
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFolic acid-conjugated magnetic mesoporous silica nanoparticles loaded with quercetin: a theranostic approach for cancer management†
Snehasis Mishraae,
Krishnendu Mannaa,
Utpal Kayalb,
Moumita Sahaa,
Sauvik Chatterjeeb,
Debraj Chandra c,
Michikazu Harad,
Sriparna Dattae,
Asim Bhaumik*b and
Krishna Das Saha*a
c,
Michikazu Harad,
Sriparna Dattae,
Asim Bhaumik*b and
Krishna Das Saha*a
aCancer Biology and Inflammatory Disorder Division, CSIR-Indian Institute of Chemical Biology, 4, Raja S. C. Mullick Road, Kolkata-700032, West Bengal, India. E-mail: krishna@iicb.res.in; Fax: +91 33 2473 5197; Tel: +91 33 2499 5810
bSchool of Materials Sciences, Indian Association of Cultivation of Science, 2A & B, Raja S. C. Mullick Road, Kolkata-700032, West Bengal, India. E-mail: msab@iacs.res.in; Fax: +91-33-2473-2805; Tel: +91-33-2473-4971
cWorld Research Hub Initiative (WRHI), Institute of Innovative Research, Tokyo Institute of Technology, Nagatsuta-cho 4259, Midori-ku, Yokohama 226-8503, Japan
dLaboratory for Materials and Structures, Institute of Innovative Research, Tokyo Institute of Technology, Nagatsuta-cho 4259, Midori-ku, Yokohama 226-8503, Japan
eDepartment of Chemical Technology, University of Calcutta, Kolkata-700009, West Bengal, India
First published on 17th June 2020
Abstract
The development of drug carriers based on nanomaterials that can selectively carry chemotherapeutic agents to cancer cells has become a major focus in biomedical research. A novel pH-sensitive multifunctional envelope-type mesoporous silica nanoparticle (SBA-15) was fabricated for targeted drug delivery to human colorectal carcinoma cells (HCT-116). SBA-15 was functionalized with folic acid (FA), and the material was loaded with the water-insoluble flavonoid, quercetin (QN). Additionally, acid-labile magnetite Fe3O4 nanoparticles were embedded over the FA-functionalized QN-loaded monodisperse SBA-15 to prepare the highly orchestrated material FA-FE-SBA15QN. The in vitro and in vivo anti-carcinogenic efficacy of FA-FE-SBA15QN was carried out to explore the pH-sensitive QN release with putative mechanistic aspects. FA-FE-SBA15QN caused a marked tumor suppression, and triggered mitochondrial-dependent apoptosis through a redox-regulated cellular signaling system. Furthermore, FA-IO-SBA-15-QN initiated the c-Jun N-terminal Kinase (JNK)-guided H2AX phosphorylation, which relayed the downstream apoptotic signal to the phosphorylate tumor suppressor protein, p53. On the other hand, the selective inhibition of heat shock protein-27 (HSP-27) by FA-FE-SBA15QN augmented the apoptotic fate through JNK/H2AX/p53 axis. The in vitro and in vivo magnetic resonance imaging (MRI) studies have indicated the theranostic perspective of the composite. Thus, the result suggested that the newly synthesized FA-FE-SBA15QN could be used as a promising chemo theranostic material for the management of carcinoma.
1. Introduction
Cancer is a major alarming matter of concern in both developed and developing countries.1 According to the GLOBOCAN 2018 report, cancer is the second leading global reason for death and is accountable for an estimated 9.6 million deaths in 2018. All over the world, colon cancer is the 4th most commonly diagnosed cancer type, after lung, breast and prostate cancer.2 Numerous medical approaches have been carried out for cancer therapy, such as chemotherapy, radiotherapy, and surgery.3 The removal of tumor tissues (malignant and benign) through a surgical procedure is a big challenge to achieve. On the one hand, the current chemotherapy strategies have been shown to be useful. The use-of-applicability limits are due to its systematic toxicity, which causes a plethora of damage to healthy tissues. On the other hand, radiotherapy also has several drawbacks due to its nonspecificity and nonselective side effects.3,4 Thus, nanomedicines have become an integral part to combat the disease progression, as well as an associated complication in cancer therapy.5,6Quercetin (3,3′,4′,5,7-pentahydroxy-2-phenylchromen-4-one, QN), a naturally occurring aglycone flavonoid, belongs to a group of polyphenols with a flavonol core, and is extracted from several plant sources, viz., fruits, leaves, roots, and barks.7 It has unique biological properties that may improve disease resistance and overall health, including antioxidant, anti-inflammatory, anti-carcinogenic, antiviral, and antibacterial activities.8 QN also can inhibit lipid peroxidation and platelet aggregation. An ample range of its different cellular effects is associated with multiple signaling pathways, which are essential in the regulation of physiological processes.9 An inhibition of protein tyrosine kinase (PTK) signaling pathways by QN reflects the primary cellular response in cancer cell survival.9 Besides the anti-cancer property, the anti-inflammatory effect of QN is consigned in inhibiting inducible nitric oxide synthase (iNOS), followed by cytokine production and prostaglandin synthesis. QN prevents several pro-inflammatory cytokine secretions by directly interacting with the components of the mitogen-activated protein kinase (MAPK) and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which are potent inducers of inflammatory gene expression.10 Additionally, QN may indirectly prevent inflammation by increasing peroxisome proliferator-activated receptor gamma (PPARγ) activity, thereby antagonizing the important redox-regulated transcription factor-guided transcriptional activation of several inflammatory genes.11 Numerous mechanisms have been portrayed as a potential approach of the antitumor action of QN. It is well known that QN is proficient at inducing apoptotic cell death in various carcinomas, including cervical, squamous, colon, pancreatic, leukemia, breast, lung, and hepatocellular cells.12 QN-guided apoptosis is primarily demonstrated through the mitochondrial pathway, which includes an imbalance of the pro/anti-apoptotic protein and the activation of selective cysteine-aspartic proteases (caspases-3, -9).12 In contempt of an extensive range of biological effects, the physicochemical properties of QN represent a limit for its application. The low water solubility, low bioavailability, low oxidative stability and intensive biotransformation confine its applicability. Therefore, different approaches have been taken, and enormous research is going on to improve its delivery and efficiency.13
Mesoporous materials, by virtue of having an exceptionally high surface area and large mesopores, can hold reactive nanoparticles at its pore surfaces.14 Mesoporous silica nanoparticles based on the 2D hexagonally ordered SBA-15 material have been used for improved and targeted drug delivery, which open up new possibilities in nanomedical research.15,16 The diffusion process generally directs the liberation of the drug from SBA-15. Thus, the release profile may be altered by modulating the pore diameter. The interaction of the encapsulated drug with the pore wall of SBA-15 may play an essential role in drug release, which is dependent on the relative amount and obeys the rule of displacement desorption. It has also been established that the folate receptor (FR), which is widely distributed in colon carcinomas, is a potentially useful target for cancer therapy and imaging purposes. Therefore, the covalent conjugation of folic acid (FA) to drug carriers could attain selective targeting to the tumor mass. It has been observed that FA can be readily attached through amide reactions with the outer surface of SBA-15.17
Magnetic resonance imaging (MRI) is a prevailing technique for capturing and analyzing tomographic images of cancers, as it offers an amalgamation of three-dimensional tumor coverage and deep tissue penetration. In spite of the extensive research that has been carried out to develop novel MRI contrast agents, the single mode contrast agents are far from being ideal. Current contrasting agents based on magnetic nanoparticles18,19 displayed fundamental limitations in terms of imaging artifacts, imaging uncertainties, and health risks. Several studies are being carrying out to develop functionalized contrasting agents, which can be useful for cancer management. The present study aimed to explore a novel theranostic platform, which includes three major components (FA, QN and Fe3O4 NPs) over the 2D-hexagonal mesoporous silica material SBA-15 (Scheme 1) to enable a “dual sword”: (i) targeted and enhanced QN loading that regulates its release using external and internal triggers (pH), (ii) provide imaging capability with a suitable contrasting agent (Fe3O4).20 The current study was also designed to explore the prolonged efficacy of QN (Scheme 1) with the evaluation of a possible molecular mechanism.
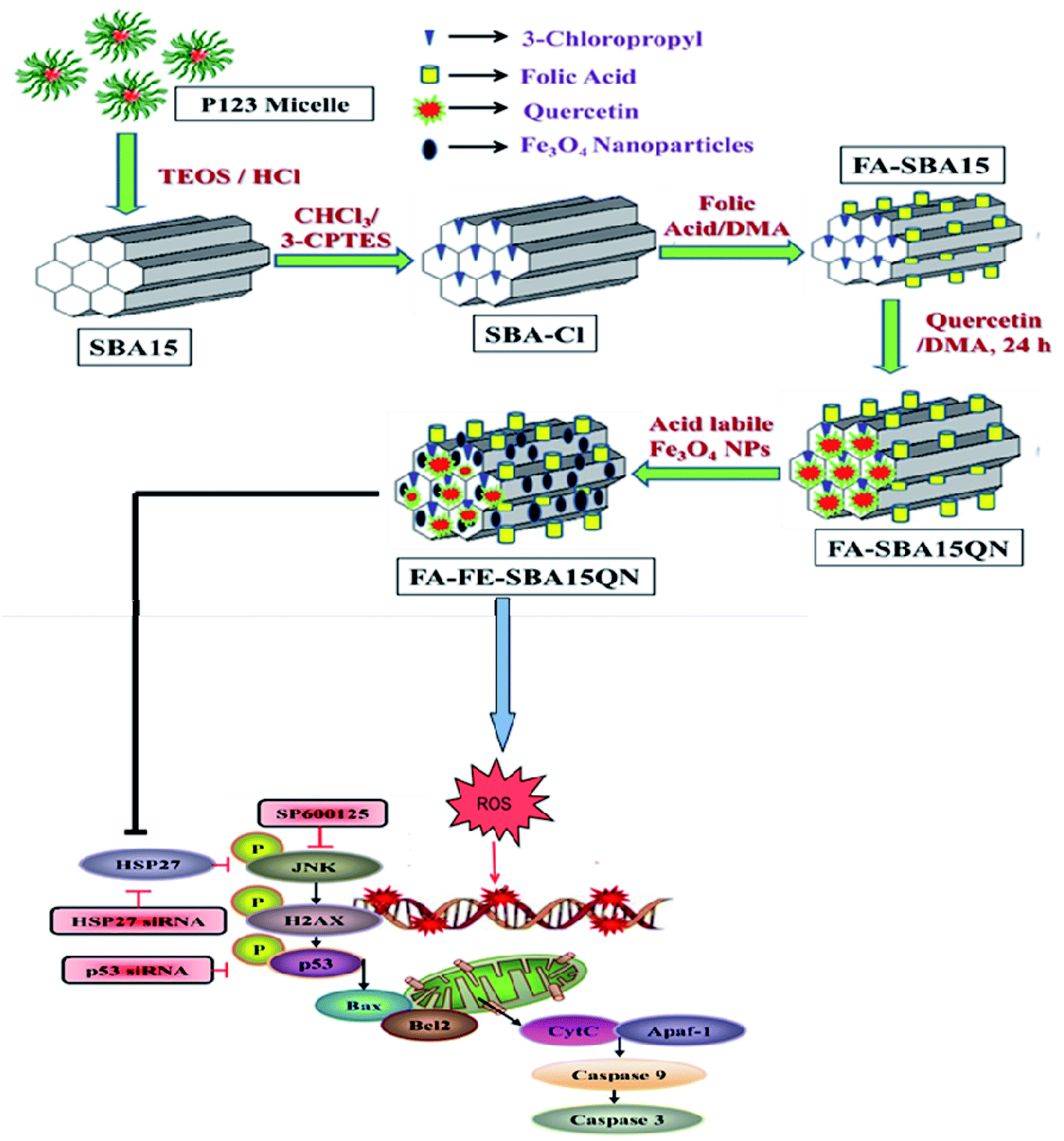 | ||
| Scheme 1 Schematic representation of FA-FE-SBA15QN synthesis and graphical representation illustrating the probable mechanism of action of FA-FE-SBA15QN in modulating the cancer microenvironment. | ||
2. Results and discussion
2.1. Characterization of FA-IO-SBA-15-QN
The FTIR spectra of pure SBA-15, FA-SBA-15, and FA-FE-SBA15QN are shown in Fig. 2A. As seen in the spectra, the additional transmittance peaks were observed in FA-IO-SBA-15-QN due to the presence of Fe3O4 at the surface of the functionalized SBA-15. The small angle powder XRD patterns of four materials, i.e., calcined SBA-15, SBA-Cl, FA-SBA-15, and FA-FE-SBA15QN are shown in Fig. 2B. As observed in Fig. 2B, the pure SBA-15 material displayed three characteristic diffraction peaks at the 2θ region (0.91, 1.58, and 1.82) for the three distinctive planes [(100) strong, (110) weak and (200) weak], respectively, which suggested the presence of the ordered 2D-hexagonal mesoporous structure.21 In the 3-chloropropyl functionalized material, SBA-Cl also displayed a similar diffraction pattern, as shown in Fig. 2B. Furthermore, FA-SBA-15 showed a similar diffraction pattern, with a 2θ value of 0.92, 1.56 and 1.83 degrees (Fig. 2B). This corresponds to the above-mentioned three characteristic planes, suggesting the retention of the ordered 2D-hexagonal mesophase in the material. The small increase in the 2θ values indicated a minor decrease in the d-spacing values, which confirmed the surface functionalization of the SBA-15 base material.21–23 In contrast, the broad peak in FA-FE-SBA15QN appears at a 2θ value of 20–23° due to the amorphous nature of the SBA-15 material. Another broad peak and a sharp diffraction peak were observed at 2θ values of 9.27–15.53 and 18.19°, respectively, corresponding to the folic acid moiety bound at the mesopore surface. In Fig. 2C, the N2 adsorption/desorption isotherms of FA-FE-SBA15QN at 77 K are shown. This isotherm could be classified as a type IV isotherm with two steep rises in the N2 uptakes.22,23 This capillary condensation is more pronounced in the desorption isotherm, which showed a large hysteresis. The BET surface area and pore volume of this FA-FE-SBA15QN material were 402 m2 g−1 and 0.0737 cm3 g−1, respectively. The pore size distribution plot was estimated from this sorption isotherm using the NLDFT model (Fig. 2D), and it displayed trimodal peak pores of 3.2, 5.0 and 7.6 nm. The presence of three peak pores could be attributed to the stepwise functionalization of the 2D hexagonal mesopores of SBA-15.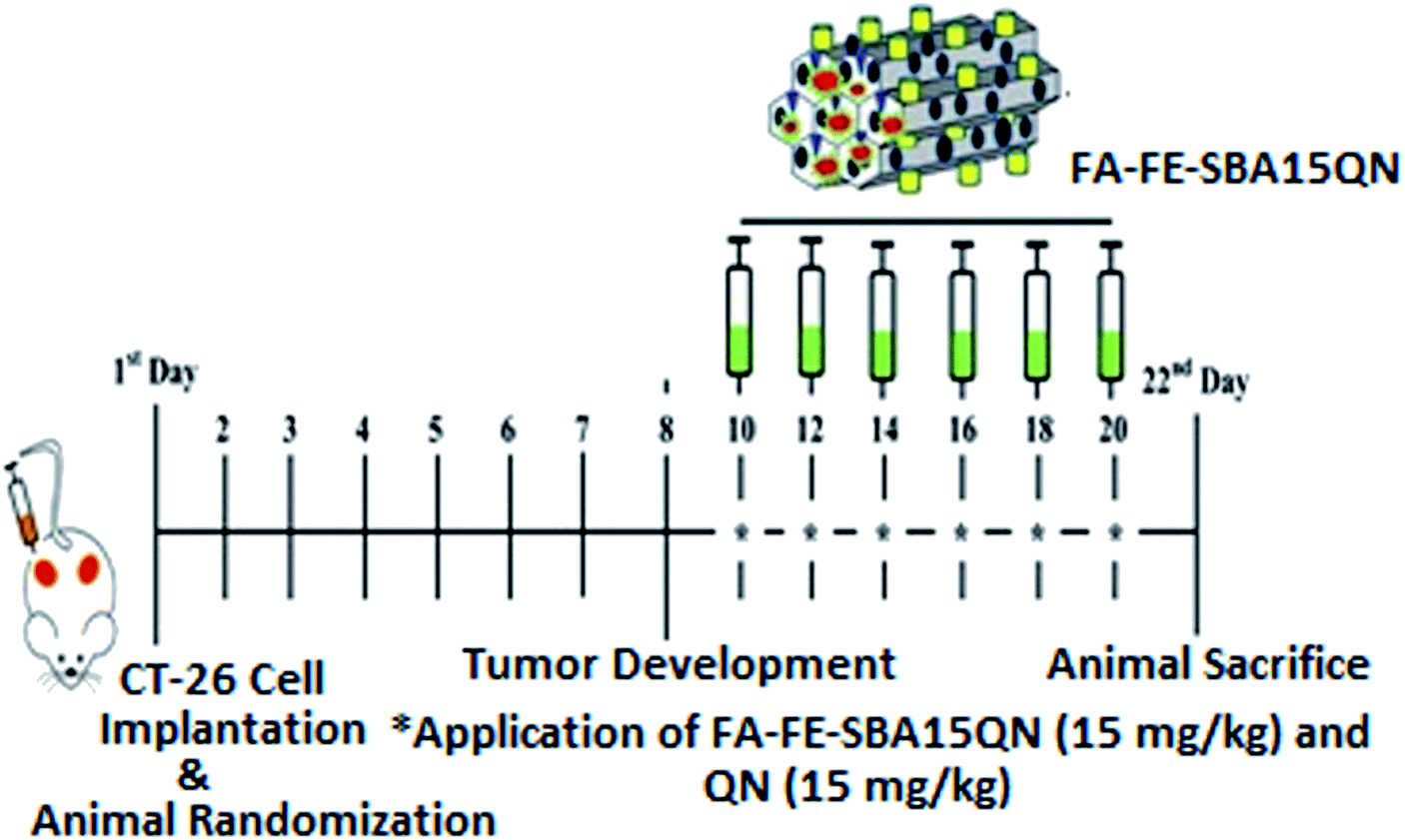 | ||
| Fig. 1 Schematic representation of the experimental design outlining the phases of the CT-26-challenge, along with the QN and FA-FE-SBA15QN application (15 mg kg−1) in the murine system. | ||
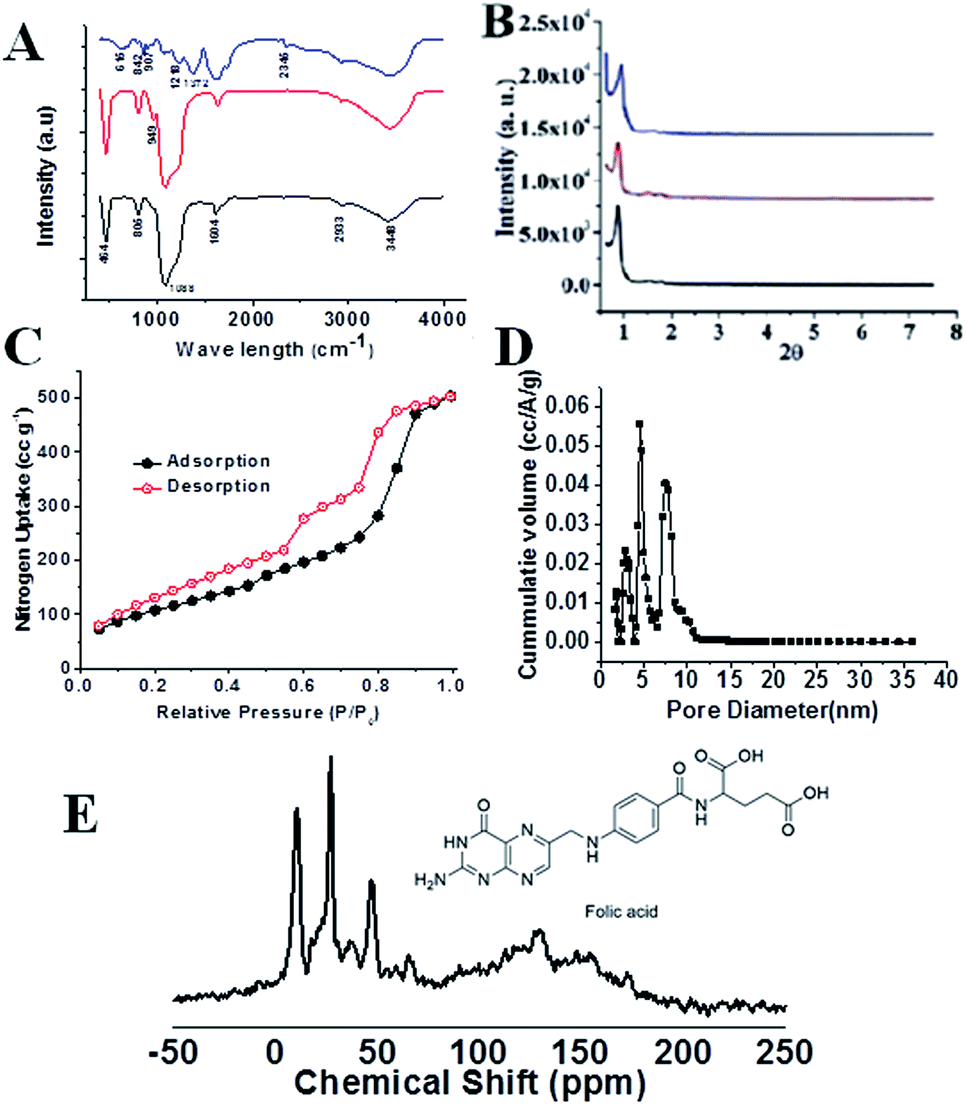 | ||
| Fig. 2 FTIR spectra (A) and small angle powder XRD patterns (B) of SBA-15 (black), FA-SBA15QN (red), and FA-FE-SBA15QN (blue). N2 adsorption/desorption isotherms of FA-FE-SBA15QN (C) at 77 K and respective pore size distribution (D). Adsorption points are marked with filled black circles and desorption points by empty red circles. 13C CP MAS NMR of folic acid-functionalized mesoporous material FA-SBA15 (E). | ||
Furthermore, the 13C CP-MAS (cross-polarization magic angle spinning) NMR for the FA-SBA15 was taken, and it is shown in Fig. 2E. Chemical shifts corresponding to the presence of aliphatic atoms of the propyl group and different aliphatic, aromatic carbons, as well as the carboxylic and amide carbons of the folic acid moiety are clearly seen in Fig. 2E. Thus, this solid-state 13C NMR data suggested the successful functionalization of the surface of SBA-15 with the FA moiety at the mesopore surface. The HRTEM images and SAED pattern of the pristine SBA-15 and Fe3O4 NPs are shown in Fig. 3. The SAED pattern of Fig. 3B suggested hexagonally order pore channels in the SBA-15 material. The iron oxide nanoparticles were observed in the TEM images (Fig. 3C and D). In the high-resolution TEM image (Fig. 3D), the 311 and 220 planes are marked. Indexing of the crystalline planes of the Fe3O4 NPs is shown in the SAED pattern in Fig. 3F. In Fig. 3E, the HRTEM image of the drug-loaded FA-conjugated magnetic mesoporous silica material FA-IO-SBA-15-QN is shown. Fig. 3E further confirms the loading of magnetic nanoparticles of iron oxide-quercetin onto SBA-15. As seen in Fig. 3A and E, the periodicity of the hexagonal mesopores were retained in the QN-loaded functionalized SBA-15 material.
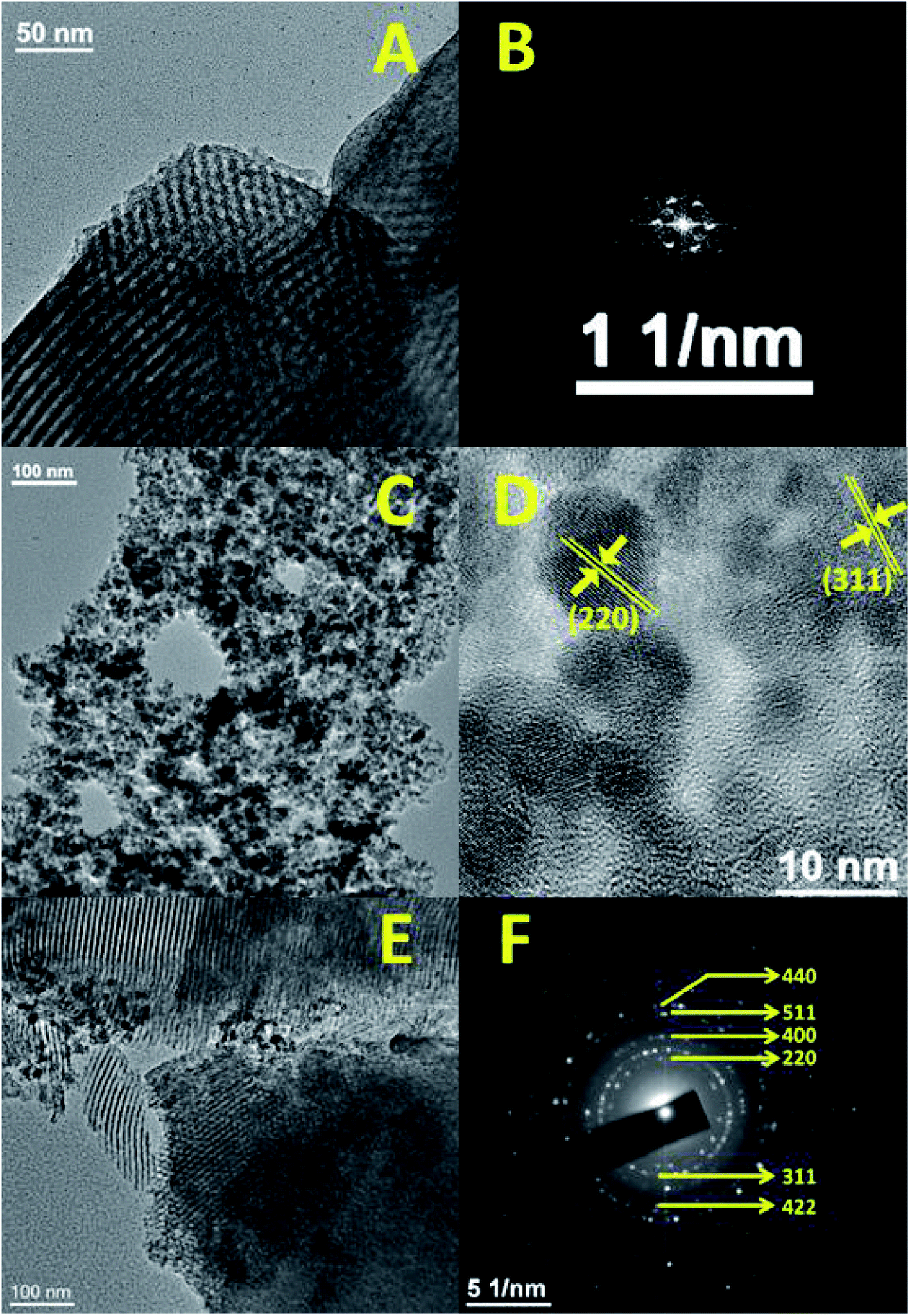 | ||
| Fig. 3 Representative TEM image and FFT pattern of SBA-15 (A and B), TEM (C), HR-TEM (D) and SAED (F) of Fe3O4 nanoparticles, and FA-FE-SBA15QN (E). | ||
2.2. pH-Dependent release of QN from FA-FE-SBA15QN in a cell-free system
The in vitro release behavior of conjugated QN from FA-IO-SBA-15-QN in response to a physiological, tumor endosomal/lysosomal change was studied using a cell-free system, and the release pattern was measured through fluorescence assessment. As shown in Fig. 4B, a gradual increase in the QN release from FA-FE-SBA15QN was evident at pH 7.4, where 30.03% of QN was discharged from the SBA-15 core at 8 h incubation. However, a slight decline in pH from 7.4 to 5.5 increased the QN liberation. 74.81% of QN was released at 4 h incubation, and the maximum peak (93.39%) was reached at 8 h. In order to determine the effect of stomach pH ranging from 1.5 to 3.5 on the QN release from the SBA-15 nanopore, the release assay was also assessed using HPLC. At a pH value of 3.5, the release of QN was not evident under the limit detection of HPLC due to the circumventing interaction with the acidic medium. Thus, most of the QN was retained in the SBA-15 pore at pH 3.5, as evident from the HPLC chromatogram (data was not shown). In contrast, at pH 7.4, most of the QN was slowly released from the SBA-15 core. On the other hand, due to the enhanced electrostatic repulsion between the positively charged nanopore decorated in SBA-15 at pH 5.5, the maximum QN moiety was released in the medium. This shows the useful cytotoxic activity in cancer cells, which was consistent with earlier scientific reports.22 As QN displayed a dominant emission maximum at ∼540 nm and two different excitation maxima (at ∼380 nm and ∼440 nm), the enhanced green emission was observed under pH 5.5 with respect to pH 7.4, confirming the maximum liberation in the medium (Fig. 4A).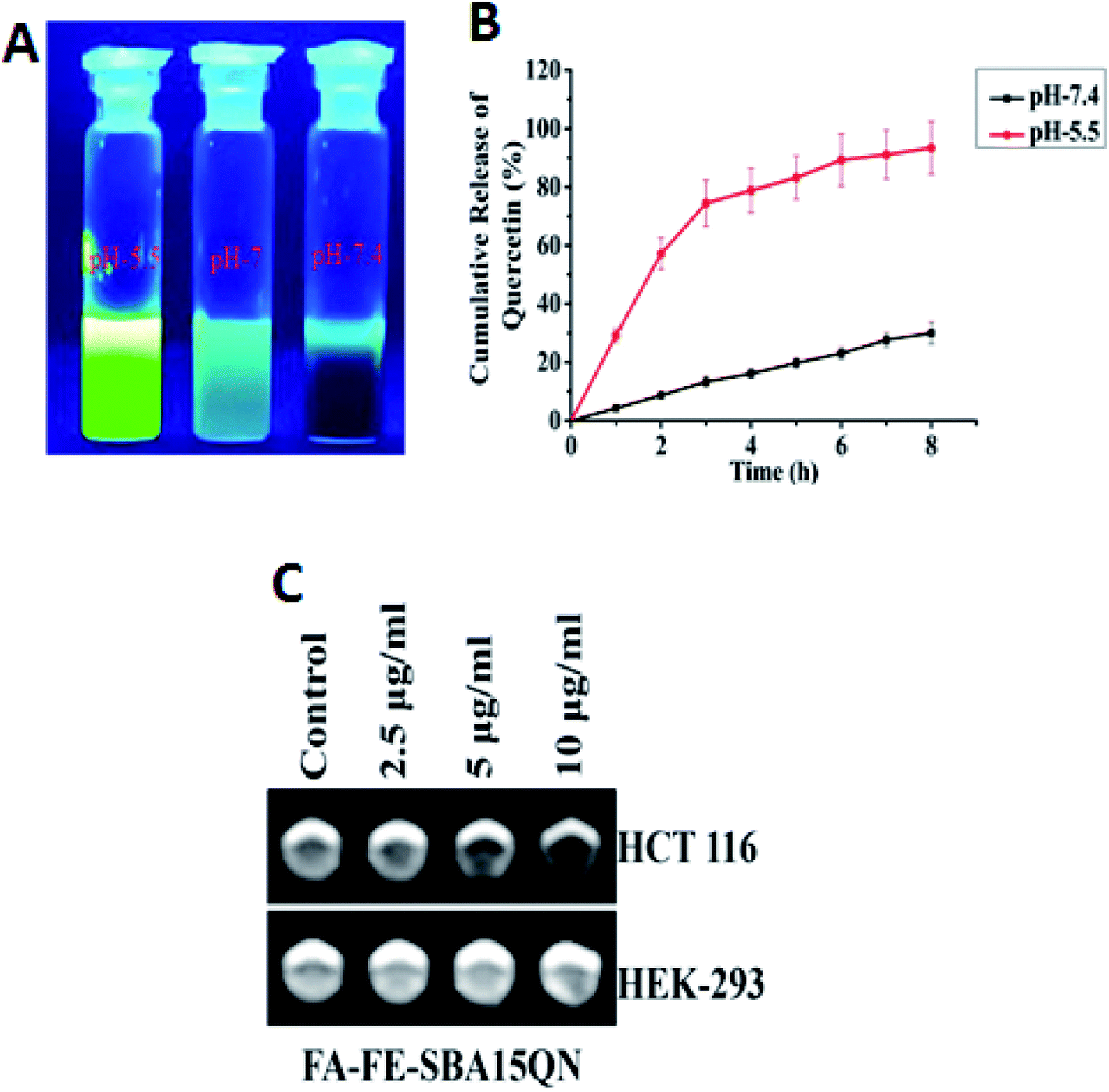 | ||
| Fig. 4 (A) FL emission released by QN after the dispersion of FA-FE-SBA15QN in PBS at different pH values (5.5, 7 and 7.4). (B) Line diagram representing the release kinetics of QN from FA-FE-SBA15QN in different pH values (7.4 and 5.5). (C) In vitro T2 weight MRI images of HCT 116 and HEK-293 cells after the treatment of FA-FE-SBA15QN at different concentrations (2.5, 5, and 10 μg ml−1). Values are represented as the mean ± SEM (n = 5). | ||
2.3. In vitro contrasting property of FA-FE-SBA15QN
The contrast effect of FA-FE-SBA15QN was assessed by measuring the transverse (T2) relaxation times of the dispersed nanostructures under the MRI equipment (3.0 T). As evident in Fig. 4C, the T2-weight MR signals were significantly enhanced with the dose-dependent increase of FA-FE-SBA15QN in HCT 116 cells. In contrast, there was no MR signal when the HEK-293 cells were treated with FA-FE-SBA15QN, suggesting the pH-sensitive contrasting property of the nanomaterial.25 The specific relaxivity value (r2) was also calculated in HCT 116 cells, which was more than enough (145.2 mM−1 s−1) to serve as a sensitive contrast agent for diagnostic purposes.2.4. QN and FA-FE-SBA15QN induced cytotoxicity in HCT 116 cells
To determine the cytotoxic activity of native QN, as well as compare the growth inhibitory potential of different components of orchestrated FA-FE-SBA15QN (including Folic Acid (FA), iron oxide (Fe3O4) and mesoporous silica nanoparticle (SBA-15)), various concentrations were assessed (3–30 μg ml−1 for QN, 10–100 μg ml−1 for Fe3O4, 5–30 μg ml−1 for SBA-15, 5–30 μg ml−1 for FA-SBA15, and 5–30 μg ml−1 for FA-FE-SBA15QN) in colorectal carcinoma cells (HCT 116), where 50% (IC50) and 70% (IC70) growth inhibitions were also evaluated on the basis of cytotoxicity. As shown in Fig. S1A,† a gradual increase in the cytotoxicity was found upon the treatment of native QN, at which the IC50 value was 15 μg ml−1. The potential activity of QN to perturb the cell viability of different cancer cells has been previously described in detail.26,27Thus, the current approach of this study was to develop a nanomaterial that could enhance the QN activity, along with its bioavailability at the target site. Scientific evidence reported that the loading of various chemotherapeutic drugs in the nanomaterial has been conserved, and sometimes potentiated the activity.24 On the one hand, Fe3O4 treatment alone did not show any promising cytotoxicity at higher concentrations, indicating little effect on the cell viability (Fig. S1B†). On the other hand, SBA-15, FA-conjugated SBA-15, and fully orchestrated SBA-15 treatment showed a variable reduction in the cell viability (Fig. S1C–S1E†). More than 50% cell death occurred at ∼14 μg ml−1 of SBA-15, and FA-SBA15 application. In contrast, the IC50 value was lower (10 μg ml−1) when the cells were treated with FA-FE-SBA15QN. This suggests the enhancement of the cytotoxic activity, which was in accordance with the previous studies.26 Based on the initial experimentation, the effective concentrations of 15 μg ml−1 of QN and 10 μg ml−1 of FA-FE-SBA15QN were chosen for the entire study for the exploration of the molecular mechanism of action.
2.5. FA-FE-SBA15QN-induced intercellular reactive oxygen species in HCT116 cells27–30
A growing body of evidence reported that QN triggers cytotoxicity in various carcinoma cells through the generation of iROS.28,29 To evaluate the role of FA-FE-SBA15QN in triggering iROS, flow cytometric evaluation was carried out using the ROS-sensitive dye, H2DCFDA. Upon the action of iROS, the acetate group of H2DCFDA is removed through the activity of intercellular esterase, following the conversion of the non-fluorescent H2DCFDA to the highly fluorescent 2′,7′-dichlorofluorescein (DCF).30 Hence, an increase in the DCF fluorescence reflects the relative amount of ROS generated within the cells.30 The results revealed an enhanced DCF fluorescence (62.9%) upon the treatment of FA-FE-SBA15QN (10 μg ml−1), which was much higher than the resultant DCF fluorescence of the control (22.3%) and QN-treated state (42.9%) (Fig. 5B). This suggested a possibility of higher release of native QN in the HCT-116 cells, which mediated the cytotoxicity. In contrast, a specific ROS scavenger, N-acetyl cysteine (NAC), significantly suppressed the ROS (34.5%) when treated along with FA-FE-SBA15QN. This indicated the direct correlation of the ROS-mediated cytotoxicity in the HCT-116 cells. The following results corroborated with the earlier demonstrated literature.27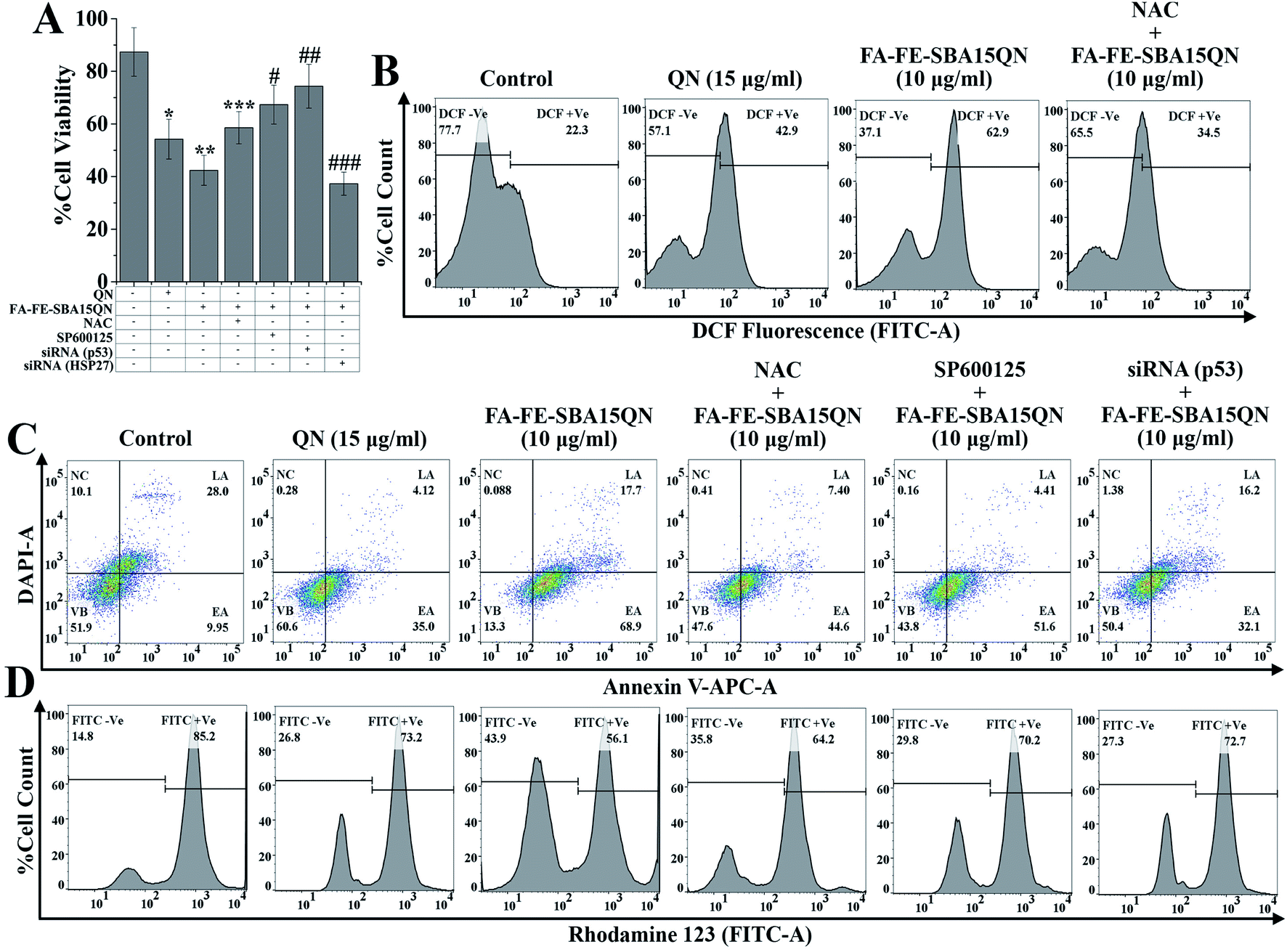 | ||
| Fig. 5 Determination of cell viability, iROS, apoptosis/necrosis, and mitochondrial membrane potential upon the treatment of QN (15 μg ml−1) and FA-FE-SBA15QN (10 μg ml−1), along with/without NAC, SP600125, p53 siRNA, and HSP27 siRNA application in HCT 116 cells. (A) Bar graph showing cell viability. Representative flow cytometric dot plot and gating hierarchy used to define (B) DCF +Ve cells and DCF −Ve cells, (B) viable cells (VB), early apoptotic cells (EA), late apoptotic cells (LA), and necrotic cells (NC), (C) FITC +Ve cells and FITC −Ve cells, (D) high ΔΨ and low ΔΨ. Values are represented as the mean ± SEM (n = 5). p < 0.05 was considered as significant. Statistical comparison: *control vs. QN (15 μg ml−1); **control vs. FA-FE-SBA15QN (10 μg ml−1); ***FA-FE-SBA15QN (10 μg ml−1) vs. FA-FE-SBA15QN (10 μg ml−1) + NAC; #FA-FE-SBA15QN (10 μg ml−1) vs. FA-FE-SBA15QN (10 μg ml−1) + SP600125; ##FA-FE-SBA15QN (10 μg ml−1) vs. FA-FE-SBA15QN (10 μg ml−1) + siRNA (p53); ###FA-FE-SBA15QN (10 μg ml−1) vs. FA-FE-SBA15QN (10 μg ml−1) + siRNA (HSP27). | ||
2.6. FA-FE-SBA15QN-mediated apoptosis and necrosis in HCT-116 cells
To evaluate the apoptosis and necrosis upon FA-FE-SBA15QN treatment, a detailed flow cytometric evaluation was carried out using annexin V/DAPI. Annexin V is a commonly used marker to detect apoptotic cells by its ability to bind phosphatidylserine of the outer leaflet of the cellular membrane.31 Fig. 5C demonstrated that the percent cell population of early (EA) and late (LA) apoptosis was markedly enhanced with the treatment of FA-FE-SBA15QN (68.9% of EA and 17.7% of LA), which was much higher than that of the native QN treatment (35.0% of EA and 4.12% of LA) with respect to the control cells (9.95% of EA and 28.0% of LA). On the other hand, for the treatment of NAC along with FA-FE-SBA15QN, the level of EA was augmented (44.6%) and the LA (7.40%) population was reduced. Conversely, the silencing of p53 (p53 siRNA) markedly reduced EA (32.1%) and LA (16.2%) in the FA-FE-SBA15QN-treated condition, which was much higher than that with the co-application of the JNK inhibitor (SP600125) (51.6% of EA and 4.41% of LA) and FA-FE-SBA15QN (10 μg ml−1). The above results indicated the involvement of JNK and p53-regulated apoptosis in FA-FE-SBA15QN-mediated cytotoxicity, which was in accordance with earlier reports.322.7. FA-FE-SBA15QN-triggered mitochondrial membrane potential change (ΔΨm) in HCT-116 cells
Numerous scientific reports demonstrated that mitochondrial dysfunction has been associated with the apoptosis phenomenon, and also involved in the regulation of crucial signaling mediators in triggering apoptosis.33 To establish the fact underlying the involvement of ΔΨm in FA-FE-SBA15QN-mediated apoptosis, a tracer dye rhodamine 123 (RH-123) was used through flow cytometry application. The rate of mitochondrial energization mediates the quenching of RH-123, which reflects a progressive reduction with the change in ΔΨm.34 Thus, the resultant fluorescence decay of RH-123 is proportional to the mitochondrial membrane potential. As depicted in Fig. 5D, the relative RH-123 +Ve cells (FITC +Ve) were decreased (73.2% and 56.1%) with the treatment of QN (15 μg ml−1) and FA-FE-SBA15QN (10 μg ml−1). In contrast, for the co-treatment of NAC, SP600125, and siRNA specific for p53 with FA-FE-SBA15QN, the percentages of RH-123 +Ve cells were gradually increased (64.2%, 70.2%, and 72.7%) with respect to the FA-FE-SBA15QN-‘treated alone’ group, representing the possible correlation between the mitochondrial-dependent apoptosis and JNK/p53-mediated downstream signaling system.262.8. FA-FE-SBA15QN induced phosphorylation H2AX in HCT-116 cells
A direct connection between DNA damage and apoptosis exists, as supported by a large body of literature.35 DNA damage, a common event of life, is responsible for triggering apoptosis by controlling numerous signaling systems.35 H2AX, a key variant of the H2A protein family, is a component of the histone octamer of nucleosomes.36 Upon DNA damage (DSB) by exogenous and endogenous sources, H2AX is phosphorylated at Ser139 to form gamma-H2AX (γ-H2AX), which recruits repair proteins at the damaged site of DNA. Reports also demonstrated that γ-H2AX formation is an indicative step for DNA damage, which provokes the activation of downstream machinery associated with apoptosis.36,37 To elucidate the role of FA-FE-SBA15QN in triggering DNA damage following the apoptosis machinery, H2AX phosphorylation was studied through immunofluorescence. As represented in Fig. S2,† the number of γH2AX foci formation was increased with the treatment of native QN. On the contrary, the foci formation in HCT 116 was markedly increased when treated with FA-FE-SBA15QN, indicating the enhancement of DNA damage. However, for the NAC treatment along with FA-FE-SBA15QN, the enhancement of DNA damage was slower. This represents a putative effect of ROS on the γH2AX formation. In contrast, SP600125 (a known JNK inhibitor) also suppressed H2AX formation over FA-IO-SBA-15-QN treatment. This suggests that JNK inhibition may inhibit H2AX phosphorylation, which was corroborated with the previous report.382.9. FA-FE-SBA15QN-induced modulation of HSPs in HCT-116 cells
Heat shock protein (HSP), a conserved superfamily of proteins, functions as a molecular chaperone to guard proteins against illicit or promiscuous interactions with other proteins.39 Activation of HSPs has been shown to increase resistance against cell death induced by various factors.40 Reports showed that HSP27, a subfamily of HSPs, is associated with the regulation of apoptosis through the inhibition of multiple signaling systems.41 Dual color flow cytometric analysis showed that FA-FE-SBA15QN markedly decreased (36.6%) HSP27 expression (HSP-AF488 +Ve cells), as compared to the QN-treated cells (70.4%). On the other hand, further decrease in HSP27 expression (30.8%) was observed in siRNA that was specific for HSP27 treatment, along with FA-FE-SBA15QN (Fig. 6A).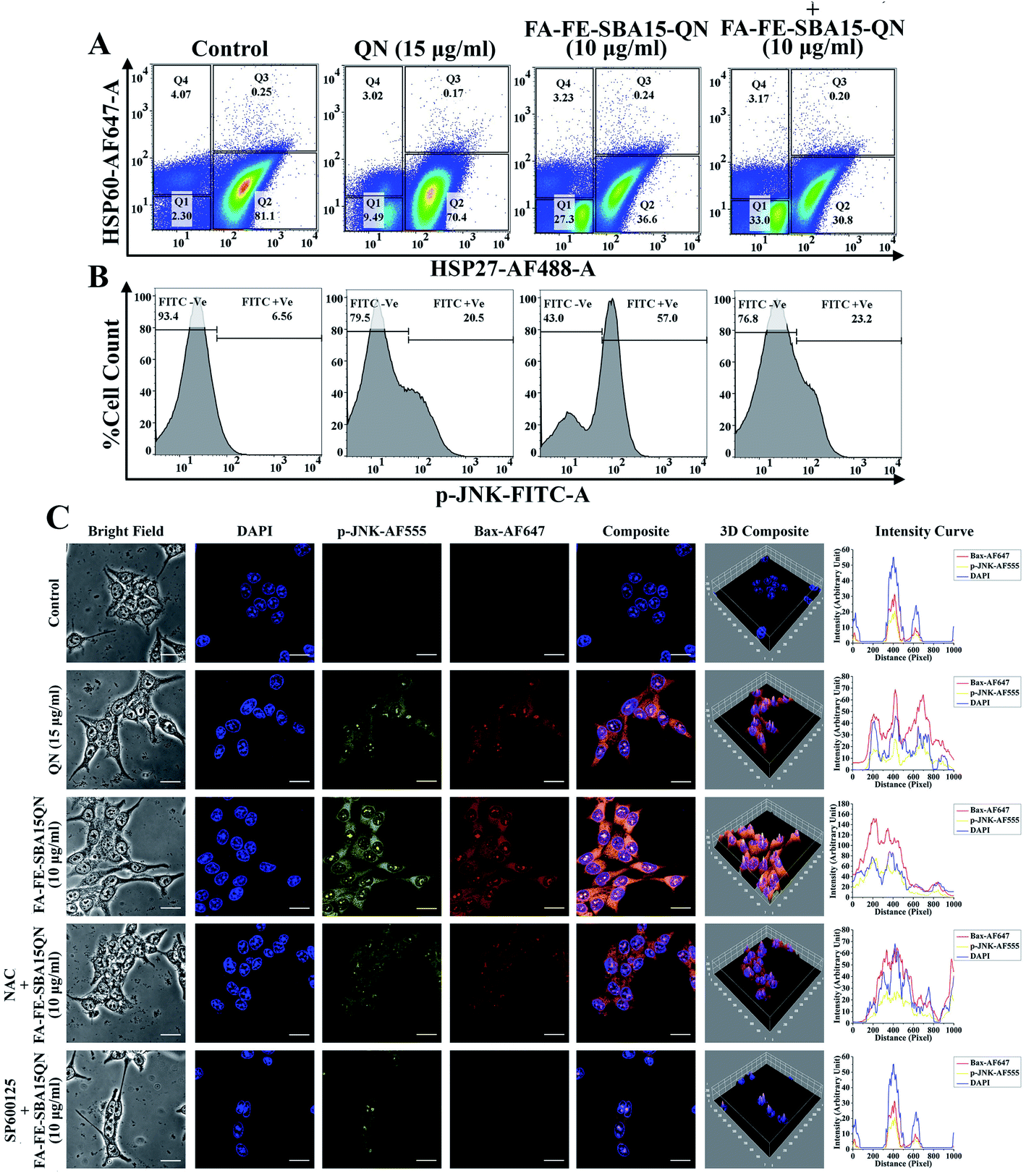 | ||
| Fig. 6 QN and FA-FE-SBA15QN-induced modulation of HSP60, HSP27, p-JNK, and Bax expression in HCT 116 cells with/without application of NAC, SP600125, and siRNA (HSP27). Representative flow cytometric dot plot and gating hierarchy used to define (A) Q1: HSP60-AF647 −Ve/HSP27-AF488 −Ve cells; Q2: HSP60-AF647 −Ve/HSP27-AF488 +Ve cells; Q3: HSP60-AF647 +Ve/HSP27-AF488 +Ve cells; Q4: HSP60-AF647 +Ve/HSP27-AF488 −Ve cells, (B) p-JNK-FITC −Ve and p-JNK-FITC +Ve cells. (C) Immunofluorescence images showing the expression of p-JNK and Bax. DAPI was used for nuclear staining. Slides were viewed using a confocal microscope (magnification 20×). Respective fluorescence intensities (p-JNK-AF555, Bax-AF647, and DAPI) were analyzed using ImageJ software through an RGB calculator. | ||
The expression of another HSP, namely HSP60 (HSP60-AF647 +Ve cells), was unchanged throughout the following treatment of QN and FA-FE-SBA15QN, suggesting that only HSP27 was inhibited with the application of QN and FA-FE-SBA15QN. Diminished cell viability (37.28%) was also detected on the use of HSP27 siRNA with FA-FE-SBA15QN treatment, which was much lower as compared to the FA-FE-SBA15QN treatment (42.38%) (Fig. 6A). This supports the fact that HSP27 inhibition was directly correlated with the cytotoxicity.
2.10. FA-FE-SBA15QN-induced phosphorylation of JNK in HCT-116 cells
One of the essential mitogen-activated protein kinase families of proteins, the Jun N-terminal Kinase (JNK) plays a decisive role in mitochondrial-assisted apoptotic pathways.42 Reports have also shown that JNK is indispensable for both cell proliferation and apoptosis.42 An intricate mechanism exists between JNK and HSP27. Inhibition of HSP27 stifles JNK activation, which also modulated apoptosis-associated protein expression.43 As observed in the immunofluorescence analysis, JNK was significantly phosphorylated when treated with FA-FE-SBA15QN. The augmented expression was markedly decreased with SP600125 treatment, which was much lower than the NAC treatment (Fig. 6C). Concomitant treatment with SP600125 and FA-FE-SBA15QN also enhanced the cell viability (67.34%) when compared with the FA-FE-SBA15QN (42.38%), indicating that the loss of the JNK function could inhibit apoptosis (Fig. 6A). In contrast, treatment with siRNA specific for HSP27 also decreased JNK phosphorylation (JNK-FITC +Ve of 23.2%) in the FA-FE-SBA15QN-treated condition, which supports the fact that HSP27 may modulate the JNK expression (Fig. 6B).2.11. FA-FE-SBA15QN induced modulation of p53-guided mitochondrial apoptosis
An important redox-assisted transcription factor, p53 (tumor suppressor protein) is involved in the regulation of mitochondrial-dependent apoptosis upon DNA damage.44 JNK phosphorylation mediates mitochondrial-dependent apoptosis through the phosphorylation of p53. Enhanced phosphorylation of p53 targets the proapoptotic members of Bcl2 family proteins, including Bax, PUMA, and NOXA, by its ability to control transcription. In addition, p53 can also transactivate several components of apoptotic effector machinery. One of these components, Apaf1 acts as a coactivator of caspase 9, which helps to initiate the caspase cascade.44 The FA-IO-SBA-15-QN treatment markedly enhanced the p53 phosphorylation (5.42%) over the control group (Fig. 7A) (0.46%), which also increased the expression of the proapoptotic protein Bax (44.5%) (Fig. 7B). On the other hand, an anti-apoptotic protein, Bcl2 (0.17-fold) was also decreased upon the FA-FE-SBA15QN treatment (Fig. 7D and E). The expression of cytochrome C (36.8%) and Apaf-1 (7.47-fold) was augmented (Fig. 7C, F and G) with the FA-FE-SBA15QN treatment, which further supported the final fate of apoptosis as observed in the activation of the caspase cascade (2.87-fold for caspase 9 and 2.47-fold for caspase 3) (Fig. 7H and I). The selective inhibition of p53 using specific siRNA remarkably diminished the p53 phosphorylation (0.78%), following the enhancement of Bcl2 (0.80-fold), as well as the suppression of Bax (22.7%), CytC (18.2%), and Apaf1 (5.82-fold) that was more pronounced than NAC (4.07% for p-p53, 38.1% for Bax, 30.9% for CytC, 7.01-fold for Apaf-1, 0.39-fold for Bcl2, 2.54-fold for caspase 9, and 2.10-fold for caspase 3)/SP600125 (3.94% for p-p53, 28.3% for Bax, 19.0% for CytC, 6.47-fold for Apaf-1, 0.57-fold for Bcl2, 1.90-fold for caspase 9, and 1.74-fold for caspase 3) treatment. Interestingly, p53 inhibition using siRNA also decreased the caspase activation (1.87-fold for caspase 9 and 1.41-fold for caspase 3), which increased the cell viability (74.35%) (Fig. 5A). This indicated the presence of a possible crosstalk mechanism between p53 phosphorylation and mitochondrial apoptosis in the FA-FE-SBA15QN-treated state.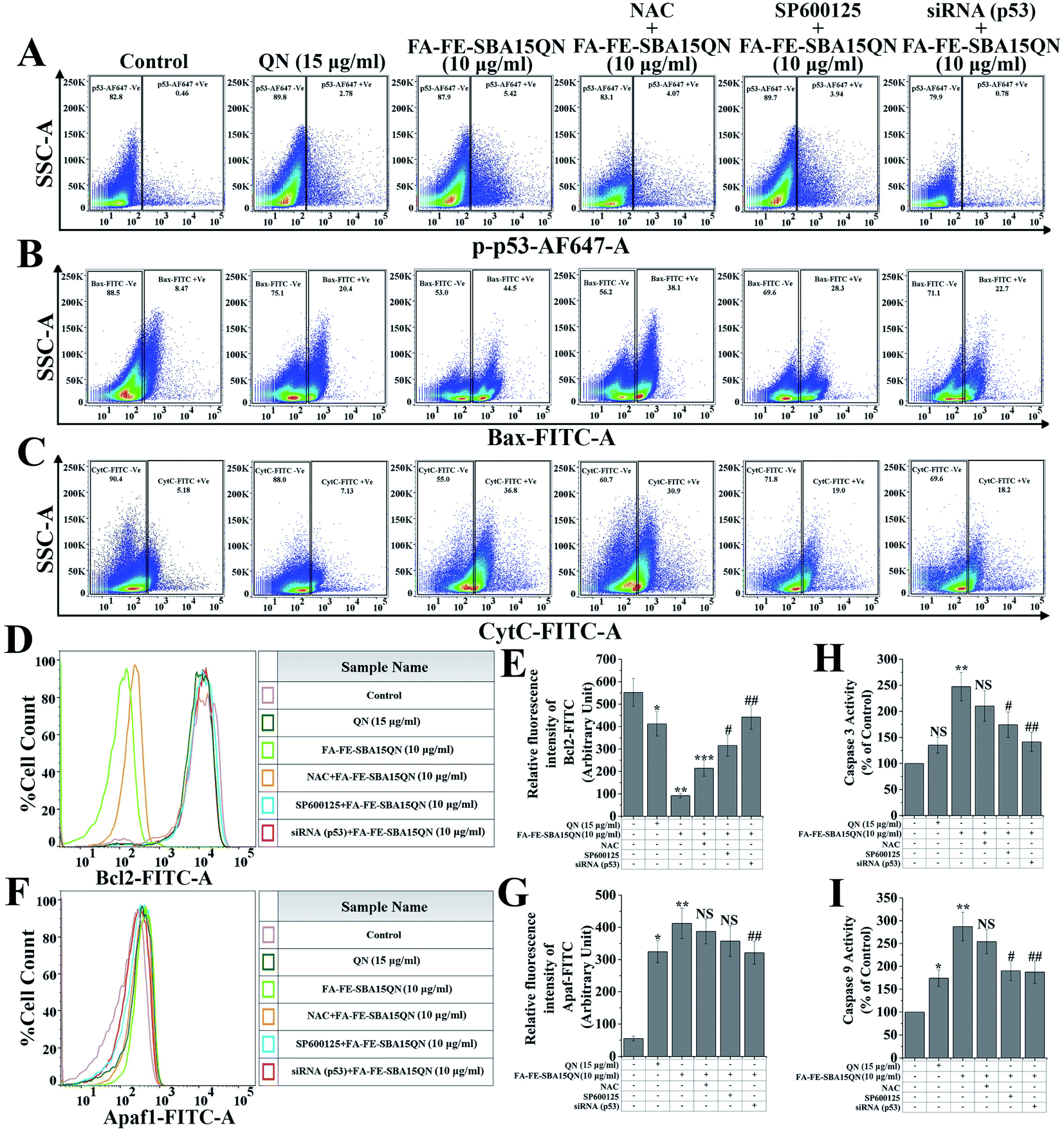 | ||
| Fig. 7 Assessment of mitochondrial-dependant apoptosis upon QN and FA-FE-SBA15QN treatment, along with/without application of NAC, SP600125, and siRNA (p53). Representative flow cytometric dot plot and gating hierarchy used to define (A) p53-AF647 +Ve and p53-AF647 −Ve cells, (B) Bax-FITC +Ve and Bax-FITC −Ve cells, (C) CytC-FITC +Ve and CytC-FITC −Ve cells. Representative flow cytometric histogram showing (D) Bcl2 and (F) Apaf1. Bar graph shows relative fluorescence intensities of (E) Bcl2-FITC and (G) Apaf1-FITC. Bar graph shows (H) caspase 3 and (I) 9 activity in the different experimental conditions in HCT-116 cells. Values are represented as the mean ± SEM (n = 6). p < 0.05 was considered as significant. Statistical comparison: *control vs. QN (15 μg ml−1); **control vs. FA-FE-SBA15QN (10 μg ml−1); ***FA-FE-SBA15QN (10 μg ml−1) vs. FA-IO-SBA-15-QN (10 μg ml−1) + NAC; #FA-FE-SBA15QN (10 μg ml−1) vs. FA-FE-SBA15QN (10 μg ml−1) + SP600125; ##FA-FE-SBA15QN (10 μg ml−1) vs. FA-FE-SBA15QN (10 μg ml−1) + siRNA (p53); NS = not significant. | ||
2.12. FA-FE-SBA15QN-induced mitochondrial apoptosis through JNK/p53 axis
To further confirm the JNK-induced activation of apoptosis in the FA-FE-SBA15QN-treated condition, immunofluorescence was carried out. The FA-FE-SBA15QN-treatment augmented Bax expression was higher than that for the natural QN treatment. On the contrary, the selective inhibition of the JNK pathway using SP600125 further diminished the Bax expression. In contrast, the NAC treatment along with FA-FE-SBA15QN did not have a significantly effect on the JNK and Bax expression. These data demonstrated that the JNK pathway might regulate p53-guided Bax expression (Fig. 6C).2.13. FA-FE-SBA15QN restored tumor-induced survival of mice
To further determine whether FA-FE-SBA15QN had substantial antiproliferative activity over native QN, a suitable tumor-bearing murine model was developed using the subcutaneous injection of CT-26 cells (Fig. 8A).45 The optimum effective dosage of FA-FE-SBA15QN and QN was also determined on the basis of survival analysis. The results corresponding to the optimum effective dosage of FA-FE-SBA15QN and QN on the survival rate of mice estimated after the CT-26 challenge is illustrated in Fig. 8C. The FA-FE-SBA15QN application alone for six alternate days did not induce any death within the 30 day observational period. The first death of the CT-26 tumor-bearing mice was observed after the 15th day of the CT-26 challenge due to the significant symptoms, including reduction of food/water intake, irritability, enhanced tumor burden, weight loss, lethargy, and roughing of hair. The results also demonstrated that 50% of the animals died within 16 days of the CT-26 challenge, and 100% mortality was reached at the 19th day. Administration of FA-FE-SBA15QN (15 mg kg−1) on the CT-26 tumor-bearing condition enhanced the survival proportion with respect to the CT-26 challenge alone. The maximum survival (50%) was evident with the treatment of 15 mg kg−1 of body weight of FA-FE-SBA15QN, while 16.67% of the animals died within the 30 day observational period upon the treatment of native QN (15 mg kg−1), validating the effectivity of FA-FE-SBA15QN over native QN.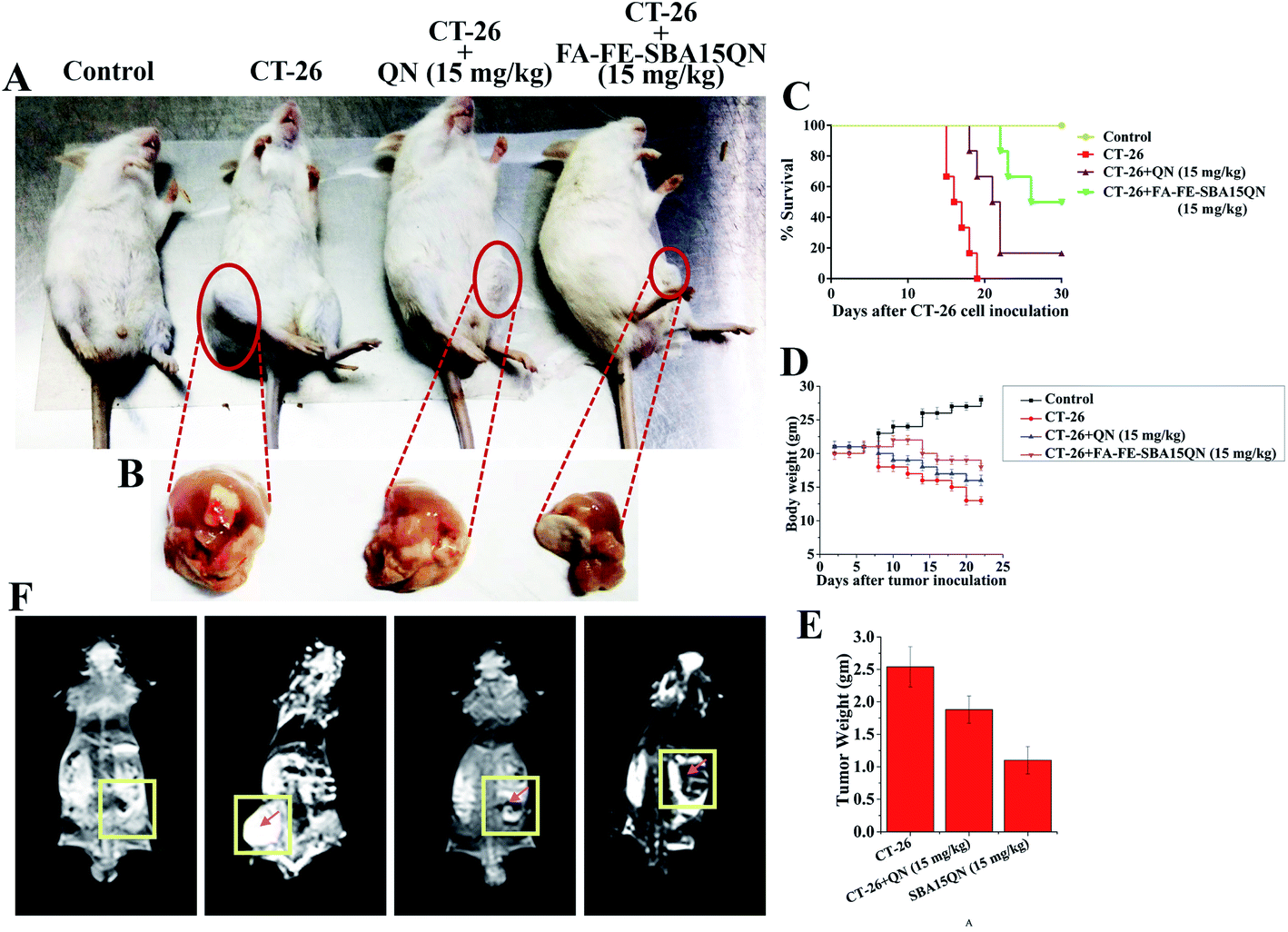 | ||
| Fig. 8 In vivo assessment of chemo theranostic potential of QN and FA-FE-SBA15QN. (A) Experimental animals showing tumor growth inhibitory activity of QN and FA-FE-SBA15QN. (B) Representative images of tumor harvested after the completion of the experiment. (C) Kaplan–Meier analysis of 30 days survival of CT-26 tumor-bearing mice post-administrated with QN and FA-FE-SBA15QN (15 mg kg−1). (D) Representative line diagram showing body weight change throughout the experimental period. (E) Bar graph showing tumor weight. (F) In vivo T2 weight MRI images of CT-26 tumor-bearing mice. Rectangular zone indicating the tumor area. Values are represented as mean ± SEM (n = 5). p < 0.05 was considered as significant. Statistical comparison: *CT-26 vs. CT-26 + QN (15 mg kg−1); **CT-26 vs. CT-26 + FA-FE-SBA15QN (15 mg kg−1). | ||
2.14. FA-FE-SBA15QN attenuated tumor-induced weight loss
A significant loss of body weight has been profoundly associated with the development of a tumor mass due to the failure of homeostatic balance.46 To examine whether the FA-FE-SBA15QN treatment was effective in reducing the cancer-induced complications, a regular assessment of body weight was monitored until the end of experimentation. A marked reduction of body weight was evident after the introduction of CT-26 cells. The average body weight was reduced to 24% on the 15th day of experimentation. Interestingly, the FA-FE-SBA15QN treatment (15 mg kg−1) markedly restored the tumor-induced body weight reduction (5%) over QN (15%), indicating that FA-FE-SBA15QN administration helped to maintain the body weight in tumor-bearing mice (Fig. 8D).2.15. FA-FE-SBA15QN-mediated reduction in tumor weight
A marked decrease in the tumor weight was observed with the application of FA-IO-SBA-15-QN (15 mg kg−1) over native QN (15 mg kg−1). The reduction was also evident in the decrease in the tumor weight, which was significantly reduced to 1.11 g and 1.88 g, compared to the untreated tumor mass at 2.54 g (Fig. 8B and E).2.16. FA-FE-SBA15QN-reduced CT-26 tumor cell proliferation
Ki-67, a cellular protein, is associated with cell proliferation, which is used for the detection of the metastatic tumor.47 Ki-67 protein expression was determined in an experimental condition in order to determine the ameliorative role of FA-FE-SBA15QN on the CT-26 tumor. Immunofluorescence analysis revealed that FA-FE-SBA15QN (15 mg kg−1) markedly reduced Ki-67 expression, as well as increased Bax expression in the CT-26 tumor, suggesting a possible inhibition on the tumor cell proliferation (Fig. 9B). On the other hand, the histopathological features of the tumor mass also confirmed a similar result (Fig. 9A). The tumors of untreated groups exhibited 70–90% viable polymorphic cells with dense and round nuclei. The tumor mass also showed an excessive microvascular compartment in an untreated condition. Interestingly, upon the treatment of FA-FE-SBA15QN (15 mg kg−1), a marked reduction of the microvascular compartment along with the viable tumor cells was observed, which substantiated the previous reports. The data also indicated that the efficacy of FA-FE-SBA15QN was more effective than the native QN (15 mg kg−1) treatment in reducing tumor cell proliferation, as observed in the Ki-67 and Bax expression.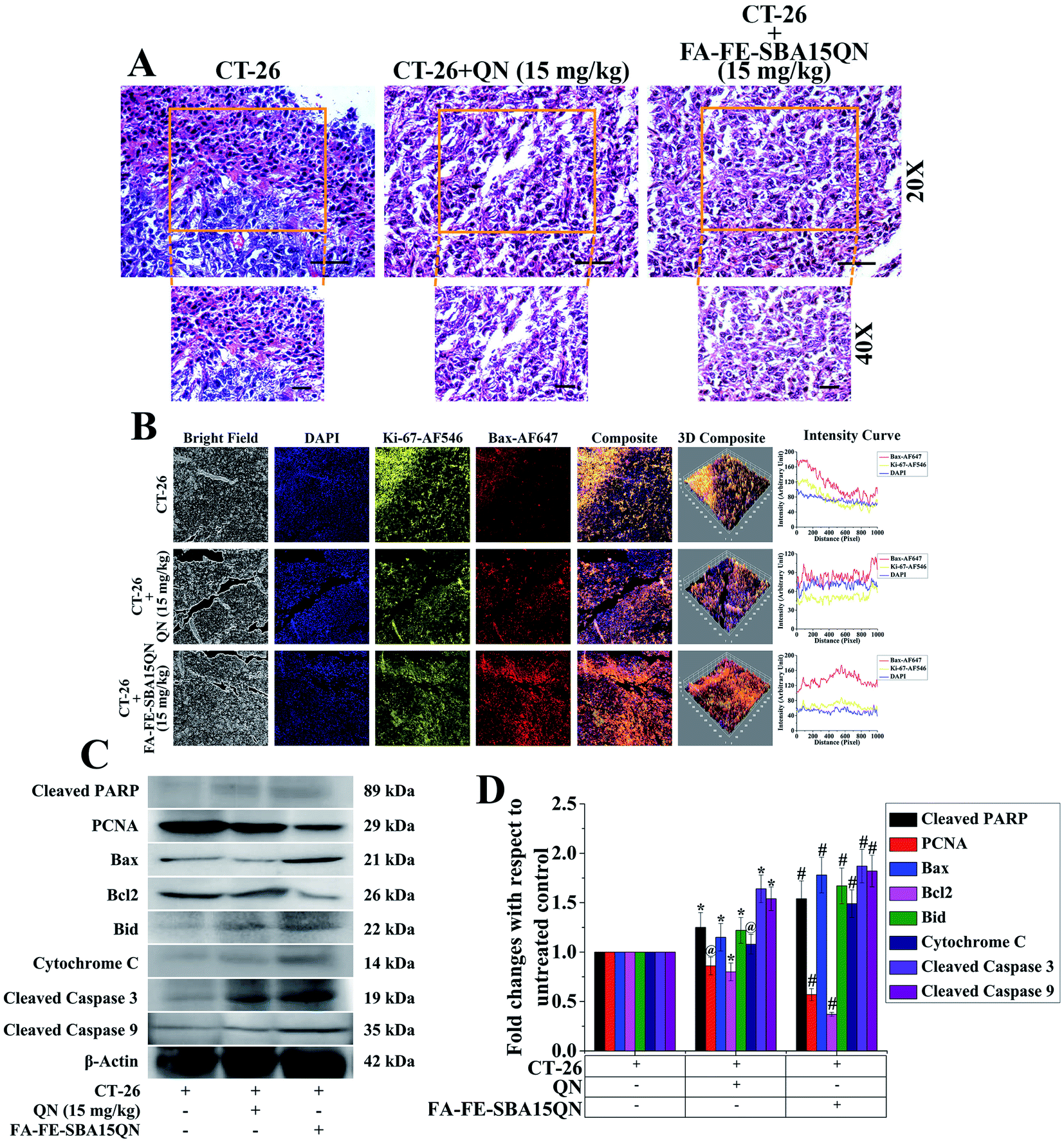 | ||
| Fig. 9 (A) Histopathological assessment of tumor microenvironment. The tumor tissue architectural assessment was done using hematoxylin & eosin staining (H&E) (nagnification: 20× and 40×). (B) Immunofluorescence images showing the expression of Ki-67 and Bax. DAPI was used for nuclear staining. Slides were viewed using a confocal microscope (magnification 20×). Respective fluorescence intensities (Ki-67-AF546, Bax-AF647, and DAPI) were analyzed using ImageJ software through an RGB calculator. (C) Representative immunoblots of cleaved PARP, PCNA, BAX, Bcl2, Bid, cytochrome C, cleaved caspase 3 and cleaved caspase 9. (D) Densitometric analysis of the relative protein expression of cleaved PARP, PCNA, BAX, Bcl2, Bid, cytochrome C, cleaved caspase 3 and cleaved caspase 9. β-Actin served as the internal control. Values are presented as mean ± SEM (n = 6). p < 0.05 was considered as significant. Statistical comparison: *CT-26 vs. CT-26 + QN (15 mg kg−1); #CT-26 vs. CT-26 + FA-FE-SBA15QN (15 mg kg−1); @ = not significant. | ||
To further confirm the antiproliferative property of FA-IO-SBA-15-QN, a plethora of protein expressions related to cellular proliferation and apoptosis was determined using immunoblot. As depicted in Fig. 9C and D, the cellular proliferation marker PCNA was significantly decreased with the treatment of FA-FE-SBA15QN, which was more effective than the native QN treatment (15 mg kg−1). On the other hand, the pro-apoptotic proteins, Bax, Bid, cytochrome C, and cleaved PARP were markedly augmented. In addition, a diminished expression of the anti-apoptotic protein, Bcl2, was evident upon the treatment of FA-IO-SBA-15-QN, suggesting the potent reductive ability of QN. The final fate of the tumor reduction was observed when the cleaved caspase 3 and 9 expression levels increased in the FA-IO-SBA-15-QN-treated condition, indicating the antiproliferative efficacy of FA-FE-SBA15QN over the QN-treated state.
2.17. Magnetic resonance imaging (MRI) of CT-26 adenocarcinoma using FA-FE-SBA15QN
MRI, a popular diagnostic tool, is capable of providing detailed information about the structure–composition relationship of tumors with spatial resolution.48 The use of exogenously administered contrasting agents allows for compartment-specific enhancement of tumors, enabling image specific functional and interstitial volumes.49 One of the critical goals of this study was to develop a particular molecular MR contrast agent, which could probe and interact with the ligand receptor-specific (FA) target molecule. It could also create changes in the local tissue proton relaxation times (longitudinal relaxation time, T1; and transverse relaxation time, T2) to sense the tumor site. Folic acid receptor (FaR) is highly expressed in the CT-26-induced adenocarcinoma. Hence, the approach was to target the folic acid receptor by folic acid-functionalized SBA-15. The unique capping material Fe2O3, having a superparamagnetic property, was used as a contrasting agent. The loaded QN was directed for its therapeutic efficacy. Thus, the contrasting feature of FA-FE-SBA15QN was determined to evaluate the targeted delivery of QN in the tumor site, as well as to define its theranostic approach. As evident in the in vivo MRI images, the CT-26-bearing adenocarcinoma had no clear zone in terms of the contrast area (T2 relaxation time) at the tumor site. However, an enhanced T2-MR signal (contrast area) was found at the tumor site when mice were treated with FA-IO-SBA-15-QN, establishing its notable contrasting character, which might be a promising approach for theranostic drug development (Fig. 8F).2.18. Biodistribution of Fe and Si of SBA-15, FA-FE-SBA-15 in tumors and hepatic tissue
The biodistribution of Si and Fe has been estimated in hepatic and tumor tissues by ICP-MS. As depicted in Fig. S3,† a sprinkled quantity of naked Fe3O4-NP was found after the 1st treatment in the liver and tumor, which was almost negligible after the 6th consecutive treatment. The rapid clearance of Fe3O4-NP was evident in all tissues due to its ultrasmall size. In the case of FA-SBA-15, a maximum concentration of Si in the liver and tumor tissue was found with respect to the SBA-15-treated group, which was also cleared after the 6th consecutive treatment. Compared with all hepatic tissues, the tumor had the maximum agglomeration of Si and Fe, justifying the targeted delivery of FA-FE-SBA15. Based on our in vitro and in vivo anti-carcinogenic efficacy results on FA-functionalized QN-loaded SBA-15 bearing Fe3O4 NPs at its surfaces in Scheme 1, we have presented a probable mechanism of action of FA-FE-SBA15QN in modulating the cancer microenvironment.3. Conclusion
The design and synthesis of Fe3O4 nanoparticle-immobilized, functionalized, ordered mesoporous silica SBA-15 loaded with flavonoid quercetin (FA-FE-SBA15QN) is presented in the current study. In vitro investigations on human colorectal carcinoma cells (HCT 116) showed a dose-dependent decrease of the cell viability, which was also in a direct relationship with the QN content in SBA-15. Surface modification by Fe3O4 helped to act as a better contrasting agent, which was useful for cancer/tumor detection by MRI. Folic acid (FA) conjugation increased the QN delivery with a more targeted fashion, which would provide better clinical outcomes with less toxicity and offer a superior treatment modality in future chemotherapeutic approaches. Furthermore, FA-FE-SBA15QN-triggered caspase-dependent apoptosis integrated with the overexpression of p53, Bax, and CytC, as well as the down-regulation of Bcl-2. Additionally, FA-FE-SBA15QN inhibited HSP27 expression, which could enhance the JNK expression. This was followed by the indirect phosphorylation of H2AX/DSBs. All of these results demonstrated the positive effects of the 2D-hexagonal mesoporous SBA-15 material as a drug carrier system with pronounced cytotoxic activity. SBA-15 amplified the activity of the QN in vitro system, and protected the active compound from the impulsive, light-directed inactivation or degradation guided by extremely acidic conditions corresponding to gastric pH. Thus, FA-FE-SBA15QN can be successfully used in cancer therapy, with the vital ability to deliver chemotherapeutic drugs intracellularly.4. Experimental section
4.1. Chemicals
P123 poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol), TEOS (tetraethyl orthosilicate) and 3-chloropropyltriethoxysilane (3-CPTES) were purchased from Sigma Aldrich, India. Folic acid (M = 441.4 g mol−1), and quercetin (M = 302.236 g mol−1) were procured from Sigma Aldrich, India. All other organic solvents were used as received without further purification.4.2. Synthesis of mesoporous 3-chloropropyl-functionalized SBA-15 (SBA-Cl)
Pure SBA-15 material was synthesized using P123 as a structure-directing agent in the presence of HCl and tetraethyl orthosilicate as a silica source.50 The as-synthesized material was calcined at 623 K to remove the template P123 molecules. Then, the calcined SBA-15 (1.0 g) was dispersed in a round bottom flask (100 ml) containing CHCl3 (50 ml). 3-Chloropropyltriethoxysilane (3-CPTES) (1.2 g) was then added to the dispersed solution. The resulting solution mixture was allowed to stir continuously at room temperature for about 36 h under nitrogen atmosphere. After completion of the reaction, the solid product was filtered through a simple filtration technique, and washed with excess chloroform. Finally, it was dried in air to obtain SBA-Cl (Scheme 1).514.3. Synthesis of folic acid-functionalized SBA-15 (FA-SBA15)
Briefly, solid white SBA-Cl (1.5 g) was further dispersed in dimethylacetamide (DMA) (25 ml), and folic acid (250 mg) was added to this reaction mixture. Then, it was allowed to reflux at 160 °C for 36 h. Finally, the pale yellow colored solid product was filtered and washed with copious amounts of distilled water to remove the unreacted folic acid from the functionalized material. The resulting solid was dried in the oven at 75 °C temperature and subjected to characterization (Fig. 2).524.4. Quercetin loading
FA-functionalized SBA-15 particles were added to a quercetin (QN) solution, and incubated in a shaker at room temperature overnight. QN molecules could diffuse in the solution into the pore channels. The QN-loaded SBA-15 was taken out by centrifugation and rinsed with water to get rid of the redundant QN. Finally, the resulting particles were washed with water and dried in vacuo at 60 °C to yield the QN-loaded folate (FA) functionalized SBA-15. The amount of QN loaded into the FA-functionalized SBA-15 was estimated by UV-Vis spectroscopy at 369 nm (λmax = 369 nm):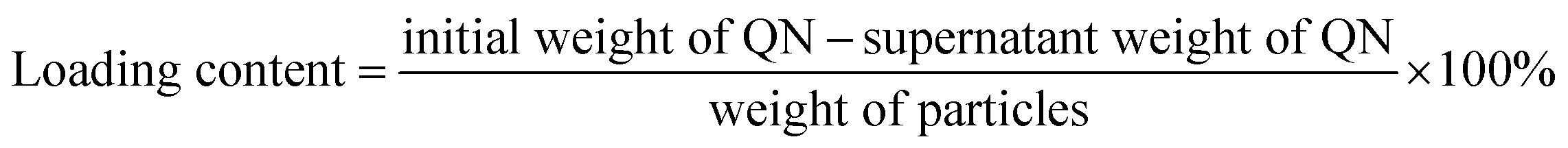
4.5. Synthesis of Fe3O4 (FE)
Iron(II) chloride tetrahydrate and iron(III) chloride hexahydrate was measured at 2.50 × 10−3 and 2.99 × 10−3 mol, respectively. They were added to a vigorously stirred deoxygenated aqueous solution (600.0 ml, pH ∼1.5). Then, ammonium hydroxide (1.50 M) was added dropwise to the aqueous solutions of iron(II) and iron(III). The addition of ammonium hydroxide was continued until the pH value reached 11.0 from 1.5. As the pH increased (from 1.5 to 11.0), a black precipitate of magnetite (Fe3O4) simultaneously formed. The black precipitate was separated by magnetic decantation, and then it was washed five times with water. Ethanol was used to collect the dried Fe3O4 power before evaporating (Fig. 2). Then, the powder Fe3O4 was collected.534.6. Embedding of Fe3O4 into the QN-loaded FA-functionalized SBA-15 composite (FA-FE-SBA15QN)
To transfer the Fe3O4 nanoparticles into an aqueous phase, the dispersion of nanoparticles (1.2 ml in chloroform) was mixed with the CTAB solution (0.8 g in 20 ml water), and then stirred at 60 °C for 2 h. Then, the mixture was filtered through a 0.44 μm syringe filter to remove large aggregates. The filtered solution and NaOH (2 M, 175 μl) solution were diluted with deionized water (22 ml), followed by heating to 70 °C. After stirring for 10 min at 70 °C, the monodispersed Fe3O4 was added to FA-FE-SBA15QN and kept overnight under magnetic stirring. Then, it was centrifuged (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min) to remove the unbound Fe3O4 and dried to evaporate the solvent (Fig. 2). The dried FA-FE-SBA15QN was characterized.
000 rpm, 10 min) to remove the unbound Fe3O4 and dried to evaporate the solvent (Fig. 2). The dried FA-FE-SBA15QN was characterized.
4.7. Characterization
Nitrogen adsorption/desorption analysis was carried out using a Quantachrome Autosorb 1-C surface area analyzer at 77 K. Prior to gas adsorption, a sample was degassed for 8 h at 403 K under high vacuum. The NLDFT pore size distribution method was employed to determine the average pore size of the samples from their respective N2 sorption isotherm using the silica/cylindrical pore model as a reference. The powder X-ray diffraction patterns were recorded on a Bruker D8 Advance SWAX diffractometer, which was operated at 40 kV voltage and 40 mA current. The instrument was calibrated with a standard silicon sample, using Ni-filtered Cu Kα (λ = 0.15406 nm) radiation. For TEM analysis, 10 mg of material was dispersed into absolute ethanol for 5 min under sonication. Then, one drop of the dispersed solution was dropped onto the carbon-coated copper grid and dried before analysis in the air. FTIR spectra of the samples were recorded using KBr pellets in a PerkinElmer IR 783 spectrophotometer. Thermogravimetric analysis (TGA) and differential thermal analysis (DTA) of the sample were performed in a thermal analyzer TA-SDT Q-600 of TA Instruments under an air flow at a temperature ramp of 10 °C min−1. The carbon, hydrogen, and nitrogen (CHN) contents of different functionalized materials were determined in a Vario EL III elemental analyzer. The 13C CP MAS NMR study was carried out on a JEOL-ECA400NB 400 MHz spectrometer at resonance frequencies of 100.6 MHz.4.8. In vitro QN release assay
The release of QN from FA-FE-SBA15QN was studied using the dialysis bag diffusion method. 2 ml of sample was placed in the dialysis bag, sealed, and suspended in a beaker containing 40 ml of PBS, pH 3.5/5.5/7.4 for different time points (1–8 h). The entire system was kept at 37 °C with vigorous stirring on a mechanical shaking bath. Samples were withdrawn from the receptor media at predetermined time intervals and replaced with fresh buffer. The relative amount of QN in the samples was determined using a UV-Vis spectrophotometer (Biorad, Hercules, California, USA).544.9. Cell line and chemicals
Human colorectal carcinoma (HCT 116) cell line was obtained from the National Centre for Cell Sciences (NCCS), Pune, India. Constituents of cell culture media, viz., Dulbecco's Modified Eagle Medium (DMEM), fetal bovine serum (FBS), trypsin, penicillin–streptomycin–neomycin (PSN) antibiotic cocktail, and ethylenediaminetetraacetic acid (EDTA) were bought from Gibco (Grand Island, NY, USA). Other raw and fine chemicals were procured from Merck (India), Sisco Research Laboratories (SRL), Mumbai, India, and Sigma-Aldrich, St. Louis, MO, USA, respectively. Antibodies were purchased from Santa Cruz Biotechnology, Dallas, Texas, USA and eBioscience, San Diego, USA.4.10. Cell culture
HCT 116 cells were cultured in DMEM (Dulbecco's Modified Eagle Medium) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic (PSN) at 37 °C in a humidified atmosphere under 5% CO2. After reaching 75–80% confluence, cells were harvested with trypsin and EDTA in phosphate buffered saline (PBS), and plated at the desired density a day before the start of the experiment to allow them to re-equilibrate.4.11. In vitro MRI
For the in vitro cellular MR imaging experimentation, HCT 116 and HEK 293 cells (6 × 103) were incubated in a 96-well plate. After 24 h, cells were treated with FA-FE-SBA15QN (2.5–10 μg ml−1) and kept for 3 h. After that, the cells were washed with PBS three times and fixed using paraformaldehyde. To avoid air exposure, low melting agarose (2%) was added to each well. Then, the plate was kept at 4 °C to freeze the cell suspensions. The MRI was performed under a 3T clinical MRI scanner (Siemens MAGNETOM Verio) using a pre-fabricated sample holder. MR phantom images were acquired by applying a spin-echo multi-section pulse sequence. The MRI signal intensity (SI) and visualization of the phantom images were analyzed using the standard software provided by the manufacturer.554.12. Measurement of intracellular ROS (iROS) generation using flow cytometry
The level of intracellular reactive oxygen species (iROS) was measured using the oxidation sensor dye 2′,7′-dichlorofluorescein diacetate (H2DCFDA), wherein an increase in the green fluorescence intensity was used to quantify the generation of intracellular ROS with respect to the untreated control. The treated cells (2 × 106) were resuspended in complete medium containing H2DCFDA and incubated at 37 °C for 30 min, following acquisition by BD LSRFortessa flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) using an argon laser at 488 nm.564.13. Quantification of apoptosis and necrosis using flow cytometry
The determination of apoptosis and necrosis were analyzed by flow cytometry using an annexin-V APC/DAPI apoptosis/necrosis detection kit (Calbiochem, CA, USA). Treated cells (1 × 106) were washed and stained with annexin-V-APC and DAPI in accordance with the manufacturer's instructions. The percentages of apoptotic (early and late) cells, viable cells and necrotic cells were estimated by flow cytometry (BD LSRFortessa, San Jose, CA, USA).574.14. Measurement of mitochondrial membrane potential (ΔΨm) using flow cytometry
Following the treatment of the desired concentration of QN and FA-IO-SBA-15-QN for 24 h, the cells were collected and washed with PBS, followed by incubation with rhodamine 123 (10 μM) at 37 °C for 20 min. The stained cells were washed and resuspended in PBS before analyzing the (ΔΨm) using flow cytometry (Becton Dickinson, Franklin Lakes, NJ, USA).584.15. Assessment of protein expression by flow cytometry
Treated cells (HCT 116 cells, isolated tumor, and colonic cells) were fixed in paraformaldehyde (4%) in PBS (pH 7.4) for 20 min at room temperature, followed by permeabilization (0.1% Triton X-100 in PBS) for 5 min with 0.1% FBS for 5 min. Then, the permeabilized cells were washed twice using PBS with FBS (3%) and incubated with their respective primary antibodies (HSP60, HSP27, p-JNK, p-p53, Bax, CytC, Bcl2, and Apaf1) overnight. After removing the unbound primary antibodies, the cells were further incubated with their particular fluorophore-tagged secondary antibody (anti-rabbit/mice/goat AF488, FITC, and AF647) for 2 h on ice. The stained cells were then subjected to flow cytometric analysis using a BD LSRFortessa flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) equipped with FlowJo software. The mean fluorescence intensities (MFI) were measured by Flow Jo (Version 10.0) software.594.16. Assessment of protein expression/localization using immunofluorescence
Briefly, the control/treated HCT 116 cells were washed twice for 10 min each in PBS (0.01 M), and incubated for 1 h in blocking solution containing 2% normal bovine serum and 0.3% Triton X-100 in PBS. After blocking, the cells were incubated overnight at 4 °C with the respective primary antibody (γH2AX, p-JNK, Bax), followed by washing and incubation with their respective fluorophore-conjugated secondary antibodies (anti-mouse/rabbit AF647, AF555) for 2 h. The slides were then counterstained with 6-diamidino-2-phenylindole (DAPI) for 10 min, and mounted with the Prolong Anti-fade Reagent (Molecular Probe, Eugene, OR, USA). Stained cells were examined using a confocal laser-scanning microscope (FV 10i, Olympus, Japan).604.17. Measurement of caspase 3 and caspase 9 activities
The caspase 3/9 activities were measured according to the manufacturer's directions with commercially existing caspase 3 and caspase 9 colorimetric assay kits (BioVision Research Products, Mountain View, CA). The absorbance was measured at 405 nm by an ELISA reader.4.18. Animals
Adult male BALB/c mice ranging from 8 to 10 weeks old with initial body weights of 20–22 g were procured from the inbred institutional facility. The animals were kept under standard laboratory conditions of 21 °C ± 2 °C, relative humidity of 55% and 12 h:12 h light:dark cycle maintained during the study. The animals were given standard rat pellets and tap water ad libitum. All experimental protocols were performed by the guideline of the Institutional Animal Ethics Committee, CSIR-Indian Institute of Chemical Biology and approved by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Environment, Forests and Climate Change, Government of India.
4.19. In vivo experimentation
On the 1st day, the animals were shaved on the back flank. Each mouse was injected subcutaneously in the shaved right flank with CT-26 (2 × 106) in PBS. Eight days after tumor implantation, the animals were randomly divided into four groups as follows (5 mice in each group) (Fig. 1):Group I: served as control and mice in this group were kept at a standard ambient temperature of 24 ± 2 °C and 60–70% relative humidity.
Group II: CT-26 tumor-bearing mice treated with normal saline.
Group III: CT-26 tumor-bearing mice treated with QN (15 mg kg−1) intraperitoneally for six alternate days.
Group IV: in this group, mice were injected with FA-IO-SBA-15-QN (15 mg kg−1) intraperitoneally for six alternate days.
The resultant tumor length, as well as the weight, was measured after the completion of the experimentation. In addition, the animal survival rate was evaluated for up to 30 days.
4.20. In vivo MRI
Tumor-bearing mice were injected with FA-FE-SBA15QN (15 mg kg−1) via a tail vein. In addition, magnetic targeting was applied to half of these mice. MRI was performed using a 3T MRI scanner (Sigma; GE Medical Systems, Milwaukee, WI, USA). The MR images were captured by a T2-weighted spin-echo sequence (TR/TE = 4000/108 ms; slice thickness, 2 mm; slice spacing, 1 mm; matrix, 256 × 256; FOV, 8 cm × 8 cm). Signal variation was observed after the injection of nanocarriers for 1 h.534.21. Tumor morphological analysis using histology
The harvested tumor tissue was dipped in 10% formalin and processed routinely in paraffin. Then, the 5 μM thick slices were prepared and stained with H&E for histological analysis according to the standard protocol.384.22. Determination of protein expression using immunofluorescence
The unstained tumor section (6 μm of thickness) of all experimental groups was analyzed through indirect immunofluorescence.61 Briefly, the sections were deparaffinized and hydrated in graded ethanol, followed by blocking (0.3% hydrogen peroxide) for 20 min to mask the endogenous peroxidase activity. A microwave oven was used to retrieve the antigen using citrate buffer (10 mM, pH 6.0) at temperatures ranging between 95 °C and 98 °C for 15 min, and then the section was further blocked with BSA (5%) for 30 min at 37 °C. The sections were incubated with the primary antibody against Ki-67 and Bax (Dilution: 1:250) at 4 °C for overnight. After washing the samples with PBS, the sections were incubated with anti-rabbit/mice-AF546, and AF647 secondary antibody for 1 h each at 37 °C, followed by nuclear staining with DAPI for 10 min. The images were observed using an Olympus FV1000 MPE SIM Laser Scanning confocal microscope (Olympus, Tokyo, Japan).61,62 The resultant intensity was analysed by ImageJ software.
4.23. Determination of protein expression using immunoblotting
A tumor tissue lysate was prepared using tissue lysis buffer containing Tris–HCl (20 mM) pH 7.5, 2-mercaptoethanol (50 mM), EGTA (5 mM), EDTA (2 mM), NP40 (1%), SDS (0.1%), deoxycholic acid (0.5%), NaF (10 mM), PMSF (1 mM), leupeptin (25 mg ml−1) and aprotinin (2 mg ml−1). This was followed by the determination of the protein concentrations using a protocol of the BCA assay. Protein fractions were reconstituted in a sample buffer containing Tris–HCl (62 mM), SDS (2%), glycerol (10%) and β-mercaptoethanol (5%), and boiled at 97 °C for 6 min for the protein denaturation. Equal amounts (25 μg) of the denatured protein samples were separated using SDS-PAGE on a polyacrylamide gel (12.5%), and transferred overnight onto polyvinylidene difluoride membranes (PVDF). The membranes were blocked with non-fat dried milk (5%) in TBS containing Tween-20 (1%) for 1 h at room temperature, followed by incubation of the respective primary antibodies (1:1000 dilution) overnight. The secondary antibody was coupled with either horseradish peroxidase (HRP)-conjugated goat anti-rabbit or anti-mouse antibodies (1:3000), and used for a second incubation for 2 h after washing with TBS containing Tween-20 (1%). The blots were detected using an enhanced chemiluminescence (ECL) reagent, and the photographs were taken using a Chemidoc XR Imager (Bio-Rad Laboratories, Hercules, CA, USA).63 The relative density of each blot was measured using ImageJ software.61,64
4.24. Statistical analysis
All values are represented as mean values with their standard errors (mean ± SEM). The statistical software, OriginPro version 8.0, was used for the data analysis. The statistical significance values were evaluated through one-way ANOVA analysis, following the Tukey method with the post-hoc test. The critical significance level was set at p < 0·05.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is funded by the National Medicinal Plant Board, Ministry of AYUSH (Project No. Z18017/187/CCS/R&D/MH-02/2016-17-NMPB-IVA). AB acknowledges the Department of Science & Technology, Science and Engineering Research Board (DST-SERB), New Delhi for a core grant (file no. CRG/2018/000230). In addition, the authors are thankful to Mr Tanmoy Dalui, Mrs Debalina Chakraborty, Sri Binayak Pal and Smt. Banasri Das of Central Instrumentation Facility, CSIR-Indian Institute of Chemical Biology for providing the flow cytometer and confocal microscope facilities.Notes and references
- N. G. Zaorsky, T. M. Churilla, B. L. Egleston, S. G. Fisher, J. A. Ridge, E. M. Horwitz and J. E. Meyer, Ann. Oncol., 2017, 28, 400–407 CrossRef CAS PubMed.
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- A. Urruticoechea, R. Alemany, J. Balart, A. Villanueva, F. Vinals and G. Capella, Curr. Pharm. Des., 2010, 16, 3–10 CrossRef CAS PubMed.
- H. Zaid, M. Silbermann, A. Amash, D. Gincel, E. Abdel-Sattar and N. B. Sarikahya, Evidence-Based Complementary and Alternative Medicine, 2017, 7952417 Search PubMed.
- N. Bertranda, J. Wub, X. Y. Xua, N. Kamaly and O. C. Farokhzadb, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef PubMed.
- L. Ding, X. B. Zhu, Y. Wang, B. Y. Shi, X. Ling, H. J. Chen, W. H. Nan, A. Barrett, Z. Guo, W. Tao, J. Wu and X. J. Shi, Nano Lett., 2017, 17(11), 6790–6801 CrossRef CAS PubMed.
- A. Massi, O. Bortolini, D. Ragno, T. Bernardi, G. Sacchetti, M. Tacchini and C. De Risi, Molecules, 2017, 22, 1270 CrossRef PubMed.
- A. Sarkar, S. Ghosh, S. Chowdhury, B. Pandey and P. C. Sil, Biochim. Biophys. Acta, Gen. Subj., 2016, 1860, 2065–2075 CrossRef CAS PubMed.
- M. L. Mansuri, P. Parihar, I. Solanki and M. S. Parihar, Genes Nutr., 2014, 9, 400 CrossRef PubMed.
- Y. Li, J. Yao, C. Han, J. Yang, M. T. Chaudhry, S. Wang, H. Liu and Y. Yin, Nutrients, 2016, 8, 167 CrossRef PubMed.
- X. Liu, Z. Yu, X. Huang, Y. Gao, X. Wang, J. Gu and S. Xue, Am. J. Transl. Res., 2016, 8, 5169–5186 CAS.
- A. Rauf, M. Imran, I. A. Khan, M. Ur-Rehman, S. A. Gilani, Z. Mehmood and M. S. Mubarak, Phytother. Res., 2018, 32, 2109–2130 CrossRef CAS PubMed.
- T. Ramasamy, H. B. Ruttala, N. Chitrapriya, B. K. Poudal, J. Y. Choi, S. T. Kim, Y. S. Youn, S. K. Ku, H. G. Choi, C. S. Yong and J. O. Kim, Acta Biomater., 2017, 48, 131–143 CrossRef CAS PubMed.
- B. Liu, T. Jiang, H. Q. Zheng, S. Dissanayke, W. Q. Song, A. Federico, S. L. Suib and J. He, Nanoscale, 2017, 9, 6380–6390 RSC.
- Z. Vargas-Osorio, M. A. Gonzalez-Gomez, Y. Pineiro, C. Vazquez-Vazquez, C. Rodriguez-Abreu, M. A. Lopez-Quintela and J. Rivas, J. Mater. Chem. B, 2017, 5, 9395–9404 RSC.
- Z. Tabia, K. El Mabrouk, M. Bricha and K. Nouneh, RSC Adv., 2019, 9, 12232–12246 RSC.
- L. B. D. Freitas, I. J. G. Bravo, W. A. D. Macedo, E. M. B. de Sousa and J. Sol-Gel, Sci. Technol., 2016, 77, 186–204 CAS.
- T. Nissinen, S. Nakki, M. Latikka, M. Heinonen, T. Liimatainen, W. J. Xu, R. H. A. Ras, O. Grohn, J. Riikonen and V. P. Lehto, Microporous Mesoporous Mater., 2014, 195, 2–8 CrossRef CAS.
- K. Sarkar, K. Dhara, M. Nandi, P. Roy, A. Bhaumik and P. Banerjee, Adv. Funct. Mater., 2009, 19, 223–234 CrossRef CAS.
- Q. Gao, W. S. Xie, Y. Wang, D. Wang, Z. H. Guo, F. Gao, L. Y. Zhao and Q. Cai, RSC Adv., 2018, 8, 4321–4328 RSC.
- W. Chen, C. A. Cheng, B. Y. Lee, D. L. Clemens, W. Y. Huang, M. A. Horwitz and J. I. Zink, ACS Appl. Mater. Interfaces, 2018, 10, 31870–31881 CrossRef CAS PubMed.
- M. S. Moorthy, J. H. Park, J. H. Bae, S. H. Kim and C. S. Ha, J. Mater. Chem. B, 2014, 2, 6487–6499 RSC.
- R. M. Clauson, M. S. Chen, L. M. Scheetz, B. Berg and B. Chertok, ACS Appl. Mater. Interfaces, 2018, 10, 20281–20295 CrossRef CAS PubMed.
- S. Tanaka, Y. V. Kaneti, N. L. W. Septiani, S. X. Dou, Y. Bando, M. S. A. Hossain, J. Kim and Y. Yamauchi, Small, 2019, 1800512 CrossRef.
- H. Kalita, S. Rajput, B. N. P. Kumar, M. Mandal and A. Pathak, RSC Adv., 2016, 6, 21285–21292 RSC.
- M. X. Ren, X. H. Deng, F. Ai, G. Y. Yuan and H. Y. Song, Exp. Ther. Med., 2015, 10, 579–583 CrossRef CAS PubMed.
- S. Y. Kim, H. C. Jeong, S. K. Hong, M. O. Lee, S. J. Cho and H. J. Cha, Oncotarget, 2017, 8, 64964–64973 CrossRef.
- R. P. Rastogi, S. P. Singh, D. P. Haeder and R. P. Sinha, Biochem. Biophys. Res. Commun., 2010, 397, 603–607 CrossRef CAS.
- B. Schutte, R. Nuydens, H. Geerts and F. Ramaekers, J. Neurosci. Methods, 1998, 86, 63–69 CrossRef CAS.
- L. T. Nguyen, Y. H. Lee, A. R. Sharma, J. B. Park, S. Jagga, G. Sharma, S. S. Lee and J. S. Nam, J. Physiol. Pharmacol., 2017, 21, 205–213 CrossRef CAS PubMed.
- M. Hashemzaei, A. Delarami Far, A. Yari, R. E. Heravi, K. Tabrizian, S. M. Taghdisi, S. E. Sadegh, K. Tsarouhas, D. Kouretas, G. Tzanakakis, D. Nikitovic, N. Y. Anisimov, D. A. Spandidos, A. M. Tsatsakis and R. Rezaee, Oncol. Rep., 2017, 38, 819–828 CrossRef CAS PubMed.
- S. R. Kumar, S. Priyatharshni, V. N. Babu, D. Mangalaraj, C. Viswanathan, S. Kannan and N. Ponpandian, J. Colloid Interface Sci., 2014, 436, 234–242 CrossRef PubMed.
- J. D. Ly, D. R. Grubb and A. J. A. Lawen, Apoptosis, 2003, 8, 115–128 CrossRef CAS PubMed.
- D. Wlodkowic, W. Telford, J. Skommer and Z. Darzynkiewicz, Methods Cell Biol., 2017, 103, 55–98 Search PubMed.
- J. Y. J. Wang, Cell Death Differ., 2001, 8, 1047–1048 CrossRef CAS PubMed.
- A. Sharma, K. Singh and A. Almasan, Methods Cell Biol., 2012, 920, 613–626 CAS.
- E. P. Rogakou, W. Nieves-Neira, C. Boon, Y. Pommier and W. M. Bonner, J. Biol. Chem., 2000, 275, 9390–9395 CrossRef CAS PubMed.
- V. Picco and G. Pagès, Genes Cancer, 2013, 4, 360–368 CrossRef PubMed.
- D. Whitley, S. P. Goldberg and W. D. Jordan, J. Vasc. Surg., 1999, 29, 748–751 CrossRef CAS PubMed.
- D. Lanneau, M. Brunet, E. Frisan, E. Solary, M. Fontenay and C. Garrido, J. Cell. Mol. Med., 2008, 12, 743–761 CrossRef CAS PubMed.
- C. G. Concannon, A. M. Gorman and A. Samali, Apoptosis, 2003, 8, 61–70 CrossRef CAS PubMed.
- D. N. Dhanasekaran and E. P. Reddy, Genes Cancer, 2017, 8, 682–694 CAS.
- J. Ruan, Z. Qi, L. Shen, Y. Jiang, Y. Xu, L. Lan, L. Luo and Z. Yin, Biochem. Biophys. Res. Commun., 2015, 456, 122–128 CrossRef CAS PubMed.
- J. S. Fridman and S. W. Lowe, Oncogene, 2003, 22, 9030–9040 CrossRef CAS PubMed.
- A. Kim, M. Im, M. J. Gu and J. Y. Ma, Sci. Rep., 2016, 6, 24214 CrossRef CAS PubMed.
- A. Bonetto, J. E. Rupert, R. Barreto and T. A. Zimmers, J. Visualized Exp., 2017, 117, 54893 Search PubMed.
- O. Fluge, K. Gravdal, E. Carlsen, B. Vonen, K. Kjellevold, S. Refsum, R. Lilleng, T. J. Eide, T. B. Halvorsen, K. M. Tveit, A. P. Otte, L. A. Akslen, O. Dahl and G. Norwegian, Br. J. Cancer, 2009, 101, 1282–1289 CrossRef CAS PubMed.
- D. A. Orringer, D. R. Vago and A. J. Golby, Semin. Neurol., 2012, 32, 466–475 CrossRef PubMed.
- G. Brix, F. Kiessling, R. Lucht, S. Darai, K. Wasser, S. Delorme and J. Griebel, Magn. Reson. Med., 2004, 52, 420–429 CrossRef PubMed.
- B. Dragoi, I. Mazilu, A. Chirieac, C. Ciotonea, A. Ungureanu, E. Marceau, E. Dumitriu and S. Royer, Catal. Sci. Technol., 2017, 7, 5376–5385 RSC.
- P. Bhanja, A. Modak, S. Chatterjee and A. Bhaumik, ACS Sustainable Chem. Eng., 2017, 5, 2763–2773 CrossRef CAS.
- J. Pang, L. Zhao, L. Zhang, Z. Li and Y. Luan, J. Colloid Interface Sci., 2013, 395, 31–39 CrossRef CAS PubMed.
- E. Li, Y. Yang, G. Hao, X. Yi, S. Zhang, Y. Pan, B. Xing and M. Gao, Nanotheranostics, 2018, 2, 233–242 CrossRef PubMed.
- P. Bhanja, S. Mishra, K. Manna, A. Mallick, K. Das Saha and A. Bhaumik, ACS Appl. Mater. Interfaces, 2017, 9, 31411–31423 CrossRef CAS PubMed.
- R. Nandi, S. Mishra, T. K. Maji, K. Manna, P. Kar, S. Banerjee, S. Dutta, S. K. Sharma, P. Lemmens, K. D. Saha and S. K. Pal, J. Mater. Chem. B, 2017, 5, 3927–3939 RSC.
- K. Manna, A. Khan, D. K. Das, S. B. Kesh, U. Das, S. Ghosh, R. Sharma Dey, K. Das Saha, A. Chakraborty, S. Chattopadhyay, S. Dey and D. Chattopadhyay, J. Ethnopharmacol., 2014, 155, 132–146 CrossRef CAS PubMed.
- K. Manna, A. Khan, S. Biswas, U. Das, A. Sengupta, D. Mukherjee, A. Chakraborty and S. Dey, RSC Adv., 2016, 6, 23058–23073 RSC.
- J. Gu, H. Xu, Y. Han, W. Dai, W. Hao, C. Wang, N. Gu, H. Xu and J. Cao, Sci. China Life Sci., 2011, 54, 793–805 CrossRef CAS PubMed.
- K. Manna, S. Mishra, M. Saha, S. Mahapatra, C. Saha, G. Yenge, N. Gaikwad, R. Pal, D. Oulkar, K. Banerjee and K. Das Saha, Int. J. Nanomed., 2019, 14, 1753–1777 CrossRef CAS PubMed.
- S. K. Das, S. Mishra, K. Manna, U. Kayal, S. Mahapatra, K. Das Saha, S. Dalapati, G. P. Das, A. A. Mostafa and A. Bhaumik, Chem. Commun., 2018, 54, 11475–11478 RSC.
- Y. Zhang, B. C. Y. Cheng, R. Xie, B. Xu, X. Y. Gao and G. Luo, RSC Adv., 2019, 9, 8912–8925 RSC.
- S. B. Kesh, K. Sikder, K. Manna, D. K. Das, A. Khan, N. Das and S. Dey, Life Sci., 2013, 92, 938–949 CrossRef CAS PubMed.
- K. Manna, A. Khan, D. K. Das, S. Bandhu Kesh, U. Das, S. Ghosh, R. Sharma Dey, K. Das Saha, A. Chakraborty, S. Chattopadhyay, S. Dey and D. Chattopadhyay, J. Ethnopharmacol., 2014, 155, 132–146 CrossRef CAS PubMed.
- K. Manna, U. Das, D. Das, S. B. Kesh, A. Khan, A. Chakraborty and S. Dey, Free Radic. Res., 2015, 49, 422–439 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00664e |
| This journal is © The Royal Society of Chemistry 2020 |