
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntioxidant activities of anastatin A & B derivatives and compound 38c's protective effect in a mouse model of CCl4-induced acute liver injury
Cen Xiang a,
Menglin Caoa,
Ai Miaoa,
Feng Gaoa,
Xuzhe Lia,
Guojun Panb,
Wenqiang Zhanga,
Yongmin Zhang*ac,
Peng Yu*a and
Yuou Teng*a
a,
Menglin Caoa,
Ai Miaoa,
Feng Gaoa,
Xuzhe Lia,
Guojun Panb,
Wenqiang Zhanga,
Yongmin Zhang*ac,
Peng Yu*a and
Yuou Teng*a
aChina International Science and Technology Cooperation Base of Food Nutrition/Safety and Medicinal Chemistry, Tianjin University of Science and Technology, Tianjin 300457, China. E-mail: tyo201485@tust.edu.cn; yupeng@tust.edu.cn
bSchool of Life Sciences, Shandong First Medical University, Shandong Academy of Medical Sciences, Changcheng Road 619, Tai'an City 271000, Shandong province, China
cSorbonne Université, Institut Parisien de Chimie Moléculaire, UMR8232 CNRS, 4 Place Jussieu, 75005 Paris, France. E-mail: yongmin.zhang@upmc.fr
First published on 14th April 2020
Abstract
Anastatins A and B, two flavonoid compounds isolated from desert plant Anastatica hierochuntica, have protective activities for primary rat hepatocytes. Anastatins A and B, and their derivatives, were synthesized by our group previously. In this study, the antioxidant activity and cytotoxicity of these compounds were studied using chemical assessment methods, cell proliferation inhibition experiments, and cell oxidative damage models. The best compound, 38c, was used to study the hepatoprotection activity and mechanism by using a CCl4-induced liver injury model in mice. The results show that most of these flavonoid compounds have good antioxidant activity and low cytotoxicity in vitro. Among them, the most potent compound was 38c, which exhibited a protective effect on CCl4-induced hepatic injury by suppressing the amount of CYP2E1. These findings indicate that anastatin flavonoid derivatives have potential therapeutic utility against oxidative hepatic injury.
Introduction
More than 4000 uniquely structured flavonoids have been identified in plants, most of which are found in vegetables, fruits, herbs, nuts, seeds, spices, stems, flowers, tea, and red wine.1–5 Most flavonoids have pharmacological activities such as antitumor, antioxidant, antiviral, and anti-inflammatory effects.6–13 Two new flavonoids, anastatins A and B (Fig. 1A and B), were isolated from Anastatica hierochuntica in Egypt by Yoshikawa.14 A preliminary biological evaluation revealed that both compounds exhibited protective effects on D-galactosamine-induced cytotoxicity in primary cultured mouse hepatocytes. Moreover, their protective activities were stronger than the well-known and commercial hepatoprotective drug, silybin.15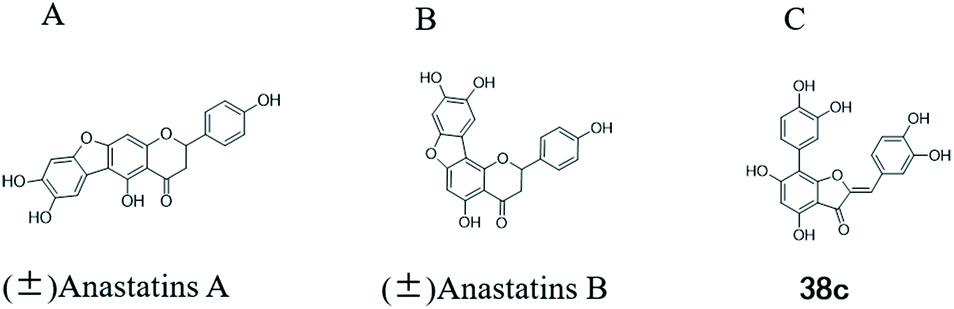 | ||
| Fig. 1 Chemical structure of anastatins A (A), anastatins B (B) and 38c (C). | ||
Liver damage is caused by a variety of pathogenic factors, such as oxidative stress, immune reactions, hepatitis viruses, ethanol, drugs, and various toxic substances.16–20 This damage can progress to liver fibrosis and cirrhosis.21,22 An accumulation of free radicals is an important pathogenic mechanism leading to liver injury. When the amount of chemical poisons and drugs ingested by the body is excessive, or genetic metabolic disorders occur, a large amount of reactive metabolites, such as electrophilic groups and free radicals, are generated. These can then damage unsaturated fatty acids in phospholipid molecules in cellular membranes causing changes in membrane structure and permeability.23 Oxidative changes in the structure and function of the membrane leads to liver cell damage.24,25 Therefore, developing new and highly effective antioxidant drugs is very important to prevent or reduce the accumulation of free radicals, and provide hepatoprotection.
The hepatoprotective effects of anastatins A and B appear to be related to their antioxidant activity. To identify new types of antioxidants and study their mechanisms of action, chemical and cell-based assessments, and in vivo studies are needed.26 The chemical antioxidant evaluation method is fast, reproducible, and convenient. Antioxidant capacity can be demonstrated by the ability of a chemical to inhibit or remove free radicals such as 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS) and 2,2-diphenyl-1-picrylhydrazyl (DPPH).
H2O2 is the main reactive oxygen species with high cell membrane permeability, and can cause intracellular lipid peroxidation and DNA damage. It is often used to mimic oxidative stress-induced injury in vitro. H2O2-induced oxidative damage in PC12 and L02 cells can be used to evaluate the properties of antioxidants in vitro.27 The PC12 cell line was derived from the adrenal medulla of a rat pheochromocytoma, and is a model system used for neurological and neurochemical studies. L02 cells were derived from human normal liver cells and are a model system for human liver disease research.28
A mouse model of CCl4-induced liver injury is often used to evaluate the in vivo effects of antioxidants.29,30 CCl4 is metabolized by cytochrome P450 2E1 (CYP2E1) to generate unstable trichloromethyl radicals, peroxyl trichloromethyl radicals, and reactive oxygen species.31,32 The main mechanism of CCl4-induced liver injury involves the trichloromethyl metabolite and other free radicals.
We have previously synthesized anastatins A and B and a series of their derivatives (including compound 38c).33 See Appendix A for their chemical structures. In our earlier research on compound 1 and 2 of anastatins B derivatives, we found that both compounds 1 and 2 had good antioxidant and radical scavenging abilities in vitro. Both compounds showed cytoprotective activity in H2O2-treated PC12 cells. Both of they were potent hepatoprotectants in a mouse model of CCl4-induced hepatotoxicity.34 The result suggests that our series of compounds from anastatins A and B are a good class of antioxidants. This study aimed to investigate the antioxidant and hepatoprotective effects of analogs of anastatins A and B by in vitro chemical evaluation experiments, H2O2-induced oxidative damage in PC12 and L02 cells, and a mouse CCl4-induced liver injury model.
Materials and methods
Materials
ABTS, DPPH, thiazolyl blue tetrazolium bromide (MTT), ferric chloride (FeCl3), vitamin C (VC), carbon tetrachloride (CCl4), were purchased from Sigma-Aldrich (Beijing, China). Glacial acetic acid, gallic acid (GA) was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China), tri-distilled water was produced by a Milli-Q system (Molsheim, France). Cereal third transaminase (ALT), aspartate transaminase (AST), lactate dehydrogenase (LDH), glutathione (GSH), CYP2E1 ELISA assay kit were produced by Nanjing Jiancheng Bioengineering Institute (Nanjing, China).Sodium acetate buffer solution was prepared as follow: accurately weighed sodium acetate 0.246 g (3 mmol) powder completely dissolved in 100 mL of water to prepare a 30 mM sodium acetate solution; accurately measured 0.172 mL (3 mmol) of glacial acetic acid and added it to 99.828 mL of water to prepare 30 mM acetic acid solution; accurately measure 7.5 mL of sodium acetate solution and 92.5 mL of acetic acid solution, adjust the pH of mixture to 3.6.
Concentrated ABTS˙+ solution was prepared as follow: accurately weighed 0.549 g (1 mmol) of ABTS powder, completely dissolved in 100 mL of sodium acetate buffer, and then added 35% H2O2 172.7 μL to it, store at 4 °C in the dark environment, overnight. This solution was kept for 6 months at 4 °C. Working solutions (50 μM) were prepared by diluting the concentrated ABTS˙+ solution in sodium acetate buffer solution.
DPPH˙ ethanol solution was prepared as follow: accurately weighed 19.7 mmol DPPH, dissolve and mix in 250 mL of absolute ethanol to obtain a 78.8 mM DPPH ethanol solution, and store at 4 °C in the dark environment.
Antioxidant activity by ABTS˙+ assay
VC and GA, two well-known potent antioxidant, were used as a positive control. In the system control group (Acontrol), added 130 μL of ABTS˙+ working solution and 5.5 μL DMSO; in the system blank group (Ablank), added 130 μL of sodium acetate buffer solution and 5.5 μL DMSO; in the sample control group (A1), added 130 μL of sodium acetate buffer solution and 5.5 μL of samples or positive control (VC or GA) to be tested at 0.02, 0.06, 0.2, 0.6, 2, 6 and 20 mM in DMSO; in the sample group (A2), added 130 μL of ABTS˙+ working solution and 5.5 μL of the same samples or positive control as group A1. All groups were shaken thoroughly for 30 s, and let stand at room temperature for 10 min. OD measured at 650 nm by Synergy H1 Hybrid Multi-Mode Microplate Reader (Winooski, USA). Each group performed three replicates and averaged. Finally, the measured values are calculated by the following formula (1) to obtain the ABTS˙+ radical scavenging rate of the test compound.
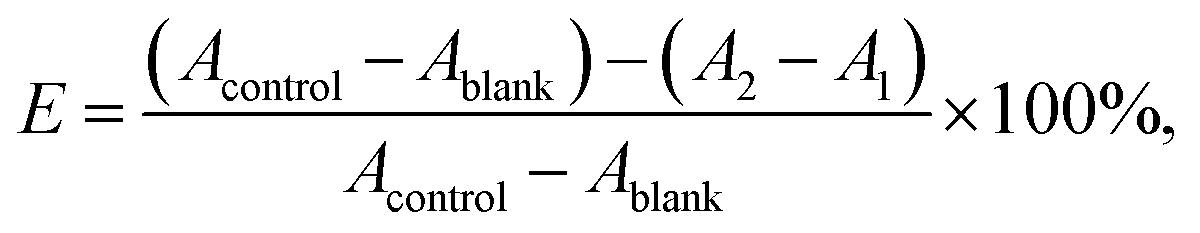 | (1) |
EC50 values for the test compound were calculated from three independent experiments by nonlinear regression using GraphPad Prism 7.0.
Antioxidant activity by DPPH˙ assay
In the system control group (Acontrol), added 50 μL DPPH˙ working solution and 50 μL DMSO; in the system blank group (Ablank), added 50 μL of absolute ethanol and 50 μL of the sample solution in DMSO; in the sample control group (A1), added 50 μL of absolute ethanol and 50 μL of sample or positive control (VC or GA) in each concentration (0.02, 0.06, 0.2, 0.6, 2, 6 and 20 mM) in DMSO; in the sample group (A2), added 50 μL of DPPH˙ working solution and 50 μL of the same samples or positive control as group A1. All groups incubation at 37 °C for 30 minutes, placed the liquid in a 96-well plate, and measure the OD value at 540 nm by Synergy H1 Hybrid Multi-Mode Microplate Reader. Each group performed three replicates and averaged. Finally, the measured values are calculated by the following formula (2) to obtain the DPPH˙ radical scavenging rate of the test compound.
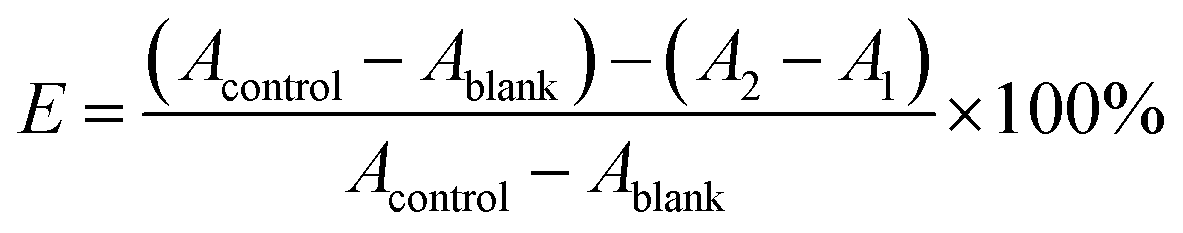 | (2) |
EC50 values for the test compound were calculated from three independent experiments by nonlinear regression using GraphPad Prism 7.0.
Cell lines and culture conditions
PC12 and L02 cell line were obtained from the Shanghai Institutes of Biological Sciences (Shanghai, China). Cells were grown at 37 °C in RPMI-1640 (Gibco; Beijing; China) supplemented with 10% fetal bovine serum, 2.05 mM glutamine, and 1% penicillin/streptomycin in a humidified atmosphere containing 5% CO2. The medium was replaced once every third day.Cytotoxic activity assay
Inhibition of cell proliferation by test compound or positive control (GA) were measured by the MTT assay. In brief, PC12 was plated in 96-well plates as 5 × 105 cells per well in 90 μL medium. After 16 h incubation, DMSO, test compound (1, 10, 100 μM), or GA (1, 10, 100 μM) were added to each well and incubated for 0.5 h. Then, 10 μL medium was added and cells incubated for another 2 h. After 2 h of incubation, 20 μL MTT (5 mg mL−1) was added to each well and the plates were further incubated for 4 h. MTT assay was performed using Synergy H1 Hybrid Multi-Mode Microplate Reader. The DMSO-treated controls were calculated as a cell viability value of 100%. The inhibitory concentrations (IC50) were obtained by nonlinear regression using GraphPad Prism 7.0. For each experiment, IC50 value was calculated from three independent assays.MTT assay for H2O2-treated PC12 or L02 cells
H2O2-induced oxidative-damage PC12 or L02 cell model were established and the cell viability was evaluated by MTT assay as a measure of the antioxidant activity of test compound GA, was used as a positive control in MTT assay. PC12 cells were plated in 96-well plates as 5 × 105 cells per mL in 90 μL medium. After 16 h incubation, 0.5 μL DMSO, test compound (20 mM in DMSO) or GA (20 mM in DMSO) were added to each well and incubated for 0.5 h. Afterwards, 10 μL H2O2 (1 mM in medium) was added to half of a 96-well plate, equal volume of medium was added to the other half of the 96-well plate for 2 h to induce cell injury. After 2 h, 20 μL MTT (5 mg mL−1) was added to each well and the plates were further incubated for 4 h. MTT assay was performed as mentioned above. In the same compound concentration group, the medium controls were assigned a cell viability value of 100%.L02 cells were plated in 96-well plates as 1 × 105 cells per mL in 90 μL medium. After 24 h incubation, 0.5 μL DMSO, test compound (20 mM in DMSO), or GA (20 mM in DMSO) were added to each well and incubated for 0.5 h. Afterwards, treatment with the same way as H2O2-induced oxidative-damage PC12 cells model.
Animal grouping and drug administration
Kunming mice (male, 4 weeks, 18–22 g) were purchased from Laboratory Animal Center of the Academy of Military Medical Sciences (Beijing, China). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Tianjin University of Science and Technology and approved by the Animal Ethics Committee of Tianjin University of Science and Technology. The mice were housed at 25 ± 2 °C under a 12 hours light/12 hours dark cycle with access to food and water libitum. After acclimation for one week, the animals were randomly divided into 5 groups (n = 8): normal (tri-distilled water without CCl4 injection), model (tri-distilled water and CCl4 injection), compound 38c (Fig. 1C) treatment groups (50, 100, and 200 mg kg−1 of compound 38c and CCl4 injection), and positive control group (200 mg kg−1 of biphenyldicarboxylate pills (BP, Beijing Union Pharmaceutical Factory, Beijing, China) and CCl4 injection). Compound 38c and BP were dissolved in tri-distilled water. Tri-distilled water, compound 38c or BP was i.g. for 10 consecutive days before CCl4 injection. 1 h after the final drug treatment, severe, acute liver damage was induced by i.p. injection with CCl4 (0.25% (v/v) peanut oil mixture; 10 mL kg−1). The normal group was intraperitoneally injected with peanut oil. All mice were starved for 20 h afterwards, and were then sacrificed for collection of whole blood and liver samples. Liver tissue was removed, immediately weighed, and then fixed or stored in 10% neutral buffered formalin, or frozen for histopathological analysis and determination of biochemical parameters.Histological analysis
For evaluation of hepatotoxicity, the tissue fixed in 10% formalin was embedded in paraffin and cut into 5 μm-thick sections for histomorphological examination. After drying, hepatic tissue section slides were stained with hematoxylin–eosin (H&E). The stained sections were visualized using a microscope.Biochemical analysis
Serum was analyzed for ALT, AST, LDH and CYP2E1 levels according to the manufacturer's instructions. GSH levels were determined in hepatic homogenates using a commercial kit. The results were corrected for their protein content.Western blot
Briefly, liver tissue homogenized in pre-cooled normal saline. Equal amount of protein were separated by 10% SDS-PAGE and electro-transferred onto a PVDF membrane. After blocking with 5% (w/v) nonfat milk, the membranes were incubated with CYP2E1 antibodies (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 diluted, Tianjin Sungene Biotech Co. Ltd.; Tianjin, China) overnight at 4 °C, and then incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody (1:2000 diluted, Tianjin Sungene Biotech Co. Ltd.; Tianjin, China) for 1 h. The immunoblots were visualized using an Odyssey infrared imaging system. An anti-tubulin antibody (1:1000 diluted, Tianjin Sungene Biotech Co. Ltd.; Tianjin, China) was used as a control for equal loading. The densities of protein bands were determined using ImageJ software.
:1000 diluted, Tianjin Sungene Biotech Co. Ltd.; Tianjin, China) overnight at 4 °C, and then incubated with a horseradish peroxidase (HRP)-conjugated secondary antibody (1:2000 diluted, Tianjin Sungene Biotech Co. Ltd.; Tianjin, China) for 1 h. The immunoblots were visualized using an Odyssey infrared imaging system. An anti-tubulin antibody (1:1000 diluted, Tianjin Sungene Biotech Co. Ltd.; Tianjin, China) was used as a control for equal loading. The densities of protein bands were determined using ImageJ software.
Statistical analysis
All data were expressed as mean ± S.D. The data were analyzed by one-way analysis of variance (ANOVA) and significant differences were determined by post hoc Tukey's test using GraphPad Prism 7.0. P value < 0.05 was considered as significant.Results
ABTS˙+ scavenging abilities
The antioxidant activity of test compound in various concentrations was evaluated using in vitro models. It was observed that the test compounds scavenged free radicals in concentration dependent manner in all the models. The antioxidant activity was expressed as EC50 (the amount of antioxidant needed to decrease the radical concentration by 50%), which is negatively related to antioxidant activity. The lower the EC50 value, the higher is the antioxidant activity of the tested sample. Table 1 shows that the ABTS˙+ scavenging capacities (EC50) of VC and GA were 2.11 and 0.56 mM, respectively. This indicated that the ABTS˙+ scavenging ability of GA was better than VC. The EC50 values of compounds 42c, 44c, 40a, 38c and 40c were 0.88 ± 0.24, 0.88 ± 0.10, 1.03 ± 0.07, 0.88 ± 0.32, and 0.91 ± 0.01 mM, respectively, and were not significantly different from GA (P > 0.05), but were better than VC (P < 0.05). These results show that compounds 42c, 44c, 40a, 38c and 40c have good antioxidant activity.| Name | ABTS˙+ scavenging activity (EC50, mM) | Name | ABTS˙+ scavenging activity (EC50, mM) |
|---|---|---|---|
| a Data are presented as means ± SD.b *P < 0.05 vs. VC, #P < 0.05 vs. GA. | |||
| 42a | 1.06 ± 0.08*,# | 38a | 1.05 ± 0.09*,# |
| 43a | 2.78 ± 0.27*,# | 39a | 2.99 ± 0.24*,# |
| 44a | 1.06 ± 0.02*,# | 40a | 1.03 ± 0.07* |
| 42b | 1.18 ± 0.05*,# | 38b | 1.08 ± 0.04*,# |
| 43b | 2.96 ± 0.44*,# | 39b | 2.62 ± 0.74# |
| 44b | 1.16 ± 0.07*,# | 40b | 1.23 ± 0.17*,# |
| 42c | 0.88 ± 0.24* | 38c | 0.88 ± 0.32* |
| 42c-1 | 1.10 ± 0.01*,# | 38c-1 | 1.13 ± 0.01*,# |
| 43c | 2.18 ± 0.65*,# | 39c | 3.34 ± 0.52*,# |
| 44c | 0.88 ± 0.10* | 40c | 0.91 ± 0.01* |
| 19 | 1.05 ± 0.09*,# | 20 | 1.05 ± 0.10*,# |
| 31 | 3.18 ± 0.18*,# | 30 | 2.99 ± 0.09*,# |
| (±) Anastatins A | 1.11 ± 0.11*,# | (±) Anastatins B | 1.19 ± 0.12*,# |
| VC | 2.11 ± 0.39# | GA | 0.56 ± 0.12* |
DPPH˙ scavenging abilities
Table 2 shows that the DPPH˙ EC50 values of VC and GA were 0.23 and 0.05 mM, respectively. This indicates that the DPPH˙ scavenging activity of GA is better than VC. The EC50 values of compounds 42c-1, 19, 38a, 38b, 38c, 38c-1, 40c, and 20 were 0.04 ± 0.01, 0.07 ± 0.02, 0.08 ± 0.02, 0.08 ± 0.01, 0.06 ± 0.01, 0.06 ± 0.01, 0.05 ± 0.01, and 0.06 ± 0.01 mM, respectively. These were equivalent to the GA value (P > 0.05), but were higher than that of VC (P < 0.05). These results show that compounds 42c-1, 19, 38a, 38b, 38c, 38c-1, 40c, and 20 have good antioxidant activity.| Name | DPPH˙ scavenging activity (EC50, mM) | Name | DPPH˙ scavenging activity (EC50, mM) |
|---|---|---|---|
| a Data are presented as means ± SD.b *P < 0.05 vs. VC, P < 0.05 vs. GA. | |||
| 42a | 0.13 ± 0.04 | 38a | 0.08 ± 0.02* |
| 43a | 0.20 ± 0.21# | 39a | >20*,# |
| 44a | 0.13 ± 0.07 | 40a | 0.13 ± 0.09 |
| 42b | 0.43 ± 0.06*,# | 38b | 0.08 ± 0.01* |
| 43b | >20*,# | 39b | 0.32 ± 0.24# |
| 44b | 0.28 ± 0.01# | 40b | 0.10 ± 0.01* |
| 42c | 0.42 ± 0.06*,# | 38c | 0.06 ± 0.01# |
| 42c-1 | 0.04 ± 0.01*,# | 38c-1 | 0.06 ± 0.01# |
| 43c | >20*,# | 39c | 0.11 ± 0.11 |
| 44c | 0.67 ± 0.11*,# | 40c | 0.05 ± 0.01* |
| 19 | 0.07 ± 0.02* | 20 | 0.06 ± 0.01* |
| 31 | >20*,# | 30 | >20*,# |
| (±) Anastatins A | 0.12 ± 0.12 | (±) Anastatins B | 0.17 ± 0.11 |
| VC | 0.23 ± 0.09 # | GA | 0.05 ± 0.01* |
Toxicity of anastatins A and B, and their derivatives, on PC12 cells
Compounds 38a, 39a, 40a, and 40b, and anastatins B were more toxic to PC12 cells than the other compounds, with IC50 values of 44.69 ± 1.38, 32.82 ± 2.64, 31.54 ± 0.97, 53.41 ± 0.85, and 49.45 ± 2.03 μM, respectively (Table 3). The IC50 values of the other compounds, and the positive control (GA), were all greater than 100 μM, indicating that their toxicity to PC12 cells was relatively low.| Name | IC50 (μM) | Name | IC50 (μM) |
|---|---|---|---|
| a Data are presented as means ± SD. | |||
| 42a | >100 | 38a | 44.69 ± 1.38 |
| 43a | >100 | 39a | 32.82 ± 2.64 |
| 44a | >100 | 40a | 31.54 ± 0.97 |
| 42b | >100 | 38b | >100 |
| 43b | >100 | 39b | >100 |
| 44b | >100 | 40b | 53.41 ± 0.85 |
| 42c | >100 | 38c | >100 |
| 42c-1 | >100 | 38c-1 | >100 |
| 43c | >100 | 39c | >100 |
| 44c | >100 | 40c | >100 |
| 19 | >100 | 20 | >100 |
| 31 | >100 | 30 | >100 |
| (±) Anastatins A | >100 | (±) Anastatins B | 49.45 ± 2.03 |
| GA | >100 | ||
Antioxidant activities of anastatins compounds at final concentrations of 10 μM in PC12 cells
H2O2 alone at 100 μM decreased the viability of PC12 cells to 16.8 ± 1.27% of control (Fig. 2). Treatment of the cells with 10 μM of the positive control, GA, increased viability to 43.9 ± 4.50%. In contrast, 10 μM of anastatins A, B and compounds 19, 38a, 39a, 40a, 38b, 39b, 40b, 38c, 39c, 20 and 30 increased PC12 cell viability from 16.8 to 87.4 ± 3.8, 74.55 ± 0.56, 107.6 ± 2.71, 99.98 ± 2.68, 70.97 ± 3.76, 81.57 ± 3.33, 70.59 ± 3.82, 83.33 ± 2.47, 81.54 ± 3.84, 85.94 ± 1.16, 83.27 ± 2.63, 63.11 ± 3.8 and 68.5 ± 3.95%, respectively. These increases were significantly greater than that of GA and indicate that these compounds can protect against H2O2-induced oxidative injury in PC12 cells.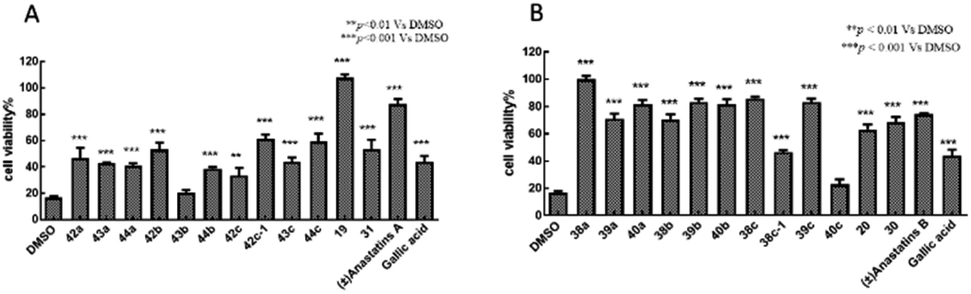 | ||
| Fig. 2 Antioxidant activities of anastatins A and B, and their derivatives in PC12 cells. PC12 cells were treated with 10 μM of each compound in DMSO then exposed to 100 mM H2O2. Cell viability was assessed with the MTT assay. DMSO was used as the control and set to 100% viability. GA was used as the positive control. (A) Anastatins A and its derivatives. (B) Anastatins B and its derivatives. Data are expressed as means ± SD. **P < 0.01, ***P < 0.001 compared to DMSO-treated control cells. | ||
Antioxidant activities of anastatins compounds at final concentrations of 10 μM in L02 cells
H2O2 alone at 100 μM decreased the viability of L02 cells to 39.9% of control (Fig. 3). Treatment of these cells with 10 μM of the positive control, GA, had no effect on the cell survival rate (37.8%). In contrast, 10 μM of anastatins B, and compounds 40b and 38c, increased viability from 39.9 to 89.5, 90.1 and 78.1%, respectively. These increases were significantly greater than that of GA and indicate that these compounds can protect against H2O2-induced oxidative injury in L02 cells.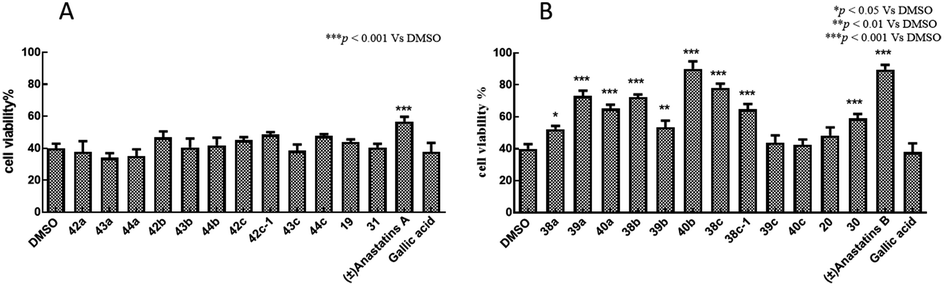 | ||
| Fig. 3 Antioxidant activities of anastatins A and B, and their derivatives in L02 cells. L02 cells were treated with 10 μM of each compound in DMSO then exposed to 100 mM H2O2. Cell viability was assessed with the MTT assay. DMSO was used as the control and set to 100% viability. GA was used as the positive control. (A) Anastatins A and its derivatives. (B) Anastatins B and its derivatives. Data are expressed as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 compared to DMSO-treated control cells. | ||
In summary, the 38c derivative of anastatins A and B showed good antioxidant activity and lower cytotoxicity in vitro. Thus, 38c was selected as the candidate compound for in vivo studies.
Compound 38c ameliorated CCl4-induced liver injury
To determine the protective effects of 38c on CCl4-induced hepatotoxicity, mice were pretreated with different doses by gavage once daily for ten days. H&E staining showed that CCl4 administration caused massive necrosis in the liver (Fig. 4). Pretreatment with 38c at 50, 100, and 200 mg kg−1 ameliorated CCl4-induced hepatic necrosis (Fig. 4). Serum levels of AST (Fig. 5A), ALT (Fig. 5B), and LDH (Fig. 5C), markers of liver injury, dramatically increased after CCl4 treatment. Consistent with the histological data, pretreatment with 38c 200, or 100 mg kg−1 significantly reduced serum AST, ALT, and LDH levels in CCl4-treated mice. Compound 38c at a dose of 50 mg kg−1 did not show good activity on this model. These results suggested that pretreatment with 38c effectively protected the liver against CCl4 toxicity.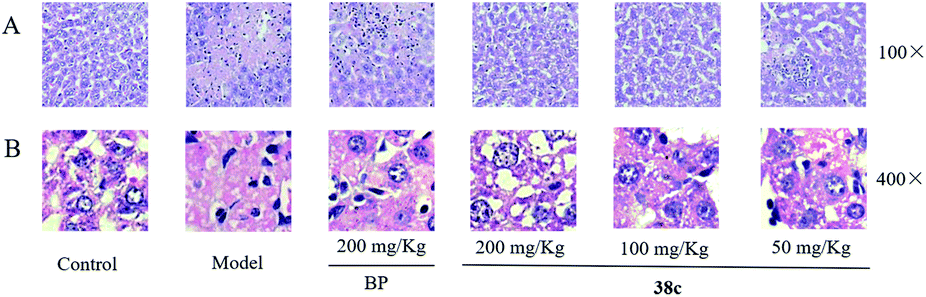 | ||
| Fig. 4 Liver histopathology after treatment of mice with 10 mL kg−1 CCl4 without and with compound 38c or biphenyldicarboxylate (BP) pretreatment. Liver slices were stained with hematoxylin and eosin. | ||
 | ||
| Fig. 5 Serum levels of (A) aspartate aminotransferase (AST), (B) alanine aminotransferase (ALT), and (C) lactate dehydrogenase (LDH), and (D) liver levels of glutathione (GSH) in mice treated with 10 mL kg−1 CCl4 without and with compound 38c pretreatment. Data are expressed as means ± SD (n = 8). ***P < 0.001 compared to control. ###P < 0.001 compared to CCl4 model mice. | ||
To investigate the mechanisms of protection from CCl4 hepatotoxicity by pretreatment with 38c, we first evaluated the liver GSH antioxidant defense system. In CCl4-treated mice, there was a significant decrease in the GSH level (11.0 μmol g−1 protein) compared to that observed for the control group (21.0 μmol g−1 protein). Pretreatment with 38c suppressed this decrease (Fig. 5D).
CCl4 toxicity results from its reductive dehalogenation by cytochrome P450 into highly reactive free radicals such as the trichloromethyl radical. To investigate whether modification of the CCl4 metabolism pathway was involved in the observed hepatoprotective effect of compound 38c, we measured the CYP2E1 level in serum using an enzyme-linked immunoassay kit, and in liver homogenates by western blotting. As shown in Fig. 6, serum and hepatic CYP2E1 were significantly upregulated in CCl4-treated mice. However, pretreatment with compound 38c effectively suppressed this increase.
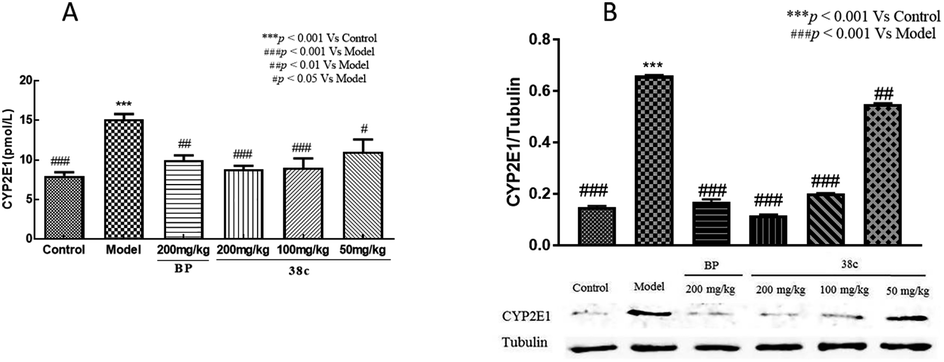 | ||
| Fig. 6 Effect of compound 38c pretreatment on (A) serum and (B) liver homogenates CYP2E1 protein levels in mice treated with 10 mL kg−1 CCl4. Data are expressed as means ± SD (n = 8). ***P < 0.001 compared to control. #P < 0.05, ##P < 0.01, ###P < 0.001 compared to CCl4 model mice. | ||
Discussion
Oxidative stress can cause cell injury and death, which may underlie many disorders including liver damage, aging, cancer, stroke, myocardial infarction, Alzheimer's disease, and Parkinson's disease.35 Anastatins A and B, flavonoid compounds isolated from the desert plant Anastatica hierochuntica, were reported to have protective activities against injury to primary rat hepatocytes.14,15 Anastatins A and B, and their derivatives, were synthesized by our group previously, and two derivatives of anastatins B were tested and found that they have good antioxidant and hepatoprotective activities.33,34The previous studies indicated that ABTS, DPPH assays as simple and efficient methods could be used to determine antioxidant activity in many compounds.36 Hence, the antioxidant capacities of compounds 38c and 40c were studied using DPPH and ABTS, assays first. In fact, VC and GA have been used as a standard antioxidant in the performed experiments,37,38 According to the data obtained from the present study, it was found that compounds 38c and 40c showed good scavenging activity against DPPH and ABTS free radicals, and suggested that both of compounds 38c and 40c had good antioxidant activity. Testing compound inhibition cell proliferation method is a common method to indicate the toxicity of a compound.39 We use the inhibition of PC12 cell proliferation to indicate the toxicity of a compound. According to the data, compounds 38c and 40c had lower cytotoxicity.
H2O2-induced PC12 or L02 oxidative damage cell model are a classic cell-level model for evaluating the antioxidant properties of compounds.40,41 Fortunately, compound 38c also had good antioxidant activity, but 40c did not show better antioxidant activity on H2O2-induced PC12 or L02 oxidative damage model.
CCl4 has been widely studied as a liver poison. CCl4-induced liver toxicity is the most commonly used model system for screening compounds,42 and more and more studies show that oxidative stress is an important CCl4-induced liver toxicity mechanism,43 CCl4 passes cytochrome P450 system biotransforms to produce trichloromethyl radicals, which in turn covalently bind to cell membranes and organelles and cause lipid peroxidation.44 BP is a drug that has been reported to have a good effect against CCl4 hepatotoxicity,45 and we used it as a positive control. Compound 38c has been shown to have good antioxidant activity in vitro. We assumed that compound 38c also has a good hepatoprotective effect in vivo. We used CCl4-induced liver injury in mice model to verify our hypothesis. The levels of serum AST, ALT and LDH activities reflects damage to hepatocytes and were considered to be highly sensitive and fairly specific preclinical and clinical biomarkers of hepatotoxicity.46 It is exciting that high dose (200 mg kg−1) and medium dose (100 mg kg−1) of compound 38c show good protection, which can reverse the increase of AST, ALT, LDH in serum caused by CCl4, the current results are also consistent with previous reports. GSH, an important part of the main defense system, can scavenge free radicals in the body.47 In this study, treatment with compound 38c can enhance the activity of antioxidant enzyme systems, including GSH, and it can also resist liver pathological changes caused by CCl4 (necrosis in hepatic lobules, vacuolization, Kupffer cells around the central vein). Cytochrome P450 (CYP) is a phase I enzyme play an important role for the metabolic activation of many procarcinogens.48 CYP2E1 represents a major CYP isoform and is expressed in the liver.49 In mice pretreated with compound 38c, the expression of CYP2E1 in serum and liver were significantly reduced. This may be one of the mechanisms of hepatoprotective effect of compound 38c.
Conclusions
In this study, the antioxidant activity and cytotoxicity of anastatins A & B, and its derivatives were studied using chemical assessment methods, cell proliferation inhibition experiments, and cell oxidative damage models. We have screened compound 38c with better activity and lower toxicity in vitro. And it was used to study the hepatoprotection activity and mechanism by using a CCl4-induced liver injury model in mice. The results suggest that the anastatins B derivative compound, 38c, has good anti-oxidative and lower cytotoxicity in vitro and in vivo in an animal model of liver injury. Decreasing CYP2E1 expression is one of the mechanisms of hepatoprotection effect of compound 38c.Appendix A
Chemical structure of anastatins A & B and derivatives| Name | Structure | Name | Structure |
|---|---|---|---|
| (±) Anastatins A | 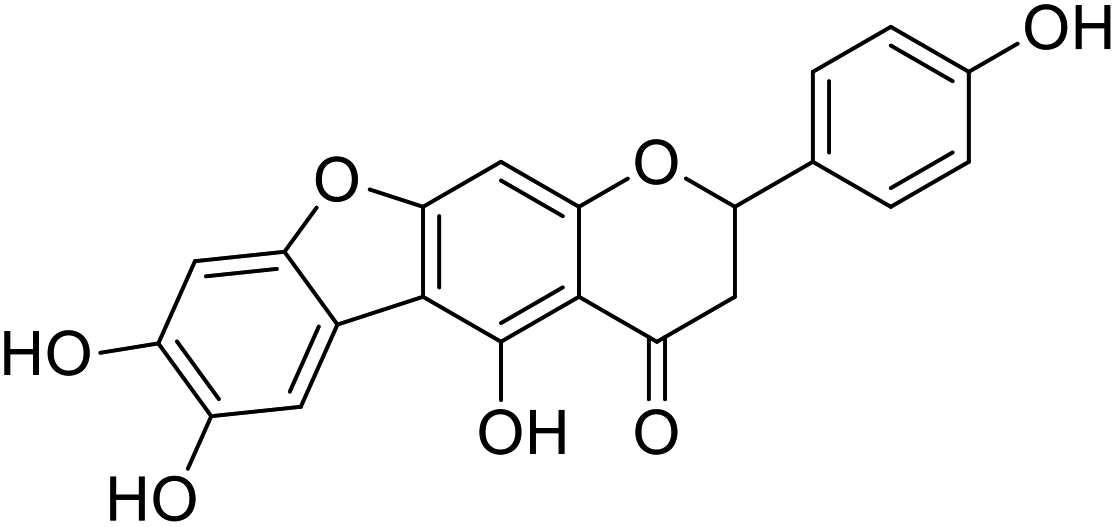 |
(±) Anastatins B | 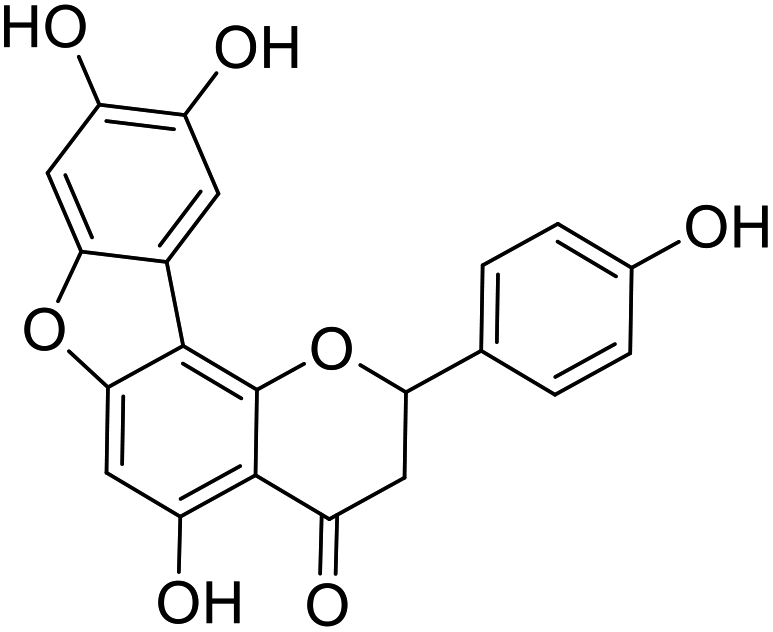 |
| 42a | 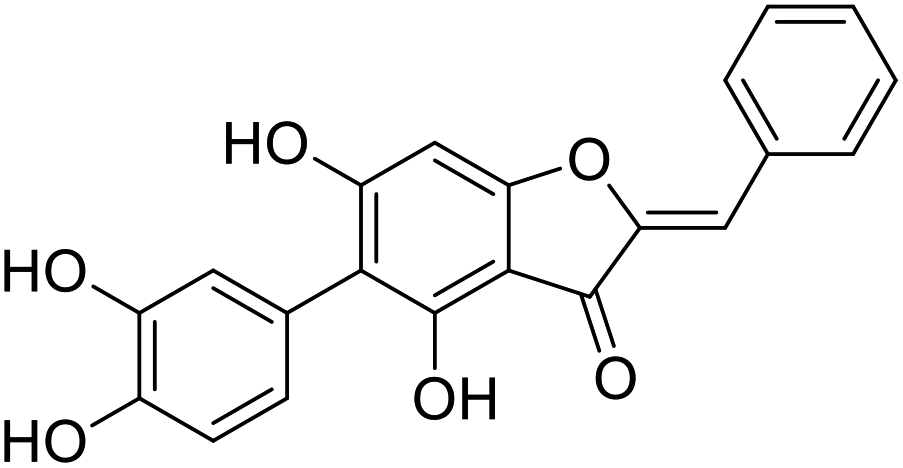 |
38a |  |
| 43a |  |
39a | 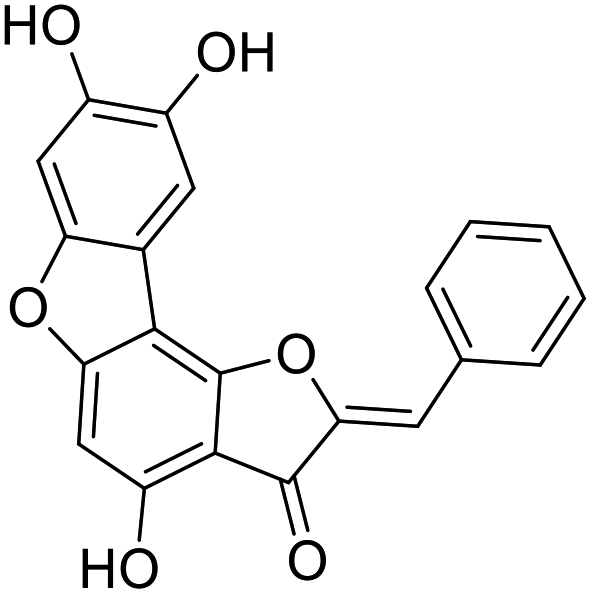 |
| 44a | 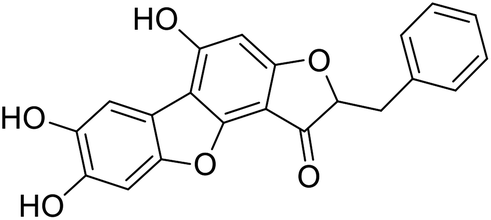 |
40a | 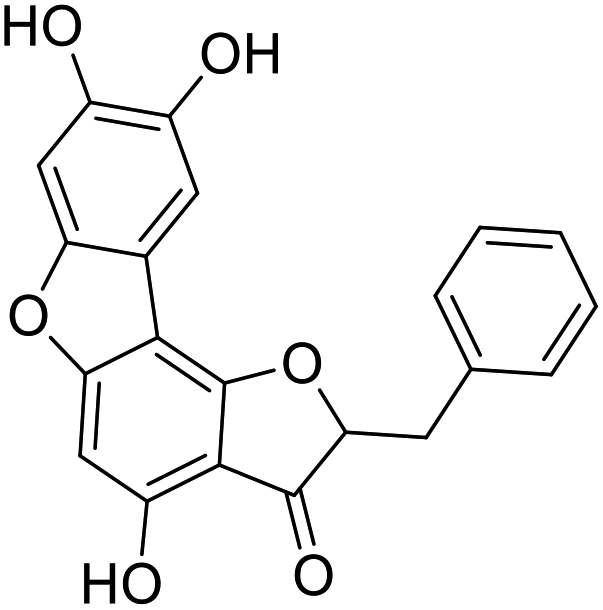 |
| 42b | 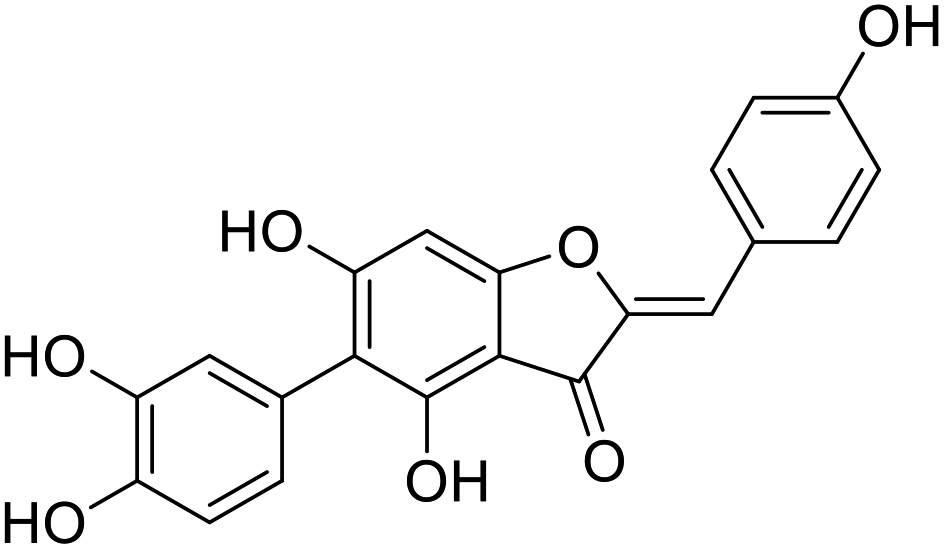 |
38b | 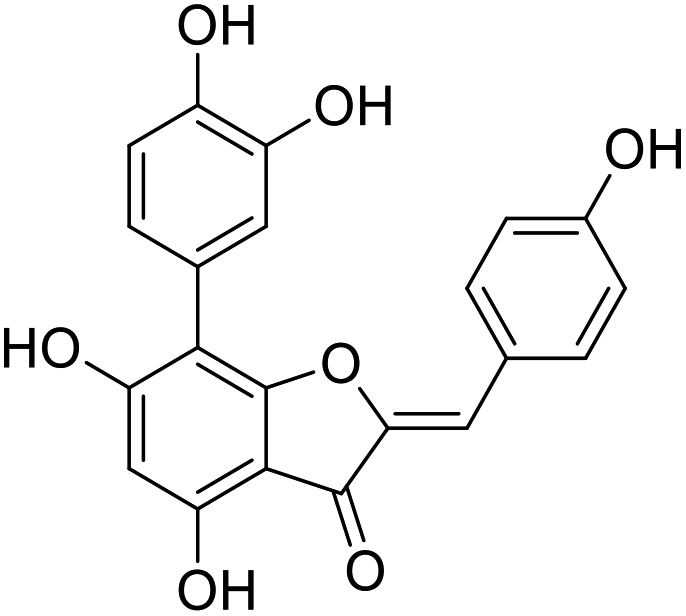 |
| 43b |  |
39b |  |
| 44b | 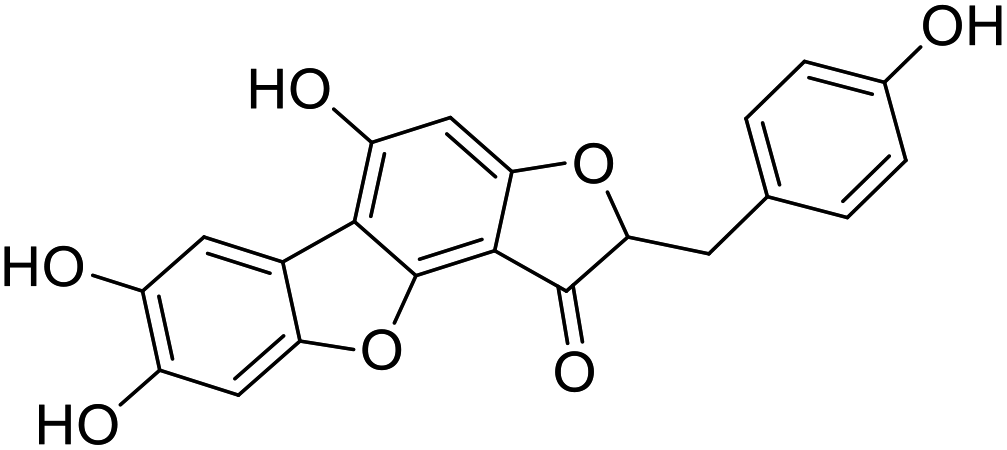 |
40b | 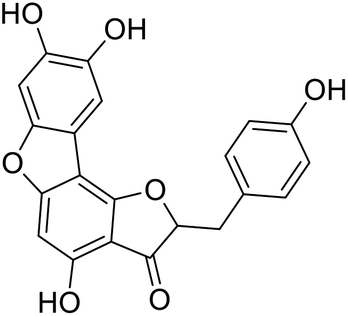 |
| 42c | 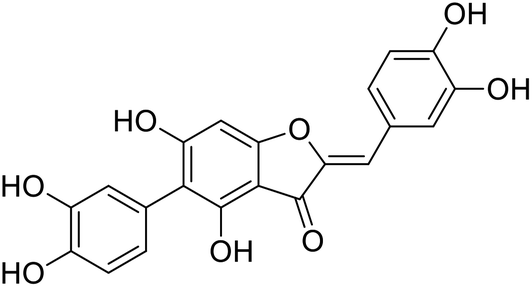 |
38c | 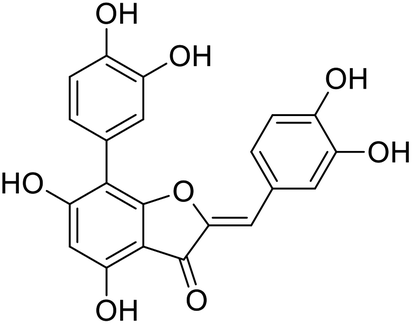 |
| 42c-1 |  |
38c-1 | 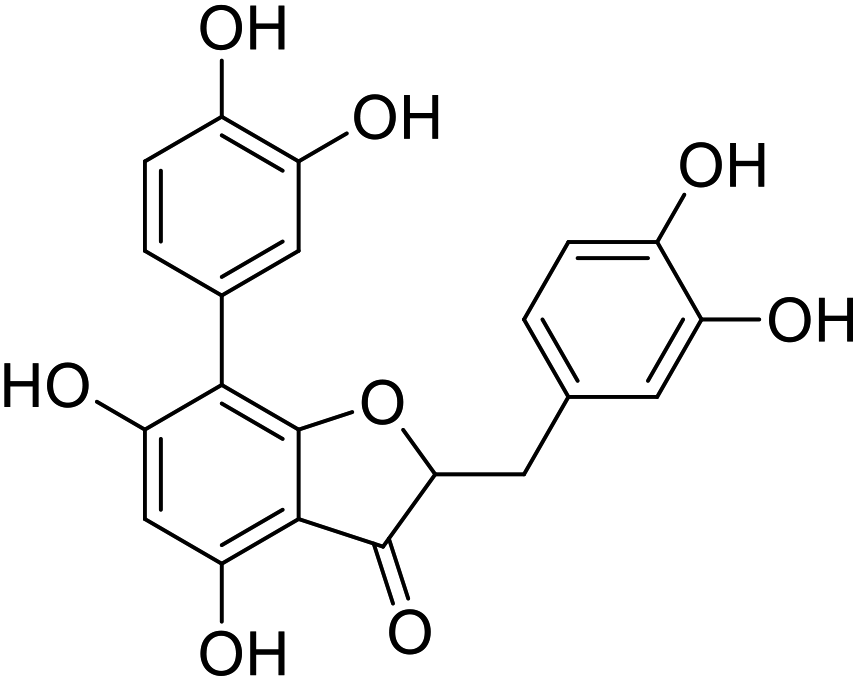 |
| 43c | 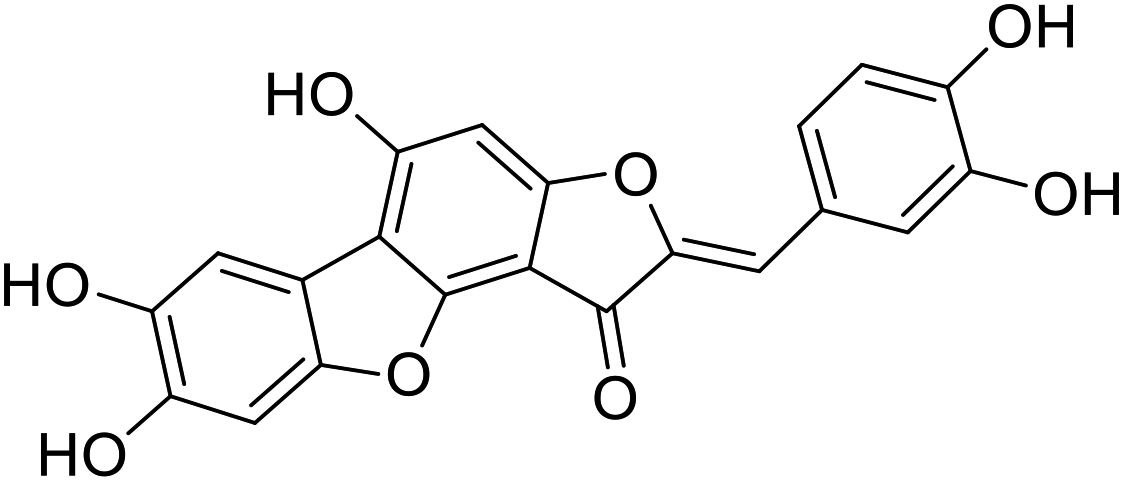 |
39c | 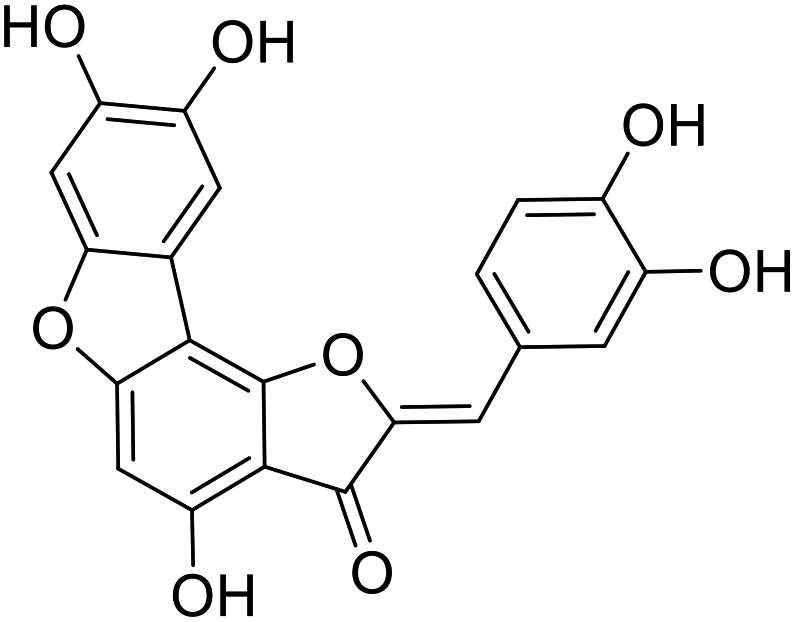 |
| 44c | 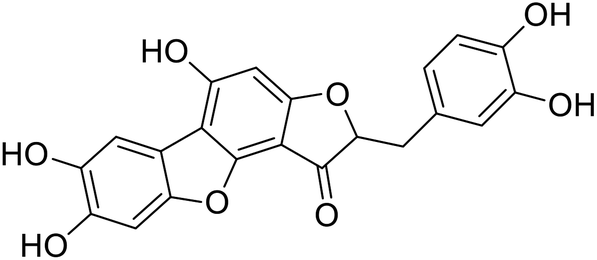 |
40c |  |
| 19 | 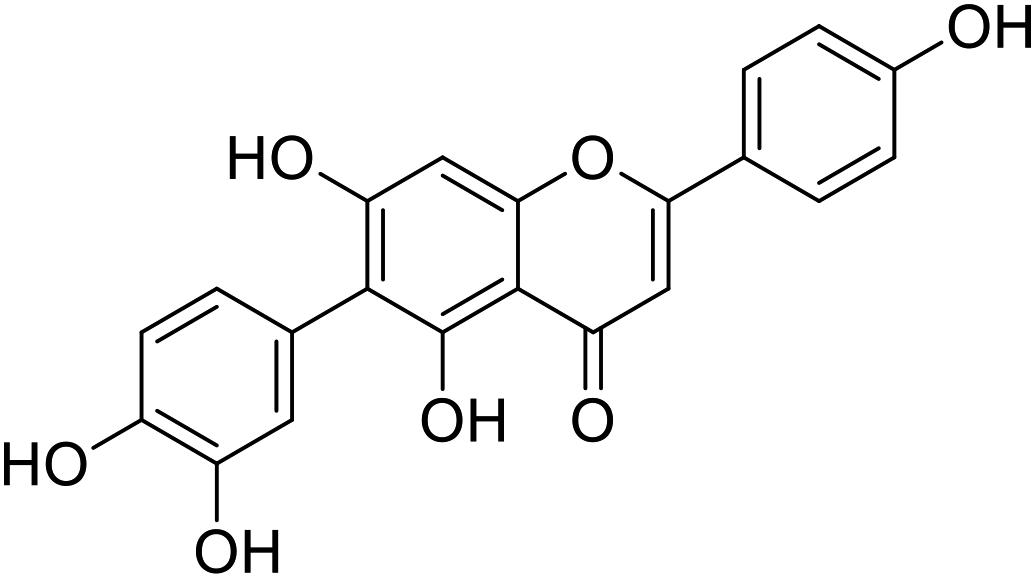 |
20 | 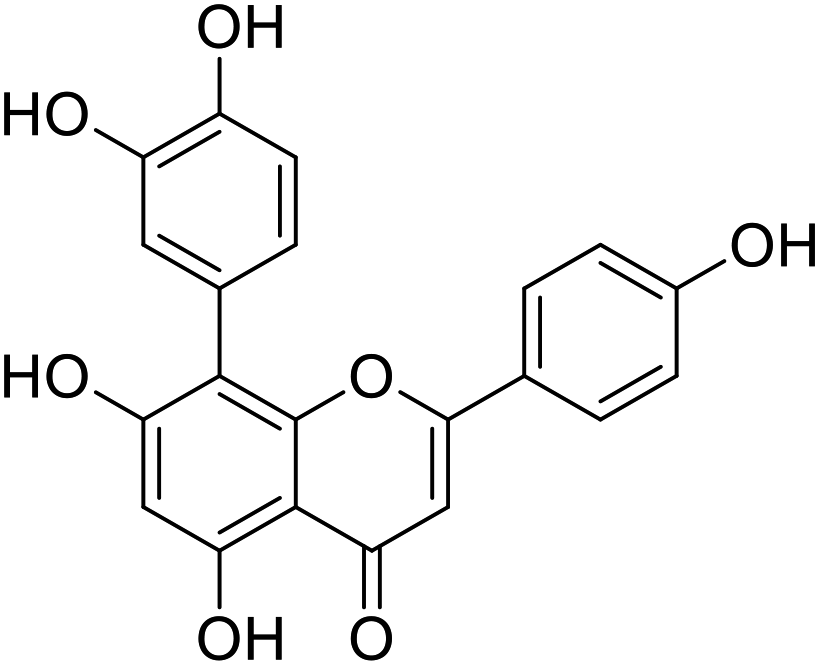 |
| 31 | 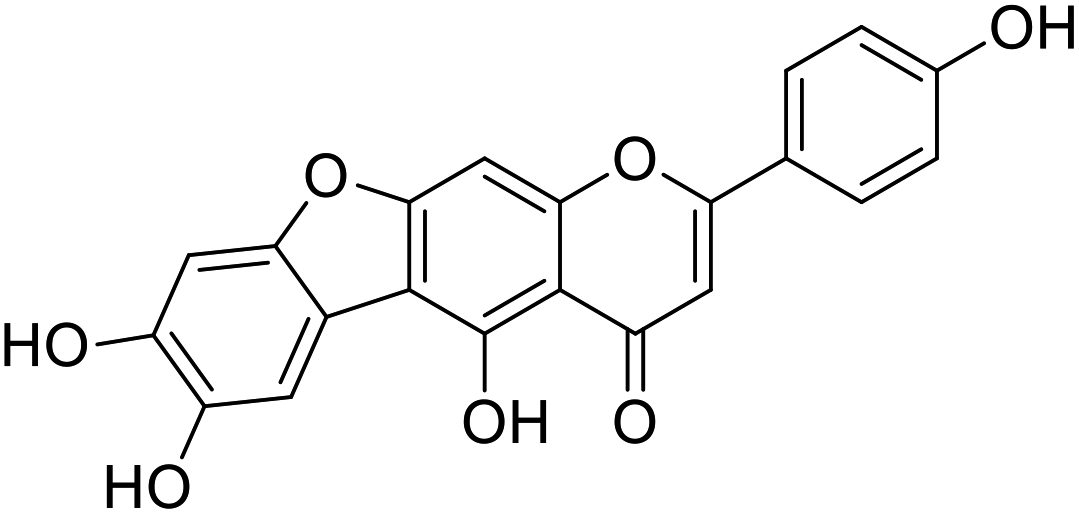 |
30 |  |
Conflicts of interest
We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work. There is no professional or other personal interest of any nature or kind in any product, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript.Acknowledgements
The authors sincerely thank the financial support from National Natural Science Foundation of China (31601203) and Natural Science Foundation of Shandong (ZR2018LB013). The authors are thankful to the Research Centre of Modern Analytical Technology, Tianjin University of Science & Technology, for NMR and HRMS analyses.References
- C. T. Sulaiman and I. Balachandran, Indian J. Pharm. Sci., 2012, 74, 258–260 CrossRef CAS PubMed.
- T. Wang, Q. Li and K. Bi, Asian J. Pharm. Sci., 2018, 13, 12–23 CrossRef PubMed.
- T. Aoki, T. Akashi and S. I. Ayabe, J. Plant Res., 2000, 113, 475–488 CrossRef.
- B. Ahmed, T. A. Al-Howiriny, J. S. Mossa and K. E. H. E. Tahir, ChemInform, 2005, 36, 400–404 Search PubMed.
- T. Iwashina, J. Plant Res., 2000, 113, 287–299 CrossRef CAS.
- H. Chen, J. Pu, D. Liu, W. Yu, Y. Shao, G. Yang, Z. Xiang and N. He, PLoS One, 2016, 11, e153080 Search PubMed.
- H. P. Kim, K. H. Son, H. W. Chang and S. S. Kang, J. Pharmacol. Sci., 2004, 96, 229–245 CrossRef CAS PubMed.
- S. Bhargava, T. Patel, R. Gaikwad, U. K. Patil and S. Gayen, Nat. Prod. Res., 2019, 33, 851–857 CrossRef CAS PubMed.
- Z. F. Bai, Y. K. Meng, H. E. Lan-Zhi, J. F. Tang, J. B. Wang and X. H. Xiao, Chin. Pharm. J., 2017, 52, 1105–1109 CAS.
- B. Wang, J. Qu, S. Luo, S. Feng, T. Li, M. Yuan, Y. Huang, J. Liao, R. Yang and C. Ding, Molecules, 2018, 23, 2513 CrossRef PubMed.
- M. Friedman, Mol. Nutr. Food Res., 2007, 51, 116–134 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, W. Shen, S. Ma, W. Chen and R. Qi, Microcirculation, 2017, 24, e12385 CrossRef PubMed.
- A. Liu, H. Wang, S. M. Lee, Y. Wang and G. Du, Bioorg. Med. Chem., 2008, 16, 7141–7147 CrossRef CAS PubMed.
- M. Yoshikawa, F. Xu, T. Morikawa, K. Ninomiya and H. Matsuda, Bioorg. Med. Chem. Lett., 2003, 13, 1045–1049 CrossRef CAS PubMed.
- S. Nakashima, H. Matsuda, Y. Oda, S. Nakamura, F. Xu and M. Yoshikawa, Bioorg. Med. Chem., 2010, 18, 2337–2345 CrossRef CAS PubMed.
- A. Ortega-Alonso, C. Stephens, M. Lucena and R. Andrade, Int. J. Mol. Sci., 2016, 17, 714 CrossRef PubMed.
- A. Yang, T. Inamine, K. Hochrath, P. Chen, L. Wang, C. Llorente, S. Bluemel, P. Hartmann, J. Xu, Y. Koyama, T. Kisseleva, M. G. Torralba, K. Moncera, K. Beeri, C. Chen, K. Freese, C. Hellerbrand, S. M. L. Lee, H. M. Hoffman, W. Z. Mehal, G. Garcia-Tsao, E. A. Mutlu, A. Keshavarzian, G. D. Brown, S. B. Ho, R. Bataller, P. Stärkel, D. E. Fouts and B. Schnabl, J. Clin. Invest., 2017, 127, 2829–2841 CrossRef PubMed.
- V. G. Jonnalagadda, H. P. Char and P. K. Ankana, Cardiovasc. Drugs Ther., 2018, 32, 311 CrossRef CAS PubMed.
- S. Petta, C. Cammà, D. Cabibi, V. Di Marco and A. Craxì, Aliment. Pharmacol. Ther., 2011, 34, 757–766 CrossRef CAS PubMed.
- V. G. Jonnalagadda and A. Shaik, Diabetes, Metab. Syndr. Obes.: Targets Ther., 2018, 11, 131 CrossRef PubMed.
- N. Chalasani and A. Regev, Gastroenterology, 2016, 151, 1046–1051 CrossRef PubMed.
- R. Liberal and C. R. Grant, World J. Hepatol., 2016, 8, 1157 CrossRef PubMed.
- B. Kalyanaraman, Redox Biol., 2013, 1, 244–257 CrossRef CAS PubMed.
- K. Dyke and M. Sacks, US Pat., US2004/0176306A1, Sep 9, 2004.
- A. Ozkaya, Z. Sahin, U. Dag and M. Ozkaraca, J. Biochem. Mol. Toxicol., 2016, 30, 243–248 CrossRef CAS PubMed.
- Z. Allameh and J. Salamzadeh, J. Res. Pharm. Pract., 2016, 5, 79–85 CrossRef CAS PubMed.
- D. Singh, W. C. Cho and G. Upadhyay, Front. Physiol., 2016, 6, 1–18 Search PubMed.
- H. Lin, Q. Mao, Y. M. Wang and L. Jiang, World J. Gastroenterol., 2008, 14, 2329–2337 CrossRef CAS PubMed.
- H. Lee, S. Kim, G. Lee, M. Choi, H. Jung, Y. Kim, H. Kwon and H. Chae, BMC Complementary Altern. Med., 2016, 16, 316–324 CrossRef PubMed.
- X. Chen, X. Gong, R. Jiang, B. Wang, G. Kuang, K. Li and J. Wan, Immunopharmacol. Immunotoxicol., 2016, 38, 61–67 CrossRef CAS PubMed.
- L. Pan, Y. Qiu, T. Chen, J. Lin, Y. Chi, M. Su, A. Zhao and W. Jia, J. Pharm. Biomed. Anal., 2010, 52, 589–596 CrossRef CAS PubMed.
- N. Dai, Y. Zou, L. Zhu, H. Wang and M. Dai, J. Med. Food, 2014, 17, 663–669 CrossRef CAS PubMed.
- G. Pan, X. Li, L. Zhao, M. Wu, C. Su, X. Li, Y. Zhang, P. Yu, Y. Teng and K. Lu, Eur. J. Med. Chem., 2017, 138, 577–589 CrossRef CAS PubMed.
- C. Xiang, Y. Teng, C. Yao, X. Li, M. Cao, X. Li, G. Pan, K. Lu, H. Galons and P. Yu, RSC Adv., 2018, 8, 15366–15371 RSC.
- B. Chhunchha, N. Fatma, E. Kubo, P. Rai, S. P. Singh and D. P. Singh, Am. J. Physiol.: Cell Physiol., 2013, 304, C636–C655 CrossRef CAS PubMed.
- G. Estefania, L. A. Puente, J. A. Pérez-Álvarez and J. Fernández-López, J. Sci. Food Agric., 2016, 12, 4235–4242 Search PubMed.
- C. Lu, C. Li, B. Chen and Y. Shen, Food Chem., 2018, 265, 111–119 CrossRef CAS PubMed.
- K. Li, M. Wang, T. Wang, D. Sun, R. Zhu, X. Sun, X. Wu and S. Wang, Photochem. Photobiol., 2013, 89, 1064–1070 CrossRef CAS PubMed.
- A. Rejhová, A. Opattová, A. Čumová, D. Slíva and P. Vodička, Eur. J. Med. Chem., 2018, 144, 582–594 CrossRef PubMed.
- X. Chen, X. Zhou, X. Yang, Z. Zhou, D. Lu, Y. Tang, Z. Ling, L. Zhou and X. Feng, Cell. Mol. Neurobiol., 2016, 36, 541–551 CrossRef CAS PubMed.
- A. Rejhová, A. Opattová, A. Čumová, D. Slíva and P. Vodička, Eur. J. Med. Chem., 2018, 144, 582–594 CrossRef PubMed.
- S. B. Ahmad, M. U. Rehman, B. Fatima, B. Ahmad, I. Hussain, S. P. Ahmad, A. Farooq, S. Muzamil, R. Razzaq, S. M. Rashid, S. Ahmad Bhat and M. U. R. Mir, Environ. Toxicol., 2018, 33, 361–369 CrossRef CAS PubMed.
- A. Khan, B. Shal, M. Naveed, F. A. Shah, A. Atiq, N. U. Khan, Y. S. Kim and S. Khan, Neurotoxicology, 2019, 72, 38–50 CrossRef CAS PubMed.
- R. A. Kepekçi, S. Polat, A. Çelik, N. Bayat and S. D. Saygideger, Food Chem., 2013, 141, 1972–1979 CrossRef PubMed.
- X. Huo, C. Liu, L. Gao, X. Xu, N. Zhu and L. Cao, Int. J. Mol. Sci., 2017, 18, 1197 CrossRef PubMed.
- Z. Huang, P. Chen, W. Su, Y. Wang, H. Wu, W. Peng and P. Li, Molecules, 2018, 23, 1188 CrossRef PubMed.
- P. Poprac, K. Jomova, M. Simunkova, V. Kollar, C. J. Rhodes and M. Valko, Trends Pharmacol. Sci., 2017, 38, 592–607 CrossRef CAS PubMed.
- F. P. Guengerich, Trends Pharmacol. Sci., 1991, 12, 281–283 CrossRef CAS PubMed.
- X. Yin, W. Xiong, Y. Wang, W. Tang, W. Xi, S. Qian and Y. Guo, Medicine, 2018, 97, e11910 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |