
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoexistence of light-induced photoluminescence enhancement and quenching in CH3NH3PbBr3 perovskite films†
Haoran Loua,
Chen Lina,
Zhishan Fanga,
Li Jianga,
Xiaofeng Chenb,
Zhizhen Yea and
Haiping He *a
*a
aState Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: hphe@zju.edu.cn
bSchool of Environmental Science and Engineering, Yangzhou University, Yangzhou 225127, China
First published on 17th March 2020
Abstract
Lead halide perovskites are promising semiconductors for various optoelectronic devices working in a wide photo-excitation density regime. However, photo-induced instability, attributed to illumination-activated mobile ions, has been an obstacle to their application. Herein, we use the time evolution of photoluminescence (PL) to investigate the light illumination effects of CH3NH3PbBr3 perovskite films under relatively high excitation (up to 4.5 W cm−2). We demonstrate that continuous illumination can lead to both PL enhancement and PL quenching simultaneously, with their weight ratios depending on the excitation density. The experimental data can be well described and interpreted by considering the coexistence of and competition between the photo-induced annihilation and the formation of long-living filled trap states. Our study may provide in-depth insight into the photo-induced instability of perovskite films and help to improve the performance of perovskite-based optoelectronic devices.
1. Introduction
Organic–inorganic lead halide perovskites have attracted growing attention on account of their great potential in the photovoltaic and light-emitting applications for the past decade.1–3 Owing to their excellent optoelectronic properties, such as balanced long-range hole and electron diffusion lengths,4 high photoluminescence (PL) quantum efficiency,5 and high defect tolerance,6 organic–inorganic lead halide perovskites have been regarded as promising materials for their unprecedented development in low-cost solar cells7–9 with increasing power conversion efficiency (PCE) and efficient light emitting diodes (LEDs).10–13 The PCEs of perovskite solar cells have rapidly exceeded 25% for the past few years,14 despite Miyasaka and co-workers reporting a CH3NH3PbI3 dye-sensitized solar cell with a PCE of only 3.81% in 2009.7 Meanwhile, highly efficient perovskite LEDs have been gradually making progress, and operate with constantly improving external quantum efficiency exceeding 20%.11Despite the remarkable progress in device efficiency, there still exists a huge obstacle in the future application. A proverbial difficulty of perovskite-based optoelectronic devices is their unstable performance when exposed to oxygen,15,16 moisture,17 heat,18,19 and light,20–24 which have gradually attracted more and more attention. Different types of environmental factors can lead to various degradation pathways, and light itself also can induce diverse illumination effects under different operational conditions, making it difficult to study the mechanism behind the instability of perovskite thin film. In general, oxygen and moisture both can be prevented from interacting with the perovskite layer by using the appropriate encapsulation.25,26 However, light illumination is inevitable in the materials processing and devices operating. Therefore, it is of high importance to investigate the features and mechanisms of the photo-induced instability.
In previous reports, a photoluminescence intensity enhancement (PLIE) under continuous light excitation has been reported in organic–inorganic lead halide perovskites as the consequence of photoinduced trap annihilation mechanism.22–24,27 In direct contradiction, a photoluminescence intensity decrease (PLID) has also been observed, which was explained by ion migration and photoinduced trap formation.28,29 The increase of defect density has been interpreted as a result of enhanced illumination-induced ion migration under light excitation. In comparison, all-inorganic perovskites exhibit superior light-stability,30 however are also slightly subject to illumination effects, such as photo-induced phase transition.31,32 For example, Xue et al.32 has observed photo-induced reversible phase transition in CsPbBr3 nanocrystals. In some reports, Cs is replaced with formamidinium (FA) to form (NH2)2CHPbI3 (FAPI) which presents an improved structural stability.33 Despite noble effort, the comprehensive consensus of the illumination effects in lead halide perovskites has not yet been available. For example, it is easy to imagine that the photo-induced instability is sensitive to the excitation density, and it is important to evaluate such effects under high excitation close to the operating regime of concentrated sunlight solar cells or high-power LEDs.
In this study, we prepare and encapsulate methylammonium lead bromide (CH3NH3PbBr3) perovskite thin films to make them suitable for studying the photo-induced instability separately. Time-dependent steady state and time-resolved PL spectroscopy are used to investigate the dependencies of PLIE and PLID effects on the excitation density. We demonstrate that both the two effects can occur and compete with each other, which can be interpreted in term of defect annihilation and formation under different excitation densities. We adopt a suitable model to explain the PL results and the competition between PLIE and PLID under different excitation densities.
2. Experimental section
We fabricate the CH3NH3PbBr3 perovskite films by virtue of chloroform-assisted process technology.34 CH3NH3Br (MABr, 99.9%) are purchased from Xi'an Polymer Light Technology Corp and lead bromide (PbBr2, 99.99%) are purchased from Sigma-Aldrich. Dimethyl sulfoxide (DMSO, 99.8%) are obtained from Alfa Aesar and chloroform (CHCI3, AR) are obtained from Sinopharm Chemical Reagent Corp. The precursor solution, prepared by mixing equal molar portions of PbBr2 and CH3NH3Br to form 40 wt% solutions in DMSO, is spin-coated with speed of 3000 rpm on a clean glass in N2 atmosphere. During the spin-coating process, a few drops of chloroform are dripped on the film to promote the crystallization. At last, CH3NH3PbBr3 films are thermally annealed at 90 °C for 10 min. All the samples are encapsulated and kept in a glove box before measurements, thus minimizing the impacts of oxygen and moisture.Time-dependent steady state and time-resolved PL measurements are performed on an MS3504i fluorescence spectrophotometer (SOL Instruments) to study the evolution of PL intensity and lifetime with illumination time under gradient increasing excitation densities, and the time resolution of the TCSPC system is around 100 ps. A semiconductor pulsed laser operating at a repeating frequency of 10 MHz and a wavelength of 405 nm is used as the excitation source. Field-emission scanning electron microscope (FE-SEM, SU-8010) is utilized to study the morphology of samples. X-ray diffraction (XRD), equipped with a PANalytical X'pert PRO diffractometer operating at 40 keV and 40 mA using Cu Kα radiation (λ = 0.15406 nm), is performed to study the crystallinity of films.
3. Results and discussion
The morphology of CH3NH3PbBr3 perovskite film is displayed in Fig. 1. From the low-magnification of SEM image shown in Fig. 1a, we find the CH3NH3PbBr3 perovskite film has a smooth and compact surface. High-magnification of SEM image shown in Fig. 1b displays the uniform distribution of large grains with an average grain size of ∼200 nm and clear grain boundaries. The XRD pattern (Fig. S1†) shows the CH3NH3PbBr3 films have a good crystallinity, consistent with previous reports.35 The good repeatability of the preparation process and high quality of CH3NH3PbBr3 film ensure the reliability of experimental results. | ||
| Fig. 1 (a) Low-magnification (b) high-magnification of SEM images of CH3NH3PbBr3 perovskite films on the glass substrate. | ||
In the following study, we compare the evolution of PL intensity and lifetime of the CH3NH3PbBr3 film with time under different excitation densities during continuous illumination. Fig. 2a displays the PL spectra of the same sample under different excitation densities. The emission peak is around 537 nm, consistent with the results reported in literature.36 It should be noted that the PL peak position and spectral lineshape almost remain unchanged with increasing excitation density up to 4.5 W cm−2, indicating that the perovskite film does not suffer degradation. Fig. 2b displays the evolution of PL intensity with time in the CH3NH3PbBr3 film at increasing power densities. During continuous illumination, the PLID and PLIE are both observed in the CH3NH3PbBr3 film, each playing a dominant role at different power densities. At a low power density of 0.3 W cm−2, the PL intensity shows a quick but slight increase, followed by a continuous decrease lasting for over 30 min. When the power density increases to 0.5 W cm−2, an obvious increase (∼39%) of PL intensity within 10 min is observed in the CH3NH3PbBr3 film, followed by a slow decrease to 87% of the maximum value. However, when we increase the power density to 0.9 W cm−2, only a monotonic PLIE occurs and the PL intensity undergoes a continuous increase to 138% of the initial value during 8 min until it reaches a stable state. Nevertheless, at a higher power density of 4.5 W cm−2, only a monotonic PLID is observed in the same CH3NH3PbBr3 film and the PL intensity drops to 71% of the initial value within 6 min. Chen20 has also reported that CH3NH3PbI3 films suffer from PLIE and PLID respectively under low and high excitation densities, which however are different from our experiments in two aspects. Firstly, in terms of experimental conditions we excite the same CH3NH3PbBr3 film with continuously increased excitation densities up to 4.5 W cm−2 lasting for 30 min. Secondly, in terms of results we observe that CH3NH3PbBr3 films undergo PLIE and PLID successively under low excitation densities, however Chen reported that CH3NH3PbI3 films only suffer from PLIE maybe due to the limit of illumination time.
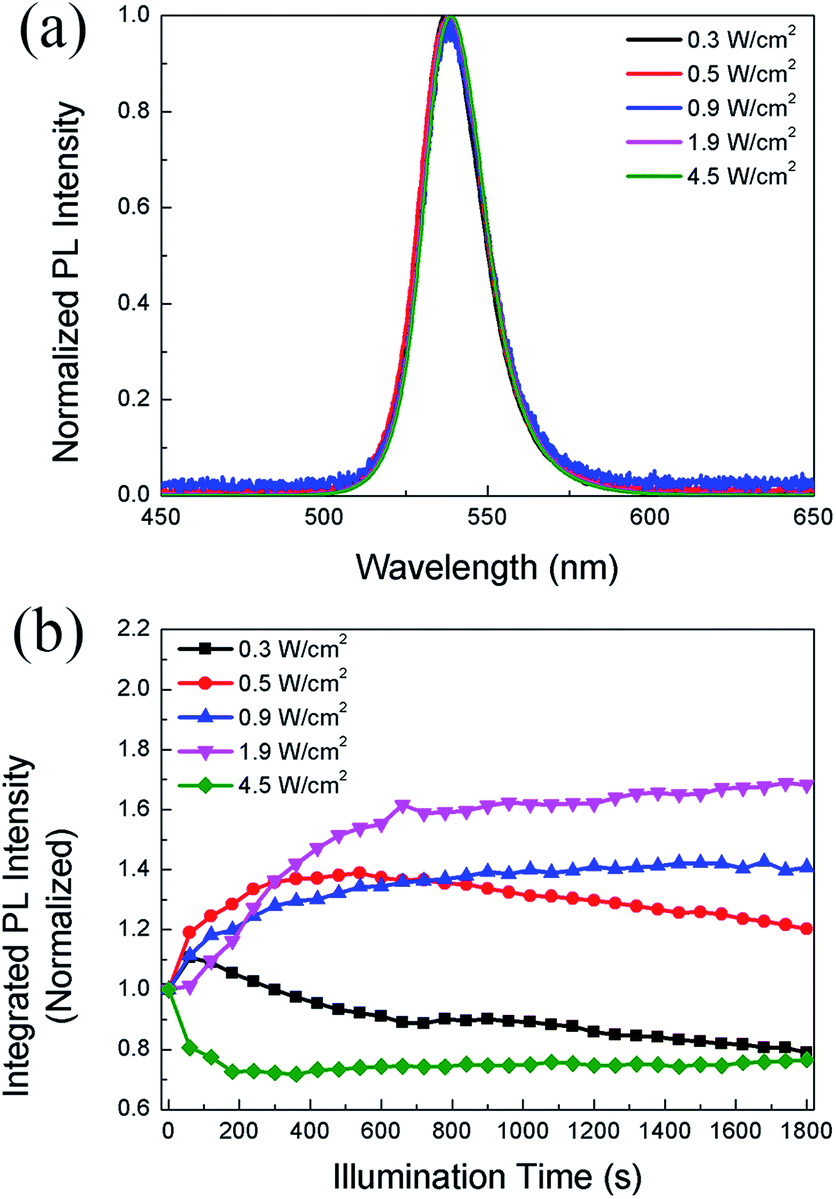 | ||
| Fig. 2 (a) The PL spectra of CH3NH3PbBr3 films under various excitation power densities. The intensities are normalized to the peak value; (b) the evolution of PL intensity with time in CH3NH3PbBr3 film under different excitation power densities. | ||
We also monitor the evolution of PL lifetime with the illumination time. Fig. 3a shows the PL decay kinetics of CH3NH3PbBr3 film under 0.5 W cm−2 excitation recorded after continuous illumination of 0, 10, 20, and 30 min, respectively. The decay clearly consists of a fast and a slow component, which can be well fitted (Fig. S2†) by a bi-exponential decay function, I(t) = I0 + A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2), where τ1 and τ2 are lifetimes for the two decay channels and A1, A2 the corresponding weight ratios. The fitting results give a short lifetime of ∼0.5 ns with a weight ratio of ∼0.3 and a long lifetime of ∼22 ns with a weight ratio of ∼0.7. The short lifetime can be assigned to excitonic recombination considering the relatively large exciton binding energy of CH3NH3PbBr3, while the long lifetime represents the trapped-assisted recombination.37 As shown in Fig. 3b, after 10 min illumination, the weight ratio of trap-assisted recombination decreases, which suggests a decrease of trap density and is consistent with the PLIE in the initial stage shown in Fig. 2b. After that, the weight ratio does not change markedly, while the excitonic lifetime decreases from 0.50 to 0.43 ns (Fig. 3c), suggesting the formation of non-radiative defects. This also agrees with the PLID observed in Fig. 2b.
exp(−t/τ1) + A2exp(−t/τ2), where τ1 and τ2 are lifetimes for the two decay channels and A1, A2 the corresponding weight ratios. The fitting results give a short lifetime of ∼0.5 ns with a weight ratio of ∼0.3 and a long lifetime of ∼22 ns with a weight ratio of ∼0.7. The short lifetime can be assigned to excitonic recombination considering the relatively large exciton binding energy of CH3NH3PbBr3, while the long lifetime represents the trapped-assisted recombination.37 As shown in Fig. 3b, after 10 min illumination, the weight ratio of trap-assisted recombination decreases, which suggests a decrease of trap density and is consistent with the PLIE in the initial stage shown in Fig. 2b. After that, the weight ratio does not change markedly, while the excitonic lifetime decreases from 0.50 to 0.43 ns (Fig. 3c), suggesting the formation of non-radiative defects. This also agrees with the PLID observed in Fig. 2b.
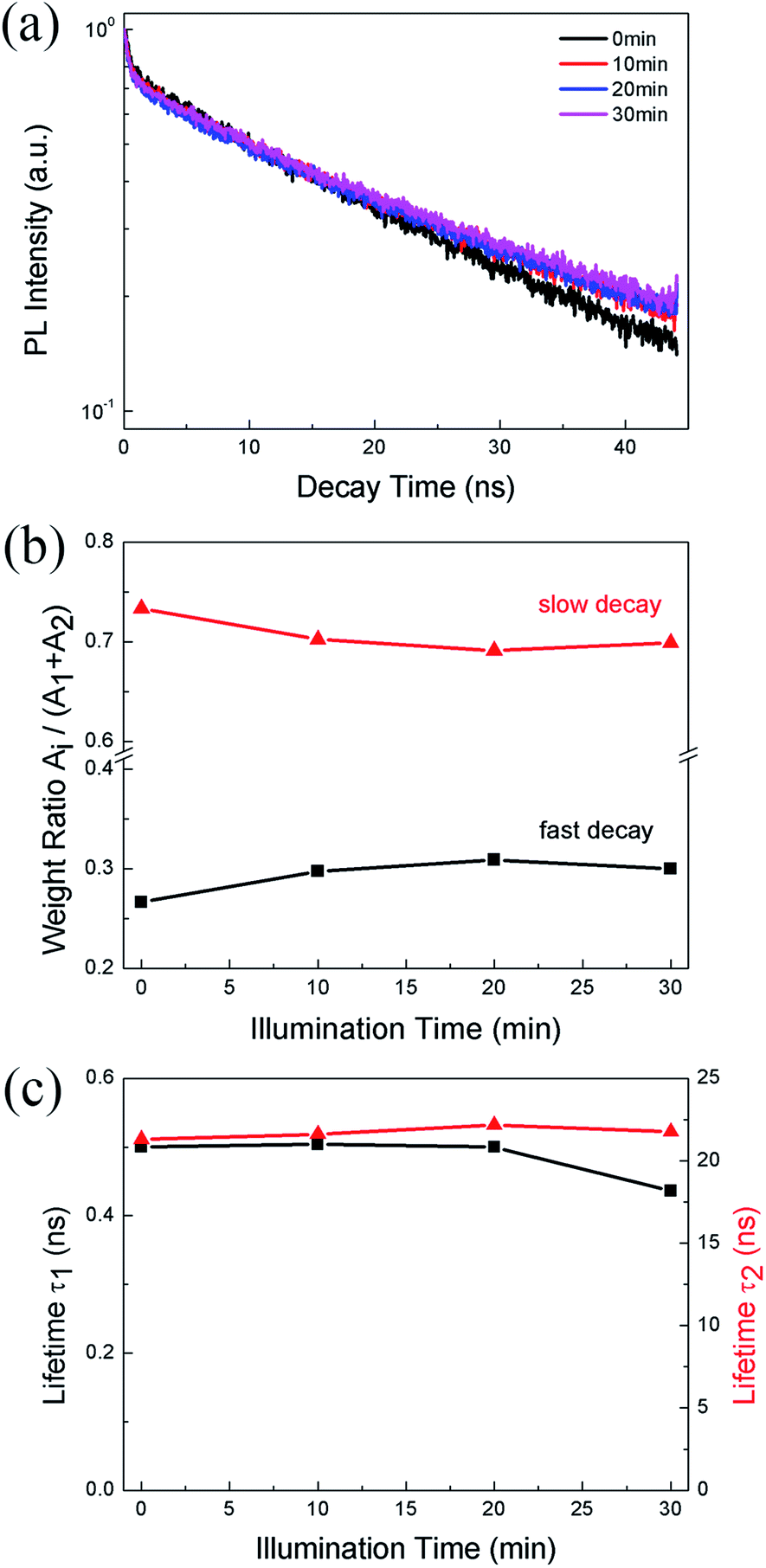 | ||
| Fig. 3 (a) The PL decay traces (b) the weight ratio of fast decay (black line) and slow decay (red line) (c) the short lifetime (black line) and long lifetime (red line) of CH3NH3PbBr3 films under 0.5 W cm−2 excitation recorded after continuous illumination of 0, 10, 20, 30 min, respectively. | ||
In the previous reports, the PLID and PLIE in the organic–inorganic lead halide perovskites under light illumination were observed and explained separately.21,23,38 In general, the defect curing has been put forward to explain the PLIE effect occurring in the organic–inorganic lead halide perovskite materials. Mosconi et al.23 has proposed an explanation in which illumination could promote the annihilation of VI+/Ii− Frenkel pairs, where VI+ and Ii− represent iodine vacancies and iodine interstitial ions respectively, accompanied by a decrease of trap density, in methylammonium lead iodide perovskite thin films. In CH3NH3PbI3 films, the lead-related defects are reported with high migration barriers ranging from 0.8 to 1.4 eV,39,40 however the iodide-related defects including VI+ and Ii− have small activation barriers (0.10 and 0.24 eV, respectively),41 thus considered to be the fasted migrating defects. However, the PLID is more likely to be attributed to illumination-activated mobile ions.28,42 Domanski et al.38 provided direct evidence of photo-driving defect migration and accumulation which are the origin of degradation occurring in the CH3NH3PbI3 film. CH3NH3PbBr3 and CH3NH3PbI3 both show a remarkably similar defect chemistry which is dominated by interstitial halogen defects and lead vacancies,24 therefore defect curing and ion migration could both apply to CH3NH3PbBr3 and CH3NH3PbI3. We propose that the evolution of PL intensity and PL lifetime in the CH3NH3PbBr3 film is the result of competition between the PLIE and PLID effects, which will eventually reach a dynamic equilibrium state. To further explore the illumination effects in the CH3NH3PbBr3 film, PL intensity could be described as a function of illumination time for 0.5, 1.9 and 4.5 W cm−2, which represent low, medium, and high excitation density respectively. Therefore, the PL intensity could be described as following:
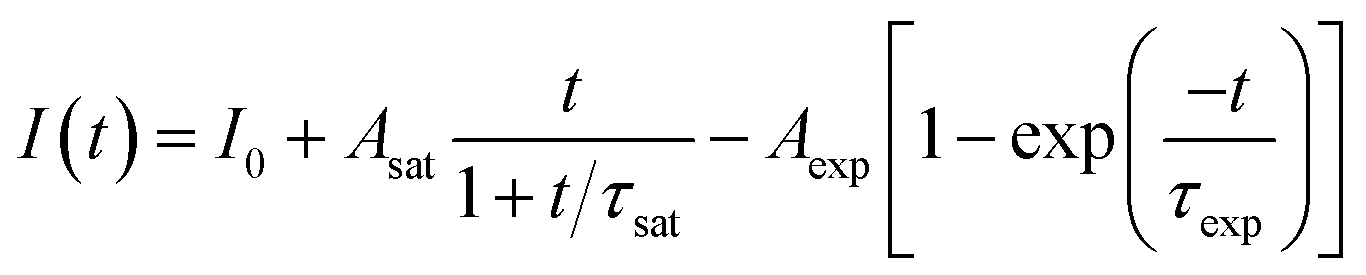 | (1) |
| Power density (W cm−2) | τsat (s) | τexp (s) |
|---|---|---|
| 0.5 | 165 | 2938 |
| 1.9 | 720 | 2593 |
| 4.5 | 1003 | 58 |
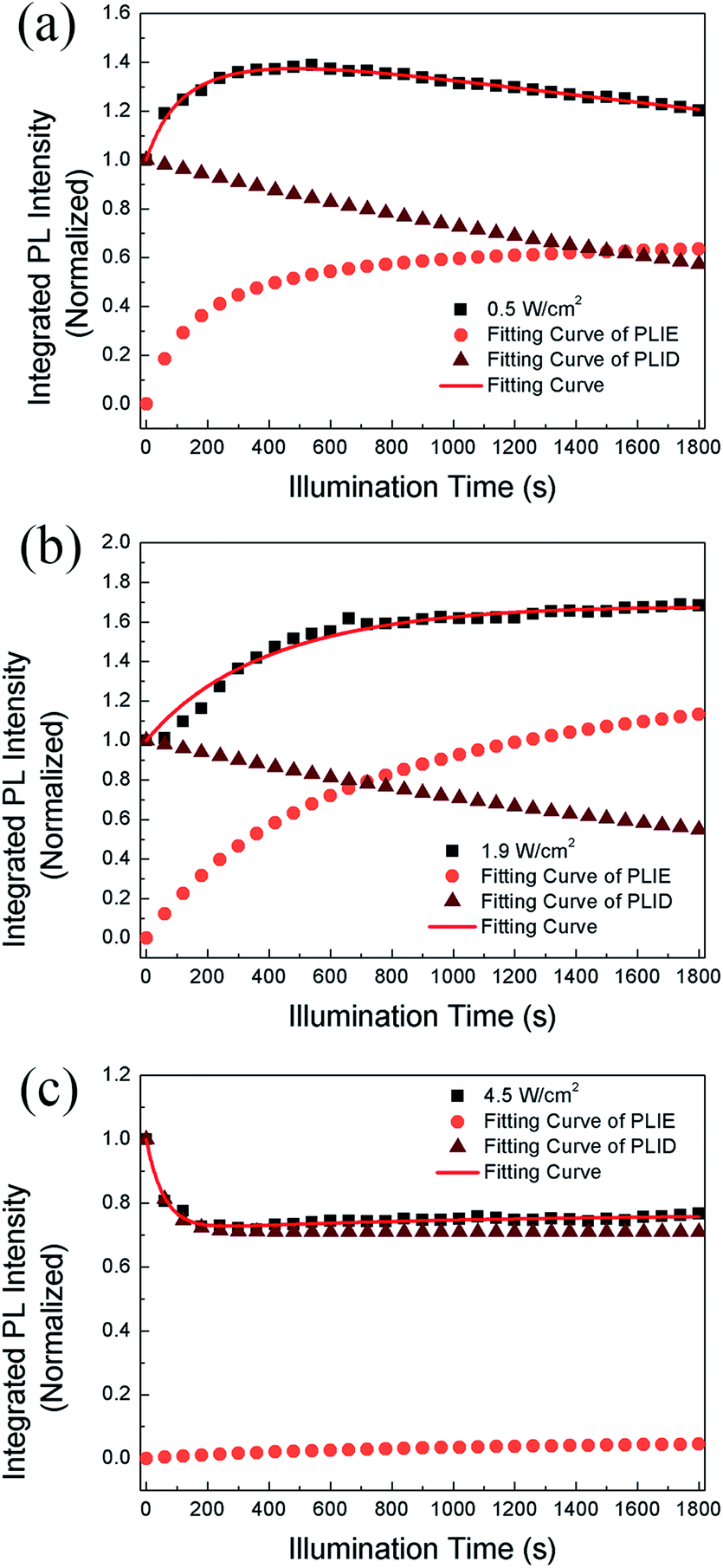 | ||
| Fig. 4 The evolution of PL intensity with time and the fitting curve based on saturation and quenching functions together and respectively in CH3NH3PbBr3 films when the power densities are (a) 0.5 W cm−2, (b) 1.9 W cm−2, (c) 4.5 W cm−2 respectively. | ||
We propose that the formation and annihilation of defects both take place in the whole illumination process, the dominant part of which would determine the occurrence of PLID or PLIE. At the low excitation density of 0.5 W cm−2, we observe a PLIE effect at first and a PLID effect in the subsequent time in CH3NH3PbBr3 films. As shown in Fig. 4, the contributions of both PLIE and PLID are plotted separately according to the fitting parameters, and they have different effect on the PL intensity during different periods. At this point, τsat is 165 s, which means PLIE gradually reaches saturation within this period, while PLID is still in the state of accumulation until it reaches saturation with a time constant of 2938 s. Since the PLID during light illumination is ascribed to the increase of defect density, the PLID occurs after the PLIE indicates that the formation of defects is suppressed at the initial stage but dominates in the subsequent process. When the power density is 1.9 W cm−2, the PL intensity shows only increase. However, the fitting results indicate that the result is still a competition between the PLIE and PLID effects, while the saturation time constant of PLIE increases significantly to 720 s. It suggests that the annihilation of defects prevails over the formation of defects, resulting in the decrease of defect density during whole illumination process. Nevertheless, when the power density is 4.5 W cm−2, PLIE has no more advantage in the competition of two illumination effects during irradiation, and only PLID is observed in the whole process. Moreover, compared with τexp when the power density is 0.5 W cm−2, τexp decreases to 58 s, which mean the PLID effect reaches saturation faster due to the increasement of excitation density. In fact, as shown in Fig. 4c, we can observe that the formation of defects takes place quickly during short period and the annihilation of defects is greatly suppressed during the whole time. Based on the experimental data and fitting results, we put forward that the annihilation and formation of defects always coexist in the CH3NH3PbBr3 films and both processes will be influenced by the excitation density.
With assistance of the model proposed by Motti et al.24, we could explain the excitation density dependent illumination effects taking place in CH3NH3PbBr3 films. Motti et al. demonstrated that PLIE and PLID coexist and compete in the CH3NH3PbBr3 and CH3NH3PbI3 films which are associated with the formation and annihilation of defects triggering illumination-induced photochemical reaction and material transformation. In general, the PLIE effect is attributed to defect curing process, specifically caused by annihilation of VBr+/Bri− Frenkel pairs where VBr+ and Bri− represent bromine vacancies and bromine interstitial ions respectively, as shown in Fig. 5. When an electron is trapped in at Bri+ to form Br0i, along with the release of energy, Bri− and VBr+ migrates to each other in the opposite direction with a small activation energy.41 In terms of result, this defect curing process would lead to the decrease of trap density. Meanwhile, the accumulation, migration and reaction of generated defect Br0i could induce a battery of compensation effects resulting in the increase of trap density. On the one hand, the Br0i could accumulate in the grain boundary acting as a new kind of nonradiative recombination center. On the other hand, the concentration gradient of Br0i decreasing from bulk to surface and sufficient energy could apply a driving force to promote the long-distance migration of Br0i from bulk to surface, in which the bimolecular reaction of Br0i to form Br2 is favored by a relatively high trap density, effectively increasing the probability of encountering between two Br0i molecules. Moreover, the migration of Br0i could evoke the unbalanced distribution of bromine and result in a series of compensation reactions in the bulk. Therefore, the new VBr+/Bri− Frenkel pairs are generated through these reactions, which further improves trap density and the proportion of nonradiative recombination. These detailed photochemical reactions are presented in Fig. S3 and ESI Note 1.† On the basis of a Boltzmann exponential function,43 fitting of the high-energy side of PL spectrum, we calculate the corresponding plasma temperatures at different excitation densities. In terms of results, the plasma temperature increases slightly from 304 to 319 K when the excitation densities are 1.9 and 4.5 W cm−2 respectively (Fig. S4†). In essence, both PLIE and PLID involve ion migration, but PLIE effect is ascribed to short-distance movement of VBr+/Bri− Frenkel pairs, while PLID effect is attributed to long-distance migration of Br0i, which requires higher activation energy. Therefore, with the increase of excitation density, PLIE effect plays a dominant role in the first stage under low or middle excitation densities, but PLID gradually dominates the whole irradiation process at higher excitation densities.
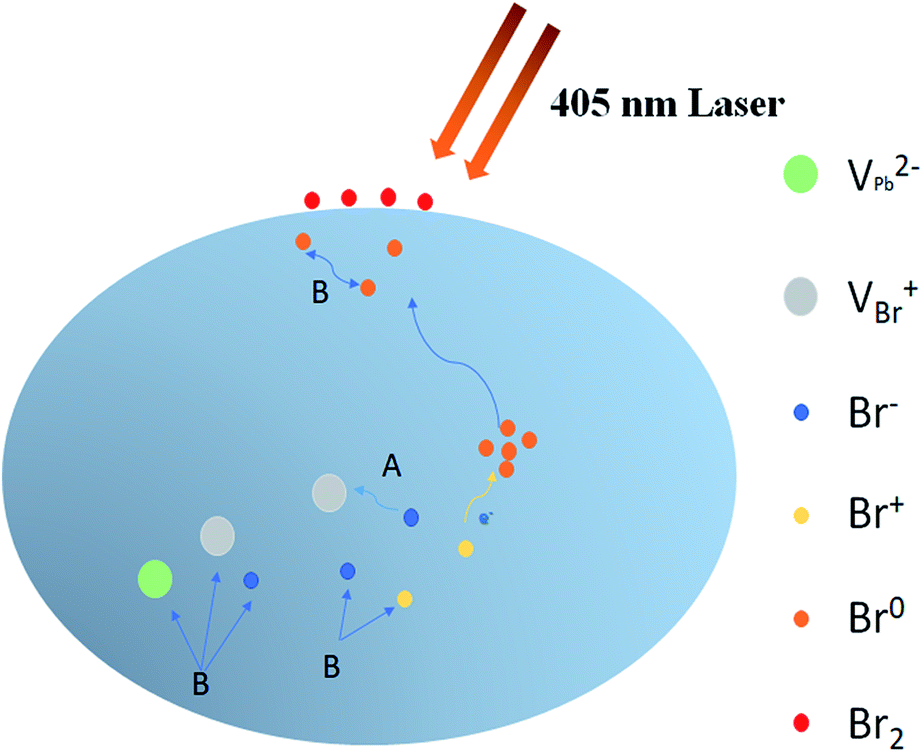 | ||
| Fig. 5 The ion migration behind PLIE and PLID process. A represents the annihilation of VBr+/Br− Frenkel pairs with an electron trapped at Br+, resulting in PLIE; B represents the formation of new defects as the compensation for migration of Br0 from bulk to surface which form Br2 through bimolecular reaction, leading to PLID. | ||
4. Conclusion
In summary, we have studied the dependence of photo-induced instability of methylammonium lead bromide perovskite films on the excitation power density. We confirm that illumination-triggered PLID and PLIE both take place and compete with each other in the perovskite. Depending on specific excitation density, one effect could prevail over the other, thus determining the variation of PL intensity and PL lifetime within illumination time. The overall PL intensity evolution can be well reproduced by a formula taking into account the two effects. A model regarding the formation and migration of long-living trapped Br0i species is adopted to explain the mechanism behind the competence between PLIE and PLID. The PLIE is associated with the annihilation of VBr+/Bri− Frenkel pairs driven by an electron trapped at Bri+. The PLID is related with a bimolecular reaction of two Br0i to form Br2 molecule, which is favored at higher excitation density.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (51772271) and the Natural Science Foundation of Zhejiang Province (LY17A040008).Notes and references
- K. Domanski, W. Tress, T. Moehl, M. Saliba, M. K. Nazeeruddin and M. Graetzel, Adv. Funct. Mater., 2015, 25, 6936–6947 CrossRef CAS.
- O. A. Jaramillo-Quintero, R. S. Sanchez, M. Rincon and I. Mora-Sero, J. Phys. Chem. Lett., 2015, 6, 1883–1890 CrossRef CAS PubMed.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- Y. Wu, C. T. Wei, X. M. Li, Y. L. Li, S. C. Qiu, W. Shen, B. Cai, Z. G. Sun, D. D. Yang, Z. T. Deng and H. B. Zeng, ACS Energy Lett., 2018, 3, 2030–2037 CrossRef CAS.
- H. Cho, S.-H. Jeong, M.-H. Park, Y.-H. Kim, C. Wolf, C.-L. Lee, J. H. Heo, A. Sadhanala, N. Myoung, S. Yoo, S. H. Im, R. H. Friend and T.-W. Lee, Science, 2015, 350, 1222–1225 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- X. Li, D. Bi, C. Yi, J.-D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, 353, 58–62 CrossRef CAS PubMed.
- Y. Cao, N. Wang, H. Tian, J. Guo, Y. Wei, H. Chen, Y. Miao, W. Zou, K. Pan, Y. He, H. Cao, Y. Ke, M. Xu, Y. Wang, M. Yang, K. Du, Z. Fu, D. Kong, D. Dai, Y. Jin, G. Li, H. Li, Q. Peng, J. Wang and W. Huang, Nature, 2018, 562, 249–253 CrossRef CAS PubMed.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang, C. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef CAS PubMed.
- Y. Liu, J. Cui, K. Du, H. Tian, Z. He, Q. Zhou, Z. Yang, Y. Deng, D. Chen, X. Zuo, Y. Ren, L. Wang, H. Zhu, B. Zhao, D. Di, J. Wang, R. H. Friend and Y. Jin, Nat. Photonics, 2019, 13, 760–764 CrossRef CAS.
- J. Li, L. Xu, T. Wang, J. Song, J. Chen, J. Xue, Y. Dong, B. Cai, Q. Shan, B. Han and H. Zeng, Adv. Mater., 2017, 29, 1603885 CrossRef PubMed.
- https://www.nrel.gov/pv/cell-efficiency.html, 2019.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., Int. Ed., 2015, 54, 8208–8212 CrossRef CAS PubMed.
- A. Alberti, I. Deretzis, G. Pellegrino, C. Bongiorno, E. Smecca, G. Mannino, F. Giannazzo, G. G. Condorelli, N. Sakai, T. Miyasaka, C. Spinella and A. La Magna, ChemPhysChem, 2015, 16, 3064–3071 CrossRef CAS PubMed.
- J. A. Christians, P. A. Miranda Herrera and P. V. Kamat, J. Am. Chem. Soc., 2015, 137, 1530–1538 CrossRef CAS PubMed.
- G. Divitini, S. Cacovich, F. Matteocci, L. Cina, A. Di Carlo and C. Ducati, Nat. Energy, 2016, 1, 15012 CrossRef CAS.
- N.-K. Kim, Y. H. Min, S. Noh, E. Cho, G. Jeong, M. Joo, S.-W. Ahn, J. S. Lee, S. Kim, K. Ihm, H. Ahn, Y. Kang, H.-S. Lee and D. Kim, Sci. Rep., 2017, 7, 4645 CrossRef PubMed.
- S. Chen, X. Wen, S. Huang, F. Huang, Y.-B. Cheng, M. Green and A. Ho-Baillie, Sol. RRL, 2017, 1, 1600001 CrossRef.
- D. W. deQuilettes, W. Zhang, V. M. Burlakov, D. J. Graham, T. Leijtens, A. Osherov, V. Bulovic, H. J. Snaith, D. S. Ginger and S. D. Stranks, Nat. Commun., 2016, 7, 11683 CrossRef CAS PubMed.
- Y. Tian, M. Peter, E. Unger, M. Abdellah, K. Zheng, T. Pullerits, A. Yartsev, V. Sundström and I. G. Scheblykin, Phys. Chem. Chem. Phys., 2015, 17, 24978–24987 RSC.
- E. Mosconi, D. Meggiolaro, H. J. Snaith, S. D. Stranks and F. De Angelis, Energy Environ. Sci., 2016, 9, 3180–3187 RSC.
- S. G. Motti, D. Meggiolaro, A. J. Barker, E. Mosconi, C. A. R. Perini, J. M. Ball, M. Gandini, M. Kim, F. De Angelis and A. Petrozza, Nat. Photonics, 2019, 13, 532–539 CrossRef CAS.
- W. Chen, Y. Wu, Y. Yue, J. Liu, W. Zhang, X. Yang, H. Chen, E. Bi, I. Ashraful, M. Graetzel and L. Han, Science, 2015, 350, 944–948 CrossRef CAS PubMed.
- J. You, L. Meng, T.-B. Song, T.-F. Guo, Y. Yang, W.-H. Chang, Z. Hong, H. Chen, H. Zhou, Q. Chen, Y. Liu, N. De Marco and Y. Yang, Nat. Nanotechnol., 2016, 11, 75–81 CrossRef CAS PubMed.
- S. Ghosh, S. K. Pal, K. J. Karki and T. Pullerits, ACS Energy Lett., 2017, 2, 2133–2139 CrossRef CAS.
- S. G. Motti, M. Gandini, A. J. Barker, J. M. Ball, A. R. S. Kandada and A. Petrozza, ACS Energy Lett., 2016, 1, 726–730 CrossRef CAS.
- J. Xing, Q. Wang, Q. Dong, Y. Yuan, Y. Fanga and J. Huang, Phys. Chem. Chem. Phys., 2016, 18, 30484–30490 RSC.
- K. M. Sim, A. Swarnkar, A. Nag and D. S. Chung, Laser Photonics Rev., 2018, 12, 1700209 CrossRef.
- M. S. Kirschner, B. T. Diroll, P. Guo, S. M. Harvey, W. Helweh, N. C. Flanders, A. Brumberg, N. E. Watkins, A. A. Leonard, A. M. Evans, M. R. Wasielewski, W. R. Dichtel, X. Zhang, L. X. Chen and R. D. Schaller, Nat. Commun., 2019, 10, 504 CrossRef CAS PubMed.
- J. Xue, D. Yang, B. Cai, X. Xu, J. Wang, H. Ma, X. Yu, G. Yuan, Y. Zou, J. Song and H. Zeng, Adv. Funct. Mater., 2019, 29, 1807922 CrossRef.
- D. Amelot, P. Rastogi, B. Martinez, C. Gréboval, C. Livache, F. A. Bresciani, J. Qu, A. Chu, M. Goyal, S.-S. Chee, N. Casaretto, X. Z. Xu, C. Méthivier, H. Cruguel, A. Ouerghi, A. Nag, M. G. Silly, N. Witkowski and E. Lhuillier, J. Phys. Chem. C, 2020, 124, 3873–3880 CrossRef CAS.
- C. Wolf, J.-S. Kim and T.-W. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 10344–10348 CrossRef CAS PubMed.
- K.-H. Wang, L.-C. Li, M. Shellaiah and K. Wen Sun, Sci. Rep., 2017, 7, 13643 CrossRef PubMed.
- S. Park, Y.-S. Seo, C. W. Ahn, W. S. Woo, J. Kyhm, S. A. Lee, I. W. Kim and J. Hwang, J. Phys. D: Appl. Phys., 2019, 52, 335302 CrossRef CAS.
- F. Zheng, W. Chen, T. Bu, K. P. Ghiggino, F. Huang, Y. Cheng, P. Tapping, T. W. Kee, B. Jia and X. Wen, Adv. Energy Mater., 2019, 9, 1901016 CrossRef.
- K. Domanski, B. Roose, T. Matsui, M. Saliba, S.-H. Turren-Cruz, J.-P. Correa-Baena, C. Roldan-Carmona, G. Richardson, J. M. Foster, F. De Angelis, J. M. Ball, A. Petrozza, N. Mine, M. K. Nazeeruddin, W. Tress, M. Gratzel, U. Steiner, A. Hagfeldt and A. Abate, Energy Environ. Sci., 2017, 10, 604–613 RSC.
- S. Meloni, T. Moehl, W. Tress, M. Franckevicius, M. Saliba, Y. H. Lee, P. Gao, M. K. Nazeeruddin, S. M. Zakeeruddin, U. Rothlisberger and M. Graetzel, Nat. Commun., 2016, 7, 10334 CrossRef CAS PubMed.
- E. Mosconi and F. De Angelis, ACS Energy Lett., 2016, 1, 182–188 CrossRef CAS.
- P. Delugas, C. Caddeo, A. Filippetti and A. Mattoni, J. Phys. Chem. Lett., 2016, 7, 2356–2361 CrossRef CAS PubMed.
- G. Y. Kim, A. Senocrate, T.-Y. Yang, G. Gregori, M. Gratzel and J. Maier, Nat. Mater., 2018, 17, 445–449 CrossRef CAS PubMed.
- M. Cadelano, V. Sarritzu, N. Sestu, D. Marongiu, F. Chen, R. Piras, R. Corpino, C. M. Carbonaro, F. Quochi, M. Saba, A. Mura and G. Bongiovanni, Adv. Opt. Mater., 2015, 3, 1557–1564 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00605j |
| This journal is © The Royal Society of Chemistry 2020 |