DOI:
10.1039/D0RA00857E
(Paper)
RSC Adv., 2020,
10, 10789-10798
Dual-mode response behavior of a graphene oxide implanted energetic system under different thermal stimuli
Received
29th January 2020
, Accepted 27th February 2020
First published on 16th March 2020
Abstract
GO, produced by the Hummers' method and characterized by scanning electron microscopy (SEM), elemental analysis (EA), Fourier-transform infrared spectroscopy (FT-IR), Fourier-transform infrared nanospectroscopy (nano FT-IR), X-ray photoelectron spectroscopy (XPS), Raman spectroscopy, and simultaneous thermal analysis combined with mass spectrometry (TG-DSC-MS), was appended to boron/potassium nitrate (B/KNO3) in different proportions, to regulate the response of B/KNO3 to thermal stimuli. The addition of GO delayed the onset temperature of the reaction between B and KNO3, and brought the second reaction stage forward, however, it did not change the reaction mechanism. The integral model functions, which were in good agreement with the values calculated using the Kissinger and Ozawa method, took the form of Jander equations for three-dimensional diffusion processes. Results showing the sensitivity to flame testing demonstrated that the higher the GO content, the more insensitive the system was to temperature, which was consistent with the conclusion of the previous thermal analysis on the onset temperature of the reaction between B and KNO3. In a closed-vessel test, as the GO content increased, the pressure peak and maximum slopes of pressure–time curves increased. Under a thermal stimulus, GO was reduced to RGO, and when the stimulation was small and slow, this helped with heat dissipation and improved safety. If the stimulation was enough to ignite the energetic materials, GO contributed to the rapid attainment of the reaction temperature and sped up the reaction process.
Introduction
Special energetic devices, such as the actuators, launchers, thrusters and igniters that are applied in aerospace, biomedicine and other fields, require high energy densities, fast rates of energy release, great stability, and high safety.1–3 Energetic devices must be designed to solve the contradictory requirements of safety and reliability for the energetic materials that they contain. It is necessary for energetic devices and energetic materials to be safely assembled, stored, and transported, and thus sensitivity testing applies a controlled level of energy to the energetic materials to investigate their sensitivity to stimuli, such as friction, impact, spark and heat. Meanwhile, an energetic device must be capable of delivering a reliable function when an ignition stimulus is provided, and must produce the desired effect, for instance, high temperature, heat, pressure or gaseous products.4 An energetic device must prevent misoperation at low energy and slow loading, while it must respond speedily with high stimulation. Thermal stimuli are common during storage and use, since electrical, mechanical and optical stimulation may influence the temperature of devices or materials. Thus, it is significant to consider how devices and materials respond to thermal stimuli and how to ameliorate these responses.
Graphene has been prominent since it was discovered owing to its outstanding thermal conductivity. Theoretical research has shown that room-temperature thermal conductivity decays monotonically with the number of layers in few-layer graphene,5,6 and defects and doping in graphene affect the thermal conductivity.7,8 In experiments at room temperature, the value of the thermal conductivity of a suspended single-layer graphene could reach 5300 W m−1 K−1,9 while the thermal conductivity of tri-layer graphene produced by a suspended microelectrothermal system was ∼1400 W m−1 K−1 at 300 K.10 Even the thermal conductivity of graphene laminate films on polyethylene terephthalate were in the range from 40 to 90 W m−1 K−1 at room temperature.11 Incorporating graphene into other material systems, may lead the thermal conductivity to advance significantly. After adding graphene which was made from the sheets of graphene oxide in a hydrothermal reduction reaction, the thermal conductivity of the resultant phase change material consisting of docosane and spongy graphene increased to 0.59 W m−1 K−1 from 0.26 W m−1 K−1 (for pure docosane) in the presence of ∼3 mg cm−3 spongy graphene.12 The thermal conductivity of a polycarbonate–graphene nanocomposite increased by more than two fold due to the addition of 5 wt% graphene.13
However, few-layer flawless graphene sheets are rarely achievable.14 In general, graphene is obtained from graphite through an oxidation–exfoliation–reduction procedure with an intermediate product named graphene oxide (GO for short) which contains abundant oxygen-rich functional groups.15 The oxygen functional groups cause the decrease of thermal conductivity of GO.16 After reduction, the reduced graphene oxide (RGO for short) has thermal conductivity of ∼900 W m−1 K−1.17 Consequently, GO offers more engineering applications than graphene to ameliorate thermal conductivity and other features of other materials.18,19 The introduction of GO into an energetic system is expected to improve the thermal response characteristics of the whole system.
Currently, graphite-like or graphene-like materials are numerously employed to enhance the safety of sensitive energetic materials,20–22 giving low energy and slow loading rates. In this work, different thermal stimuli at different loading rates were considered. GO was produced and used as an additive in boron/potassium nitrate mixtures (B/KNO3 for short) which are used extensively as propellant igniters and gas generators.23,24 Several different percentages of GO were appended to B/KNO3, and the thermal decomposition, critical ignition distance and pressure–time curves for each sample were used to analyse the changes achieved under diverse thermal stimuli.
Experimental
Materials
Flake graphite (D90 = 40 μm, 99.99% purity) was purchased from Nanjing Xianfeng Nano Technology Co., Ltd. Concentrated sulfuric acid (H2SO4, 98 wt%), potassium permanganate (KMnO4), sodium nitrate (NaNO3), hydrogen peroxide (H2O2, 30 wt%), concentrated hydrochloric acid (HCl, 37 wt%), barium chloride (BaCl2, used in solution at 1 mg mL−1 to check for SO42− in the GO products after washing) and potassium nitrate (KNO3, >99% purity, grain size < 5 μm) were provided by Beijing Tongguang Fine Chemicals Company. Deionized water was made in our own laboratory. Boron powder (average particle size 25 μm, 95% purity) was purchased from Baoding Pengda New Materials Technology Co., Ltd. Viton (Fluororubber, FE2601) was provided by Shanghai 3F New Materials Technology Co., Ltd.
Preparation of samples
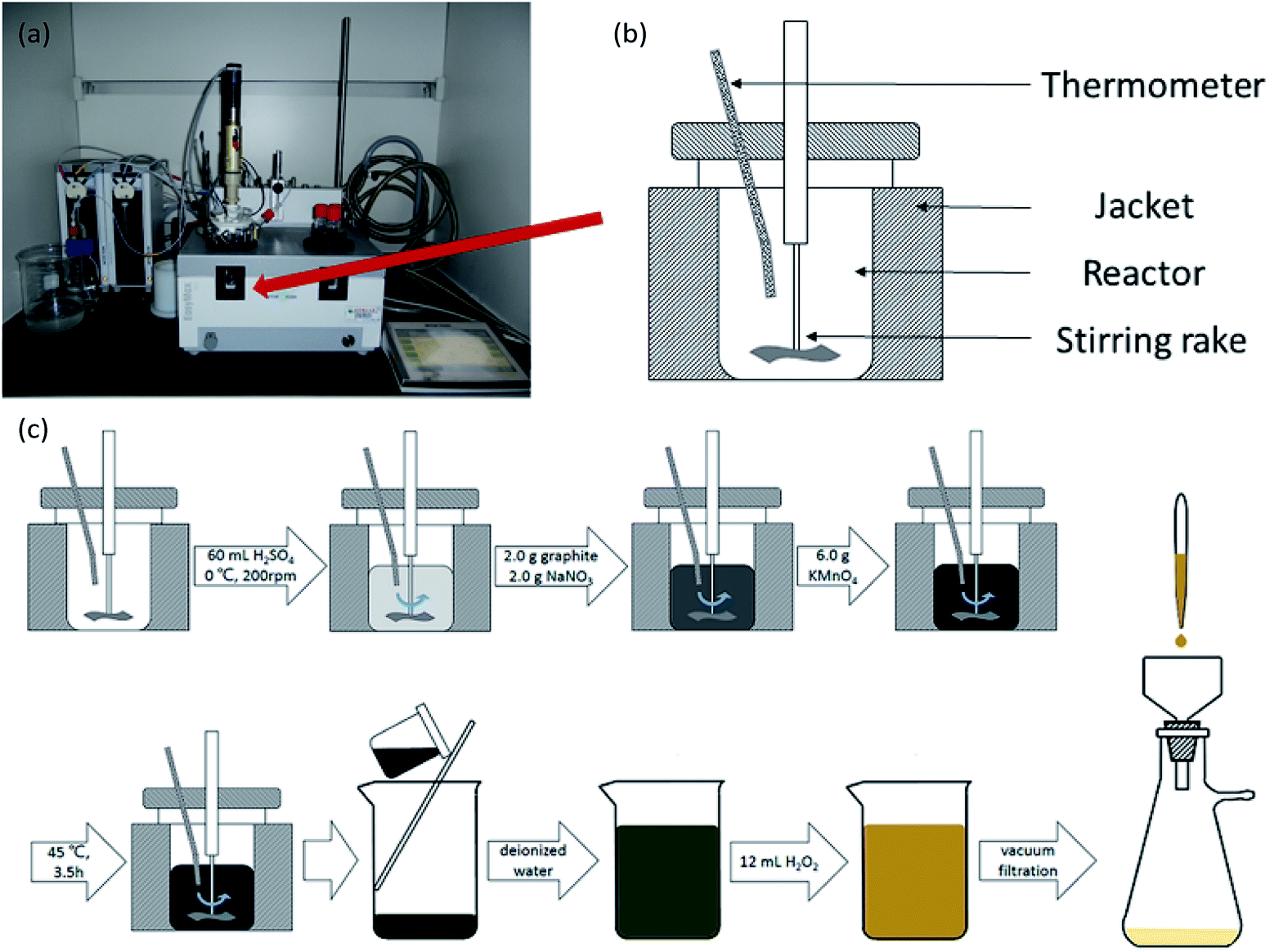
GO was produced using a modified Hummers' method.25 The oxidation took place in a Mettler-Toledo EasyMax 102 reactor, and the temperature and rotation rate were controlled by software (as shown in Fig. 1(a) and (b)). Under conditions of 0 °C and 200 rpm, 60 mL of H2SO4, 2.0 g of graphite, 2.0 g of NaNO3 and 6.0 g of KMnO4 were mixed in the reactor. Then, the temperature in the reactor was raised to 45 °C for 3.5 h. After the reaction, the mixture was poured into a 2000 mL glass beaker and diluted with deionized water. Lastly, 12 mL of 30 wt% H2O2, was added and the color of the solution turned light yellow indicating successful oxidation. The solution was filtered, and the graphite oxide product was washed and dried. By means of ultrasonication and centrifugation, graphite oxide was transformed to GO. The process of oxidation is shown in Fig. 1(c).
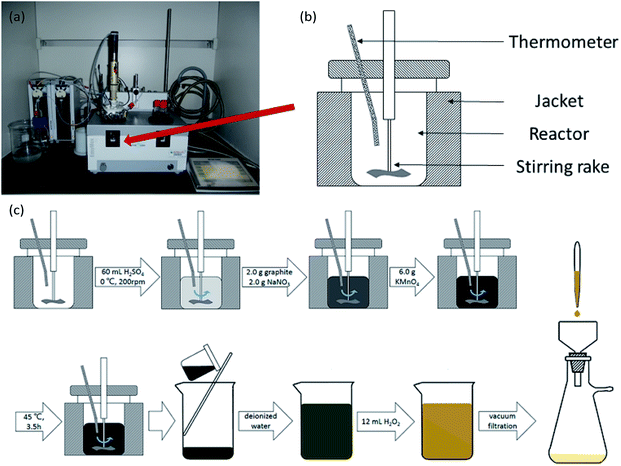 |
| | Fig. 1 (a) A physical picture of a Mettler-Toledo EasyMax 102 reactor. (b) A schematic drawing of the reactor. (c) The process of oxidation of graphite. | |
As previously reported, B/KNO3 is composed of boron powder (B, 26.60 wt%), potassium nitrate (KNO3, 67.40 wt%) and Viton (6.00 wt%).26 GO (1.00 wt%, 3.00 wt% and 5.00 wt%) were appended to B/KNO3 (see Table 1; data for B/KNO3 were adjusted to sum to 100 percent in each formula).
Table 1 Compositions of B/KNO3 samples
| Sample |
Amount of boron/wt% |
Amount of KNO3/wt% |
Amount of Viton/wt% |
Amount of GO/wt% |
| S1 |
26.60 |
67.40 |
6.00 |
0.00 |
| S2 |
26.34 |
66.73 |
5.94 |
0.99 |
| S3 |
25.82 |
65.44 |
5.83 |
2.91 |
| S4 |
25.33 |
64.19 |
5.72 |
4.76 |
After thermal stimulation, GO changes to RGO.27 In order to compare the characteristics of the samples, we used a crude device formed by two Petri dishes to thermally reduce GO simply and efficiently. The larger dish was inverted to cover the smaller one containing the samples. The device was heated to 180 °C rapidly. After several minutes, the samples expanded and the colour turned to black.
Characterizations
Microphotographs were taken using a scanning electron microscope (SEM, Hitachi) to observe the morphology of graphene oxide flakes and B/KNO3. To analyse the distributions of the elements and functional groups, a range of techniques were used, namely elemental analysis (EA, Elementar, Vario EL cube, Germany), Fourier-transform infrared spectroscopy (FT-IR, Bruker Optics, Vertex 70, Germany), X-ray photoelectron spectroscopy (XPS, Thermo Fisher Scientific, Thermo ESCALAB 250Xi, US), Raman spectroscopy (Renishaw plc, Renishaw inVia, UK) and Fourier-transform infrared nanospectroscopy (nano FT-IR based on a scattering-type scanning near-field optical microscope s-SNOM; Neaspec GmbH, Germany).
Thermal decomposition
A simultaneous thermal analyzer (TG-DSC, STA 449 F3 Jupiter®, NETZSCH, Germany) was used to analyze the thermal decomposition of the GO and B/KNO3 samples. GO was tested from room temperature to 300 °C under an argon atmosphere at a heating rate of 10 °C min−1 combined with mass spectrometry (MS, QMS 403 Aëolos® Quadro, NETZSCH, Germany), and at a heating rate of 40 °C min−1. RGO was tested at a heating rate of 10 °C min−1 from room temperature to 300 °C under an argon atmosphere. Samples S1 to S4 were tested at heating rates of 5, 10, 15 and 20 °C min−1 from room temperature to 600 °C under an air atmosphere. Each sample mass was approximately 2.0 mg.
Critical ignition distances
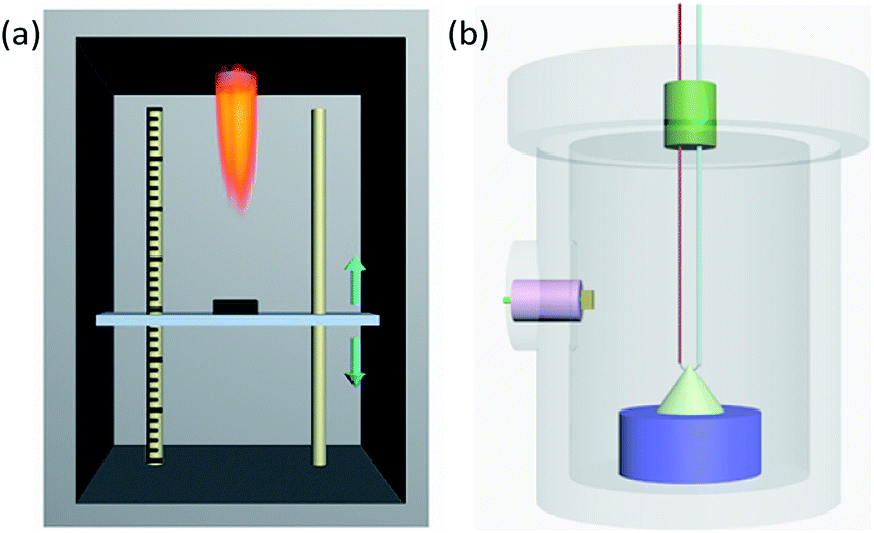
Sensitivity to flame testing reflects the difficulty of ignition of energetic materials under the action of a flame and is evaluated from the critical ignition distance,28 the distance at which 50% of a sample can be ignited. Energetic materials with a short critical ignition distance have high safety. As shown in Fig. 2(a), the test is conducted in a protective box, with a movable vertical pallet on which the sample mold is placed at a readable distance to the ignition source of the testing device. When the ignition source is on fire, the sample is in a temperature field of thermal radiation which simulates a small external energy stimulus and slow loading speed. In detail, 20.0 ± 5.0 mg samples were weighed and loaded into a vessel at 58.8 MPa pressure. Then, the vessel was placed in the mold and the sample was ignited. According to the up-down method,29 the height which gave 50% ignition (H50) and the standard deviation were calculated after the test was repeated 30 times.
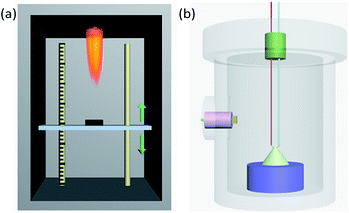 |
| | Fig. 2 (a) Diagrammatic sketch of the device used for flame sensitivity testing. (b) Schematic perspective drawing of the closed vessel used for the testing of burning performance. | |
Pressure–time curves
Pressure–time curves produced from a closed-vessel test are usually used to study the burning performance of energetic materials.30 The closed-vessel test is conducted in a closed vessel of known capacity with good impact resistance, as shown in Fig. 2(b). The sample is ignited by an electric wire buried in the vessel, and the pressure signal is collected by a pressure sensor plug in the side of the vessel. The pressure–time curve is simultaneously recorded on an oscilloscope. The temperature of the electric wire is very high and is increased rapidly. In this work, we used a closed vessel with 100 mL volume and 1.0 ± 0.05 g powder for each test.
Results and discussion
Thermal decomposition of graphene oxide
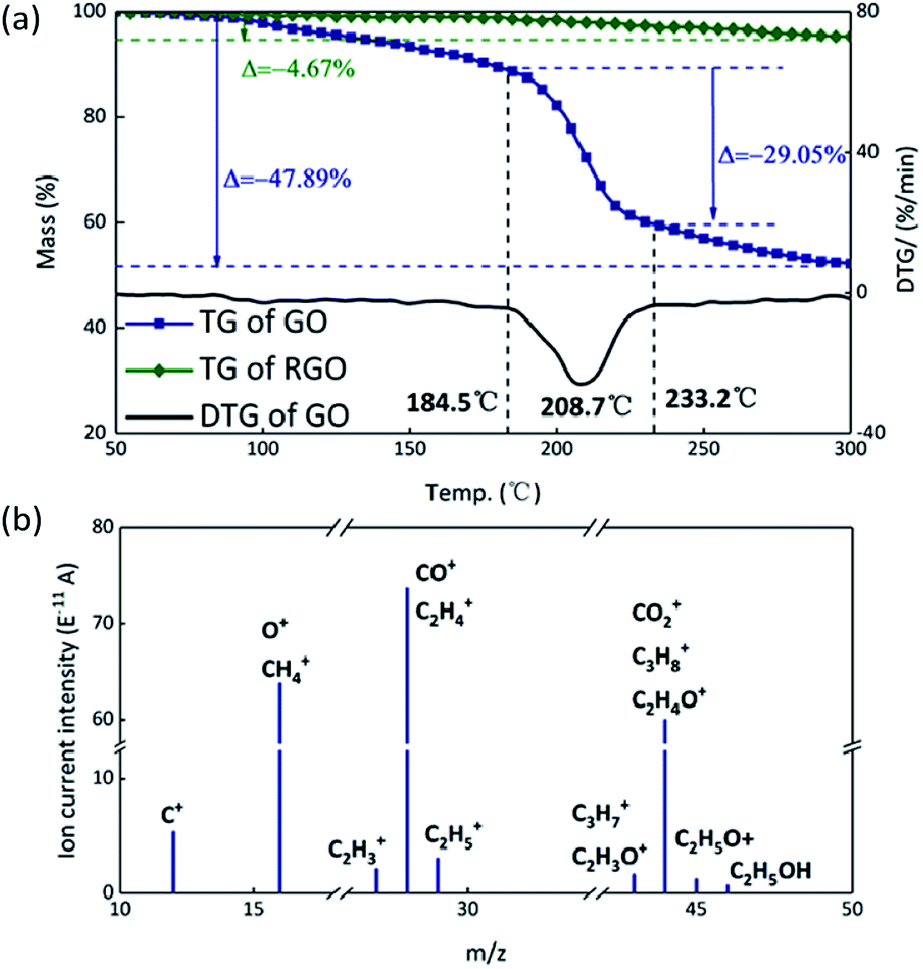
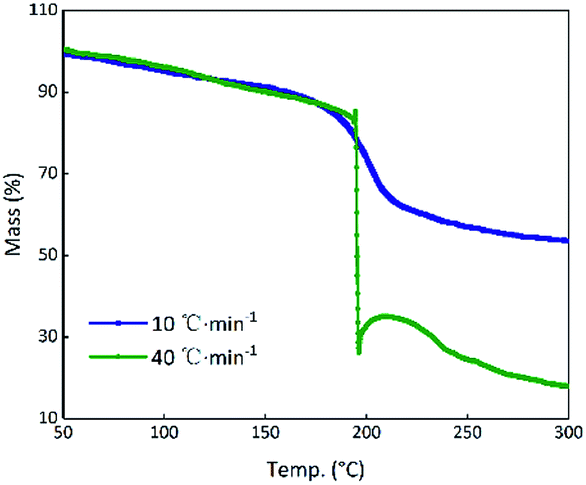
Theoretically, GO is enriched with a large number of functional groups such as hydroxyl, carboxyl and epoxy groups on the graphene sheet skeleton, and these functional groups are stripped from the skeleton and form several gaseous products after they are subjected to certain thermal stimuli.31 The GO and RGO were decomposed under an argon atmosphere at a heating rate of 10 °C min−1. It can be seen from Fig. 3(a) that a weight loss of GO corresponding to about 29% started at 184.5 °C and stopped at 233.2 °C, whereas the RGO was practically thermally stable.32 The main possible gaseous products of GO are O+, CO, CO2 and CxHy as seen from the peak values of the MS curves in the vicinity of 208.7 °C (Fig. 3(b)).33 In order to compare the change of state of GO at the higher thermal stimulus loading speed, the same mass of GO was tested at a heating rate of 40 °C min−1 under an argon atmosphere. The result is shown in Fig. 4 in comparison to the curve for the heating rate of 10 °C min−1. At a rate of 40 °C min−1, GO decomposed rapidly and the huge impact disturbed the balance of the thermal analyzer, leading to an anomalous TG curve. This appearance indicates that, at a low thermal stimulus loading rate, GO responds slowly and gently, while at a fast loading rate, the GO responds reliably and rapidly.
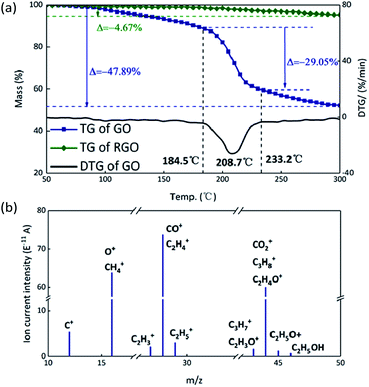 |
| | Fig. 3 (a) TG curves of GO and RGO. (b) MS spectrum of GO; the peaks are assigned to the possible gaseous products formed during decomposition. | |
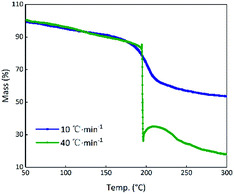 |
| | Fig. 4 TG curves of GO at heating rates of 10 °C min−1 and 40 °C min−1. | |
Characterization of graphene oxide
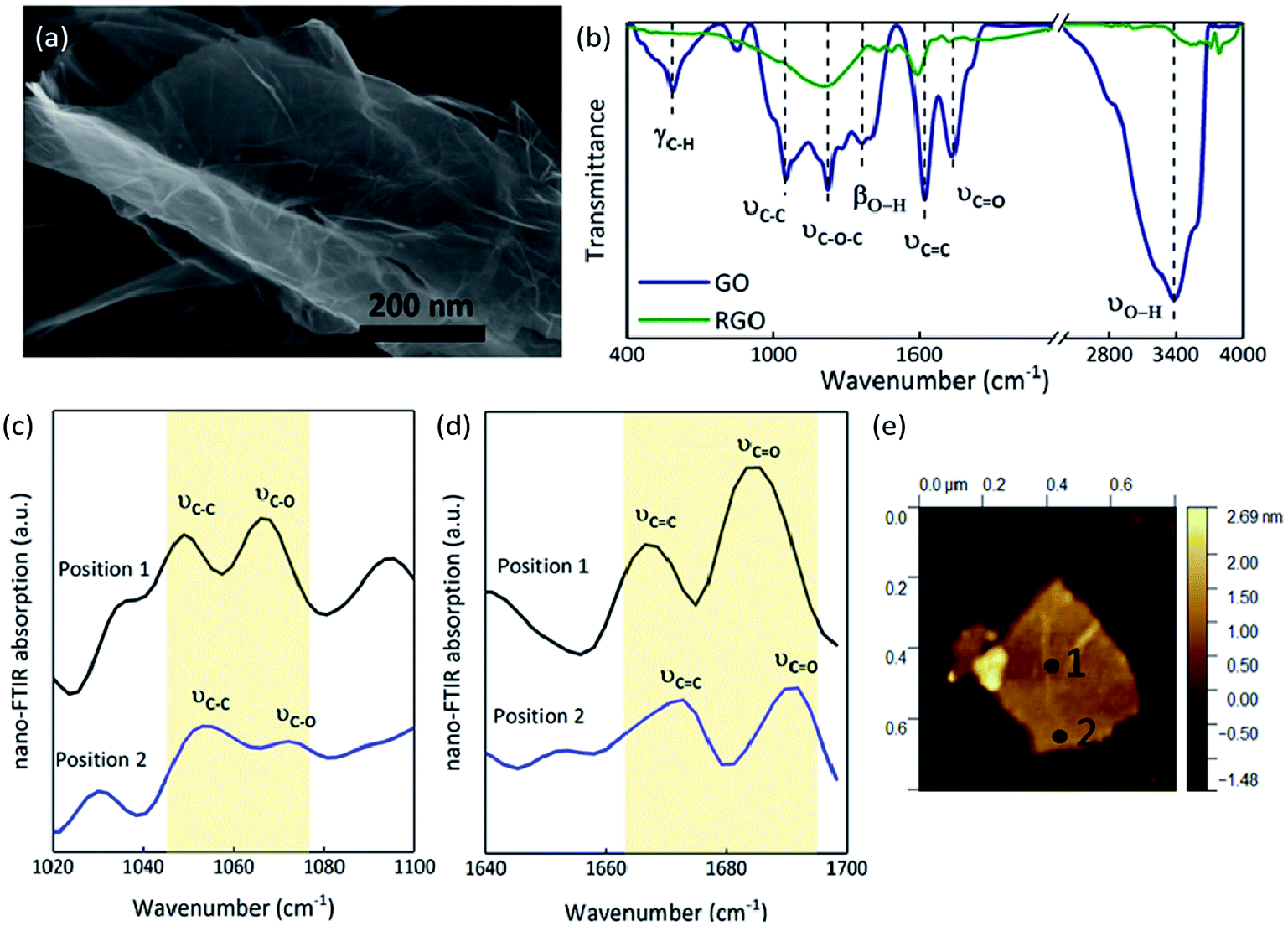
The SEM image of GO shows a 2D nanosheet morphology, in which the GO sheet is folded on the plate and is curling at its edge (Fig. 5(a)).34 EA analysis determined that the GO sample consists of 46.78% carbon, 50.43% oxygen and 2.79% hydrogen (molecular formula C3.90H2.79O3.15), while the RGO contains 86.40% carbon, 13.28% oxygen and 0.32% hydrogen (molecular formula C7.20H0.83O0.32). As we know, GO sheets have a large number of functional groups attached on their surfaces and edges, which are an important source of the gaseous products released after thermal stimulation. The FT-IR spectrum of GO (Fig. 5(b)) illustrates the presence of O–H (at 3377 and 1369 cm−1), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (at 1736 cm−1), CC (at 1622 cm−1), C–O–C (at 1227 cm−1), C–C (at 1055 cm−1), and C–H (at 586 cm−1); after thermal reduction, the transmittances are all weaker, meaning that the oxygen functional groups have been released and remain only in minute quantities.35–38 The FT-IR spectra confirmed that there are many oxygen functional groups on GO films, but that there are differences in the distribution of these groups between the center and edges of the sheets. The absorption at two positions on a GO film were contrasted through nano FT-IR. The absorption at the middle of the GO film (position 1, Fig. 5(e)) is stronger than that at the edge (position 2, Fig. 5(e)). The C–O stretching vibration around 1070 cm−1 is weak at the edge while the CO stretching vibrations at around 1690 cm−1 are strong in both positions (Fig. 5(c) and (d)).39 The C–C skeleton vibration at around 1050 cm−1 and the CC stretching vibration at around 1670 cm−1 at the middle of GO film are stronger than those at the edge (Fig. 5(c) and (d)). The blue shift seen in position 2 with respect to position 1 can be attributed to the higher sp3/sp2 character of the carbon at the edge than in the plane.40 Thus, there may be certain advantages when a thermal stimulus passes through the plane of a GO film.
O (at 1736 cm−1), CC (at 1622 cm−1), C–O–C (at 1227 cm−1), C–C (at 1055 cm−1), and C–H (at 586 cm−1); after thermal reduction, the transmittances are all weaker, meaning that the oxygen functional groups have been released and remain only in minute quantities.35–38 The FT-IR spectra confirmed that there are many oxygen functional groups on GO films, but that there are differences in the distribution of these groups between the center and edges of the sheets. The absorption at two positions on a GO film were contrasted through nano FT-IR. The absorption at the middle of the GO film (position 1, Fig. 5(e)) is stronger than that at the edge (position 2, Fig. 5(e)). The C–O stretching vibration around 1070 cm−1 is weak at the edge while the CO stretching vibrations at around 1690 cm−1 are strong in both positions (Fig. 5(c) and (d)).39 The C–C skeleton vibration at around 1050 cm−1 and the CC stretching vibration at around 1670 cm−1 at the middle of GO film are stronger than those at the edge (Fig. 5(c) and (d)). The blue shift seen in position 2 with respect to position 1 can be attributed to the higher sp3/sp2 character of the carbon at the edge than in the plane.40 Thus, there may be certain advantages when a thermal stimulus passes through the plane of a GO film.
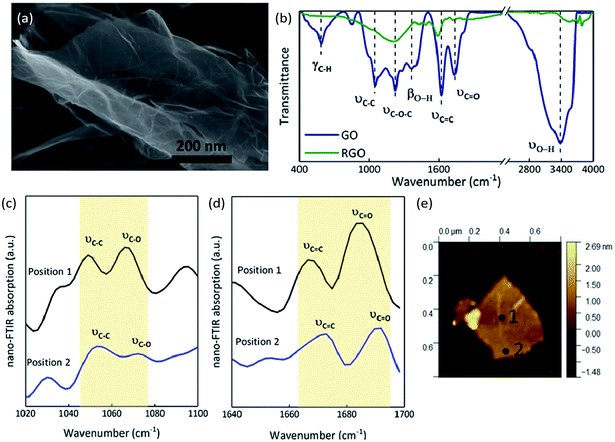 |
| | Fig. 5 Morphology of GO and the nano FT-IR curves of GO compared to the FT-IR curves: (a) SEM image of GO. (b) FT-IR transmittance spectra of GO and RGO. (c) Nano FT-IR absorption from 1020 cm−1 to 1100 cm−1. (d) Nano FT-IR absorption from 1640 cm−1 to 1700 cm−1. (e) AFM image showing the positions at which the spectra in parts (c and d) were recorded. | |
XPS and Raman spectroscopy were used to analyse and compare the surface valence bond states of GO and RGO. The C 1s XPS spectra of GO and RGO are shown in Fig. 6 after peak fitting. The binding energies of sp2 and sp3 carbon are assigned at 284.5 and 285.3 eV, and binding energies of 286.3, 287.1, 288.2 and 289.4 eV are assigned to the C–OH, C–O–C, CO and HO–CO functional groups, respectively.41,42 The proportions of each functional group are shown in Table 2 for GO and RGO. After thermal reduction, the content of oxygen-containing functional groups is reduced, with C–O–C, CO and HO–CO removed in particular. On the other hand, the decrease in the ratio of sp3 carbon to sp2 carbon reveals that defects are reduced.
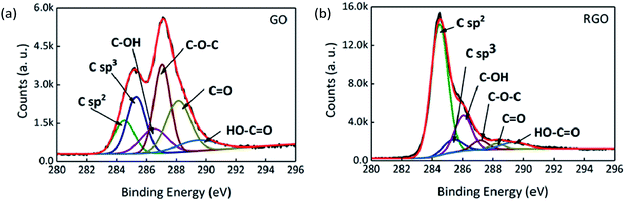 |
| | Fig. 6 C 1s XPS spectra of (a) GO and (b) RGO. | |
Table 2 The proportions of functional groups determined by C 1s XPS
| Functional groups |
C sp2 |
C sp3 |
C–OH |
C–O–C |
CO |
HO–CO |
| Binding energies/eV |
284.5 |
285.3 ± 0.1 |
286.3 ± 0.2 |
287.1 ± 0.1 |
288.2 ± 0.2 |
289.4 ± 0.1 |
| GO |
11.89% |
19.33% |
11.84% |
26.43% |
22.85% |
7.66% |
| RGO |
64.09% |
6.93% |
17.44% |
4.20% |
2.32% |
5.02% |
Raman spectroscopy can indicate the presence and level of impurities in carbon hybridization;43 the D-band is associated with the disorders or defects in graphitic structures and the G-band is attributed to the presence of graphitic carbon.44,45 As shown in Fig. 7, the relative intensity ID/IG ratios (where ID and IG are the D-band and G-band Raman intensities46) of the D band (∼1329 cm−1) and G band (∼1584 cm−1) for GO and RGO are 1.37 and 0.96, implying that the proportion of defects in RGO is less than that in GO.
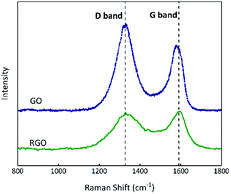 |
| | Fig. 7 Raman spectra of GO and RGO. | |
In summary, a large number of hydrophilic oxygen-containing functional groups such as hydroxyl groups and carboxyl groups are suspended at the plane and edges of GO, and intermolecular hydrogen bonding forces cause the lamellar edges to curl, resulting in the appearance seen in the SEM image (Fig. 5(a)).47 Under thermal stimulation, GO is reduced to RGO releasing gaseous products, and the proportion of defects is decreased, which could cause the improvement in thermal conductivity.
Effect of GO on B/KNO3 at slow heating rates
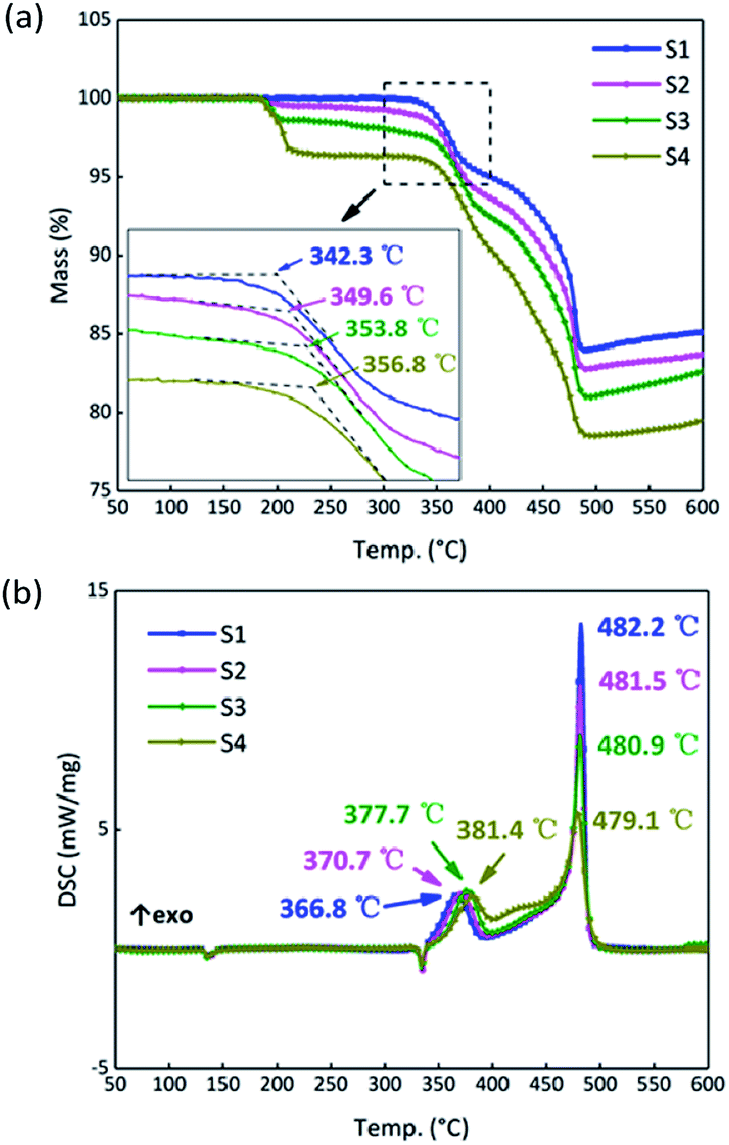
To analyze the effects of GO on the thermal properties of B/KNO3, the thermal decomposition processes were considered. Samples S1–S4 were monitored at a heating rate of 5 °C min−1 under an air atmosphere to simulate the application environment. As a whole, the curve trends were basically the same with two endothermic peaks and one two-stage exothermic peak. The curves of S1 in Fig. 8 are given as an example for detailed analysis of the processes: ① the endothermic peak at around 135 °C corresponds to the crystal transition of KNO3 (from an orthorhombic structure to a trigonal structure); ② the endothermic peak at around 327 °C corresponds to the melting of KNO3; ③ the exothermic peaks from 350 °C to 490 °C correspond to the two-step reaction of B and KNO3; the temperature of the maximum exothermic peak is 481 °C.48
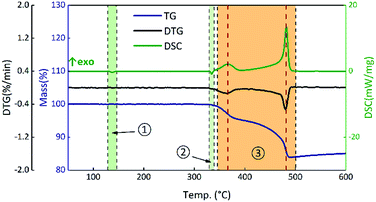 |
| | Fig. 8 TG-DSC curves of S1 at a heating rate of 5 °C min−1. | |
The results for S1–S4 are shown in Fig. 9. The TG onset temperatures and DSC peak temperatures were calculated using NETZSCH analysis software. Notably, the onset temperature in the TG curves is delayed after the addition of GO, and the two peaks in period ③ of the DSC curves tend to become closer as the GO content increases. Samples S2–S4 have different degrees of weight loss at around 200 °C due to the thermal reduction of GO. After that, GO turns to RGO and only a carbon skeleton remains. The carbon skeleton shows different responses to different thermal stimuli. During a slow heating process, the existence of the carbon skeleton allows the whole system to dissipate heat quickly and uniformly at an approximately steady state temperature equilibrium, and it is hard to accumulate heat, which delays the initiation of the reactions between B and KNO3. However, when the reactions begin, the carbon skeleton is propitious in helping the samples to reach the required reaction temperature and this accelerates the reactions, reducing the peak temperature of the second stage.
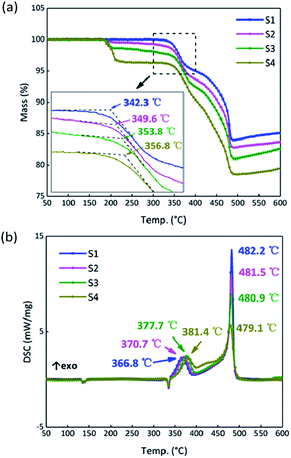 |
| | Fig. 9 (a) TG curves of samples S1–S4 at a heating rate of 5 °C min−1. (b) DSC curves of S1–S4 at a heating rate of 5 °C min−1. | |
Reaction kinetics of B/KNO3 with appended GO at slow heating rates
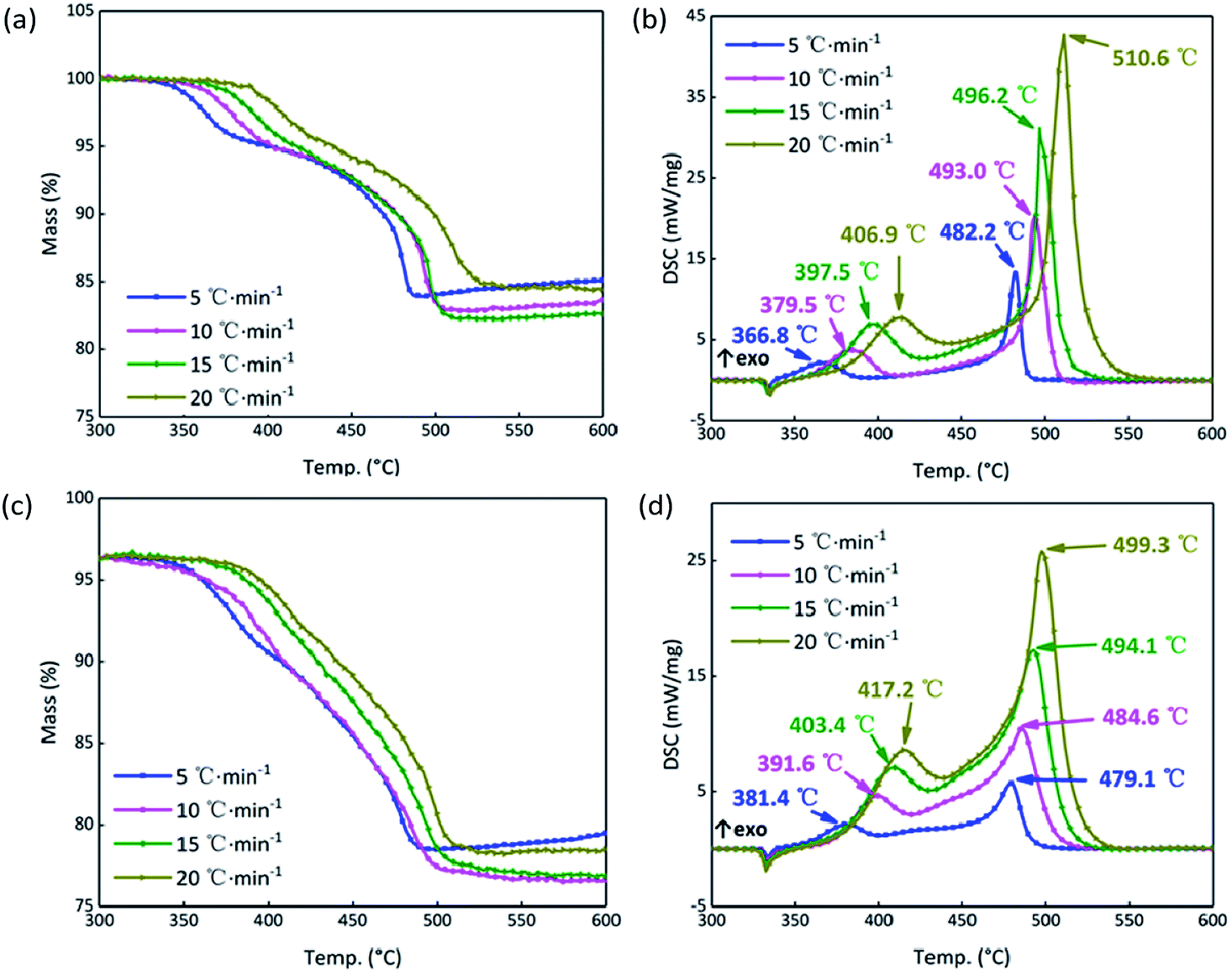
In order to further analyze the response mechanism of the B/KNO3 composited with GO in the continuous heating system at high temperature, the thermal decomposition integral model functions were fitted using non-isothermal chemical reaction dynamics. To highlight the contrasts, the system containing 5% GO (S4) was compared with the system without GO (S1). The TG and DSC curves of S1 and S4 at the heating rates of 5, 10, 15 and 20 °C min−1 in Fig. 10 were analyzed using the Kissinger and Ozawa method to obtain the kinetic parameters shown in Table 3, and forty-one types of kinetic model functions and α–T (α–T data is the conversion degree (α) varies with temperature (T)) data were calculated.49 The apparent activation energy Ea (EK by the Kissinger method and EO by the Flynn–Wall–Ozawa method) increased for the first stage of the reaction and fell for the second stage, meaning that the first stage of the reaction was more difficult to start and that the second stage proceeded more easily, which matches the results shown in Fig. 9. Because the samples were prepared by prilling, the integral model functions (Gα), which are consistent with the values in Table 3, all take the form of Jander equations for three-dimensional diffusion processes (see Table 4) and were calculated using the Satava–Sestak method.50,51
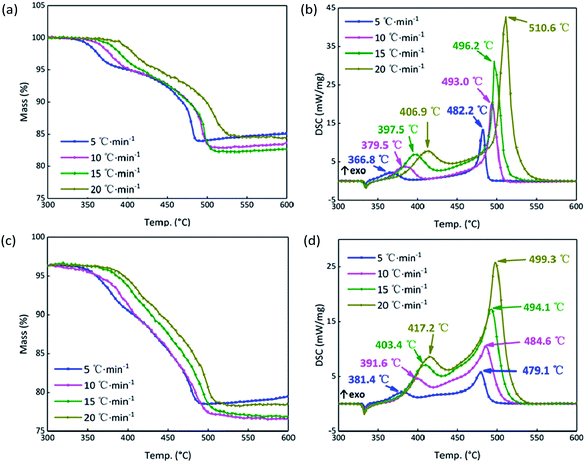 |
| | Fig. 10 (a) TG curves of S1 at 5, 10, 15, 20 °C min−1. (b) DSC curves of S1 at 5, 10, 15, 20 °C min−1. (c) TG curves of S4 at 5, 10, 15, 20 °C min−1. (d) DSC curves of S4 at 5, 10, 15, 20 °C min−1. | |
Table 3 Kinetic parameters for S1 and S4
| Sample |
EKa/kJ mol−1 |
lg(Ab) |
rKc |
EO/(kJ mol−1) |
rO |
| E is the apparent activation energy calculated by the Kissinger or Ozawa method. A is the pre-exponential factor. r indicates the linearly dependent coefficient used in data fitting. |
| S1 |
1st |
120.19 |
7.35 |
0.9973 |
124.58 |
0.9968 |
| 2nd |
215.84 |
12.55 |
0.9950 |
217.42 |
0.9943 |
| S4 |
1st |
135.05 |
8.38 |
0.9985 |
138.90 |
0.9982 |
| 2nd |
213.81 |
12.61 |
0.9972 |
215.41 |
0.9969 |
Table 4 Parameters of the reaction mechanism for S1 and S4
| Sample |
Gαa |
Es/kJ mol−1 |
lg(As) |
Qsb |
rs |
| Gα is the integral model function. Qs is the average deviation calculated by the Satava–Sestak method. |
| S1 |
1st |
[1 − (1 − α)1/3]2 |
134.58 |
10.69 |
0.0257 |
0.9932 |
| 2nd |
238.18 |
21.99 |
0.0304 |
0.9918 |
| S4 |
1st |
[1 − (1 − α)1/3]2 |
135.24 |
12.05 |
0.0209 |
0.9923 |
| 2nd |
206.73 |
16.65 |
0.0316 |
0.9825 |
From the SEM images of S4 in Fig. 11(a) and (b), we suppose that KNO3 and GO are located at the surface of B particles spliced by Viton. A schematic illustration is shown in Fig. 11(c). GO responds to the thermal stimulus and conducts heat, meanwhile, B and KNO3 are connected by the GO film and can react.
 |
| | Fig. 11 (a) and (b) SEM images of S4. (c) Schematic illustration of B/KNO3 with appended GO. | |
Critical ignition distance analyses
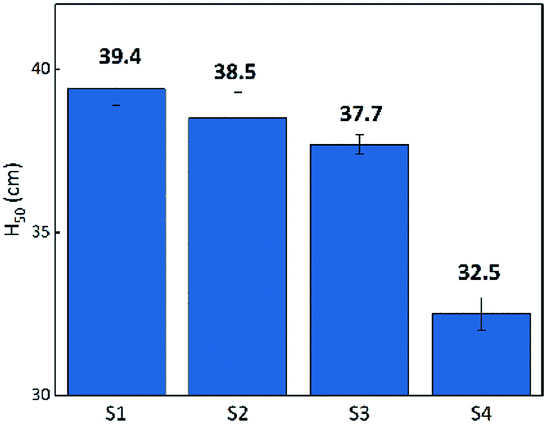
Flame testing allows a clear comparison of the sensitivity of the B/KNO3 complex to thermal stimulation before and after the addition of GO. When testing, the ignition flame of the device causes a temperature field which affects the samples within it. Samples mounted at the same distance experience the same temperature from the ignition source. It can be seen from Fig. 12 that the H50 of the B/KNO3 powder decreases after the addition of GO, and the greater the amount of GO, the lower the critical ignition distance becomes, reducing from 39.4 ± 0.5 cm (S1) to 38.5 ± 0.8 cm (S2), 37.7 ± 0.3 cm (S3) and 32.5 ± 0.5 cm (S4).
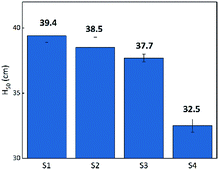 |
| | Fig. 12 Critical ignition distances of S1 to S4. | |
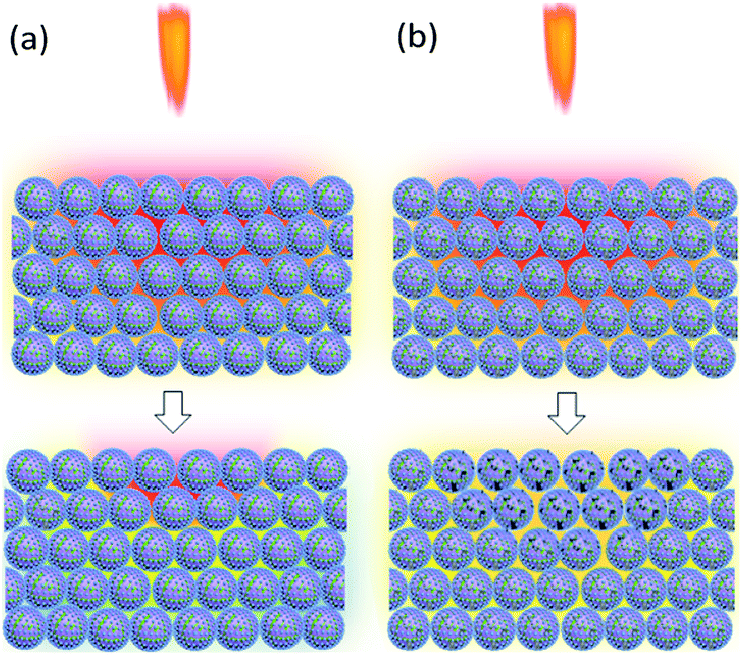
After ignition of the device, samples experience the temperature field. Because of the low heat conduction, B/KNO3 particles close to the temperature source become hot and it is difficult for them to transfer heat to other particles and to the environment. Hot spots form easily and can be ignited, as shown in Fig. 13(a). For the samples with added GO, the thermal radiation causes the slow reduction of the GO, consuming heat and also conducting heat to adjacent particles and to the environment. Thus, the overall temperature of the sample fails to reach the ignition temperature, as shown in Fig. 13(b). Only by moving the samples closer to the heat source can the ignition probability be increased. As the content of GO increases, the system becomes less sensitive, which is consistent with the conclusion of the previous thermal analysis on the initial temperature of the reaction for the GO ignition system. The results all indicate that energetic materials containing GO will be more thermally stable.
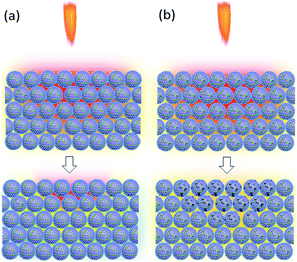 |
| | Fig. 13 Schematic diagrams of the reaction mechanism behind the sensitivity to flame testing. In the upper pictures, the samples are in the temperature field caused by the flame of the ignition device. The lower pictures indicate the temperatures of the samples: (a) B/KNO3 without GO. (b) B/KNO3 with GO. | |
Pressure–time curve analyses
On the one hand, adding GO can reduce the thermal sensitivity of energetic materials. On the other hand, according to the non-isothermal reaction kinetics, when the energy of a stimulus is greater than the activation energy required for the reaction, the carbon skeleton of GO will cause the whole system to rapidly warm up and trigger the reaction. The closed-vessel test was performed using sufficient energy to rapidly ignite the energetic materials. The pressure–time (p–t) curves of B/KNO3 are shown in Fig. 14. The addition of GO increased the pressure peaks (pmax) by 5.7%, 7.3%, and 12.2%, for S2, S3 and S4, respectively. Moreover, the maximum slopes of the p–t curves ((dp/dt)max) were increased (see in Table 5), indicating that the reaction rates were increased.
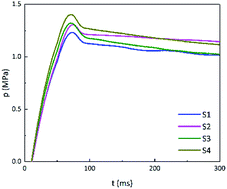 |
| | Fig. 14 P–t curves of S1 to S4. | |
Table 5 Pressure peaks and maximum slopes of p–t curves
| Sample |
pmax/MPa |
(dp/dt)max/MPa s−1 |
| S1 |
1.23 |
20.7 |
| S2 |
1.30 |
21.3 |
| S3 |
1.32 |
22.3 |
| S4 |
1.38 |
22.7 |
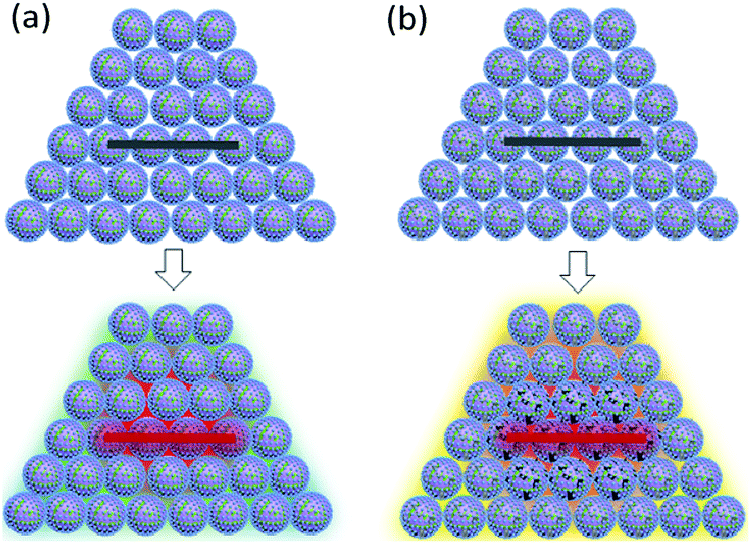
The closed vessel can be regarded as a constant-capacity adiabatic system. Under a high-power current input, the electric igniter quickly ignites the samples within. B/KNO3 reaches the ignition temperature in the local area under sufficient thermal stimulation, and the energy generated by the reaction of this area heats up adjacent areas, so as to transfer the reaction (Fig. 15(a)). For the samples containing GO, GO is reduced to RGO instantly and conducts the heat rapidly, facilitating the reaction and shortening the delay time, as shown in Fig. 15(b). In addition, GO releases a certain amount of hot gas during decomposition, which enhances the ignition pressure in the fixed-volume cavity.
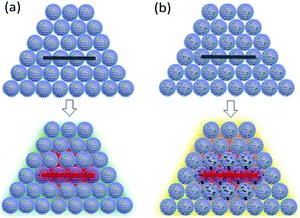 |
| | Fig. 15 Schematic diagrams of the reaction mechanism of the closed-vessel test. In the upper pictures, the electric wire is not ignited. In the lower pictures, the electric wire is ignited and the temperatures of the samples are changed: (a) B/KNO3 without GO. (b) B/KNO3 with GO. | |
Conclusions
In this work, we have discussed the response mechanisms of energetic materials with appended GO under different energetic stimuli and different loading rates. Previous studies have shown that GO can not only improve safety with regards to thermal stimulation, but also improve the response and speed up the reaction of energetic materials under high-energy stimuli. Thus energetic materials with appended GO embody dual-mode response behavior.
In this work, GO produced by the Hummers' method and characterized by SEM, EA, FT-IR spectroscopy, nano FT-IR spectroscopy, XPS, Raman spectroscopy, and TG-DSC-MS, was appended to B/KNO3 in different proportions, to regulate the response of B/KNO3 to thermal stimuli. The results of TG-DSC-MS indicated that at temperatures above 250 °C, GO was reduced to RGO, and, at a high heating rate, the reduction was more thorough.
According to non-isothermal reaction kinetics, GO does not change the reaction mechanism of B/KNO3. In the first stage of the reaction, RGO causes the temperature to dissipate, delaying the onset temperature of the reaction. After the reaction has begun, RGO facilitates energy transfer throughout the sample, so that the activation energy for the second stage is reduced.
The sensitivity to both flame testing and closed-vessel testing was used to analyze the behavior of the B/KNO3 samples. The sensitivity to flame testing indicates the behavior of the sample to a small external energy stimulus and slow loading speed, while the closed-vessel test creates a strong and rapid energy stimulus. According to the results of the flame sensitivity testing, the higher the GO content, the more insensitive the system became, which proves that the addition of GO could improve safety. In the closed-vessel test, as the GO content increased, both the pressure peak and maximum slopes of the p–t curves increased, demonstrating an increase of work capability after the addition of GO.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The work was supported by the National Natural Science Foundation of China (U1530262, 21975024).
Notes and references
- D. A. Benson, M. E. Larsen, A. M. Renlund, W. M. Trott and R. W. Bickes Jr, J. Appl. Phys., 1987, 62, 1622 CrossRef.
- H. P. Cheng and H. S. Chuang, ACS Sens., 2019, 4, 1754 CrossRef CAS.
- C. Rossi, K. Zhang, D. Esteve, P. Alphonse, P. Tailhades and C. Vahlas, J. Microelectromech. Syst., 2007, 16, 919 CAS.
- J. A. Conkling and C. Mocella, Chemistry of pyrotechnics: basic principles and theory, 3rd edn, 2019 Search PubMed.
- W. R. Zhong, M. P. Zhang, B. Q. Ai and D. Q. Zheng, Appl. Phys. Lett., 2011, 98, 113107 CrossRef.
- Z. Wei, Z. Ni, K. Bi, M. Chen and Y. Chen, Carbon, 2011, 49, 2653 CrossRef CAS.
- S. K. Jaćimovski, M. Bukurov, J. P. Šetrajčić and D. I. Raković, Superlattices Microstruct., 2015, 88, 330 CrossRef.
- S. Hu, J. Chen, N. Yang and B. Li, Carbon, 2017, 116, 139 CrossRef CAS.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902 CrossRef CAS PubMed.
- J. Wang, L. Zhu, J. Chen, B. Li and J. T. Thong, Adv. Mater., 2013, 25, 6884 CrossRef CAS PubMed.
- H. Malekpour, K. H. Chang, J. C. Chen, C. Y. Lu, D. L. Nika, K. S. Novoselov and A. A. Balandin, Nano Lett., 2014, 14, 5155 CrossRef CAS PubMed.
- J. F. Li, W. Lu, Y. B. Zeng and Z. P. Luo, Sol. Energy Mater. Sol. Cells, 2014, 128, 48 CrossRef CAS.
- G. Gedler, M. Antunes, T. Borca-Tasciuc, J. I. Velasco and R. Ozisik, Eur. Polym. J., 2016, 75, 190 CrossRef CAS.
- S. Hong, S. S. Yoo and P. J. Yoo, J. Mater. Chem. C, 2019, 7, 9380 RSC.
- S. Gadipelli and Z. X. Guo, Prog. Mater. Sci., 2015, 69, 1 CrossRef CAS.
- S. Lin and M. J. Buehler, Carbon, 2014, 77, 351 CrossRef CAS.
- P. Kumar, F. Shahzad, S. Yu, S. M. Hong, Y. H. Kim and C. M. Koo, Carbon, 2015, 94, 494 CrossRef CAS.
- Y. Yao, X. Zeng, F. Wang, R. Sun, J. B. Xu and C. P. Wong, Chem. Mater., 2016, 28, 1049 CrossRef CAS.
- M. Santiago-Calvo, V. Blasco, C. Ruiz, R. París, F. Villafañe and M. Á. Rodríguez-Pérez, J. Appl. Polym. Sci., 2019, 136, 47474 CrossRef.
- C. Zhang, J. Phys. Chem. B, 2007, 111, 6208 CrossRef CAS PubMed.
- Y. Su, Y. Sun and J. Zhao, Nanomaterials, 2019, 9, 1290 CrossRef CAS PubMed.
- V. Pelletier, S. Bhattacharyya, I. Knoke, F. Forohar, M. Bichay and Y. Gogotsi, Adv. Funct. Mater., 2010, 20, 3168 CrossRef CAS.
- J. Sivan and Y. Haas, J. Phys. Chem. A, 2013, 117, 11808 CrossRef CAS PubMed.
- J. Sivan and Y. Haas, Propellants, Explos., Pyrotech., 2015, 40, 755 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- Q. Wang, J. Liu, H. Ren and Q. Jiao, Chin. J. Energ. Mater., 2018, 26, 875 Search PubMed.
- S. F. Pei and H. M. Cheng, Carbon, 2012, 50, 3210 CrossRef CAS.
- G. P. Pan, Principles of fireworks and firecrackers, 2013 Search PubMed.
- H. Levitt, J. Acoust. Soc. Am., 1971, 49, 467 CrossRef.
- A. Bougamra and H. Lu, J. Therm. Sci. Eng. Appl., 2016, 8, 021005 CrossRef.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS PubMed.
- J. Shen, Y. Hu, M. Shi, X. Lu, C. Qin, C. Li and M. Ye, Chem. Mater., 2009, 21, 3514 CrossRef CAS.
- Y. Ou, Q. Zhao, W. Zhang, B. Zhang, S. Yan and Q. Jiao, Mater. Lett., 2019, 237, 152 CrossRef CAS.
- J. Xu, K. Wang, S. Z. Zu, B. H. Han and Z. Wei, ACS Nano, 2010, 4, 5019 CrossRef CAS PubMed.
- S. Stankovich, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Carbon, 2006, 44, 3342 CrossRef CAS.
- Y. Si and E. T. Samulski, Nano Lett., 2008, 8, 1679 CrossRef CAS PubMed.
- H. L. Guo, X. F. Wang, Q. Y. Qian, F. B. Wang and X. H. Xia, ACS Nano, 2009, 3, 2653 CrossRef CAS PubMed.
- S. D. Perera, R. G. Mariano, K. Vu, N. Nour, O. Seitz, Y. Chabal and K. J. Balkus Jr, ACS Catal., 2012, 2, 949 CrossRef CAS.
- Z. Liu, K. Nørgaard, M. H. Overgaard, M. Ceccato, D. M. Mackenzie, N. Stenger and T. Hassenkam, Carbon, 2018, 127, 141 CrossRef CAS.
- A. Esmaeili and M. H. Entezari, J. Colloid Interface Sci., 2014, 432, 19 CrossRef CAS PubMed.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner and R. S. Ruoff, Carbon, 2009, 47, 145 CrossRef CAS.
- S. H. Shim, K. T. Kim, J. U. Lee and W. H. Jo, ACS Appl. Mater. Interfaces, 2012, 4, 4184 CrossRef CAS PubMed.
- L. A. Lyon, C. D. Keating, A. P. Fox, B. E. Baker, L. He, S. R. Nicewarner and M. J. Natan, Anal. Chem., 1998, 70, 341 CrossRef PubMed.
- Z. S. Wu, W. Ren, L. Xu, F. Li and H. M. Cheng, ACS Nano, 2011, 5, 5463 CrossRef CAS PubMed.
- X. Wang, Z. Feng, J. Huang, W. Deng, X. Li, H. Zhang and Z. Wen, Carbon, 2018, 127, 149 CrossRef CAS.
- P. Pachfule, D. Shinde, M. Majumder and Q. Xu, Nat. Chem., 2016, 8, 718 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim and B. H. Hong, Nature, 2009, 457, 706 CrossRef CAS PubMed.
- Y. Yano, Propellants, Explos., Pyrotech., 1989, 14, 187 CrossRef CAS.
- Y. Sun, H. Ren, F. Du, S. Yan and Q. Jiao, J. Alloys Compd., 2018, 759, 100 CrossRef CAS.
- P. Demir and İ. Y. Düşükcan, Polym. Bull., 2018, 75, 4037 CrossRef CAS.
- N. B. Allou, P. Saikia, E. F. Assanvo and R. L. Goswamee, Polym. Compos., 2018, 39, E1606 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Tao Yan,
Yaru Li,
Hui Ren*,
Qian Wang,
Fayang Guan and
Qingjie Jiao
,
Tao Yan,
Yaru Li,
Hui Ren*,
Qian Wang,
Fayang Guan and
Qingjie Jiao