
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReal-time detection of mRNA splicing variants with specifically designed reverse-transcription loop-mediated isothermal amplification†
Fengxia Su ,
Guanhao Wang,
Jianing Ji,
Pengbo Zhang,
Fangfang Wang and
Zhengping Li*
,
Guanhao Wang,
Jianing Ji,
Pengbo Zhang,
Fangfang Wang and
Zhengping Li*
School of Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing, 100083, P. R. China. E-mail: lzpbd@ustb.edu.cn
First published on 10th February 2020
Abstract
Alternative splicing is a ubiquitous and crucial process in cellular processes and has a specific linkage with diseases. To date, developing cost-effective methods with high sensitivity and specificity for detection of splicing variants has been needed. Herein, we report a novel splicing variant assay based on specifically designed reverse-transcription loop-mediated isothermal amplification. After reverse transcribing the splicing variant into cDNA, four DNA primers are specifically designed to recognize six distinct regions. The four DNA primers can hybridize with corresponding sequences for extension and strand displacement DNA synthesis to form stem-loop DNA and then LAMP amplification is started. The proposed method can detect as low as 100 aM splicing variants in real-time fashion with high specificity, showing great potential in biological function and clinical studies.
Introduction
In eukaryotes, one DNA gene is firstly transcribed into pre-mRNA, and the pre-mRNA can be alternatively spliced in different arrangements into different isoforms subsequently encoding for a variety of proteins with diverse structures and functions.1–4 Therefore, mRNA alternative splicing is widely recognized to be a ubiquitous and crucial modulating process during gene expression and a major contributor to mammalian transcriptomic and proteomic diversification, and thus is closely implicated in many cellular processes, including cell development and differentiation, which can explain the specific linkage between some deficient or aberrant splicing patterns with specific diseases.5–9 To better understand the role of alternative splicing in biological processes and disease diagnosis, appreciable efforts have been directed to identify new alternative splicing and detect specific known splicing variants.Up to now, the high throughput RNA sequencing and high-density DNA microarrays have provided powerful tools to profile the splicing variants transcriptome-wide.10–12 High throughput next-generation sequencing (NGS)13 technology is particularly helpful to identify splice events at the transcriptome scale, to reveal the combinatorial splicing diversity of exons in different cell lines or tissues and to discover the correlation between human diseases and specific splicing variants. It is estimated that up to 95% of human RNAs undergo alternative splicing.14,15 Although methods based on high throughput RNA sequencing and high-density DNA microarrays can give abundant transcriptome scale information, the complex steps, high consumption of reagents and expensive instrumentations seriously restrict their applications in ordinary laboratories to detect specific splicing variants. Therefore, simple and cost-effective methods with high sensitivity and specificity to detect specific splicing isoforms are urgently required for studies of the biological functions of mRNA alternative splicing and disease diagnosis.
Various technologies, including surface-enhanced Raman scattering (SERS),16 hyperspectral imaging17 and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry,18 have been explored for quantitative detection of known splicing variants. However, the expensive instruments limit the routine use in clinical diagnosis and ordinary laboratories. Reverse transcription-polymerase chain reaction (RT-PCR) is most commonly recruited for analysis of alternative splicing in different human tissues or cell lines due to high sensitivity and wide detection dynamic range.19,20 In the RT-PCR method, boundary-spanning primer (BSP) is usually designed to cover the exons joint for specific PCR amplification; however, partial hybridization of the BSP to other splicing isoforms may lead to false amplification and low specificity of detection.20,21 Our group has also reported a ligation-based PCR method to circumvent the issue of mispriming to improve the specificity of splicing variant detection.22 However, this PCR method suffers the instinctive problem of nonspecific ligation. Thus, a specific and sensitive method for quantitative detection of alternative variants is desirable in routine clinical diagnosis and ordinary laboratories.
Loop-mediated isothermal amplification (LAMP)23 is one of the most sensitive amplification techniques with high specificity and low background, which is capable of amplifying a few copies of targets to a detectable level under isothermal conditions. Due to these valuable advantages, LAMP has been adapted in various ways for identification of bacteria and viruses,24–26 detection of telomerase activity,27 single nucleotide polymorphism,28 DNA methylation29 and microRNA.30–32 Herein, by specifically designing DNA primers, we propose a LAMP-based method for real-time detection of splicing variants with high specificity, sensitivity and low cost. In the proposed assay, four DNA primers, including forward inner primer (FIP), backward inner primer (BIP), outer primers F3 and B3, are designed to specifically recognize six distinctive regions in specific splicing variant, ensuring the formation of a stem-loop DNA, the initial material of LAMP. Based on the specific primer design and the nature of high amplification efficiency and isothermal reaction of LAMP, the proposed method is well suited for the detection of splicing variants in ordinary laboratories and hospital clinical diagnosis.
Experimental
Reagents and instruments
The recombinant plasmids containing target sequences were obtained from Genscript Biotechnology Co., Ltd (Nanjing, China). RNA Clean Kit was purchased from TIANGEN Biotech Co., Ltd. (Beijing, China). SmaI, ProtoScript II Reverse Transcriptase and Bst 2.0 WarmStart DNA polymerase were purchased from New England Biolabs Inc. (USA). T7 in vitro transcription kit was obtained from Biomics Biotechnologies (Nantong, China). HPLC purified DNA primers (Table S1, ESI†) and the sequencing data were obtained from Shanghai Sangon Biotech (Shanghai, China). Recombinant RNase inhibitor, Recombinant DNase I, dNTPs, TaKaRa Taq™ Hot Start Version and Ribonuclease H (RNase H) were obtained from Takara Biomedical Technology Co., Ltd. (Beijing, China). Betaine was purchased from Sigma-Aldrich (Shanghai, China). SYBR Green I (20× stock solution in DMSO, 20 μg mL−1) was gained from Zhishan Biotechnology Co., Ltd. (Xiamen, China). A549, 293T, HL-60 and HeLa cells were purchased from the cell bank of Chinese Academy of Sciences (Shanghai, China).The concentration of mRNA transcribed in vitro and total RNA extracted from cells were determined by NanoDrop One (Thermo Fisher Scientific, USA). The plasmid linearization, in vitro transcription and reverse transcription were carried out in a 2720 thermal cycler (Applied Biosystems, USA). The real-time fluorescence of the LAMP reaction was monitored with StepOne Real-Time PCR System (Applied Biosystems, USA).
Preparation of splicing variants of FGFR 2 gene
FGFR 2-IIIc represents the splicing variant containing exon 7, exon 9 and exon 10, while FGFR 2-IIIb represents the splicing transcript containing exon 8 and exon 10. The FGFR 2-IIIc corresponding DNA with T7 promoter and FGFR 2-IIIb corresponding DNA with T7 promoter (Fig. S1, ESI†) were cloned in pUC57 (2710 bp) at the site of EcoRI by Genscript, respectively. The electrophoresis images of constructed plasmids were shown in Fig. S2a and S2b (ESI†). 5 μg constructed plasmids were linearized with 100 U SmaI in 1× cut smart buffer (5 mM KAc, 2 mM Tris–Ac, 1 mM Mg(Ac)2, 10 μg mL−1 BSA, pH 7.9, 25 °C) at 25 °C for 15 min. The SmaI was then inactive at 65 °C for 20 min. The linearized plasmids were transcribed into RNA with T7 in vitro transcription kit. The plasmids were degraded with 50 U Recombinant DNase I at 37 °C for 30 min. The remaining RNA was purified with RNA Clean Kit and quantified with NanoDrop One. Therefore, FGFR 2-IIIc (590 nt, sequence between the recognition site of EcoRI and SmaI) and FGFR 2-IIIb (402 nt) of known concentration were obtained (Fig. S2c, ESI†).Extraction of total RNA from cells
A549, 293T, HL-60 and HeLa cells were cultured in Roswell park memorial institute (RPMI)-1640 medium with 10% fetal bovine serum and 1% penicillin/streptomycin. All cells were maintained in a 100% humidified atmosphere containing 5% CO2 at 37 °C. Total RNA was extracted from the above cells, respectively, with TRIzol reagent according to the manufacture's protocol.Standard protocols for detection of mRNA splicing variants
The reverse transcriptions were conducted in 9 μL of mixture containing 1× ProtoScript II Reverse Transcriptase reaction buffer (50 mM Tris–HCl, 3 mM MgCl2, pH 8.3, 25 °C), 10 nM RT primer, dNTPs (250 μM each), 8 U Recombinant RNase inhibitor, 40 U of ProtoScript II Reverse Transcriptase and different amounts of mRNA splicing variant or total RNA extracted from cells. The mixture was subjected to 42 °C for 45 min to complete the reverse transcription and then incubated at 65 °C for 15 min to inactive the enzyme.1 μL RNase H (6 U) was added to the solution of reverse transcription to digest the RNA hybridized with cDNA under 37 °C for 20 min. Inactivate RNase H at 65 °C for 15 minutes.
The LAMP reaction mixture was prepared with 1× isothermal amplification buffer (20 mM Tris–HCl, 50 mM KCl, 10 mM (NH4)2SO4, 2 mM MgSO4 and 0.1% Triton X-20, pH 8.8, 25 °C), 250 μM dNTPs, 1 M betaine, 1.6 μM each of IIIc-FIP and IIIc-BIP (for FGFR 2-IIIc target) or 1.2 μM each of IIIb-FIP and IIIb-BIP (for FGFR 2-IIIb target), 0.2 μM each of IIIc-B3 and IIIc-F3 (for FGFR 2-IIIc target) or 0.15 μM each of IIIb-B3 and IIIb-F3 for (FGFR 2-IIIb target), 4 ng SYBR Green I and 2.4 U (for FGFR 2-IIIc target) or 0.8 U (for FGFR 2-IIIb target) Bst 2.0 WarmStart DNA Polymerase. 1 μL RNase H digestion product was added to the mixture and immediately put into the StepOne Real-Time PCR System to perform the LAMP reaction at 65 °C. The real-time fluorescence intensity was simultaneously monitored at intervals of 1 min.
Sequencing of the FGFR 2 mRNA in cells
The total RNA isolated from 293T and A549 cells was reverse transcribed into cDNA, respectively, with the same procedures in the standard protocols. 60 μL of mixture was prepared with 1× PCR buffer (Mg2+ plus), 250 μM dNTPs, 500 nM forward and reverse primers each, 1 U TaKaRa Taq™ Hot Start Version and 12 μL reverse transcription product. The mixture was experienced 95 °C for 2 min to denature the hybridized RNA/DNA double strands and 40 cycles containing 95 °C for 20 s, 65 °C for 20 s and 72 °C for 30 s to complete the PCR amplification. The PCR products were sequenced by Sangon Biotech.Results and discussion
Principle of the proposed method for detection of splicing variants
The primer design and mechanism of the proposed LAMP-based method for detection of splicing variants are illustrated in Fig. 1. Alternative splicing variants of fibroblast growth factor receptor 2 (FGFR 2) gene are selected as the model targets, in which exon 8 and exon 9 are mutually exclusive.6,33 FGFR 2-IIIc represents the splicing variant containing exon 7, exon 9 and exon 10, while FGFR 2-IIIb represents the splicing isoform containing exon 8 and exon 10. In the specific detection route of FGFR 2-IIIc illustrated in Fig. 1a, RT primer firstly hybridizes to exon 10 of FGFR 2-IIIc and undergoes reverse transcription with the catalysis of Protoscript II reverse transcriptase, producing IIIc-cDNA. The RNA strand in the RNA–DNA hybrid is then specifically hydrolysed with RNase H and the remaining IIIc-cDNA is amplified with specifically designed DNA primers. IIIc-FIP, containing two sequences of IIIc-F1c (the same as IIIc-F1c in exon 9 of IIIc-cDNA) and IIIc-F2 (complementary to IIIc-F2c in exon 7 of IIIc-cDNA, c means the complementary sequence), hybridizes to exon 7 of IIIc-cDNA and then undergoes elongation by the action of Bst 2.0 WarmStart DNA polymerase. IIIc-F3, in lower concentration than IIIc-FIP, then hybridizes with IIIc-F3c in exon 7 of IIIc-cDNA and initiates the strand displacement DNA synthesis, releasing IIIc-FIP elongated DNA strand, which can form a DNA strand with a loop at 5′-end. IIIc-BIP contains two specific sequences of IIIc-B1c (the same as IIIc-B1c in exon 9 of IIIc-FIP elongated DNA strand) and IIIc-B2, which is complementary to IIIc-B2c in exon 9 of IIIc-FIP elongated DNA strand. IIIc-B3 is designed complementary with IIIc-B3c in exon 10 of IIIc-FIP elongated DNA strand. The IIIc-FIP elongated DNA strand acts as template for the IIIc-BIP triggered polymerase reaction and the following IIIc-B3 initiated strand displacement DNA synthesis. The newly generated IIIc-BIP extension strand is displaced by the extension of IIIc-B3 and quickly forms a dumb-bell DNA, which is subsequently converted into a stem-loop DNA though self-primed DNA synthesis. Through the specific primer design, only in the presence of FGFR 2-IIIc (exon 7, 9, 10), the stem-loop DNA, the key initial material of LAMP, can be produced and the subsequent LAMP reaction can be carried out.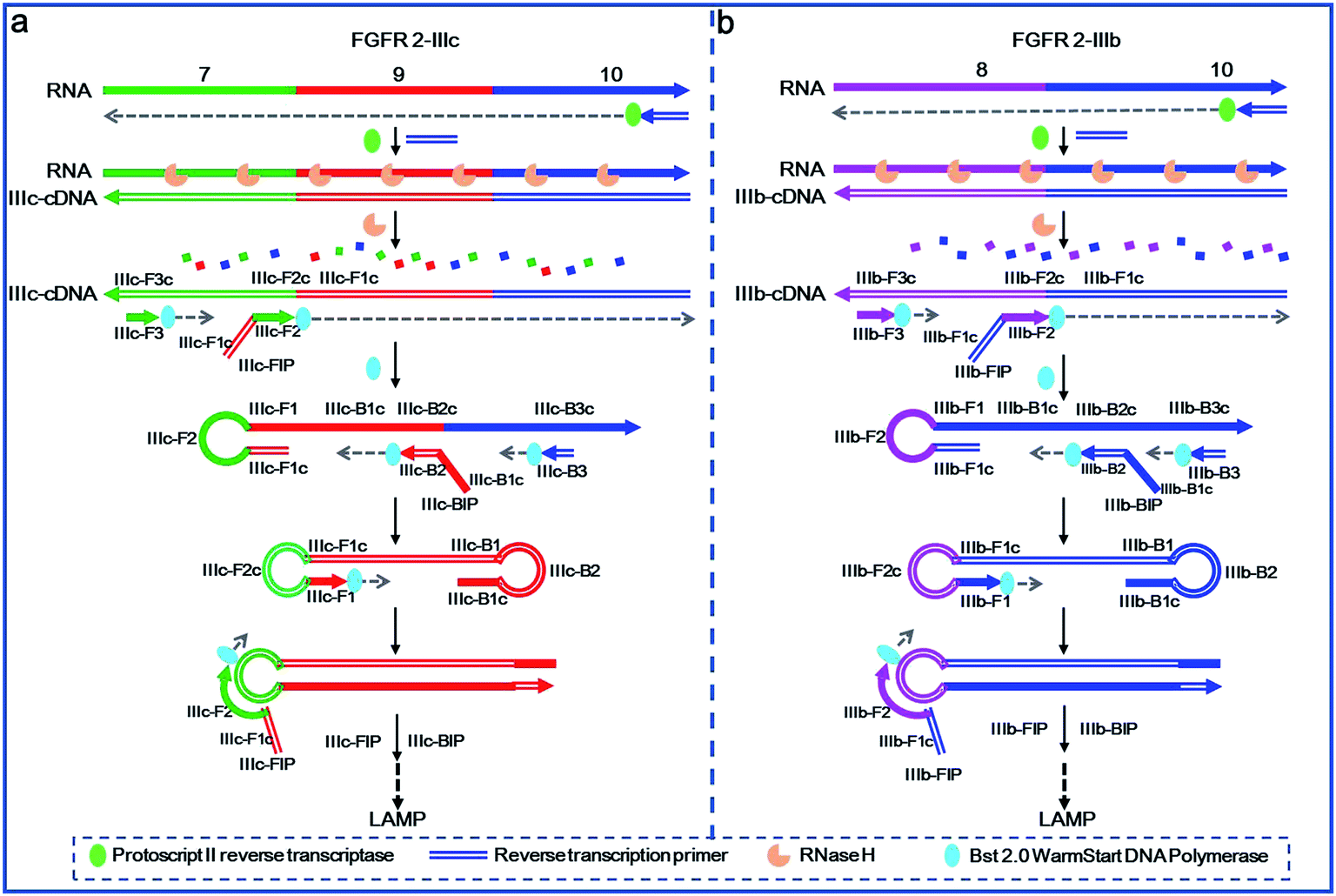 | ||
| Fig. 1 Schematic representation of the proposed method for detection of mRNA splicing variants. The complementary sequences are indicated with solid and hollow lines, respectively. The 3′-end of the sequences are indicated with arrows. | ||
In the specific detection path of FGFR 2-IIIb (Fig. 1b), IIIb-cDNA is synthesized through a similar approach of IIIc-cDNA synthesis as mentioned above. IIIb-FIP, consisting of two specific sequences of IIIb-F1c (the same as IIIb-F1c in exon 10 of IIIb-cDNA) and IIIb-F2, which is complementary to IIIb-F2c located in exon 8 of IIIb-cDNA. IIIb-F3 is designed complementary to IIIb-F3c in exon 8 of IIIb-cDNA. Therefore, IIIb-cDNA can serve as the template for the elongation of IIIb-FIP and the following strand displacement reaction of IIIb-F3, producing IIIb-FIP elongated strand with a loop at 5′-end. According to the three specific sequences of IIIb-B1c, IIIb-B2c and IIIb-B3c in exon 10 of IIIb-FIP elongated strand, IIIb-BIP (containing IIIb-B1c and IIIb-B2) and IIIb-B3 are specifically designed. Therefore, the extension of IIIb-BIP and the following displacement reaction of IIIb-B3 are carried out by using IIIb-FIP elongated strand as a template, forming a stem-loop DNA to initiate LAMP. Such the primer design allows the production of the stem-loop structure only in the presence of FGFR 2-IIIb. Once the stem-loop structure is formed, the subsequent process of auto-cycling strand displacement DNA synthesis is the same as the elongation and recycling steps in LAMP demonstrated in the previous literature.23 The FIP firstly hybridizes with the loop DNA and primes the strand displacement DNA synthesis. The newly generated DNA then initiates self-loop DNA structures, in which the BIP can hybridize with the loop DNA and primes the strand displacement DNA synthesis to generate more stem-loop DNA structures, and thus form the exponential DNA amplification with faster and faster strand displacement synthesis. Finally, the LAMP reaction produces a large number of stem-loop DNAs with longer and longer stem of double strand DNAs, which can be sensitively and cost effectively detected by using SYBR Green I as the double-strand-specific dye.
The sensitivity and detection range of the proposed assay
According to the principle demonstrated in Fig. 1, the important experimental parameters including the concentration of DNA primers and the amount of DNA polymerase, were systematically optimized for specific detection of FGFR 2-IIIc (Fig. S3 and S4, ESI†) and FGFR 2-IIIb (Fig. S5 and S6, ESI†). The sensitivity and dynamic range of the LAMP-based method were subsequently tested under optimal conditions. A series of concentrations of synthetic FGFR2-IIIc and FGFR 2-IIIb were detected by the proposed method and well-defined real-time fluorescence curves were produced from 100 aM to 10 pM of specific targets (Fig. 2a and c). The blank fluorescence signal of FGFR2-IIIc and FGFR 2-IIIb kept near-zero within 80 min, indicating almost no nonspecific amplification occurred, which was mainly ascribed to the ingenious primer design. The point of inflection (POI) value, the time when the slope of the real-time fluorescence curve was the largest, was used as a criterion for quantification. The POI value of the fluorescence curves decreased as increasing of the concentration of target. It can be observed that the POI value of the real-time fluorescence curves has a good linear correlation with the negative logarithm of the concentrations of FGFR 2-IIIc (Fig. 2b) and FGFR 2-IIIb (Fig. 2d) in the range of five orders of magnitude from 100 aM to 10 pM. The linear correlation equations were POI = −20.02 to 4.38 lg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) CFGFR 2-IIIc and POI = −16.88 to 3.70 lgCFGFR 2-IIIb with the corresponding correlation coefficient R2 of 0.9979 and 0.9985, respectively.
CFGFR 2-IIIc and POI = −16.88 to 3.70 lgCFGFR 2-IIIb with the corresponding correlation coefficient R2 of 0.9979 and 0.9985, respectively.
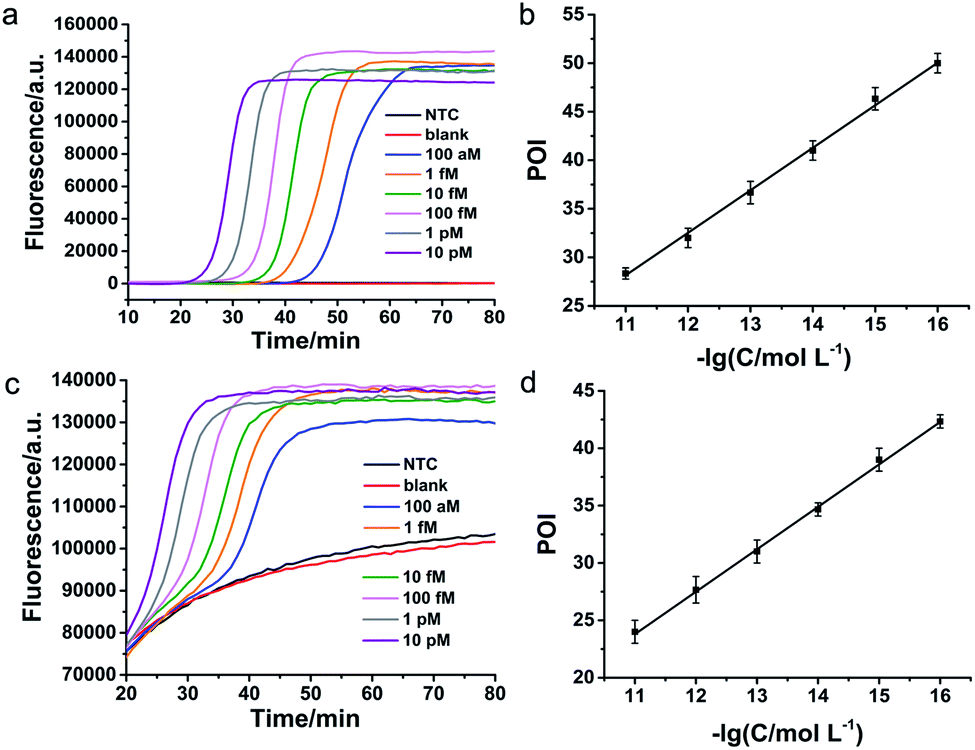 | ||
| Fig. 2 Detection sensitivity and dynamic range of the proposed method for FGFR 2 splicing variants. Real-time fluorescence curves were produced from different concentrations of FGFR 2-IIIc (a) and FGFR 2-IIIb (c). Linear relationship between POI values of the real-time fluorescence curves and the negative logarithm (−lg) of the FGFR 2-IIIc (b) and FGFR 2-IIIb (d) concentrations. The error bars in (b) and (d) were derived from three parallel experiments. NTC represents the reaction without the reverse transcription components and RNA target. Blank represents the reaction only without RNA target. | ||
Specificity of the proposed assay
To verify the specificity of the proposed method to clearly distinguish between different splicing variants of the same gene, FGFR 2-IIIc and FGFR 2-IIIb were detected with FGFR 2-IIIc specific primers and FGFR 2-IIIb specific primers, respectively. As shown in Fig. 3a, by using FGFR 2-IIIc specific primers, FGFR 2-IIIc at 10 fM and 100 fM were able to produce well-defined real-time fluorescence curves by the proposed method, while FGFR 2-IIIb at 10 fM and 100 fM generated the same responding signal as blank. As shown in Fig. 3b, by using FGFR 2-IIIb specific primers, the perfectly matched FGFR 2-IIIb produced well-defined real-time fluorescence curves, while FGFR 2-IIIc at 10 fM and 100 fM generated the same fluorescent signal as blank. These results demonstrated the high specificity of the proposed method for the splicing variants detection.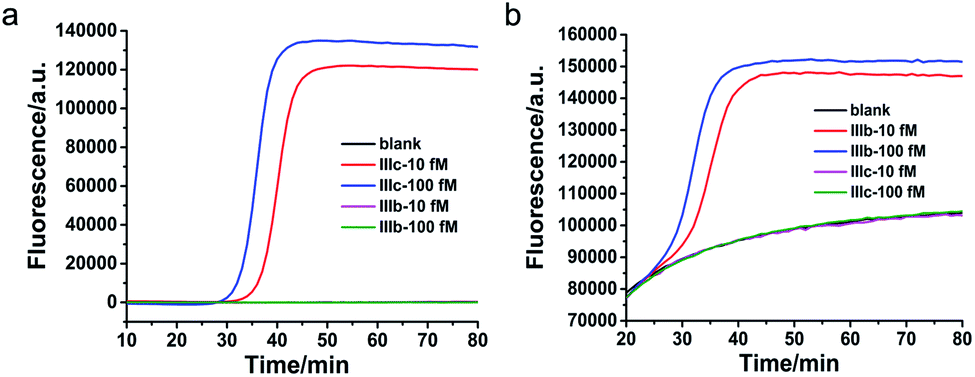 | ||
| Fig. 3 Specificity of the LAMP-based method for FGFR 2 splicing variant detection. (a) Real-time fluorescence curves were originated from FGFR2-IIIc and FGFR2-IIIb with FGFR2-IIIc specific primers. (b) Real-time fluorescence curves were produced from FGFR2-IIIc and FGFR2-IIIb with FGFR2-IIIb specific primers. | ||
Quantification of FGFR 2 splicing variants in cell lines
In view of the high sensitivity and specificity of the established method for detection of splicing variants, alternative splicing variants of FGFR 2 gene in total RNA from different cells were analysed with the proposed method. The two mutually exclusive transcripts of FGFR 2 gene can give rise to cell-surface receptors with different ligand specificity, showing cell specific splicing. As shown in Fig. 4, the copy number of FGFR 2-IIIc was quantified as 7.629 × 105 and 2.59 × 104 in 100 ng total RNA extracted from A549 cells and 293T cells, respectively. The existence of the FGFR 2-IIIc is in the two cell lines were demonstrated by sequencing (Fig. S7 and S8, ESI†). Meanwhile, FGFR 2-IIIb was undetectable in the two cell lines, indicating well defined cell-specific splicing of FGFR 2 gene. Both of the two splicing variants of FGFR 2 were undetectable in HL-60 and HeLa cells, which was consistent with the previous reports.34 The transcript containing exon 8 encodes receptors termed K-SAM, while isoforms containing exon 9 encodes BEK.35 Therefore, this method can not only be applied to the quantification of splicing variants, but also have potential for indirectly evaluation of the expression of the relative proteins, providing valuable information for further exploration of biological functions.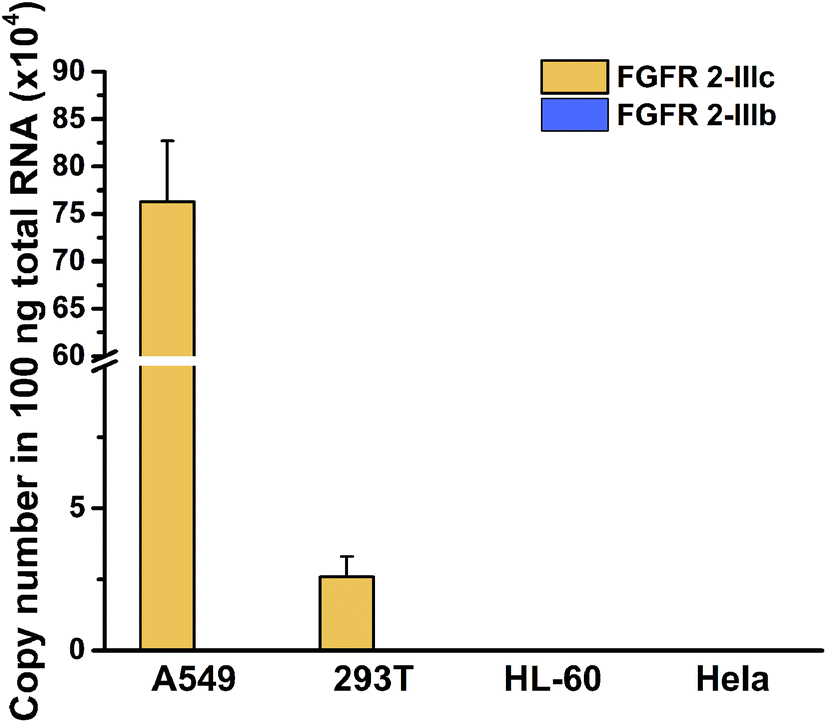 | ||
| Fig. 4 Quantification of FGFR 2 splicing variants in 100 ng total RNA from different cells. The ordinate was the copy number of the specific target in 100 ng total RNA and the abscissa indicated the cell lines. Error bars were calculated from three parallel experiments. | ||
Conclusions
In summary, we have developed a simple and low-cost RT-LAMP method for detection of alternative splicing variants with high sensitivity and specificity. The four primers were specifically designed to recognize six distinct sequences in the target, which made the method just suitable for detection of splicing variants and achieved high specificity. Although four primers were required in this method, the primers were employed in μM level in the experiments with low cost. On the other hand, the use of SYBR Green I for monitoring the double strand DNA avoided the requirement of any fluorescent modification on primers, thus the proposed assay can be cost-effectively performed. Due to the high amplification efficiency and very low background signal of LAMP, the proposed method can detect as low as 100 aM splicing variants. Just because of the high sensitivity, the proposed method could be successfully applied to splicing variant assay in real samples.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21775012), the China Postdoctoral Science Foundation (2018M641183) and the Fundamental Research Funds for the Central Universities (FRF-TP-18-052A1).Notes and references
- C. W. Smith and J. Valcárcel, Trends Biochem. Sci., 2000, 25, 381–388 CrossRef CAS PubMed.
- K. W. Lynch, Nat. Rev. Immunol., 2004, 4, 931 CrossRef CAS PubMed.
- Y. Barash, J. A. Calarco, W. Gao, Q. Pan, X. Wang, O. Shai, B. J. Blencowe and B. J. Frey, Nature, 2010, 465, 53–59 CrossRef CAS PubMed.
- B. R. Graveley, Trends Genet., 2001, 17, 100–107 CrossRef CAS PubMed.
- G. S. Wang and T. A. Cooper, Nat. Rev. Genet., 2007, 8, 749–761 CrossRef CAS PubMed.
- M. A. Garcia-Blanco, A. P. Baraniak and E. L. Lasda, Nat. Biotechnol., 2004, 22, 535–546 CrossRef CAS PubMed.
- N. A. Faustino and T. A. Cooper, Genes Dev., 2003, 17, 419–437 CrossRef CAS PubMed.
- B. M. Brinkman, Clin. Biochem., 2004, 37, 584–594 CrossRef CAS PubMed.
- M. J. Pajares, T. Ezponda, R. Catena, A. Calvo, R. Pio and L. M. Montuenga, Lancet Oncol., 2007, 8, 349–357 CrossRef CAS PubMed.
- T. C. Mockler and J. R. Ecker, Genomics, 2005, 85, 1–15 CrossRef CAS PubMed.
- Q. Xu, B. Modrek and C. Lee, Nucleic Acids Res., 2002, 30, 3754–3766 CrossRef CAS PubMed.
- J. M. Johnson, J. Castle, P. Garrett-Engele, Z. Kan, P. M. Loerch, C. D. Armour, R. Santos, E. E. Schadt, R. Stoughton and D. D. Shoemaker, Science, 2003, 302, 2141–2144 CrossRef CAS PubMed.
- M. Sultan, M. H. Schulz, H. Richard, A. Magen, A. Klingenhoff, M. Scherf, M. Seifert, T. Borodina, A. Soldatov, D. Parkhomchuk, D. Schmidt, S. O'Keeffe, S. Haas, M. Vingron, H. Lehrach and M.-L. Yaspo, Science, 2008, 321, 956–960 CrossRef CAS PubMed.
- Q. Pan, O. Shai, L. J. Lee, B. J. Frey and B. J. Blencowe, Nat. Genet., 2008, 40, 1413 CrossRef CAS PubMed.
- E. T. Wang, R. Sandberg, S. Luo, I. Khrebtukova, L. Zhang, C. Mayr, S. F. Kingsmore, G. P. Schroth and C. B. Burge, Nature, 2008, 456, 470–476 CrossRef CAS PubMed.
- L. Sun and J. Irudayaraj, Biophys. J., 2009, 96, 4709–4716 CrossRef CAS PubMed.
- K. Lee, Y. Cui, L. P. Lee and J. Irudayaraj, Nat. Nanotechnol., 2014, 9, 474–480 CrossRef CAS PubMed.
- R. M. McCullough, C. R. Cantor and C. Ding, Nucleic Acids Res., 2005, 33, e99 CrossRef PubMed.
- I. I. Vandenbroucke, J. Vandesompele, A. D. Paepe and L. Messiaen, Nucleic Acids Res., 2001, 29, e68 CrossRef CAS PubMed.
- J. P. Brosseau, J. F. Lucier, E. Lapointe, M. Durand, D. Gendron, J. Gervais-Bird and S. A. Elela, RNA, 2010, 16, 442–449 CrossRef CAS PubMed.
- H. S. Walton, F. M. Gebhardt, D. J. Innes and P. R. Dodd, J. Neurosci. Methods, 2007, 160, 294–301 CrossRef CAS PubMed.
- H. Wang, H. Wang, X. Duan, Y. Sun, X. Wang and Z. Li, Chem. Sci., 2017, 8, 3635–3640 RSC.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS PubMed.
- H. Kim, J. Lee, B. Lee, J. Choi, K. Kim, N. Song, S. Y. Lee, S. Yun, H.-S. Jeon and J. Park, Dyes Pigm., 2019, 170, 107618 CrossRef CAS.
- J. Dong, Q. Xu, C.-C. Li and C.-Y. Zhang, Chem. Commun., 2019, 55, 2457–2460 RSC.
- J. Chen, X. Xu, Z. Huang, Y. Luo, L. Tang and J.-H. Jiang, Chem. Commun., 2018, 54, 291–294 RSC.
- H. Wang, H. Wang, C. Liu, X. Duan and Z. Li, Chem. Sci., 2016, 7, 4945–4950 RSC.
- Y. Mitani, A. Lezhava, Y. Kawai, T. Kikuchi, A. Oguchi-Katayama, Y. Kogo, M. Itoh, T. Miyagi, H. Takakura, K. Hoshi, C. Kato, T. Arakawa, K. Shibata, K. Fukui, R. Masui, S. Kuramitsu, K. Kiyotani, A. Chalk, K. Tsunekawa, M. Murakami, T. Kamataki, T. Oka, H. Shimada, P. E. Cizdziel and Y. Hayashizaki, Nat. Methods, 2007, 4, 257–262 CrossRef CAS PubMed.
- H. Wen, H. Wang, H. Wang, J. Yan, H. Tian and Z. Li, Anal. Methods, 2016, 8, 5372–5377 RSC.
- W. Du, M. Lv, J. Li, R. Yu and J. Jiang, Chem. Commun., 2016, 52, 12721–12724 RSC.
- C. Li, Z. Li, H. Jia and J. Yan, Chem. Commun., 2011, 47, 2595–2597 RSC.
- Y. Sun, H. Tian, C. Liu, Y. Sun and Z. Li, Chem. Commun., 2017, 53, 11040–11043 RSC.
- T. Ishiwata, Front. Biosci., 2018, 23, 626–639 CrossRef CAS PubMed.
- J. M. Yeakley, J. B. Fan, D. Doucet, L. Luo, E. Wickham, Z. Ye, M. S. Chee and X. D. Fu, Nat. Biotechnol., 2002, 20, 353 CrossRef CAS PubMed.
- Y. A. Luqmani, G. S. Bansal, C. Mortimer, L. Buluwela and R. C. Coombes, Eur. J. Cancer, 1996, 32, 518–524 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00591f |
| This journal is © The Royal Society of Chemistry 2020 |