DOI:
10.1039/D0RA00836B
(Paper)
RSC Adv., 2020,
10, 16515-16525
Ru/g-C3N4 as an efficient catalyst for selective hydrogenation of aromatic diamines to alicyclic diamines†
Received
28th January 2020
, Accepted 15th April 2020
First published on 25th April 2020
Abstract
A series of Ru/g-C3N4 materials with highly dispersed Ru were firstly prepared by an ultrasonic impregnation method using carbon nitride as a support. The catalysts were characterized by various techniques including BET and elemental analysis, ICP-AES, XPS, XRD, CO2-TPD and TEM. The results demonstrated that Ru/g-C3N4 materials with a mesoporous structure and highly dispersed Ru were successfully prepared. The chemo-selective hydrogenation of p-phenylenediamine (PPDA) to 1,4-cyclohexanediamine (CHDA) over Ru/g-C3N4 as a model reaction was investigated in detail. PPDA conversion of 100% with a CHDA selectivity of more than 86% could be achieved under mild conditions. It can be inferred that the carbon nitride support possessed abundant basic sites and the Ru/g-C3N4-T catalysts provided suitable basicity for the aromatic ring hydrogenation. Compared to the N-free Ru/C catalyst, the involvement of nitrogen species in Ru/g-C3N4 remarkably improved the catalytic performance. In addition, the recyclability of the catalyst demonstrated that the aggregation of Ru nanoparticles was responsible for the decrease of the catalytic activity. Furthermore, this strategy also could be expanded to the selective hydrogenation of other aromatic diamines to alicyclic diamines.
1. Introduction
Diamines, including aromatic diamines, aliphatic diamines and alicyclic diamines, are important fine chemicals and intermediates. Compared with aromatic and aliphatic diamines, alicyclic diamines are considered to be special amines, because of their high flexibilities, rapid curing, high reactivity, excellent weathering resistance and low toxicity. These value-added diamines have attracted great and growing interest as valuable feedstocks for manufacturing light stable polyurethane resins, curing agents, adhesives and coatings.1 Specifically, 1,4-cyclohexanediamine (CHDA) as an organic alicyclic amine with symmetrical structure is often used in textiles, paper making, artificial leather, plastic processing, liquid crystal material production, etc. CHDA is also an important chemical intermediate, which could be used to prepare alicyclic polyurethane from 1,4-dicyclohexyl diisocyanate (CHDI) and polyamide resin.2,3 Compared to other methods such as Hofmann rearrangement, amination of 1,4-cyclohexanedio4 and reduction of 1,4-dinitrocyclohexane,5 hydrogenation of PPDA aromatic diamine has attracted more attention in recent years, because of its high efficiency, easy availability, simple process and low cost.6,7 The development of highly active and selective catalysts is one of the key problems in the hydrogenation of aromatic diamines to alicyclic diamines.
Transition metals, such as active components of catalysts, including Ni,8 Co,9 Ru,10 Rh,11,12 Pd13 and Pt,14,15 are often applied to the hydrogenation of aromatic amines,16–19 and it can catalyse the hydrogenation of aromatic-ring group of PPDA with PPDA conversion of 100% and CHDA selectivity of 71.6%, respectively.3 However, undesired side reactions such as deaminization or condensation usually occur severely during the hydrogenation by using the aforementioned catalysts, which decrease the selectivity of CHDA through formation of deamination by-products or secondary amines. To overcome this problem, alkaline additives such as liquid ammonia, LiNO3, LiOH,20 NaNO2 (ref. 21) or NaNO3 (ref. 22) were inevitably added to improve the selectivity of target products. However, the introduction of liquid ammonia or alkaline metal salt in the system complicates the reaction system as well as the separation operation, making this process produce large amount of pollutants and increase production cost. Similarly, the hydrogenation of other aromatic diamines also suffered these problems. Thus, the development of green and efficient catalytic system for the chemo-selective hydrogenation of aromatic amines to produce alicyclic diamines without involvement of other liquid alkali inhibitor is highly desired.
Graphitic carbon nitride, as a kind of fascinating carbon materials with high nitrogen content, not only has excellent chemical stability, but also can stabilize the active site of metal easily.23–25 Some researchers have applied carbon nitride materials to synthesize of primary amines with high selectivity.26,27 Liu26 et al. reported that basic N species have significant effect on catalytic performance in the synthesis of amines catalyzed by Co–N–C/CNT@AC. Zhang27 et al. used nitrogen-doped mesoporous carbon (MC)-supported for the synthesis of primary amines, showing excellent catalytic performance. Notably, compared to the N-free carbon support, g-C3N4 matrix possess higher N content, H-bonding motif, abundant Lewis and Brønsted basic sites and could regulate the metal sites finely.28–31 In principle, the surface property of catalyst played a crucial role in regulating the catalytic performance.32–34 The reactants with benzene moieties to be hydrogenated and intermediate formed during the hydrogenation process should adsorb to the catalyst surface with appropriate strength. Actually, g-C3N4 could be used as versatile metal-free basic support. In this account, Ru-based catalyst with moderate adsorption and activation ability of aromatic ring becomes the ideal candidate. Therefore, the development of g-C3N4 supported Ru catalyst and in-depth study the catalytic performance of the rationally designed Ru/g-C3N4 catalyst in the hydrogenation reaction has great significance.
Herein, a series of Ru/g-C3N4 were firstly prepared by ultrasonic impregnation method using carbon nitride as support, which were employed in the hydrogenation of PPDA to prepare CHDA as a model reaction. The Ru/g-C3N4 catalysts were systematically characterized by N2 physisorption, elemental analysis, ICP-AES, XRD, CO2-TPD, TEM and XPS. The influence of urea polymerization temperature on the C3N4 support property, and the Ru/g-C3N4-T textures, structures and individual catalytic activity was investigated. In addition, the catalytic performances of the as-prepared catalysts, effects of reaction parameters and catalyst reusability were also studied in details. Moreover, the universality of this catalytic system was also expanded to other aromatic diamines.
2. Experimental
2.1 Materials
Tetrahydrofuran (THF, 99.8% purity), aniline (>99%) and ethanol (99.5% purity) were purchased from Sinopharm Chemical Reagent Co., Ltd., China. Ruthenium (5%) on carbon, cyclohexylamine (99.18%) and 1,4-cyclohexanediamine (>98%) were purchased from Shanghai Macklin Biochemical Co., Ltd. p-Phenylenediamine (>97%) and ruthenium(III) chloride (RuCl3, Ru = 45%–55%) were purchased from Shanghai Aladdin biochemical technology Co., Ltd. Urea (>99%) was purchased from Xilong Chemical Co., Ltd., China. Sodium borohydride (98%) was purchased from Tianjin Fuchen Chemical Reagent Co., Ltd. Other reagents were of analytical grade and used as received.
2.2 Catalyst preparation
2.2.1 Synthesis of carbon matrix. Typical methods of preparing g-C3N4 as reported in the article,35 50 g of urea was directly calcinated in a 300 mL crucible at a muffle furnace. The crucible was semi-sealed, heated with a rate of 5 °C min−1 and kept at the terminal calcination temperature (T = 450 °C, 500 °C, 550 °C and 600 °C) for 4 h. After cooling, the yellowish powders were successively washed in deionized water twice and ethanol once, then filtered and dried at 100 °C for 16 h. After that, the final products of g-C3N4-T (T = 450 °C, 500 °C, 550 °C and 600 °C) were obtained.
2.2.2 Synthesis of Ru nanoparticles supported on g-C3N4. The Ru/g-C3N4 catalyst was synthesized by a simple method of ultrasonic impregnation. Typically, 2.85 g g-C3N4-T was dispersed in 100 mL deionized water in a beaker by ultrasound, then 30 mL RuCl3 aqueous solution with a concentration of 0.01 g mL−1 was added into the beaker. Subsequently, after treatment in ultrasound for 60 minutes, 15 mL deionized water containing 300 mg NaBH4 was added into the solution, and kept the solution in ultrasound for another 120 minutes. Later, the solution was filtrated and the residue (black solids) was washed with deionized water for several times, NaBH4 has been washed out completely. Then the black solid was dried at 110 °C for 16 h and collected for catalytic test. With the same procedure, 5%Ru/g-C3N4-600, 5%Ru/g-C3N4-550, 5%Ru/g-C3N4-500 and 5%Ru/g-C3N4-450 were prepared, respectively. The Ru and g-C3N4-600 carriers were weighed and measured according to different loadings, x%Ru/g-C3N4-600 (x = 0.1, 0.5, 1, 3, 5) were prepared in accordance with the process described above.
2.2.3 Preparation of 5%Ru/C. Ruthenium (5%) on carbon catalyst (Ru/C) was dried in vacuum at 100 °C drying oven for 8 hours to eliminate the moisture adsorbed on the catalyst.
2.3 Catalyst characterization
N2 physisorption measurements were carried on Quantachrome Autosorb at liquid nitrogen temperature (−196 °C) after the catalysts were outgassed at 200 °C for 4 h in vacuum with the purpose of removing physically adsorbed impurities. The special surface area was calculated by the Brunauere–Emmette–Teller (BET) method. Total pore volume (Vpore) was performed from the adsorbed N2 volume with a relative pressure of 0.99. Preliminary estimates of pore size distribution (Dpore) were obtained by the Barrett, Joyner and Halenda (BJH) method on the basis of the desorption branch of the isotherm.
Elemental analysis (C/H/N) was performed on a Vario EL Cube elemental analyser made in Germany.
Ruthenium loadings of the samples were determined from inductively coupled plasma-atomic emission spectroscopy (ICP-AES) by the United States Agilent ICPOES730.
X-ray diffraction (XRD) patterns of catalysts were obtained from a PANalytical Empyrean diffractometer with copper Kα radiation at the 2θ range of 5°–90°.
Transmission electron microscopy (TEM), HR-TEM and EDX mapping images were obtained on a microscope (JEOL, JEM-2100F) equipped with a CCD camera (ORIUS SC1000), which was operated at an acceleration voltage of 200 kV. The catalysts were ultrasonically dispersed in ethanol at room temperature for 60 min. Then drops of the suspension were deposited on transparent lacey support films containing copper and dried prior to measurement. The particle statistics were measured by Nano Measurer software, and the normal distribution is used to indicate the distribution of Ru particles.
CO2-TPD was carried out to determine the basicity of samples using an AutoChem II 2920 equipped with a thermal conductivity detector (TCD). About 50 mg of samples was heated in situ in 100 mL min−1 of helium gas at 200 °C for 60 min in order to remove adsorbed impurities. After that, the sample was cooled to 50 °C and saturated with 100 mL min−1 of 10%vol CO2/He for 90 min. Weakly adsorbed CO2 was eliminated by flushing with a 100 mL min−1 of helium gas at 50 °C for 30 min, and the temperature was increased to 450 °C with a ramping rate of 10 °C min−1.
X-ray photoelectron spectroscopy (XPS) was used to examine the surface chemical states of samples, which were performed on an ultrahigh vacuum using an ESCALAB 250Xi spectrometer with Al Kα radiation (hv = 1486.6 eV) and a multichannel detector.
2.4 Catalytic activity test
The catalytic activities and selectivities of the catalysts were firstly investigated for the hydrogenation of p-phenylenediamine (PPDA) in a 50 mL stainless steel autoclave. Typically, 50 mmol PPDA, 20 mL THF and 25 wt% catalyst dosage (based on the weight of PPDA) were added into the autoclave. After being sealed and flushed five times with N2 and H2 successively, the autoclave was charged with 5 MPa H2 at ambient conditions. The autoclave was then heated to 130 °C for 2.5 h under vigorous magnetic agitation. After completion of the hydrogenation reaction, the autoclave was cooled to room temperature with water and depressurized to atmospheric pressure. A certain amount of reaction liquid after centrifugation was mixed with an appropriate amount of diphenyl as the internal standard in a 50 mL volumetric flask to prepare a standard sample. The sample was quantitatively analyzed on Shimadzu GC-2014 with a GsBP−1 column (30 m × 0.32 mm × 1.0 μm), a flame ionization detector (FID), and an autosampler, and qualitatively analyzed on GCMS-QP2012.
The calculation method of the conversion of PPDA, selectivity and yield of CHDA are described as follows:
| |
 | (1) |
| |
 | (2) |
| |
 | (3) |
The abbreviations are as follows:
n0,PPDA: mol of PPDA charged initially,
nPPDA: mol of PPDA unreacted,
n1,4-CHDA: mol of CHDA generated (sum of
trans and
cis isomers).
3. Results and discussion
Supporter of g-C3N4 can be prepared in a variety of ways; common methods are direct thermal polymerization using cyanamide, dicyandiamide, and melamine as precursor. However, these precursors are expensive and venomous, more importantly, the g-C3N4 materials obtained usually exhibit poor hole structure and lower specific surface areas. Alternatively, some scholars increased the specific surface areas of g-C3N4 by the way of introducing a soft or hard template in the synthesis process, but the overall synthesis procedure would become relatively tedious and more ineffective.36–41 Templateless method can form porous structure in g-C3N4 by utilizing the gas released during the synthesis of g-C3N4, which is one of the more ideal methods to prepare g-C3N4. In this study, we produced g-C3N4 support by the method of directly pyrolyzing urea without templates at high temperature, and the Ru active component was loaded to prepare a multiphase catalyst for the hydrogenation of p-phenylenediamine to prepare CHDA.
Four different catalysts were prepared by changing the treatment temperature of the precursor, 5%Ru/g-C3N4-T (T = 450, 500, 550, 600).
3.1 Characterization of the supports and catalysts
3.1.1 N2 physisorption, ICP-AES analysis and elemental analysis. Fig. 1(a) presents N2 adsorption–desorption isotherms of a series of Ru/g-C3N4 catalysts, the corresponding textural properties are also summarized in Table 1. The N2 physisorption isotherms of Ru/g-C3N4 in Fig. 1(a) is identified as typical type IV35,42 and type H1 hysteresis loop according to IUPAC nomenclature,43 which indicated that the catalysts have mesoporous structures and formed by lamellar particle accumulation. What's more, the trend of nitrogen adsorption isotherm of catalyst with and without Ru was the same, indicating the pore structure of the carrier was not obviously destroyed by loading Ru active metal. As exhibited in Fig. 1(b), the temperature of catalyst prepared had great impact on the pore size distributions of Ru/g-C3N4. When the catalyst prepared at relatively lower temperature 450 and 500 °C, the pore sizes were uniformly distributed in a range of 0–100 nm. While, as the calcination temperature increased to 550 °C, and further to 600 °C, the pore size distributions at about 2.7 nm was advantageous. The results of ICP-AES in Table 2 show that the actual amount of Ru is slightly lower than the theoretical amount 5%wt. It is speculated that part of ruthenium existed in the liquid phase during solid–liquid separation in preparation. Results from BET are also listed in Table 2. With increasing the pyrolysis temperature from 450 to 600 °C, specific surface area increase from 40.31 to 85.12 m2 g−1, total pore volume increase from 13.01 to 24.21 nm, and average pore size increase from 0.13 to 0.52 cm3 g−1. In addition, It can be seen that without Ru loading, the surface area, total pore volume and average pore size of g-C3N4-600 is 94.89 m2 g−1, 20.44 nm, and 0.49 cm3 g−1, respectively. On the contrast, 5% loading of Ru slightly decreased the surface area to 85.12 m2 g−1, and increased total pore volume and average pore size to 24.21 nm, and 0.52 cm3 g−1, respectively. This phenomenon could be attributed to some of the micropores in the catalyst collapsed at the process of loading Ru.
 |
| | Fig. 1 (a) N2 adsorption desorption isotherm (a) and pore size distribution (b) of g-C3N4. | |
Table 1 Physico-chemical properties of 5%Ru/g-C3N4 prepared at different temperatures
| Catalyst |
ICPa |
BETb |
Elemental analysisc |
Catalytic performanced |
| Ru loading |
Surface area (m2 g−1) |
Dpore (nm) |
Vpore (cm3 g−1) |
C/N molar ratio |
H (wt%) |
Con. (%) PPDA |
Yield (%) CHDA |
Sel. (%) CHDA |
| Determined by ICP-AES. Obtained by N2 physisorption. Obtained by elemental analysis. Reaction conditions: 5 mmol PPDA, 5 MPa H2, 20 mL THF, 25 wt% catalyst dosage (based on the weight of PPDA), 130 °C and 1 h. |
| 5%Ru/g-C3N4-450 |
4.11 |
40.31 |
13.01 |
0.13 |
0.61 |
2.4 |
53.96 |
45.46 |
84.25 |
| 5%Ru/g-C3N4-500 |
4.07 |
42.27 |
13.66 |
0.14 |
0.62 |
2.1 |
67.09 |
58.74 |
87.55 |
| 5%Ru/g-C3N4-550 |
4.52 |
62.82 |
20.06 |
0.32 |
0.64 |
1.9 |
72.51 |
62.64 |
86.39 |
| 5%Ru/g-C3N4-600 |
4.06 |
85.12 |
24.21 |
0.52 |
0.65 |
1.8 |
80.18 |
70.50 |
87.93 |
| 5%Ru/g-C3N4-600-5th |
|
83.15 |
18.26 |
0.43 |
— |
— |
— |
— |
— |
| 5%Ru/C |
4.1 |
1039.74 |
1.75 |
0.455 |
— |
— |
53.77 |
31.25 |
58.63 |
| g-C3N4-600 |
0 |
94.89 |
20.44 |
0.49 |
0.65 |
1.8 |
2.94 |
0 |
0 |
Table 2 The component distribution of nitrogen species on the surface of catalysts according to the relative magnitude of peak area from Fig. 3
| Sample |
397.8 |
399.5 |
400.7 |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–C N–C |
C–N–(–C) or C–N–(–H)–C |
C–N–H2 or C–N–O |
| 5%Ru/g-C3N4-450 |
47.16 |
36.32 |
16.5 |
| 5%Ru/g-C3N4-500 |
56.82 |
30.11 |
13.07 |
| 5%Ru/g-C3N4-550 |
58.88 |
30.01 |
11.11 |
| 5%Ru/g-C3N4-600 |
62.11 |
29.19 |
8.7 |
The elemental composition of the catalyst was analyzed by elemental analysis, and the results of the prepared catalysts at different temperatures are shown in Table 1. The molar ratios of C to N of the g-C3N4 increases from 0.61 to 0.65 with temperature, less than 0.75 (theoretical value of g-C3N4), indicating incomplete crystallization. Moreover, the lower molar ratio of C to N, together with a larger amount of hydrogen from the uncondensed amino groups are found in samples prepared at lower temperatures, which preliminarily shown that the degree of polymerization enhanced with the temperature rising.44
3.1.2 XRD analysis. The XRD patterns of prepared Ru/g-C3N4 and Ru/C catalysts were shown in Fig. 2. Both g-C3N4 support and Ru loaded catalysts exhibited characteristic peaks at 27.40°, which is assigned to the dense interlayer-stacking (002) peak of aromatic segments of g-C3N4 materials. The (0 0 2) diffraction of the carbon in Ru/c indicates it has the quasi-graphitic structure. The reflection at 13.08° is indexed as (100) peak that arises from the in-plane ordering of tri-s-triazine attributed to units of g-C3N4.45,46 It meant that the loading of Ru did not change the basic structure of g-C3N4. In addition, these two patterns display a slight narrowing width and an increasing intensity of the overall peaks with the preparation temperature increasing, showing that the crystallinity of the g-C3N4 increased at higher temperatures and the arrangement structure of graphite phase stacking is more regular. In contrast, catalysts prepared at low temperatures formed more edge defects.47 It's reasonable to speculate that edge defects lead to accumulation of ruthenium at carrier edges, which is one of the reasons for the decreased activity of catalysts prepared at lower temperatures. There is no obvious characteristic peak attributed to Ru either on Ru/g-C3N4 or Ru/C, indicating that Ru has good dispersion on the surface of the support.48,49
 |
| | Fig. 2 XRD patterns of the prepared g-C3N4, Ru/C and Ru/g-C3N4 powders. | |
3.1.3 XPS analysis. In order to investigate the valence states of different species of Ru and N in the catalysts, XPS measurements were conducted. In terms of the overlapping between Ru 3d and C 1s signals, Ru 3p core-level spectra were used accordingly. Fig. S1† presents the XPS of the Ru 3p 5%Ru/C catalyst, which shown two peaks for the Ru 3p3/2 and Ru 3p1/2 spin orbit state at BE values around 484.5 eV and 462.2 eV, indicating that Ru is dominantly existed in the presence of Ru0 species.50,51 This result is similar to 5%Ru/g-C3N4-600 according to XPS spectra of Ru/g-C3N4 shown in Fig. 3. It can be observed that the binding energy of Ru 3p in Ru/g-C3N4-T gradually shifted toward lower value with the increase of preparation temperature of g-C3N4. In comparison with Ru/g-C3N4-450, Ru 3p in Ru/g-C3N4-600 shifted about 0.5 eV toward lower binding energy region, indicating the electron enrichment around Ru0 environment due to the electron transfer from nitrogen to Ru.31,52 These results indicated that the nitrogen species in g-C3N4 has a profound influence on the electronic property of Ru. Consequently, the metallic characteristic of Ru was enhanced along with the increment of preparation temperature for g-C3N4. The N 1s spectra can be deconvoluted into three individual peaks at about 397.8, 399.5 and 400.7 eV. The peak at about 397.8 eV correspond to CN–C in the triazine or heptazine rings. The peak at 399.5 eV is attributed to C–N(–C)–C or C–N(–H)–C, and the peak at 400.7 eV is assigned to C–N–H2 or C–N–O of the heptazine rings.53,54 The component distribution of N 1s spectra is displayed in Table 2. The percentage of CN–C increased with the treatment temperature of precursor. In contrast, the percentage of C–N–H2 (C–N–O) gradually decreased, indicating that the catalyst prepared at higher temperature had better polymerization degree. These results are consistent with those of elemental analysis and XRD. With the increase of the preparation temperature, the polymerization degree of urea was increased, and the reduction of edge defects was favourable for the dispersion of ruthenium.
 |
| | Fig. 3 XPS spectra of N 1s (a) and Ru 3p (b) for Ru/g-C3N4-T (T = 450, 500, 550, 600). | |
3.1.4 TEM analysis. The TEM images of g-C3N4-600 and 5%Ru/g-C3N4 catalysts were illustrated in Fig. 4. Particle size distribution of Ru was shown in Fig. S4.† It can be clearly seen that morphology of g-C3N4 support (Fig. 4(a) and (b)) prepared by polymerization is lamellar structure. Fig. 4(c) and (d) illustrated that the catalyst active component Ru exists in the form of nanoparticles (NPs) on the support, and the particle sizes of Ru decreased with the increase of polymerization temperatures for g-C3N4 support.
 |
| | Fig. 4 TEM images of g-C3N4 (a and b) and 5%Ru/g-C3N4-T catalysts of TEM, (c) T = 450, (d) T = 500, (e) T = 550, (f) T = 600. | |
Obviously, the better dispersity of Ru NPs could be obtained on the g-C3N4 support prepared at the higher polymerization temperature. For Ru/g-C3N4-450 and 500 (Fig. 4(c) and (d)), most of the Ru agglomerate at the edge of the catalyst. On contrast, with the temperature rising to 550 °C, the dispersion of Ru could be further improved, and the Ru mean particle size could be reduced to 2.8 nm. Especially, 5%Ru/g-C3N4-600 with a narrow size distribution centered at around 2.5 nm could be obtained. In addition, the HR-TEM images of Ru/g-C3N4 catalysts shown in Fig. S3† and EDX mapping shown in Fig. S2,† images were used to further illustrate the dispersion of ruthenium on the catalyst surfaces. It should be noted, the characteristic peak of Na at 1.04 eV is absent in the EDX spectra, suggesting the washing out of Na during the catalyst preparation was successful. The lattice fringe spacing about 0.22 nm was attributed to Ru (002). Compared with the ruthenium particles in 5%Ru/g-C3N4-550 and 5%Ru/g-C3N4-600 (Fig. S3(e1) and (f1)†) catalyst, 5%Ru/g-C3N4-450 and 5%Ru/g-C3N4-500 (Fig. S3(c1) and (d1)†) catalysts with lower preparation temperatures for g-C3N4 supports possessed obviously darker ruthenium particles at the edge of C3N4 supports. It's deduced that partial aggregation at edges of support was prone to occur at lower temperature during the catalyst preparation. From the results aforementioned, Ru/g-C3N4-600 with larger surface area possessed well dispersed Ru nanoparticals, which is consistent with the results obtained by XRD and TEM.
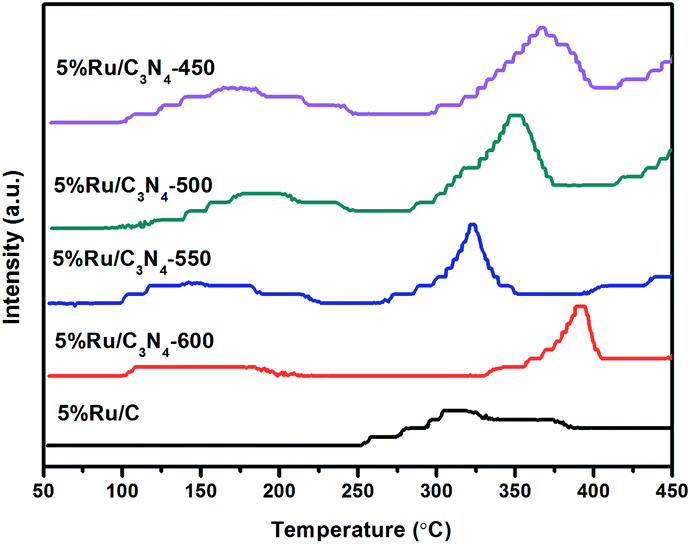
3.1.5 CO2-TPD analysis. In order to test the surface basicities of the catalysts, the CO2-TPD was conducted and the corresponding profiles of the various catalysts are shown in Fig. 5. For comparison, the basic property of N-free 5%Ru/C was also tested. It can be seen that the first CO2 desorption peak, which mainly located in the range of 100–250 °C, could be ascribed to the weak basicities for these Ru/g-C3N4-T catalysts. On the contrary, there is no obvious peak observed for Ru/C in this desorption region. It should be noted all of Ru/g-C3N4 catalysts shown a strong desorption peak of CO2 at 320 °C–400 °C, which was attributed to the strong base active site of the catalyst. The second CO2 desorption peak initially shifted toward lower temperature region along with the g-C3N4 prepared temperature increased from 450 to 550 °C. However, g-C3N4-600 exhibited desorption peak in much higher temperature region than those of the rest g-C3N4 samples. The basicity of the catalyst is mainly determined by the comprehensive influences of Lewis and Brønsted basic sites. According to the type of nitrogen in the g-C3N4 support obtained from XPS, as the polymerization degree of g-C3N4 increases, Lewis basic sites decreases. On the contrary, the distribution of Brønsted basic sites increases. The comprehensive effects may be the reason why different catalysts have different CO2 desorption temperatures in CO2-TPD. The strong Ru–N bond at the interface was considered to be beneficial for the good dispersion of Ru. On the contrast, N-free Ru/C only shown a weak desorption peak of CO2 at around 300 °C. Obviously, the abundant nitrogen could serve as Lewis or Brønsted basic sites24 to enhance the basic strength of g-C3N4. Hence, appropriate basicities are provided for Ru/g-C3N4-T catalysts, especially strong for Ru/C3N4-600 catalyst.
 |
| | Fig. 5 CO2-TPD of Ru/C and Ru/g-C3N4-T (T = 450, 500, 550, 600). | |
3.2 Catalyst evaluation
Four catalysts with different preparation temperatures (5%Ru/g-C3N4-600, 5%Ru/g-C3N4-550, 5%Ru/g-C3N4-500 and 5%Ru/g-C3N4-450) and 5%Ru/C were applied in the hydrogenation of PPDA, and the results are listed in Table 1. The by-products of the liquid phase are mainly aniline and cyclohexylamine (shown in Fig. S5†). It is obvious that Ru/g-C3N4 catalysts has advantages in selectivity compared with catalyst 5%Ru/C, and there are more by-products being formed by N-free 5%Ru/C. Clearly, all the g-C3N4 supported Ru catalysts exhibit higher catalytic activity than the N-free catalyst of Ru/C. The selectivity could be remarkably increased from 58.6% to 87.9% for Ru/C and Ru/g-C3N4-600. Clearly, the catalytic performance of Ru/g-C3N4 outperforms that of N-free Ru/C, and the difference could be attributed to the specific basicity originated from nitrogen functionality of g-C3N4. As determined by TPD results, the basicity strength of 5%Ru/C is much lower than that of Ru/g-C3N4 catalysts, especially in weak basicity region. This mean that the basicity of the catalyst imposed great influence on the catalytic activity. Besides the Ru dispersion enhanced by nitrogen functionality, and the modified electronic property, the basicity of the g-C3N4 support is also considered to play an important role in influencing the hydrogenation performance. According to the viewpoint,20,55 the stronger basicity could inhibit the excessive adsorption of amino group of the diamines on the g-C3N4 matrix to a certain extent, thus could effectively reduce the undesired deamination or condensation reaction. In account of this reason, the hydrogenation selectivity of the aromatic ring could be effectively regulated through tuning the surface basicity of the catalyst. In addition, when Ru/g-C3N4-T with the g-C3N4 support preparation temperature increased from 450 °C to 600 °C, the conversion of PPDA increased from 54% to 80.2%. Meanwhile, the yield of CHDA increased from 35.17% to 70.50%, respectively. At the same time, the selectivity did not change obviously in Ru/g-C3N4 catalysts with temperature. The reason for the improved catalytic activity could be attributed to that 5%Ru/g-C3N4-600 catalyst had the bigger surface area, better dispersion of Ru and stronger basicity strength, as illustrated in Table 1, which led to the higher catalytic activity. For comprehensive consideration, the g-C3N4 prepared at 600 °C was chosen as the preferential support.
3.3 Optimization of the reaction conditions
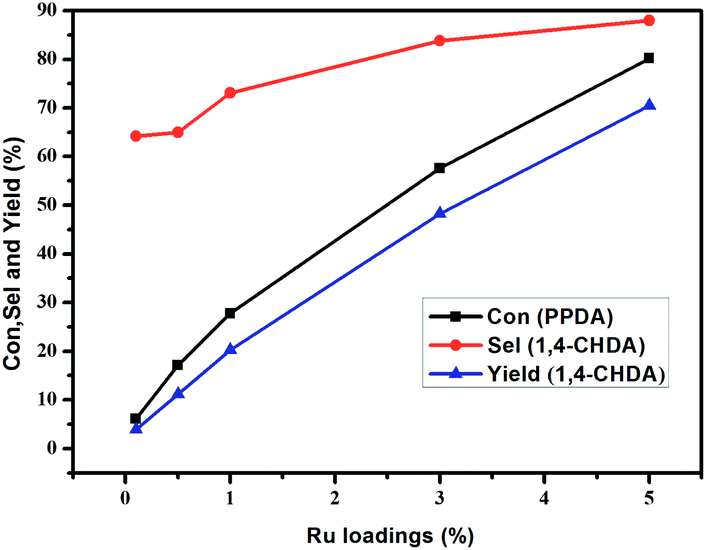
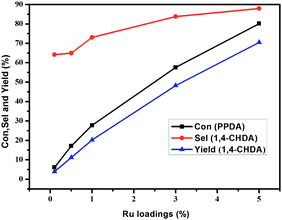
3.3.1 Effect of the different Ru loadings. Ru/g-C3N4-600 catalysts with different loadings of 0.1%, 1%, 3% and 5% were prepared by impregnation method and used for the hydrogenation of PPDA. The results in Fig. 6 shows that with increasing the Ru loading, the conversion of PPDA increased fast from 6.1% to 80.2%. The explanation is that the higher loading of Ru on g-C3N4, the more amount of Ru active sites exposed and more opportunities for the contact with the substance of PPDA, leading to an increase in the conversion of PPDA. While the selectivity of product CHDA increased slowly from 64.2% to 83.8% with Ru loading increasing from 0.1% to 3%, and with further increasing the Ru loading to 5%, the selectivity of CHDA increased to 87.9%. Conclusively, the catalyst with 5% Ru loading was more favorable for retaining higher CHDA productivity, and this content was chosen as the preferential loadings.
 |
| | Fig. 6 The effects of Ru loads for PPDA hydrogenation over the xRu/g-C3N4-600 catalyst. Reaction conditions: PPDA, 5 mmol; THF, 20 mL; reaction temperature, 130 °C; initial hydrogen pressure, 5 MPa; reaction time, 1 h; 25 wt% catalyst dosage (based on the weight of PPDA). | |
3.3.2 Effect of the reaction temperature. The influence of reaction temperature on PPDA hydrogenation was then explored. As shown in Fig. 7, the conversion of PPDA increased linearly from 31.1% to 96.2% with the reaction temperature initially rising from 100 °C to 130 °C. This result suggested that the reaction temperature has a remarkable influence on the catalytic behaviour. However, the CHDA selectivity decreased slightly from 91.1% to 85.6%, which indicated that higher reaction temperature was unfavourable to obtain higher product selectivity. As also verified, the declined selectivity at higher reaction temperature could be attributed to the greater risky side reaction of PPDA deamination to aniline at higher temperature region. Subsequently, aniline produced could be further hydrogenated to CHA, resulting in reduced CHDA selectivity. In comprehensive consideration of atom economy and productivity during the process, 130 °C is chosen as the preferred reaction temperature.
 |
| | Fig. 7 The effects of reaction temperature for PPDA hydrogenation over the 5%Ru/g-C3N4-600 catalyst. Reaction conditions: PPDA, 5 mmol; THF, 20 mL; initial hydrogen pressure, 5 MPa; reaction time, 1 h; 25 wt% catalyst dosage (based on the weight of PPDA). | |
3.3.3 Effect of the reaction time. The influence of reaction time on PPDA hydrogenation was explored using 5%Ru/g-C3N4-600 as catalyst. As shown in Fig. 8, with increasing the reaction time from 1 to 2 h, the conversion of PPDA increased from 80.2% to 98.45% with selectivity of CHDA slowly decreased from 87.9% to 86.1%, respectively. That is also because that during the PPDA hydrogenation process. That is also because that during the PPDA hydrogenation process, side reactions of PPDA deamination to aniline, and further hydrogenation to CHA. With continuous prolonging the reaction time to 2.5 h, side reaction was not detected probably due to there is no unreacted PPDA existed in the reaction system, thus the selectivity of CHDA could keep almost unchanged at about 86.3% (Fig. 9).
 |
| | Fig. 8 The effects of reaction time for PPDA hydrogenation over the 5%Ru/g-C3N4-600 catalyst. Reaction conditions: PPDA, 5 mmol; THF, 20 mL; reaction temperature, 130 °C; initial hydrogen pressure, 5 MPa; 25 wt% catalyst dosage (based on the weight of PPDA). | |
 |
| | Fig. 9 The effects of reaction pressure for PPDA over the 5%Ru/g-C3N4-600 catalyst. Reaction conditions: PPDA, 5 mmol; THF, 20 mL; reaction temperature, 130 °C; reaction time, 1 h; 25 wt% catalyst dosage (based on the weight of PPDA). | |
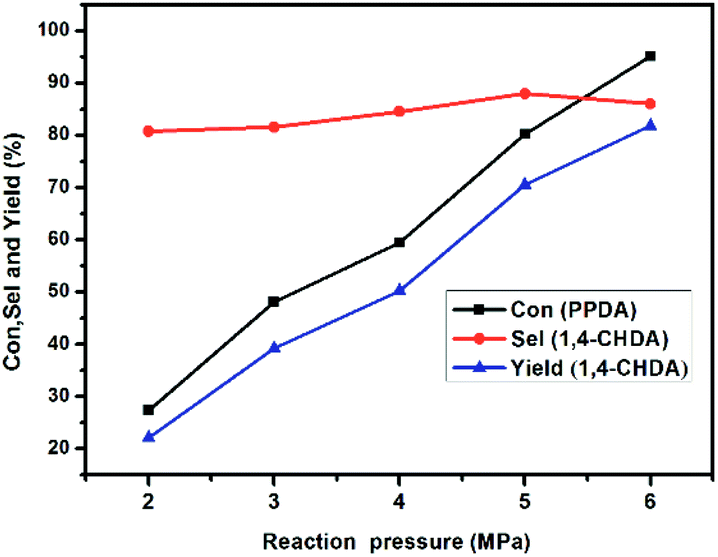
3.3.4 Effect of the reaction pressure. The reaction pressure has great effect on the conversion of PPDA according to the Fig. 10. As the pressure increased from 2 to 6 MPa, the conversion of PPDA increased sharply and almost linearly from 27.3% to 95.1%. However, the variation of CHDA selectivity is gentle in duration, and shortly increased from 80.7% to 87.9% when reaction pressure initially increased from 1 to 5 MPa, and then slightly decreased to 86.0% with further increase the reaction pressure to 6 MPa. The possible reason could be attributed to the increase of the pressure is beneficial to inhibit the side effects of deamination, but when the pressure was too high, excessive polymerization of PPDA was enhanced, leading to a decrease in the selectivity of CHDA products.
 |
| | Fig. 10 The reusability of 5%Ru/g-C3N4-600 catalysts. Reaction conditions: PPDA, 5 mmol; THF, 20 mL; reaction temperature, 130 °C; initial hydrogen pressure, 5 MPa; reaction time, 2.5 h; 25 wt% catalyst dosage (based on the weight of PPDA). | |
3.4 Catalyst recyclability
The recyclability of the Ru/g-C3N4-600 was tested and the reusability results are shown in Fig. 10. After the reaction, the catalyst of Ru/g-C3N4-600 was separated by centrifugation, then directly used in the next run with fresh reactants. 93.6% of PPDA conversion with 83.5% CHDA selectivity could be obtained during the second run. With the further increase of cycle numbers, the conversion of PPDA gradually decreased. For the catalyst used in fifth cycle, 72.2% PPDA conversion with 83.3% CHDA selectivity was obtained, indicating the catalytic activity decreased to some extent during recycling experiments. It should be noted that the selectivity of CHDA did not change a lot, and kept almost stable at about 83%. In order to examine the texture and structure changes of the catalyst, the catalyst after the five cycle (denoted as 5%Ru/g-C3N4-600-5th) was characterized by N2 physisorption and TEM and compared with the fresh one. The corresponding results are summarized in Fig. 11, S6† and Table 1. There are not obvious differences in the catalyst texture between the fresh and used catalyst of 5%Ru/g-C3N4-600-5th catalyst. The surface area, pore diameter, and pore volume were 83.17 m2 g−1, 18.26 nm and 0.43 (cm3 g−1), respectively, which did not change a lot compared with the fresh one. This result indicated that the catalyst retained its original mesoporous structure. The TEM results of the used 5%Ru/g-C3N4-600-5th catalyst are also shown in Fig. 11. TEM result Fig. 11(b) and (c) shown that the average size of Ru species in the used 5%Ru/g-C3N4-600-5th catalyst was about 3.95 nm, which was larger than that of the fresh catalyst (2.5 nm, Fig. 4(f)). Based on the above experimental results, the decreased catalytic activity of 5%Ru/g-C3N4-600 catalyst along with recycling experiments was probably attributed to the agglomeration of Ru nanoparticles.
 |
| | Fig. 11 N2 adsorption desorption isotherm (a) of the fresh 5%Ru/g-C3N4-600 catalyst (denoted as 5%Ru/g-C3N4-600-fresh) and the used 5%Ru/g-C3N4-600 catalyst after the five cycle (denoted as 5%Ru/g-C3N4-600-5th). Particle identification (b) and TEM image (c) of 5%Ru/g-C3N4-600-5th catalyst. | |
3.5 Substrate scope of the hydrogenation of aliphatic amine
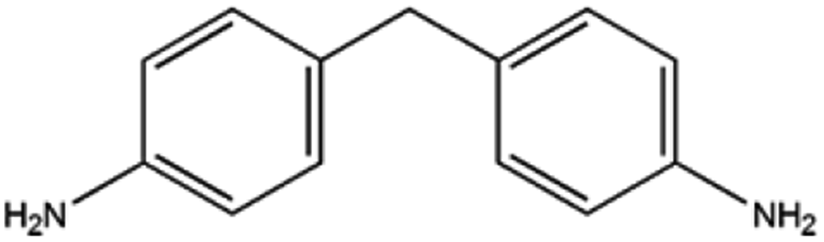
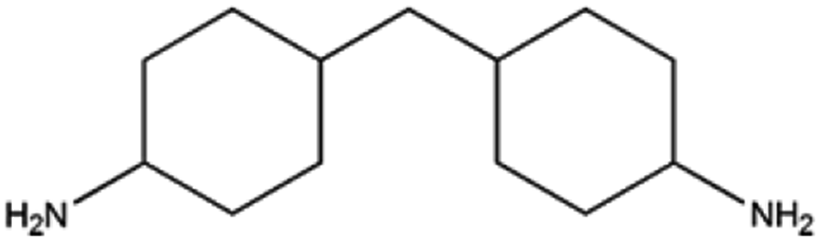
Alicyclic amines are widely used organic chemicals or intermediate compounds, which have important applications in pharmaceuticals, curing agent, rubber chemicals, etc. To illustrate the general applicability of 5%Ru/g-C3N4-600, the catalyst was also used to catalyse the hydrogenation of other aromatic monoamine or diamines, as shown in Table 3. As can be seen, 99% conversions of m-phenylene dimethylamine and 4,4-diaminodiphenylmethane were achieved in the hydrogenation process, entries 1, 2, and >99% excellent product selectivities of cyclohexylamine and 4,4′-diaminodicyclohexyl methane (H12MDA) could be obtained. It should be noted that, H12MDA is important raw materials for the synthesis of 4,4′-diisocyanato dicyclohexylmethane (H12MDI) via phosgene free route. In comparison with the conventional reaction system, this catalytic system has abandoned liquid alkali salt or liquid ammonia.10,27,55 Therefore, this study provided good candidate catalytic system for this important process. For the hydrogenation of xylylene diamine (XDA), entry 3, the conversion of selectivity of XDA could reach 99% with 73% selectivity of 1,3-cyclohexanedimethylamine. Furthermore, 5%Ru/g-C3N4-600 was also active in the hydrogenation of m-phenylene diamine.
Table 3 Substrate scope of the hydrogenation of Aromatic amine over 5%Ru/g-C3N4-600a
| Entry |
Substrate |
Product |
T (h) |
Conv. (%) |
Sel. (%) |
| Reaction conditions: 130 °C, 5 MPa H2, THF, 20 mL, the reactants: 5 mmol, catalyst dosage: 25 wt% (based on the weight of the reactants). |
| 1 |
 |
 |
2.5 |
>99 |
>99 |
| 2 |
 |
 |
2.5 |
>99 |
>99 |
| 3 |
 |
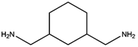 |
2.5 |
>99 |
73 |
| 4 |
 |
 |
24 |
96 |
82 |
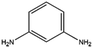
| 5 |
 |
 |
24 |
9 |
97 |
| 6 |
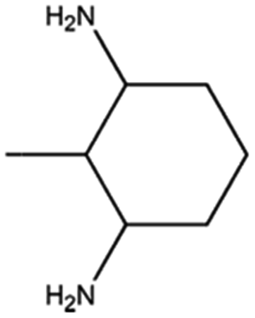
 |
 |
16 |
78 |
57 |
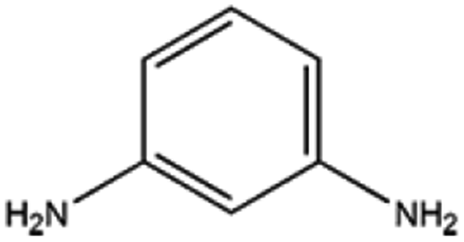
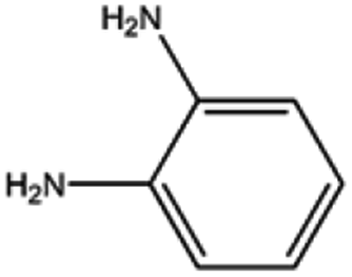
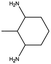
The catalyst exhibited 96% conversion with 82% of high product selectivity, entry 4. As reported in previous literature, 1,2-cycloheanediamine (1,2-DACH) is a promising chiral ligand. However, harsh reaction conditions are generally required and the selectivity of the desired product is relatively low over Ru/C catalyst. Interestingly, although conversion is relatively low, 5%Ru/g-C3N4-600 catalyst shown excellent selectivity of 97% toward the hydrogenation of o-phenylendiamine, entry 5. It's conjectured that the too strong coordination between Ru and vicinal amine groups of ortho diamines was considered to reduce the amount of Ru active sites, thus impose negative influence on the catalytic performance. For the substance of 2,6-diaminotoluene (TDA), entry 6, 78% TDA conversion with 57% moderate selectivity of 2,6-diamino-1-methylcyclohexane could be obtained. Based on the above results, it was found that the position of diamino group in the aromatic diamines have a great influence on the catalytic hydrogenation behaviour. These results demonstrated the unique behaviour of Ru NPs intimately associated to a basic support of g-C3N4 have generality for preparing alicyclic amines from aromatic amines, especially for the non ortho diamines.
4. Conclusions
In summary, a series of Ru/g-C3N4 catalysts using g-C3N4 support from different preparation temperatures were successfully prepared by ultrasonic impregnation method, which was firstly applied in hydrogenation of PPDA to 1,4-CHDA. These catalysts had mesoporous structures and formed by lamellar particle accumulation. Compared to Ru/C catalyst without skeleton nitrogen gradient, Ru/g-C3N4 catalyst with abundant N species exhibited superior catalytic performance on the target products selectivity in hydrogenation of PPDA. The involvement of nitrogen species in g-C3N4 is beneficial for Ru dispersion, and enriching electron around Ru0 environment. Ru is highly dispersed on g-C3N4 support for Ru/g-C3N4 catalysts, especially Ru with about 2.5 nm particle size could be obtained for 5%Ru/g-C3N4-600. Meanwhile, the basicity enhanced by nitrogen species in C3N4 improved the catalytic activity remarkably. The catalysts prepared at different temperatures were investigated and evaluated, and 5%Ru/g-C3N4-600 catalysts with the higher ruthenium dispersion and stronger basicity exhibited higher catalytic activity. In optimized reaction conditions over 5%Ru/g-C3N4-600 catalyst, PDA conversion of 100% with the CHDA selectivity of more than 86% were achieved under relatively mild reaction conditions, i.e. reaction temperature of 130 °C and the pressure of 5 MPa H2 within 2.5 h. Additionally, the catalyst recyclability demonstrated that the aggregation of Ru nanoparticals was responsible for the decrease of the catalytic activity. In addition, to some extent, Ru/g-C3N4-600 have universality for preparing alicyclic amines from aromatic amines. This work has provided green and efficient catalytic system for the hydrogenation of aromatic diamines to synthesize value-added alicyclic diamines. Meanwhile, this strategy of regulating the catalytic performance via tuning the support property has provided insights for the rational design of emerging hydrogenation catalyst without the involvement of additional alkali salt in the system.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the financial support from “Transformational Technologies for Clean Energy and Demonstration”, Strategic Priority Research Program of the Chinese Academy of Sciences, Grant No. XDA 21030600. We also acknowledge the projects from the National Natural Science Foundation of China (21576272), and Science and Technology Service Network Initiative, Chinese Academy of Sciences (KFJ-STS-QYZD-138) for financial supports.
Notes and references
- Y. J. Kim, J. H. Lee, V. T. Widyaya, H. S. Kim and H. Lee, Bull. Korean Chem. Soc., 2014, 35, 1117–1120 CrossRef.
- B. Vanhaecht, B. Rimez, R. Willem, M. Biesemans and C. E. Koning, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1962–1971 CrossRef.
- H. Ma and J. Cai, Russ. J. Appl. Chem., 2014, 87, 397–403 CrossRef.
- A. Fischer, T. Mallat and A. Baiker, J. Catal., 1999, 182, 289–291 CrossRef CAS.
- A. T. Nielsen, J. Org. Chem, 1962, 27, 1998–2001 CrossRef CAS.
- C. Liu, Y. Zhang and J. Cai, Can. J. Chem. Eng., 2018, 97, 917–923 CrossRef.
- L. Zuo, J. X. Cai, C. Yang, C. Hao, Y. C. Fu and J. Y. Shen, Ind. Eng. Chem. Res., 2019, 58, 19456–19464 CrossRef CAS.
- X. Lu, J. He, R. Jing, P. Tao, R. Nie, D. Zhou and Q. Xia, Sci. Rep., 2017, 7, 2676 CrossRef.
- R. Jagadeesh, K. Murugesan, A. Alshammari, H. Neumann, M. Pohl, J. Radnik and M. Beller, Science, 2017, 358, 326–332 CrossRef CAS PubMed.
- H. Li, Z. Li, D. Ji and G. Li, Catal. Commun., 2018, 104, 91–95 CrossRef CAS.
- M. Chatterjee, M. Sato, H. Kawanami, T. Ishizaka, T. Yokoyama and T. Suzuki, Appl. Catal., A, 2011, 396, 186–193 CrossRef CAS.
- K. Hindle, S. Jackson, D. Stirling and G. Webb, J. Catal., 2006, 241, 417–425 CrossRef CAS.
- L. Jia, Q. Zhang, Q. Li and H. Song, Nanotechnology, 2009, 20, 385601 CrossRef PubMed.
- A. Suzuki, H. Miyamura and S. Kobayashi, Synlett, 2019, 30, 387–392 CrossRef CAS.
- P. Tomkins, E. Gebauer-Henke and T. E. Müller, ChemCatChem, 2016, 8, 546–550 CrossRef CAS.
- X. J. Cui, A. E. Surkus, K. Junge, C. Topf, J. Radnik, C. Kreyenschulte and M. Beller, Nat. Commun., 2016, 7, 11326 CrossRef.
- Z. H. Chen, H. J. Sun, Z. K. Peng, J. Gao, B. J. Li, Z. Y. Liu and S. C. Liu, Ind. Eng. Chem. Res., 2019, 58, 13794–13803 CrossRef CAS.
- P. Tomkins, E. Gebauer-Henke, W. Leitner and T. E. Muller, ACS Catal., 2015, 5, 203–209 CrossRef CAS.
- V. Mishra, J. K. Cho, S. H. Shin, Y. W. Suh, H. S. Kim and Y. J. Kim, Appl. Catal., A, 2014, 487, 82–90 CrossRef CAS.
- P. Tomkins and T. E. Müller, ChemCatChem, 2018, 10, 1438–1445 CrossRef CAS.
- E. Gebauer-Henke, P. Tomkins, W. Leitner and T. E. Müller, ChemCatChem, 2014, 6, 2910–2917 CrossRef CAS.
- V. Mishra, J. K. Cho, S.-H. Shin, Y.-W. Suh, H. S. Kim and Y. J. Kim, Appl. Catal., A, 2014, 487, 82–90 CrossRef CAS.
- J. Huang, X. Guo, G. Yue, Q. Hu and L. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 44403–44414 CrossRef CAS PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Y. Wang, J. Yao, H. Li, D. Su and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef CAS PubMed.
- D. Liu, P. Yang, H. Zhang, M. Liu, W. Zhang, D. Xu and J. Gao, Green Chem., 2019, 21, 2129–2137 RSC.
- Y. Zhang, H. Yang, Q. Chi and Z. Zhang, ChemSusChem, 2019, 12, 1246–1255 CrossRef CAS PubMed.
- R. F. Nie, H. Z. Jiang, X. H. Lu, D. Zhou and Q. H. Xia, Catal. Sci. Technol., 2016, 6, 1913 RSC.
- T. J. Li, H. F. Lin, X. P. Ouyang, X. Q. Qiu and Z. C. Wan, ACS Catal., 2019, 9, 5828–5836 CrossRef CAS.
- X. Y. Liu, G. J. Lan, Y. Boyjoo, L. H. Qian, S. Gu, C. A. H. Price, L. Wang, Y. Li and J. Liu, Chem. Eng. J., 2019, 374, 895–903 CrossRef CAS.
- P. Chen, F. Liu, S. Chen, J. K. Guo, S. Shen, L. Chen, C.-T. Au and S. F. Yin, Chem. Eng. Sci., 2019, 207, 271–279 CrossRef CAS.
- L. M. Martinez-Prieto, M. Puche, C. Cerezo-Navarrete and B. Chaudret, J. Catal., 2019, 377, 429–437 CrossRef CAS.
- Y. J. Wu, T. Wang, H. L. Wang, X. Z. Wang, X. C. Dai and F. Shi, Nat. Commun., 2019, 10, 2599 CrossRef PubMed.
- C. S. Lu, X. J. Zhang, Y. N. Qi, H. K. Ji, Q. W. Zhu, H. Wang, Y. B. Zhou, Z. L. Feng and X. N. Li, ChemistryOpen, 2019, 8, 87–96 CrossRef CAS.
- S. Min and G. Lu, J. Phys. Chem. C, 2012, 116, 19644–19652 CrossRef CAS.
- H. Li, L. Wang, Y. Liu, J. Lei and J. Zhang, Res. Chem. Intermed., 2015, 42, 3979–3998 CrossRef.
- S. N. Talapaneni, G. P. Mane, D.-H. Park, K. S. Lakhi, K. Ramadass, S. Joseph, W. M. Skinner, U. Ravon, K. Al-Bahily and A. Vinu, J. Mater. Chem. A, 2017, 5, 18183–18192 RSC.
- Y. Guo, Q. Liu, Z. Li, Z. Zhang and X. Fang, Appl. Catal., B, 2018, 221, 362–370 CrossRef CAS.
- S. Sun and S. Liang, Nanoscale, 2017, 9, 10544–10578 RSC.
- M. Peer, M. Lusardi and K. F. Jensen, Chem. Mater., 2017, 29, 1496–1506 CrossRef CAS.
- M. Peer, M. Lusardi and K. F. Jensen, Chem. Mater., 2017, 29, 1496–1506 CrossRef CAS.
- F. Li, L. Wang, X. Han, Y. Cao, P. He and H. Li, Int. J. Hydrogen Energy, 2017, 42, 2144–2156 CrossRef CAS.
- X. Wang, D. Li and Z. Nan, Sep. Purif. Technol., 2019, 224, 152–162 CrossRef CAS.
- A. B. Jorge, D. J. Martin, M. T. S. Dhanoa, A. S. Rahman, N. Makwana, J. Tang, A. Sella, F. Corà, S. Firth, J. A. Darr and P. F. McMillan, J. Phys. Chem. C, 2013, 117, 7178–7185 CrossRef CAS.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed. Engl., 2014, 53, 9240–9245 CrossRef CAS.
- X. Wang and Z. Nan, Sep. Purif. Technol., 2020, 233, 116023 CrossRef CAS.
- Q. Su, J. Sun, J. Wang, Z. Yang, W. Cheng and S. Zhang, Catal. Sci. Technol., 2014, 4, 1556–1562 RSC.
- E. Qu, J. Luo, X. Di, C. Li and C. Liang, J. Nanosci. Nanotechnol., 2020, 20, 1140–1147 CrossRef PubMed.
- L. Li, Z. H. Zhu, Z. F. Yan, G. Q. Lu and L. Rintoul, Appl. Catal., A, 2007, 320, 166–172 CrossRef CAS.
- W. Xu, M. Dong, L. Di and X. Zhang, Nanomaterials, 2019, 9, 1432–1443 CrossRef CAS PubMed.
- Z. Sun, Z. Liu, B. Han, S. Miao, Z. Miao and G. An, J. Colloid Interface Sci., 2006, 304, 323–328 CrossRef CAS PubMed.
- S. Armenise, L. Roldan, Y. Marco, A. Monzon and E. Garcia-Bordeje, J. Phys. Chem. C, 2012, 116, 26385–26395 CrossRef CAS.
- D. Mitoraj and H. Kisch, Angew. Chem., Int. Ed. Engl., 2008, 47, 9975–9978 CrossRef CAS PubMed.
- F. Dong, L. Wu, Y. Sun, M. Fu, Z. Wu and S. C. Lee, J. Mater. Chem., 2011, 21, 15171–15174 RSC.
- Y. K. Zhong, F. Feng, C. S. Lu, Q. F. Zhang, J. H. Lv, X. L. Xu, L. Ma and X. N. Li, J. Chem. Eng. Chin. Univ., 2017, 6, 1379–1388 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00836b |
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bcde,
Xiaoyu Huangab,
Yan Caob,
Peng Heb,
Huanhuan Yangbcd,
Jiaqiang Chenb and
Huiquan Li
*bcde,
Xiaoyu Huangab,
Yan Caob,
Peng Heb,
Huanhuan Yangbcd,
Jiaqiang Chenb and
Huiquan Li