume and specific surface area were reduced in G@PI/RGO, because graphene were stacked together by filtration to form a dense sheet structure.34–36 The reduction in surface area, volume and diameter of the pores after the introduction of graphene means that the electroactive molecular PI particles are well stored in the graphene sheets.24,25,37
O bond. It declares the successful polymerization of PI on the graphene surface in G@PI/RGO composites. X-ray photoelectron spectroscopy (XPS) can also clarify the composition of G@PI/RGO (Fig. S1a–d, ESI†). Due to the carbon atoms of the graphitic carbon, the peak CI corresponding to the C–C coordination is 284.6 eV. The peak CII at 285.3 eV corresponds to the CO double bond, and the peak CIII at 285.3 eV is associated with the C–N bond. The N1s peak shown at 399.3 eV corresponds to the N–C bond of pyridine N in PI. The O1s XPS peak corresponding to the CO bond was observed at 74.4 eV. Raman spectra of RGO, G@PI, G@PI/RGO were shown in Fig. 3c. Two peaks located near 1353 and 1591 cm−1 correspond to the disordered carbon (D-bond) and ordered graphitic carbon (G-bond), respectively. As an indicator of the degree of disorder of carbon materials, the ID/IG intensity ratio (1.11) of G@PI is slightly higher than RGO (1.07), indicating the successful attachment of PI to the graphene. Similarly, ID/IG of the G@PI/RGO was 0.97, which is lower than others, indicating that the G@PI/RGO composite combines the carbon structure in both RGO and G@PI.32,33 The G@PI and G@PI/RGO composites were investigated by thermogravimetric analysis (TGA, Fig. 3d). As could be calculated, the content of graphene and PI in G@PI is about 12% and 88%. Since the amount of GO added twice is the same, the mass content of graphene and PI in G@PI/RGO is 22% and 78%, respectively, according to the results of G@PI. The N2 adsorption/desorption isotherms and the pore size distributions of G@PI/RGO and G@PI are shown on Fig. S2 (ESI†). The pore volume and surface area of the self-assembled composite G@PI/RGO was 0.3695 cm3 g−1 and 27.68 m2 g−1. The corresponding G@PI showed a pore volume of 0.6557 cm3 g−1 and a surface area of about 151.2 m2 g−1. The pore volume and specific surface area were reduced in G@PI/RGO, because graphene were stacked together by filtration to form a dense sheet structure.34–36 The reduction in surface area, volume and diameter of the pores after the introduction of graphene means that the electroactive molecular PI particles are well stored in the graphene sheets.24,25,37
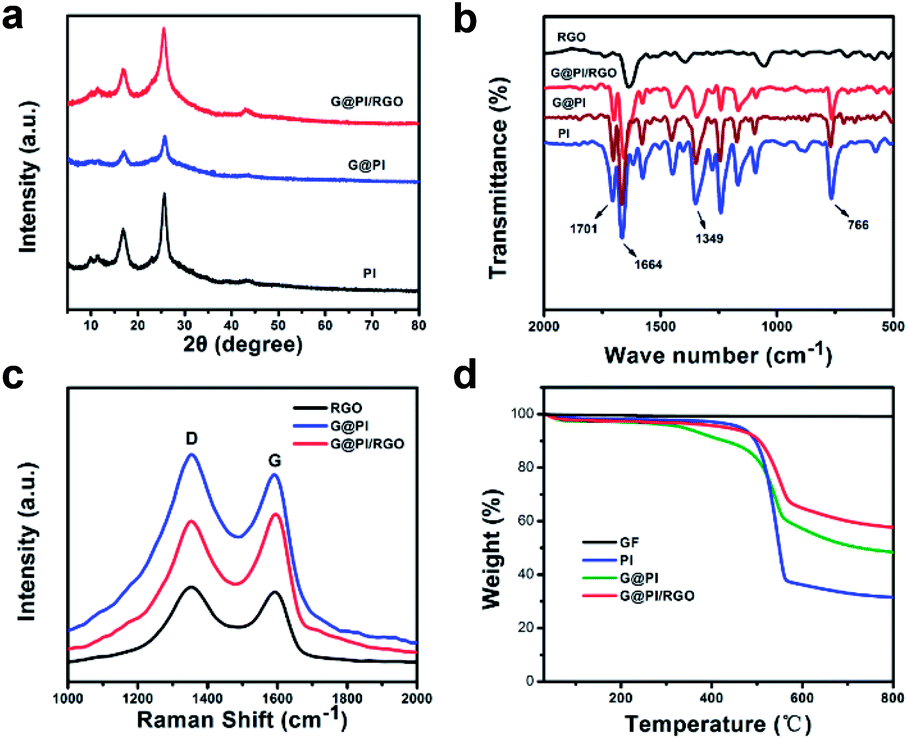 | ||
| Fig. 3 (a) XRD patterns of G@PI, G@PI/RGO and PI; (b) FTIR spectrum of the RGO, PI, G@PI and G@PI/RGO; (c) Raman of RGO, G@PI, G@PI/RGO; (d) TGA of PI, G@PI, G@PI/RGO and RGO. | ||
Due to the dense layer structure assembled by the graphene sheets and the superior mechanical stability, the prepared flexible G@PI/RGO film has a density of ∼1 mg and thickness of 50 μm, which could be used directly on the cathode electrode without any additional conditions. For comparison, G@PI electrodes were fabricated by mixing G@PI, Super P and PVDF in a weight ratio of 7:2:1 in NMP solution. The mass loading of the G@PI/RGO and G@PI electrodes were 1 mg cm−2 and 0.9 mg cm−2. In addition, the cyclic voltammograms (CV) of G@PI/RGO and G@PI were measured at a scan rate of 0.1 mV s−1 in the range of 1.5 to 3.5 V (Fig. 4a). For the G@PI/RGO film, redox peak occurred at 2.1 V, the peak was reduced to 0.4 V. For G@PI, these peaks ramped down to a more negative potential with a center voltage of 1.9 V and a widely reduced peak of 0.5 V, indicating a decrease in the polarization of the G@PI/RGO film. By measuring the CV cycle diagrams of G@PI/RGO in the 1st, 2nd, and 3rd cycles (Fig. S3†), it can be observed that the positions of the redox peaks are the same during the second and third cycles close to 2.1 V, which illustrates the reversibility of the charge and discharge process. Similarly, in the charge–discharge curves of the G@PI/RGO film and G@PI for the he 1st, 2nd, 5th, 10th, 30th and 50th cycles (Fig. 4b and c), they have the average discharge platform voltage (∼2.1 V) and charging platform voltage (2.5 V), which matched well with CV data. In the first cycle, the discharge and charge capacity of G@PI/RGO and G@PI were 193.5, 208.4 mA h g−1 and 200.6, 234.7 mA h g−1, respectively. Notably, G@PI/RGO discharge capacity can be maintained at 183.6 mA h g−1, while for G@PI composites, the capacities were sharply decreased to only 123.2 mA h g−1 after 50 cycles. The cycle performance measurement of G@PI/RGO and G@PI at 30 mA g−1 are shown in Fig. 4d. It is worth noting that after 300 cycles, the discharge capacity can be stabilized at 171.2 mA h g−1 for G@PI/RGO and the retention rate is close to 87.5%. In contrast, the G@PI complex has lower capacity stability (113.6 mA h g−1, capacity retention rate 49%) than G@PI/RGO at the same condition. The result of the difference is that the graphene sheet in G@PI/RGO can still protect the activity of PI well after multiple cycles, slowing down the rate of activity decay. The rate performance of the two electrodes were evaluated at current densities of 100, 400, 800, 1000, 2000, 4000, 6000, and 8000 mA g−1 (Fig. 4e). The G@PI/RGO flexible film could deliver an excellent revisable capacity of 135.8, 121.6, 112.8, 110.9, 101.8, 99.9, 97.6, and 86.1 mA h g−1. When the current density was restored to 100 mA g−1, a discharge capacity of 133.8 mA h g−1 remained stable in the subsequent cycle. However, the capacity of G@PI was decreased from 134.3 to 67.8, 55.6, 52.1, 40.7, 31.2, 21.7, 17.8 mA h g−1. Owing to structural features, the as-prepared G@PI/RGO flexible film exhibited more superior cycle stability and higher rate capacity than G@PI composites. It should be noted that the excellent rate performance of G@PI/RGO achieved in this work is better than most of other PI-based cathode electrodes for LIBs.20,26,38
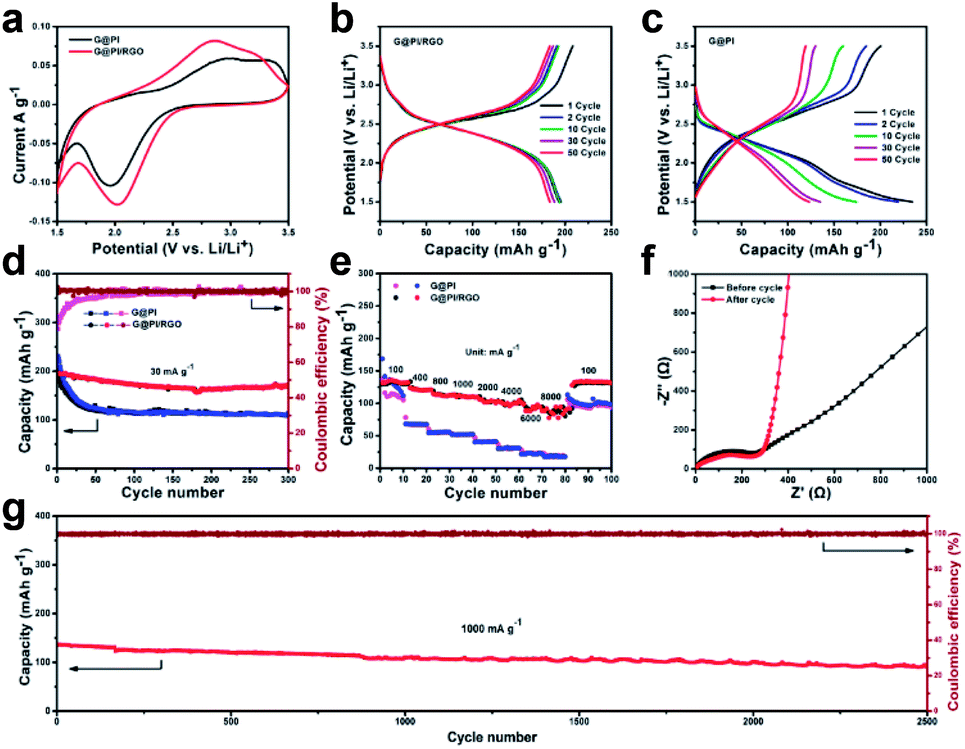 | ||
| Fig. 4 Electrochemical performances of the G@PI/RGO composites. (a) The CV curves of G@PI and G@PI/RGO at a scan rate of 0.1 mV s−1; (b) the charge–discharge profiles of G@PI/RGO at 30 mA g−1; (c) the charge–discharge profiles of G@PI at 30 mA g−1; (d) the cycling performances of the G@PI/RGO and G@PI at 30 mA g−1; (e) the rate performances of the G@PI/RGO and G@PI at various current rates; (f) Nyquist pl/ots for the G@PI/RGO electrode before and after the cycling test; (g) the cycling performance of the G@PI/RGO electrode at a current density of 1000 mA g−1. | ||
The ultra-long cycle and excellent rate performance are primarily due to the efficient integration of graphene and PI particles to form a layer film structure. The oriented layer structure should be much facilitated for the ion diffusion. The excellent performance was further confirmed by electrochemical impedance spectroscopy (EIS). According to the Nyquist diagram (Fig. S4a, ESI†), the semicircular diameter of the G@PI/RGO film is much smaller than the diameter of the G@PI composite, indicating that the G@PI/RGO film has a lower contact and charge transfer resistance.39,40 The equivalent circuit model of the studied system was also shown in Fig. S4b.†39 The solution resistance (RΩ) and charge transfer resistance (Rct) of the G@PI/RGO electrode were 6.866 and 173.2 Ω, which were obviously lower than G@PI (9.718 and 458.7 Ω) (Table S1, ESI†). Furthermore, AC impedance measurement of the G@PI/RGO before and after cycling were also performance (Fig. 4f). The RΩ and Rct of G@PI/RGO after cycling are 2.881 Ω and 45.95 Ω, respectively, indicating that the addition of sheet graphene greatly improved the electrical conductivity, which reduces the resistance of the active material after compounding. Similarly, SEM images can be observed that after 300 cycles, the PI particles in G@PI/RGO are still well coated in graphene (Fig. S5a†). However, PI particles in G@PI were significantly exposed (Fig. S5b, ESI†), indicating that RGO sheets in G@PI/RGO have a good protective effect on PI active ingredients. This result indicated that the G@PI/RGO was with the more efficient Li+ diffusion than G@PI. In addition to high capacity and excellent fixed speed capability, G@PI/RGO also exhibits good cycle stability. Even at the higher current densities of 1000 mA g−1, G@PI/RGO still provides 70% capacity retention and high reversible capacity in 2500 deep cycles (Fig. 4g). Previous PI-based LIB cathode electrodes have never experienced such exceptionally long cycle stability and excellent rate performance of G@PI/RGO.18–22,41 It is worth noting that G@PI/RGO is a binder-free electrode and the displayed capacity was calculated directly from the total weight of the electrode. According to the results above, the superior electrochemical performance of G@PI/RGO could be due to its unique flexible graphene-assist layered structure. On one hand, the PI polymer is tightly embedded in graphene based as G@PI, which can provide rapid transfer of electrons between graphene to PI particles for rapid redox reactions. On the other hand, the RGO cover on the G@PI sheet surface can further enhance the electron conductivity of the composites, thus guaranteeing of the superior rate capability and superlong stability. Benefiting from the synergetic effect between PI particles and both graphene matrix and RGO, the G@PI/RGO film with the high flexibility shows excellent rate performance and super long cycle stability.
Conclusions
In conclusion, we have developed a high-performance flexible G@PI/RGO film electrode through solvothermal in situ polymerization, followed by the vacuum filtration and thermal reduction process. The resulting G@PI/RGO was directly used as the cathode for LIB without any other current collectors and binder. The highly flexible cathode exhibits excellent rate performance with a capacity of 97.6 mA h g−1 at a high current density of 6000 mA g−1, and an ultra-long cycle life of nearly 70% capacitance retention after 2500 cycles at 1000 mA g−1. Superior lithium storage properties include excellent rate performance and long-term stability attributable to in situ polymerization of graphene sheets (G@PI) and graphene layers on the G@PI surface. We believe that the present synthetic scheme can be further applied to the design of organic electrodes for flexible LIBs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the supports from the National Natural Science Foundation of China (21707092 and 21878188), the Science and Technology Commission of Shanghai Municipality Project (18090503800), Shanghai Association for Science and Technology Achievements Transformation Alliance Program (LM201977 and LM201851), Shanghai Education Development Foundation and Shanghai Municipal Education Commission of Shuguang Program (18SG52), Shanghai Natural Science Foundation of Shanghai (Project Number 17ZR1441700 and 14ZR1440500), Shanghai Gaofeng & Gaoyuan Project for University Academic Program Development. Young Teachers Talent Development Fund of SIT (CHJJ-4), Collaborative Innovation Fund of SIT (YJ2019-20), and Young Teachers Talent Development Fund of Shanghai (ZZyyx19006).Notes and references
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167 CrossRef CAS PubMed.
- Y. Tang, Y. Zhang, W. Li, B. Ma and X. Chen, Chem. Soc. Rev., 2015, 44, 5926 RSC.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695 CrossRef CAS PubMed.
- M. G. Kim and J. Cho, Adv. Funct. Mater., 2009, 19, 1497 CrossRef CAS.
- L. Nie, Y. Zhang, W.-W. Xiong, T.-T. Lim, R. Xu, Q. Yan and Q. Zhang, Inorg. Chem. Front., 2016, 3, 111 RSC.
- J. Bai, X. Li, G. Liu, Y. Qian and S. Xiong, Adv. Funct. Mater., 2014, 24, 3012 CrossRef CAS.
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribiere, P. Poizot and J. M. Tarascon, Nat. Mater., 2009, 8, 120 CrossRef CAS.
- N. Wang, D. Hou, Q. Li, P. Zhang, H. Wei and Y. Mai, ACS Appl. Energy Mater., 2019, 2, 5816–5823 CrossRef CAS.
- J. Xie and Q. Zhang, J. Mater. Chem. A, 2016, 4, 7091 RSC.
- X. Han, C. Chang, L. Yuan, T. Sun and J. Sun, Adv. Mater., 2007, 19, 1616 CrossRef CAS.
- C. Guo, M. Wang, T. Chen, X. Lou and C. Li, Adv. Energy Mater., 2011, 1, 736 CrossRef CAS.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742 CrossRef CAS.
- L. Qie, W. Chen, Z. Wang, Q. Shao, X. Li, L. Yuan, X. Hu, W. Zhang and Y. Huang, Adv. Mater., 2012, 24, 2047 CrossRef.
- L. He, J.-H. Cao, T. Liang and D.-Y. Wu, Electrochim. Acta, 2020, 337, 135838 CrossRef CAS.
- J. Wu, X. Rui, G. Long, W. Chen, Q. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7354 CrossRef CAS.
- Z. Song, H. Zhan and Y. Zhou, Angew. Chem., Int. Ed., 2010, 49, 8444 CrossRef CAS.
- H. Wu, K. Wang, Y. Meng, K. Lu and Z. Wei, J. Mater. Chem. A, 2013, 1, 6366 RSC.
- N. Wang, H. Tian, S.-Y. Zhu, D.-Y. Yan and Y.-Y. Mai, Chin. J. Polym. Sci., 2017, 36, 266–272 CrossRef.
- Y. Meng, H. Wu, Y. Zhang and Z. Wei, J. Mater. Chem. A, 2014, 2, 10842–10846 RSC.
- G. Zhang, Z. Xu, P. Liu, Y. Su, T. Huang, R. Liu, X. Xi and D. Wu, Electrochim. Acta, 2018, 260, 598 CrossRef CAS.
- Z. Song, T. Xu, M. L. Gordin, Y. B. Jiang, I. Bae, Q. Xiao, H. Zhan, J. Liu and D. Wang, Nano Lett., 2012, 12, 2205 CrossRef CAS PubMed.
- L. Ma, D. Lu, P. Yang, X. Xi, R. Liu and D. Wu, Electrochim. Acta, 2019, 319, 201–209 CrossRef CAS.
- Y. Su, Y. Liu, P. Liu, D. Wu, X. Zhuang, F. Zhang and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 1812 CrossRef CAS PubMed.
- K. Amin, Q. Meng, A. Ahmad, M. Cheng, M. Zhang, L. J. Mao, K. Lu and Z. Wei, Adv. Mater., 2018, 30, 1703868 CrossRef.
- Q. Cheng, M. Wu, M. Li, L. Jiang and Z. Tang, Angew. Chem., Int. Ed., 2013, 52, 3750 CrossRef CAS.
- N. Li, Z. Chen, W. Ren, F. Li and H.-M. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17360 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- X. Yang, J. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833 CrossRef CAS PubMed.
- Z. Chen, R. Wu, M. Liu, H. Wang, H. Xu, Y. Guo, Y. Song, F. Fang, X. Yu and D. Sun, Adv. Funct. Mater., 2017, 27, 1702046 CrossRef.
- D. Wang, F. Li, J. Zhao, W. Ren, Z. Chen, J. Tan, Z. Wu, I. Gentle, G. Lu and H. Cheng, ACS Nano, 2009, 3, 1745 CrossRef CAS PubMed.
- Y. Yang, C. Wang, B. Yue, S. Gambhir, C. O. Too and G. G. Wallace, Adv. Energy Mater., 2012, 2, 266 CrossRef CAS.
- S. Yoon, S. Lee, S. Kim, K.-W. Park, D. Cho and Y. Jeong, J. Power Sources, 2015, 279, 495 CrossRef CAS.
- H. Zhao, J. Ren, X. He, J. Li, C. Jiang and C. Wan, Solid State Sci., 2008, 10, 612 CrossRef CAS.
- S. Cao, X. Feng, Y. Song, H. Liu, M. Miao, J. Fang and L. Shi, ACS Appl. Mater. Interfaces, 2016, 8, 1073 CrossRef CAS PubMed.
- Y. Xie, Y. Chen, L. Liu, P. Tao, M. Fan, N. Xu, X. Shen and C. Yan, Adv. Mater., 2017, 29, 1702268 CrossRef PubMed.
- A. Ahmad, H. P. Wu, Y. F. Guo, Q. H. Meng, Y. N. Meng, K. Lu, L. W. Liu and Z. X. Wei, RSC Adv., 2016, 6, 33287 RSC.
- A. Li, Z. Feng, Y. Sun, L. Shang and L. Xu, J. Power Sources, 2017, 343, 424 CrossRef CAS.
- S. Wang, Q. Wang, P. Shao, Y. Han, X. Gao, L. Ma, S. Yuan, X. Ma, J. Zhou, X. Feng and B. Wang, J. Am. Chem. Soc., 2017, 139, 4258 CrossRef CAS PubMed.
- Y. Liang, P. Zhang, S. Yang, Z. Tao and J. Chen, Adv. Energy Mater., 2013, 3, 600 CrossRef CAS.
- Y. Huang, K. Li, J. Liu, X. Zhong, X. Duan, I. Shakir and Y. Xu, J. Mater. Chem. A, 2017, 5, 2710 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00884b |
| This journal is © The Royal Society of Chemistry 2020 |