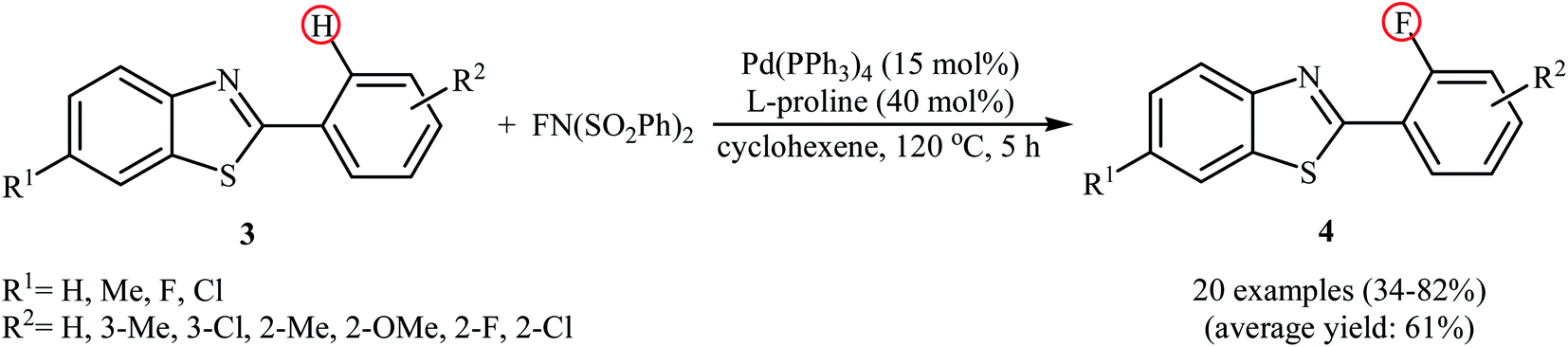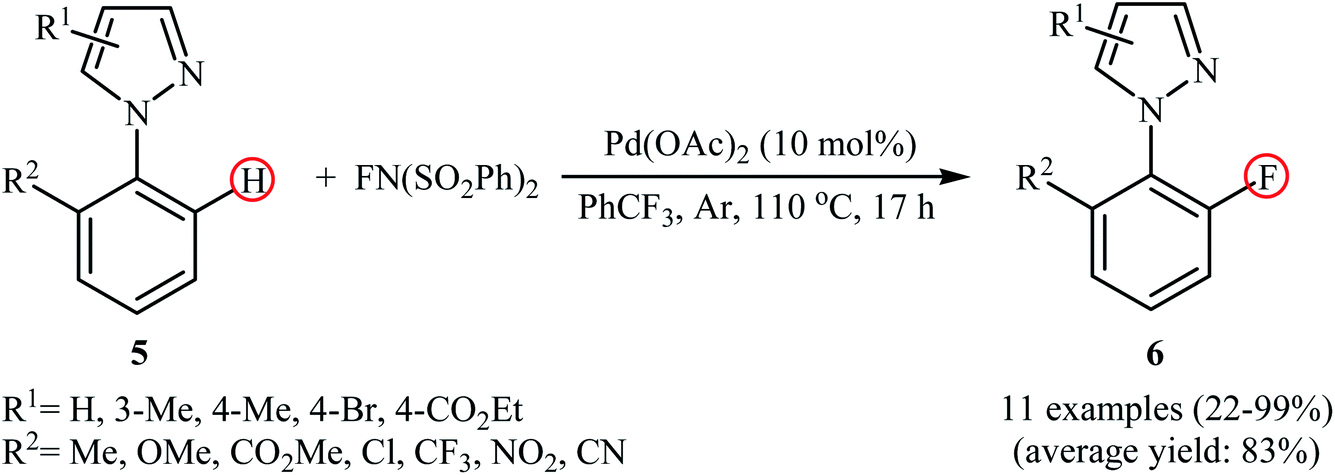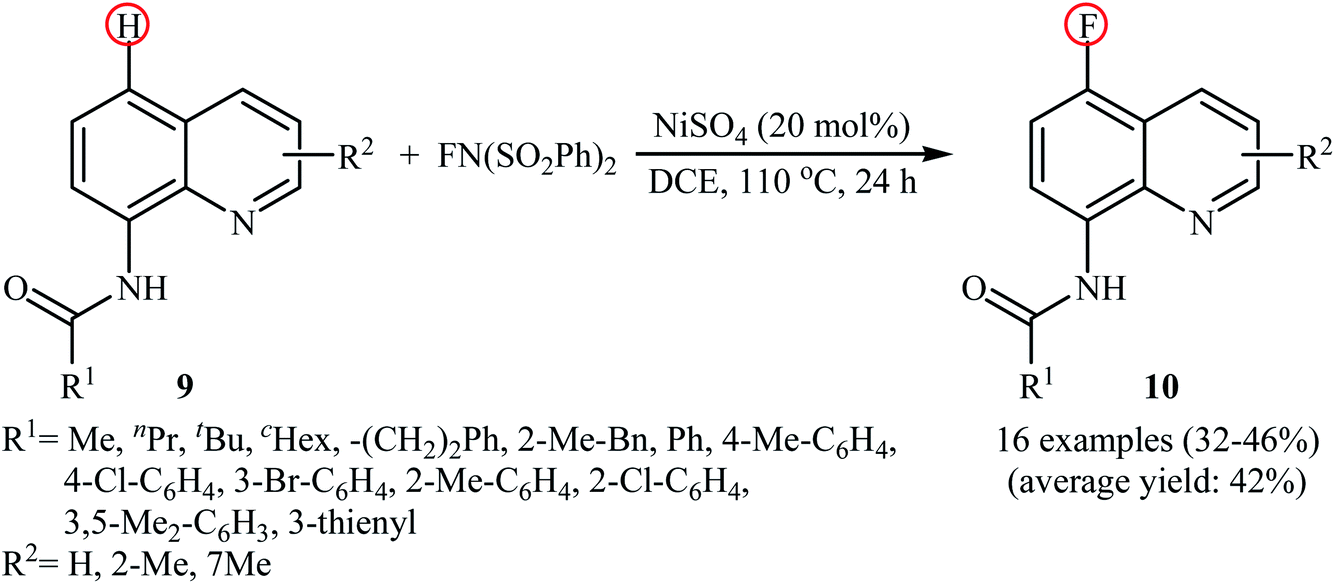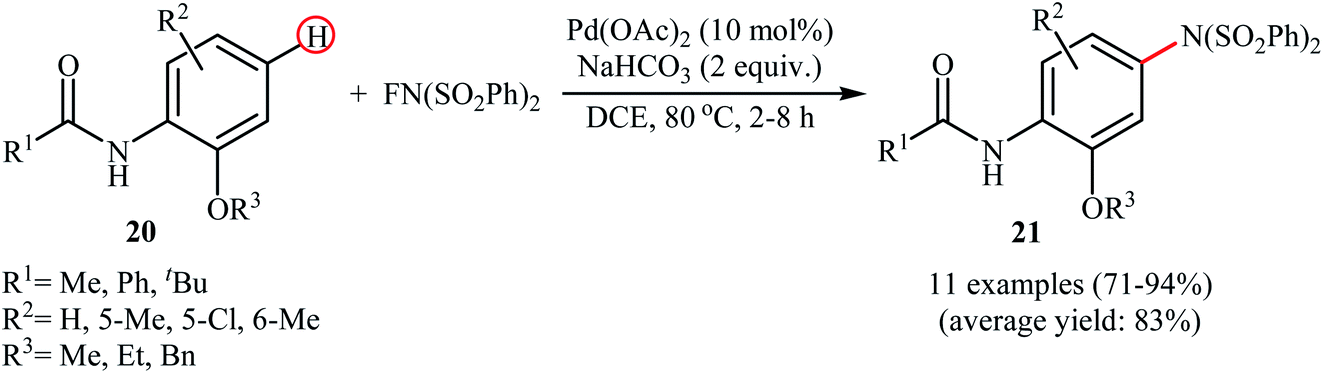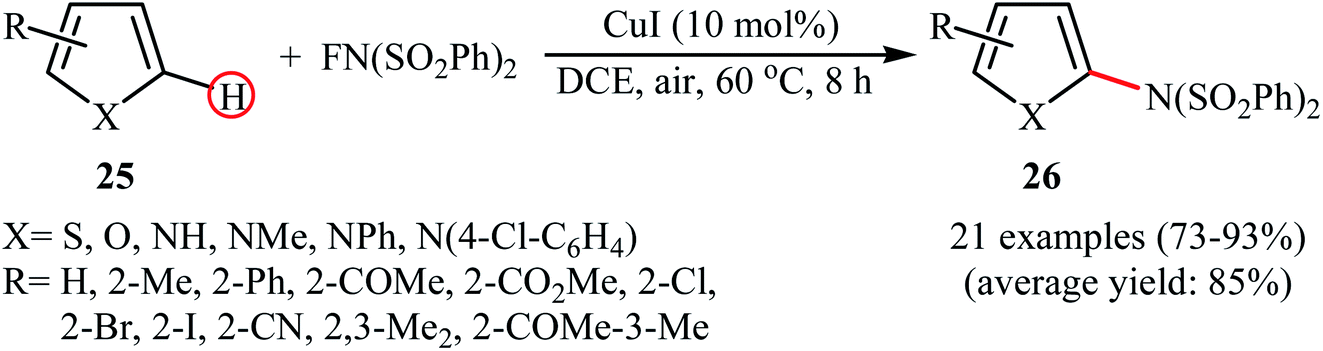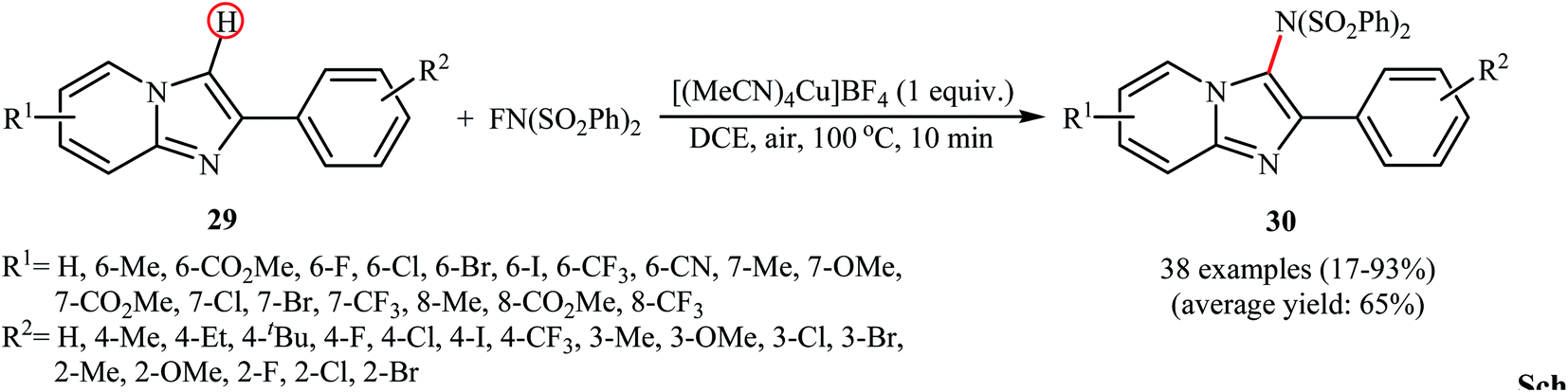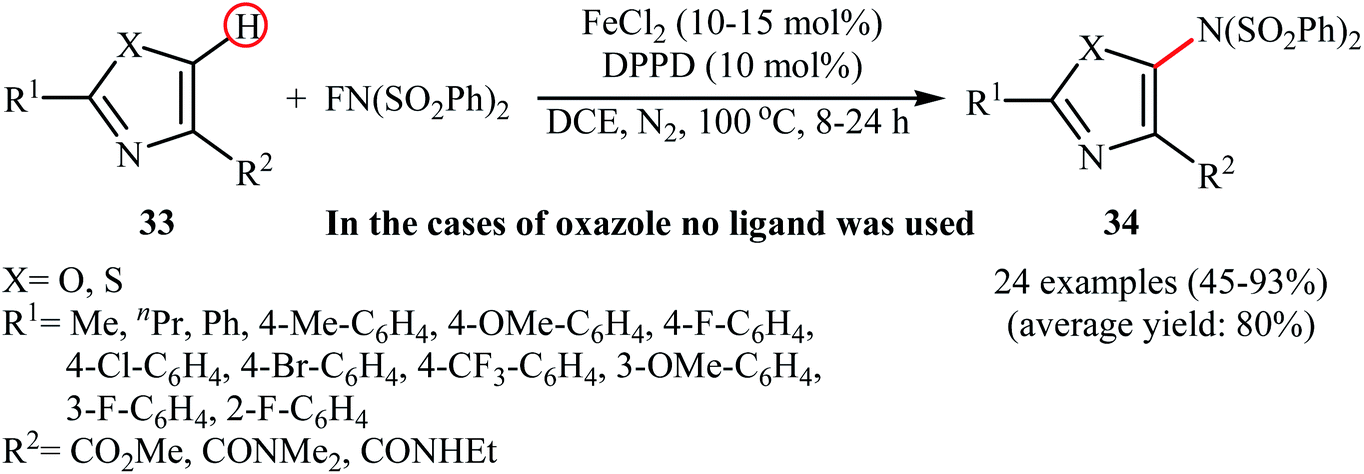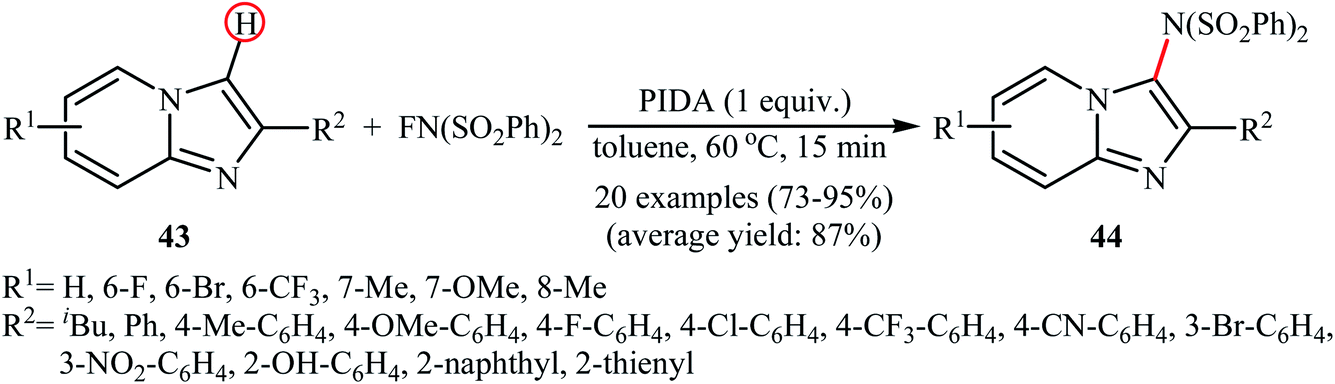DOI:
10.1039/D0RA00324G
(Review Article)
RSC Adv., 2020,
10, 16756-16768
N-Fluorobenzenesulfonimide: a useful and versatile reagent for the direct fluorination and amination of (hetero)aromatic C–H bonds
Received
12th January 2020
, Accepted 17th April 2020
First published on 29th April 2020
Abstract
This review updates recent advances and developments in the direct fluorination and amination of (hetero)aromatic C–H bonds utilizing N-fluorobenzenesulfonimide, classified according to the type of catalyst.
1. Introduction
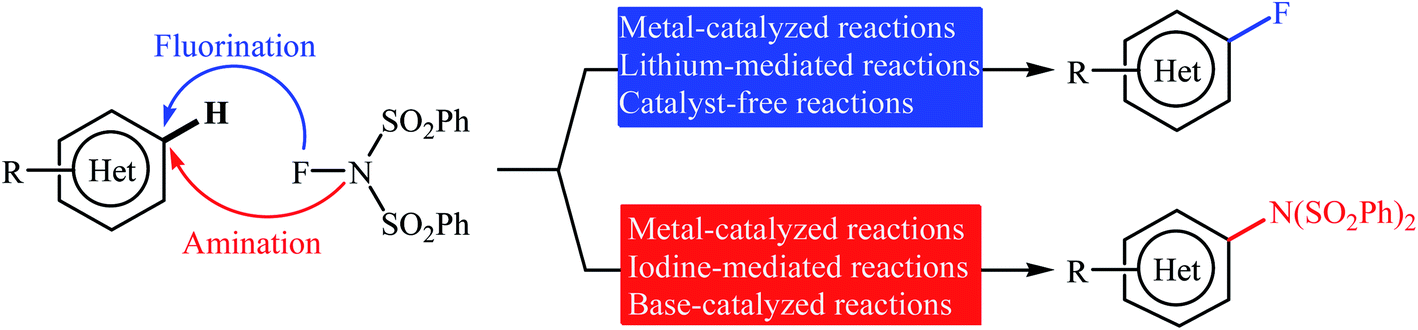
Needless to say, fluoro-organic compounds are prominent in all areas of the chemical sciences, particularly in agricultural,1 pharmaceutical,2 and materials chemistry.3 Interestingly, up to one third of all agrochemicals and one fourth of all pharmaceuticals on the market contain at least one fluorine atom in their structure.4 Among the various fluorine containing organic compounds, fluoroarenes (ring-fluorinated aromatics) are one of the most important structural motifs in medicinal chemistry that display a broad spectrum of biological and pharmacological activities including anticancer, antibacterial, antidepressant, anticonvulsant, antipsychotic, and antiemetic activities.5 As a result of the unique biological activities of fluoroarenes, considerable attention has been devoted toward their preparation.6 Arguably, direct fluorination of inert C(aryl)–H bonds in a regioselective manner is the best practical and economical method as it does not require any pre-functionalized starting materials. Traditionally, molecular fluorine (F2) was used as the sole source of fluorine in direct fluorination of (hetero)aromatic C–H bonds, but high toxicity and difficult handling of this gas limited its range of applications.7 As a result of these drawbacks, many alternative sources of electrophilic fluorine were developed. Those are HF,8 CF3OF,9 BF3,10 CsCoF4,11 XeF2 (ref. 12) and many more. However, most of these reagents, if not all, suffer from low stability, high toxicity and high corrosivity. In 1986, N-fluoropyridinium salts were introduced by Umemoto and his co-workers as the first electrophilic N–F fluorinating reagents.13 In comparison to the previous reagents, these compounds were more stable, safer, milder, easier to handle, cheaper, and selective sources of electrophilic fluorine.14 Now this class of fluorinating reagents encounters more than 100 representatives and many of them have been commercialized.15,16 N-Fluorobenzenesulfonimide (NFSI) is one of the most popular commercially available N–F reagents which was first introduced by Differding and co-workers in 1991.17,18 This mild, stable, and highly soluble crystalline solid was not only utilized as fluorination but also as amination reagent in various transformations of aliphatic and aromatic compounds.19 In continuation of our ongoing reviews on the synthesis of fluorine-containing organic compounds20 and direct functionalization reactions,21 herein we will highlight the most representative reports on the direct fluorination and amination of aromatic C–H bonds employing NFSI as a versatile fluorine and amine source until December 2019 (Fig. 1), by hoping that it will stimulate researchers for further thinking and serve as inspiration in their future work.
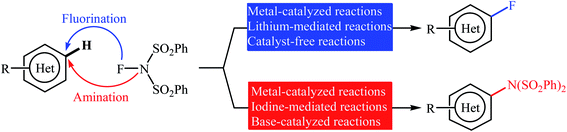 |
| | Fig. 1 Direct fluorination and amination of (hetero)aromatic C–H bonds with NFSI. | |
2. NFSI as the fluorine source
This section provides a brief overview of the direct construction of Caryl–F bonds using NFSI as the fluorine source. For clarity, the reactions are classified by the type of catalysts.
2.1 Transition-metal-catalyzed reactions
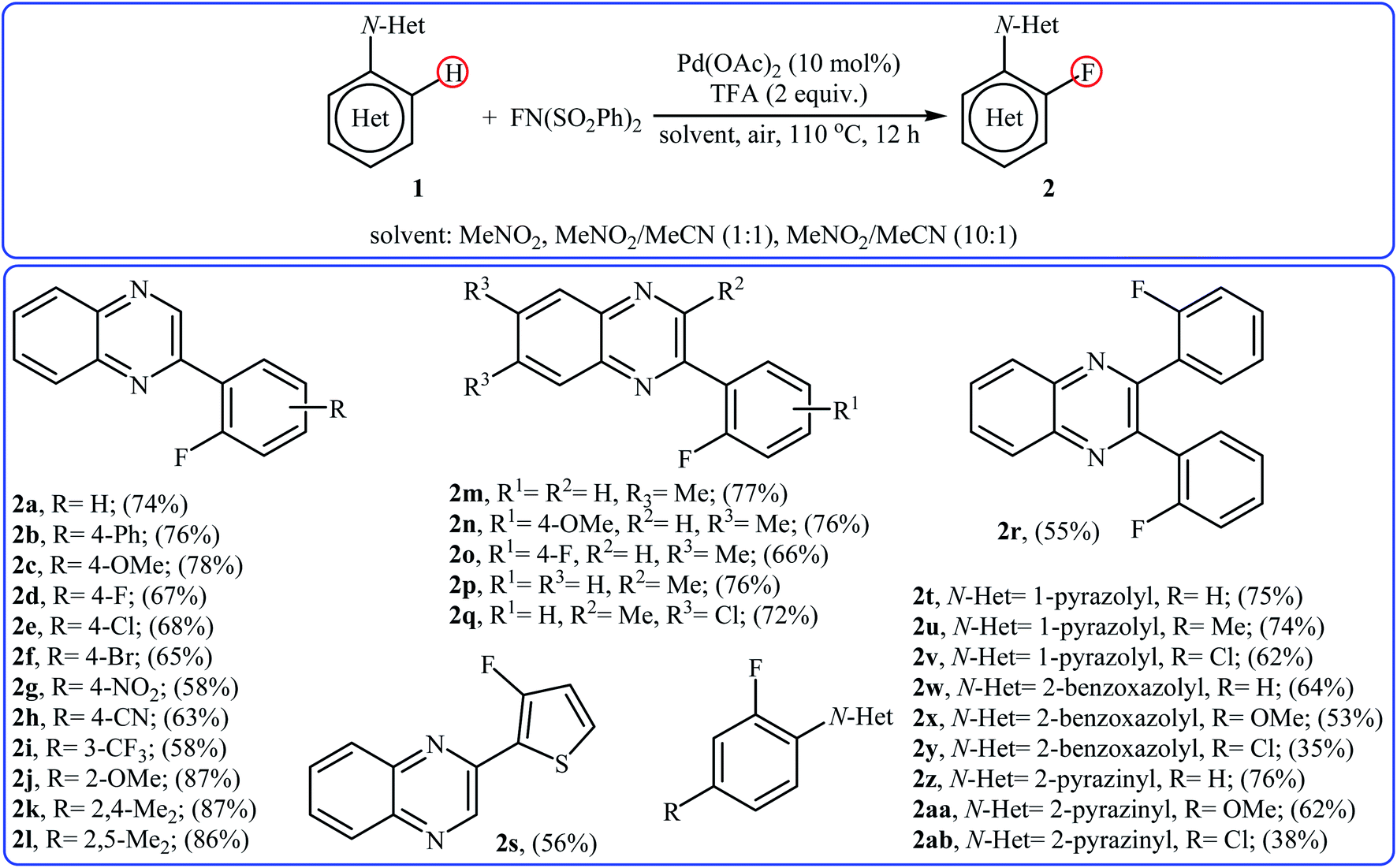
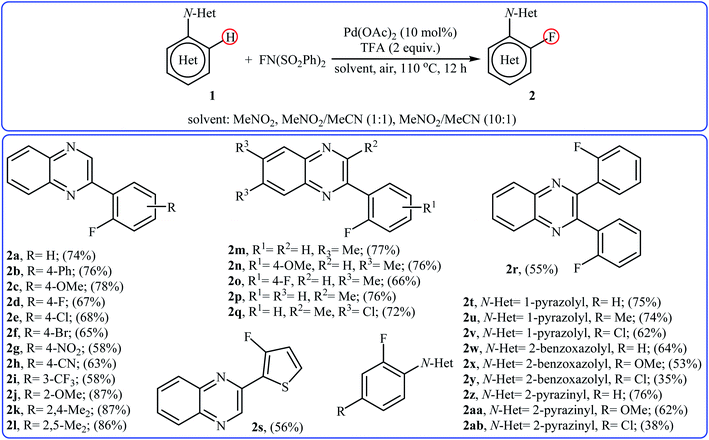
2.1.1 Palladium. In 2013, Xu and co-workers disclosed for the first time the usefulness of palladium catalysts for the regioselective C–H bond fluorination of (hetero)aromatic compounds with NFSI.22 Thus, in the presence of Pd(OAc)2/TFA (trifluoroacetic acid) combination as a catalytic system in a mixed solvent of MeNO2 and MeCN (with different ratio) at 110 °C, fluorination of (hetero)arenes 1 that bear N-heterocyclic directing groups (e.g., quinoxaline, pyrazole, benzo[d]oxazole, and pyrazine) with NFSI furnished the corresponding ortho-monofluorinated products 2 in moderate to good yields (Scheme 1). The results demonstrated that the substrates bearing electron-donating groups were relatively more favorable in this transformation and the relative activation rates of directing groups followed the order: quinoxaline ≈ pyrazole > pyrazine > benzo[d]oxazole. The authors also were able to synthesize a difluorinated product in high yield by utilizing 3 equiv. of NFSI under the standard conditions. It should be mentioned that other electrophilic N–F fluorinating reagents like selectfluor, N-fluoropyridinium triflate, and N-fluoropyridinium tetrafluoroborate are not effective to afford the target products by this reaction.
 |
| | Scheme 1 Pd-catalyzed regioselective C–H bond fluorination of (hetero)arenes 1 with NFSI. | |
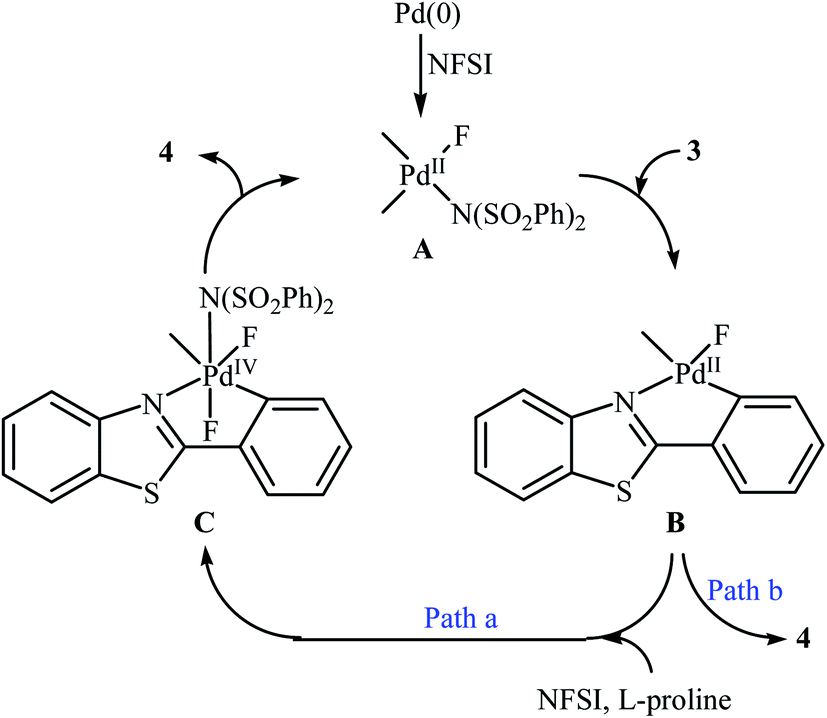
Subsequently, the group of Pu demonstrated an efficient palladium-catalyzed methodology for the regioselective ortho-C–H fluorination of 2-arylbenzo[d]thiazoles using NFSI as the F+ source and L-proline as the crucial promoter.23 After screening various palladium complexes such as Pd(OAc)2, Pd2(dba)3, Pd(PPh3)4, PdCl2, Pd(PPh3)2Cl2, Pd(PhCN)2Cl2; Pd(PPh3)4 was found to be more effective catalyst for this transformation. Solvent has a dramatic effect on this process and among the several common organic solvents (e.g., DCM, DCE, DMF, DMA, DMSO, MeCN, PhCl, dioxane, cyclohexane, toluene); cyclohexane was the most effective solvent. Under the optimized reaction conditions, various 2-arylbenzo[d]thiazoles 3 underwent ortho-selective mono-fluorination to give the corresponding fluorinated products 4 in almost fair to good yields (Scheme 2). 2-(Furan-2-yl)benzo[d]thiazole did not take part in the reaction and therefore no other 2-heteroarylbenzo[d]thiazoles were examined in this fluorination strategy. Moreover, nitro-substituted arenes were incompatible in this protocol. Of note, the reaction of 2-arylbenzo[d]thiazoles without substituents at the ortho-positions offered a mixture of mono- and di-fluorinated products. The mechanism of this interesting reaction was proposed to initiate with the formation of Pd(II) fluoride complex A through oxidation of Pd(0) by NFSI which is followed by C–H activation to form the palladacycle intermediate B. Oxidative addition of intermediate B by NFSI in the presence of L-proline provides the Pd(IV) intermediate C. Subsequent reductive elimination results in the target fluorination product 4 (Path a, Scheme 3). In another possibility, reductive elimination of intermediate B affords the final product 4, which involves a Pd(0)/Pd(II) mechanism (Path b, Scheme 3).
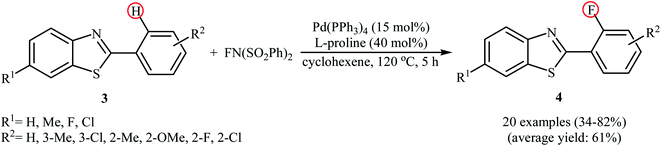 |
| | Scheme 2 Pd-catalyzed direct ortho-fluorination of 2-arylbenzothiazoles 3 with NFSI reported by Pu. | |
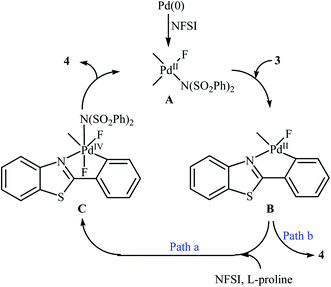 |
| | Scheme 3 Plausible mechanism for the ortho-selective mono-fluorination of 2-arylbenzo[d]thiazoles 3 with NFSI. | |
In a related investigation, Lessard and Hierso along with their co-workers informed a Pd-catalyzed regioselective ortho-C–H fluorination of arylpyrazoles 5 with NFSI.24 Low-cost and readily available Pd(OAc)2 appeared to be the most effective catalyst and compared to other solvents (DCE, DMF, Ph(CF3)2, MeCN, EtOAc), trifluoromethylbenzene (PhCF3) was the best choice for the conversion. The process was performed under an inert atmosphere in the absence of any additive or ligand, tolerated various electron-withdrawing and -donating functionalities in the both phenyl and pyrazole rings periphery, and provided the desired ortho-fluorinated products 6 in low to excellent yields (Scheme 4). However, the substitution pattern on the pyrazole directing group strongly influenced the yield of the transformation. The results revealed that the reaction was strongly dependent on the substitution pattern on the pyrazole directing groups. While 2-(4-methyl-1H-pyrazol-1-yl)benzonitrile gave the desired products in 76% yield, its 3-methyl-substituted analog afford the product in only 22% yield and 2-(5-methyl-1H-pyrazol-1-yl)benzonitrile failed to undergo the fluorination. Drawing inspiration from these works, Li's research team developed a method for the C–H fluorination of arenes on the use of oxazoline as a removable directing group in the presence of Pd(NO3)2/AgNO3 combination as a catalytic system.25 This fluorination strategy is effective at relatively high temperature (80 °C) to afford the target ortho-fluorinated products in 13–100% yields.
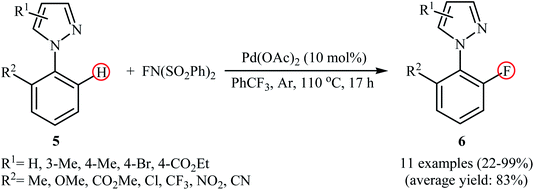 |
| | Scheme 4 Pd-catalyzed pyrazole-directed C–H fluorination of ortho-substituted arenes 5 with NFSI. | |
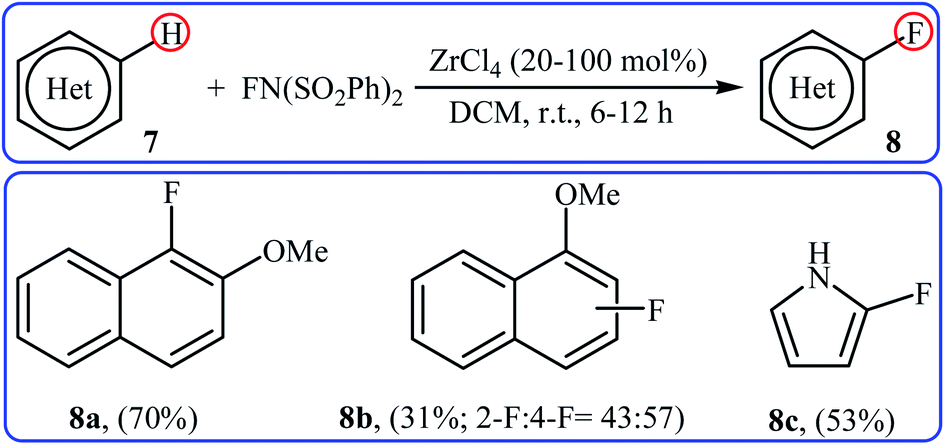
2.1.2 Zirconium. In 2005, Yamamoto and co-workers demonstrated an efficient methodology for directed C–H fluorination of (hetero)arenes 7 under zirconium-catalyzed conditions (Scheme 5).26 NFSI was employed as the fluorine source and ZrCl4 was found to be the most effective Lewis acid catalyst. The reactions were done in DCM at room temperature without consuming any additional chemical and provided the corresponding mono-fluorinated products 8 in moderate to good yields. However, only three examples were demonstrated for such fluorination process. The protocol was also compatible for chlorination, bromination, and iodination reactions when NCS, NBS, and NIS were used, respectively.
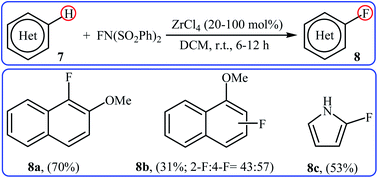 |
| | Scheme 5 Fluorination of arenes 7 with NFSI catalyzed by ZrCl4. | |
2.1.3 Nickel. In 2017, Ding, Zhang, and Li demonstrated an interesting Ni-catalyzed fluorination of 8-amidoquinolines in a highly regioselective manner at the C5 position by using NFSI as the oxidant and fluorine source.27 The method only made use of NiSO4 and neither additive nor ligand was required for this transformation. As shown in the Scheme 6, a variety of quinolines 9 containing aromatic, heteroaromatic, aliphatic, long-chain, and sterically bulky amides were applied to afford the corresponding 5-fluorinated quinolines 10 with yield ranging from 32% to 46%. The results revealed that the presence of free N–H of the amide and the N atom in the quinoline skeleton were indispensable for this transformation. The authors showed that the amido groups could be efficiently converted to the free amino (–NH2) group by hydrolysis under alkaline conditions without damaging the C–F bond. The control experiments testified that this fluorination reaction underwent a free radical cross-coupling pathway. To the best of our awareness, this is the first and only example in which a nickel catalyst has been used in direct fluorination of C(aryl)–H bonds using NFSI.
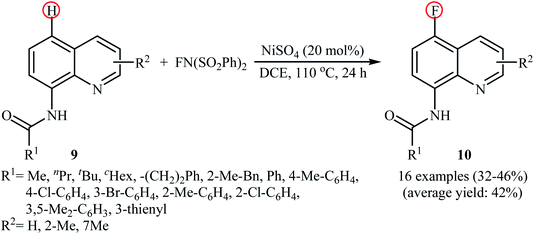 |
| | Scheme 6 Ni-catalyzed C5-selective fluorination of 8-amidoquinolines 9 with NFSI. | |
2.2 Transition-metal-free reactions
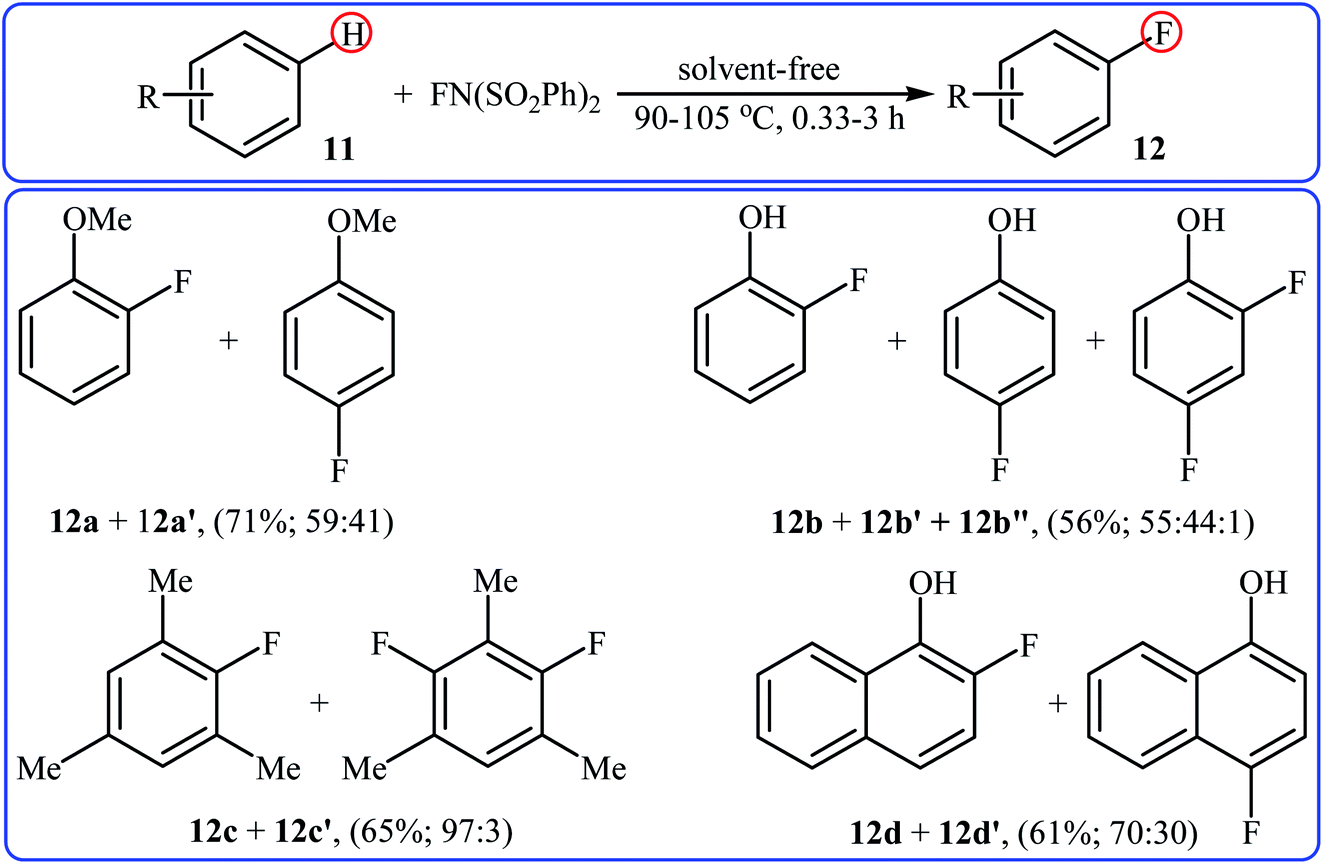
After pioneering works by Kirk and colleagues on tBuLi-mediated regioselective C4-fluorination of (5-methoxy-indol-3-yl)-N,N-dimethylmethanamine with NFSI28 and O'Neill's research group on catalyst-free C5-fluorination of 6-methoxy-8-nitroquinoline,29 the first general report of the transition-metal-free fluorination of aromatic C–H bonds using NFSI as a fluorinating reagent was published in 2009 by Andreev et al.30 They showed that in the absence of any catalyst or additive, the reaction of a variety of electron-rich arenes 11, including anisoles, phenols, toluenes, and naphthalenes with NFSI under solvent-free conditions furnished the mono-fluorinated-products 12 in poor to good yields. However, the fluorinations occurred not only on one C–H bond but resulted in mixtures of regioisometric mono-fluorides in some case, e.g. for m-xylene and 1-naphthol. Moreover, selectivity for mono- versus di-fluorination could not be controlled for substrates without ortho and para substituents. Product yields were found to be highly time-dependent, increased reaction duration resulted in increased products yield but the ratio of products almost did not change. Some reported examples are shown in the Scheme 7. Later on, an extension of the substrate scope to include aromatic amines was investigated by Ozeryanskii and co-workers.16 Upon treatment with 1.0 equiv. of NFSI in refluxing MeCN, a small series of N,N-dimethylaniline derivatives underwent C–H fluorination to give a mixture of 2-fluoro and 4-fluoro derivatives in unsatisfactory yields along with the corresponding biaryls, biarylmethanes and N-demethylated products.
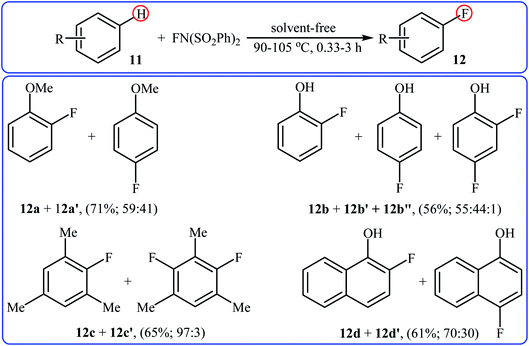 |
| | Scheme 7 Selected examples of catalyst- and solvent-free fluorination of arenes 11 with NFSI reported by Andreev. | |
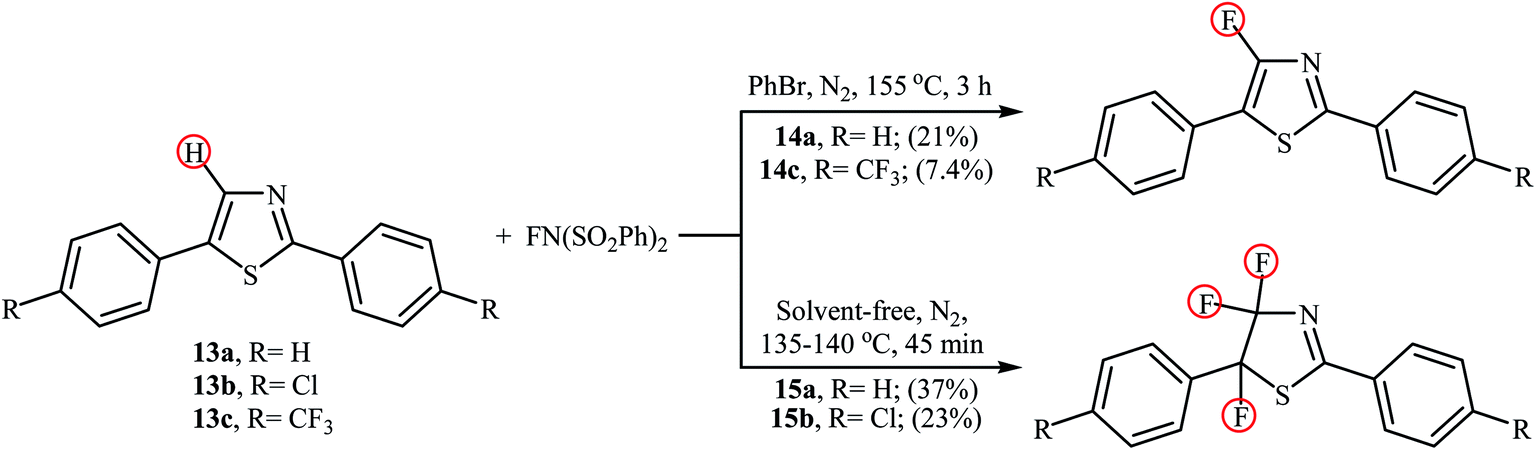
In 2013, the group of Stephens further expanded the catalyst-free aromatic C–H bond fluorination to thiazoles.31 Thus, upon reaction of 2,5-diphenylthiazoles 13 with NFSI in refluxing bromobenzene (PhBr), the corresponding mono-fluorinated products 14 were obtained in poor yields within 3 h (Scheme 8). Interestingly, when the reactions were performed under solvent-free conditions, the parent 4,4,5-trifluorothiazoles 15 were formed after just 45 min by heating at 135–140 °C. Of note, fairly dilute conditions were required for mono-fluorination reactions.
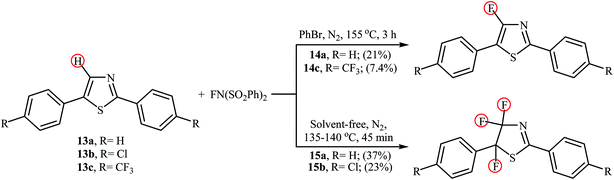 |
| | Scheme 8 Mono- and trifluorination of thiazoles 13 using NFSI. | |
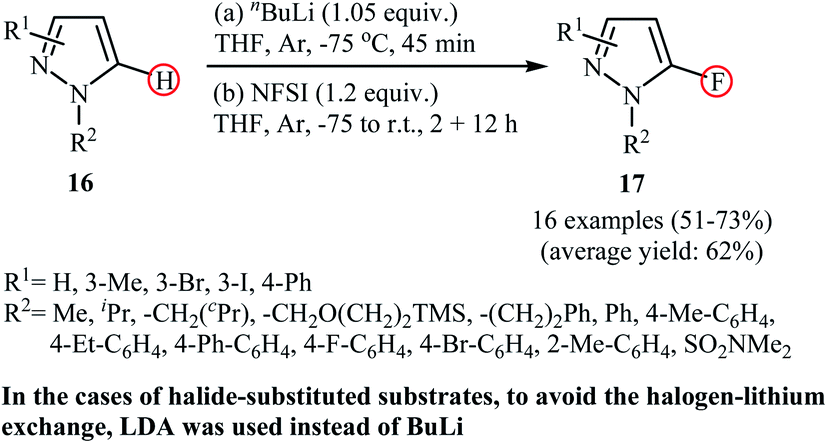
Following these works, Mykhailiuk et al. reported a one-pot synthesis of biologically important 5-fluoropyrazoles 17 by direct lithiation/fluorination of the corresponding pyrazoles 16 employing nBuLi as the lithium and NFSI as the fluorine source.32 The method was general and, as shown in Scheme 9, under the optimized conditions, various N-aryl and N-alkyl substituted pyrazole derivatives were selectively fluorinated at the C5-position with moderate to good yields. However, N-alkyl pyrazoles bearing sterically bulky substituents (e.g., N-tert-butyl-pyrazole) failed to give the desired fluorinated products. Furthermore, N-aryl substituted pyrazoles containing substituents on the meta-position of the aryl ring (e.g., N-(3-bromophenyl)-pyrazole) underwent fluorination at the C2-position of the aryl ring instead of C5-position of the pyrazole core. The authors nicely showed the synthetic application of their methodology for the high yielding preparation of the commercialized fungicide Penflufen. In addition, a large-scale (20 g) reaction was also successfully performed.
 |
| | Scheme 9 Mykhailiuk's synthesis of 5-fluoropyrazoles 17. | |
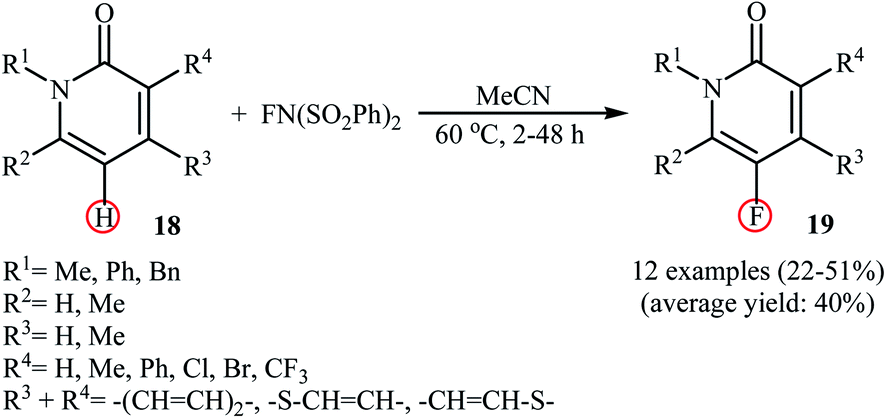
Very recently, Borodkin and co-workers found that synthesis of C5-fluorinated naproxen through the direct C–H fluorination of naproxen, an anti-inflammatory drug, with NFSI is possible even in the absence of any additional catalyst or additive.33 Thus, treatment of naproxen with 1.1 equiv. of NFSI in MeCN at 80 °C afforded 2-(5-fluoro-6-methoxynaphthalen-2-yl)propionic acid in 62% yield along with an small amount of 2-(5,5-difluoro-6-oxo-5,6-dihydronaphthalen-2-yl)propionic acid side product. In a closely related study, Sakurai's research team reported the direct monofluorination of N-protected pyridone derivatives 18 at the C5-position with NFSI under catalyst-free conditions.34 The reactions were carried out in MeCN at 60 °C, tolerated various N-alkyl, -benzyl, and -aryl-substituted mono- and bi-cyclic 2-pyridones, and afforded the desired C5-fluorinated products 19 in moderate yields (Scheme 10). However, NH-free pyridones did not furnish any product under the standard reaction condition. Unfortunately, the authors do not propose a reaction mechanism for the transformation.
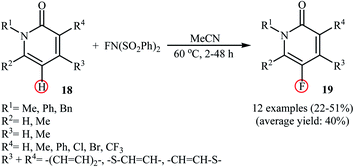 |
| | Scheme 10 Regioselective monofluorination of N-protected pyridones 18 with NFSI under catalyst-free conditions. | |
3. NFSI as the amine source
Although the direct amination of Calkyl–H bonds with NFSI has been the subject of a number of papers in recent years, much less reports are available for the amination of Caryl–H bonds by this reagent. In 2014, in a review article entitled “N-fluorobenzenesulfonimide: an efficient nitrogen source for C–N bond formation”, Li and Zhang19 nicely highlighted the strength of this synthetic process for the construction of Caryl–N bonds, albeit in some small paragraphs. Since a number of advances in the direct amination of (hetero)aromatic C–H bonds utilizing the titled reagent have occurred over the past few years, a comprehensive review on this novel and fast-growing research topic seems to be timely. In this section, we will try to summarize findings and developments on the direct C–H amination of (hetero)aromatic compounds with NFSI with special attention to the mechanistic aspects of the reactions.
3.1 Transition-metal-catalyzed reactions
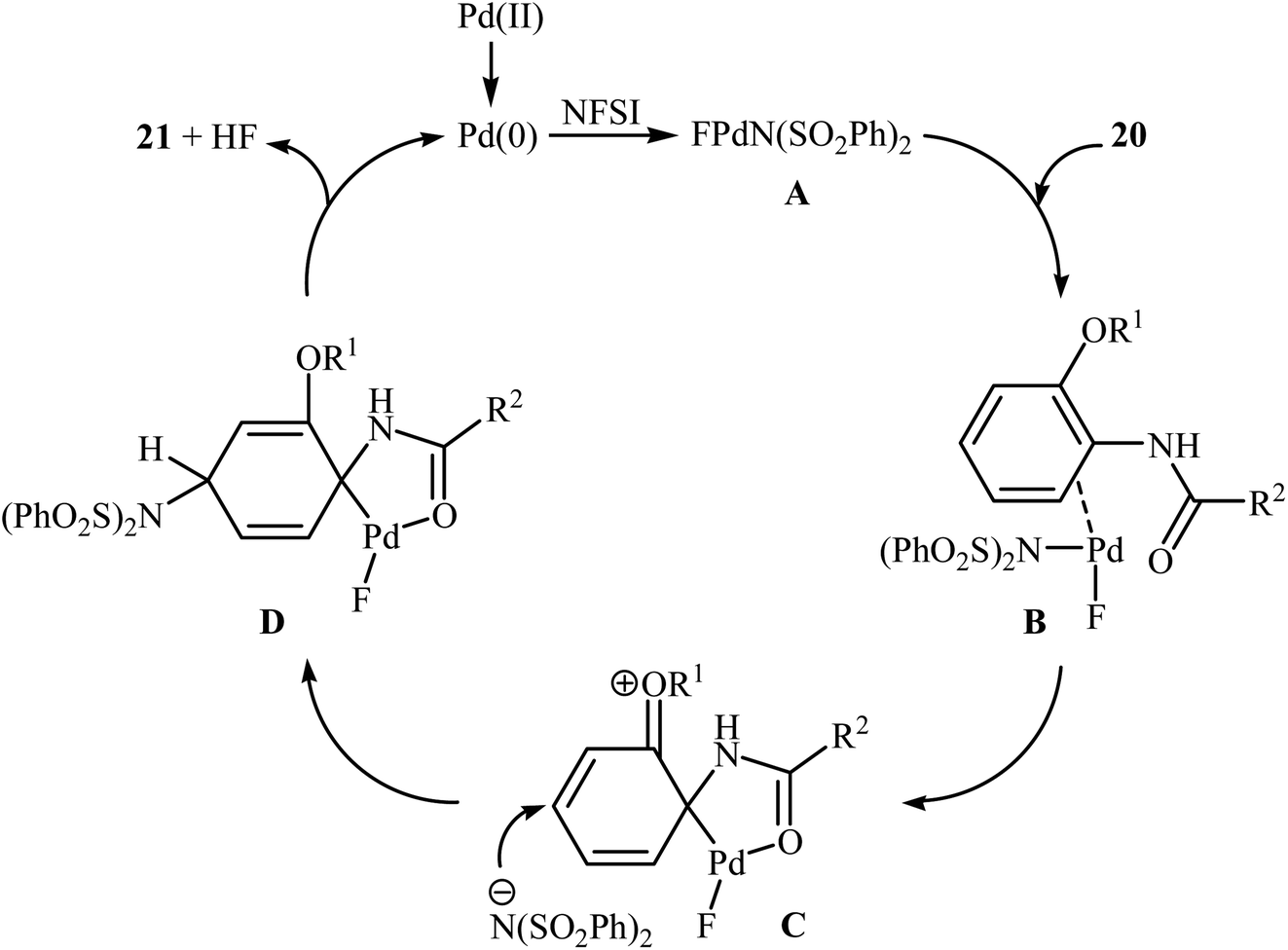
3.1.1 Palladium. The first example of the C–N bond forming reactions through palladium-catalyzed direct amination of aromatic C–H bonds employing NFSI as an aminating agent has been reported by Zhang and co-workers in 2011, when a range of 2-OR-substituted anilides 20 underwent para-selective direct C–H amination with NFSI in the presence of 10 mol% of Pd(OAc)2 as a catalyst and 2.0 equiv., of NaHCO3 as an additive in DCE to form the corresponding C(sp2)–N bearing adducts 21 in moderate to excellent yields (Scheme 11).35 Interestingly, when para-substituted substrates were applied under the standard conditions, the ortho-amination products were obtained as sole products. In this investigation, the authors reported some limitation in their protocol when they tested strongly electron-withdrawing and alkyl-substituted substrates. In these cases, no desired product was formed. Mechanistic studies suggested that radical intermediates were not involved in this reaction. In Scheme 12, a reasonable mechanistic pathway for this C–N bond formation reaction is proposed by the authors. It consists of the following steps: (i) oxidative addition of Pd(0) to the N–F bond of NFSI to form intermediate A; (ii) coordination of the Pd(II) complex A to the anilide 20 to provide intermediate B; (iii) electrophilic palladation of intermediate B to give dearomatized spiro-palladacycle intermediate C; (v) amination of intermediate C with –N(SO2Ph)2 to form intermediate D; and (vi) hydrogen elimination of intermediate D to give the target product 21.
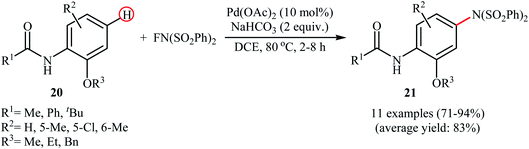 |
| | Scheme 11 Pd-catalyzed p-C–H amination of anilides 20 with NFSI. | |
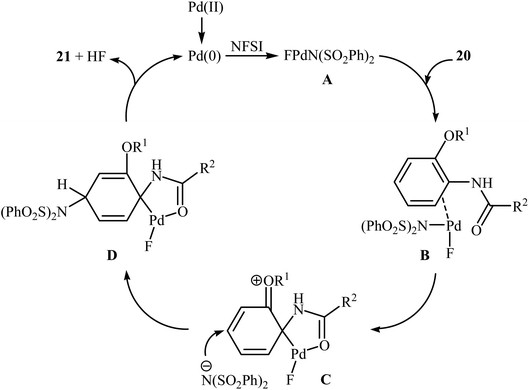 |
| | Scheme 12 Mechanistic proposal for the formation of 21. | |
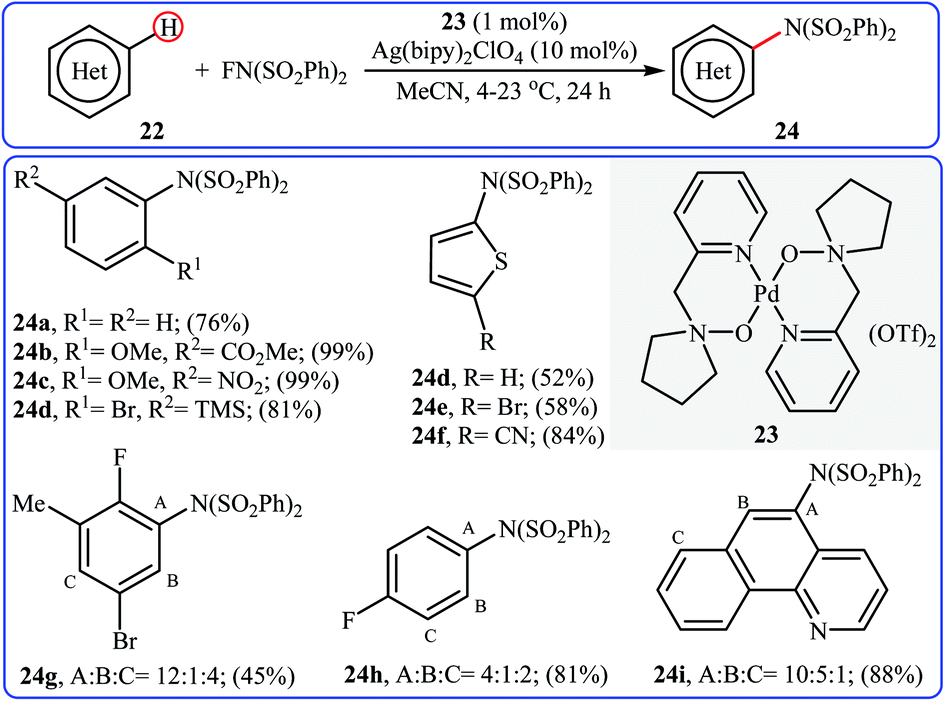
Inspired by this work, Ritter and colleagues devised a novel palladium-based catalytic system enabling direct C–H amination of a diverse range of arenes 22, including N- and S-heteroarenes, with NFSI at or below room temperature.36 Thus, by using Pd(II) complex 23 bearing 1-(pyridin-2-ylmethyl)pyrrolidine 1-oxide as the catalyst, in conjunction with Ag(bipy)2ClO4 as the co-catalyst in MeCN, the desired mono-aminated products 23 were obtained in moderate to quantitative yields without employing directing groups. Although amination preferably occurred at the most acidic C–H bond of arenes, in some cases a mixture of possible products was obtained. Some of the reported examples are shown in Scheme 13.
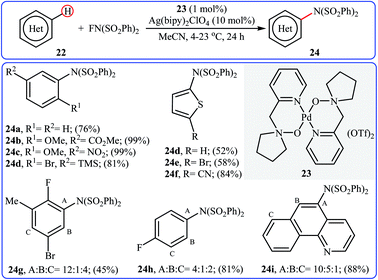 |
| | Scheme 13 Selected examples of amine-N-oxide-ligated palladium complex-catalyzed amination of arenes 22 with NFSI. | |
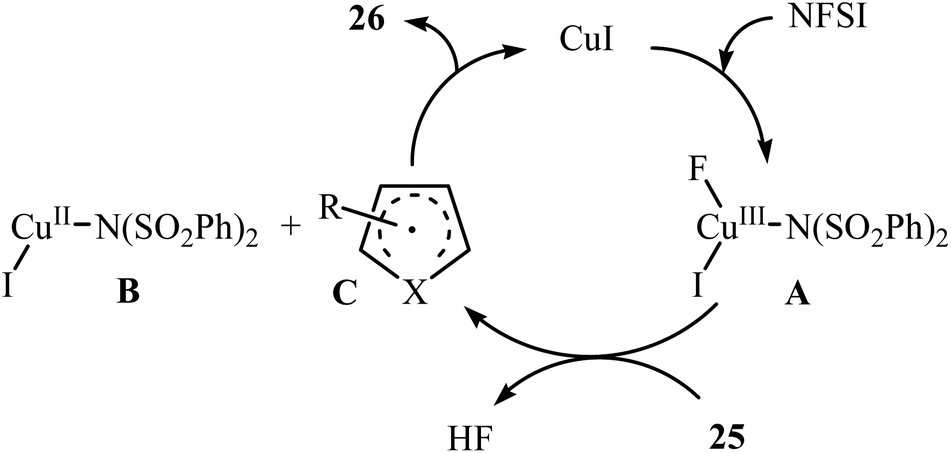
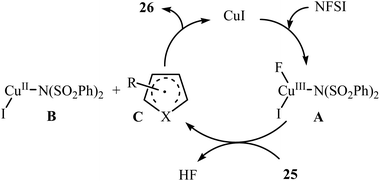
3.1.2 Copper. In 2014, Pan's research group communicated the first example of copper-catalyzed direct amination of aromatic C–H bonds with NFSI.37 By employing thiophene as the model substrate, the reaction variables such as catalyst, solvent, and temperature were carefully investigated. The best conversion efficiency was obtained for the reaction performed in the presence of 5 mol% of CuI in DCE at 60 °C. Under the optimized condition, various five-membered heterocycles 25 (pyrrole, thiophene and furans) reacted well and afforded the corresponding α-aminated heterocycles 26 in good to excellent yields (Scheme 14). The reaction tolerated a variety of functional groups such as chloro, bromo, iodo, cyano, ester, and ketone functionalities and promised its potential applications for further modifications of the end products. However, this procedure was not applicable for formyl- and methoxy-substituted substrates. The following mechanistic pathway was proposed by the authors for this C–N bond forming reaction (Scheme 15): initially, NFSI oxidizes CuI to form a Cu(III) species A. Subsequently, the reaction of this intermediate with heterocycle 25 gives the heterocycle radical intermediate C and the Cu(II) species B. Finally, the oxidation of heterocycle radical C by the Cu(II) species B affords the amination product 26.
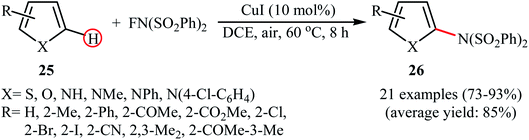 |
| | Scheme 14 α-Selective direct amination of five-membered heterocycles 25 with NFSI catalyzed by CuI. | |
 |
| | Scheme 15 Suggested mechanism for the formation of α-aminated heterocycles 26. | |
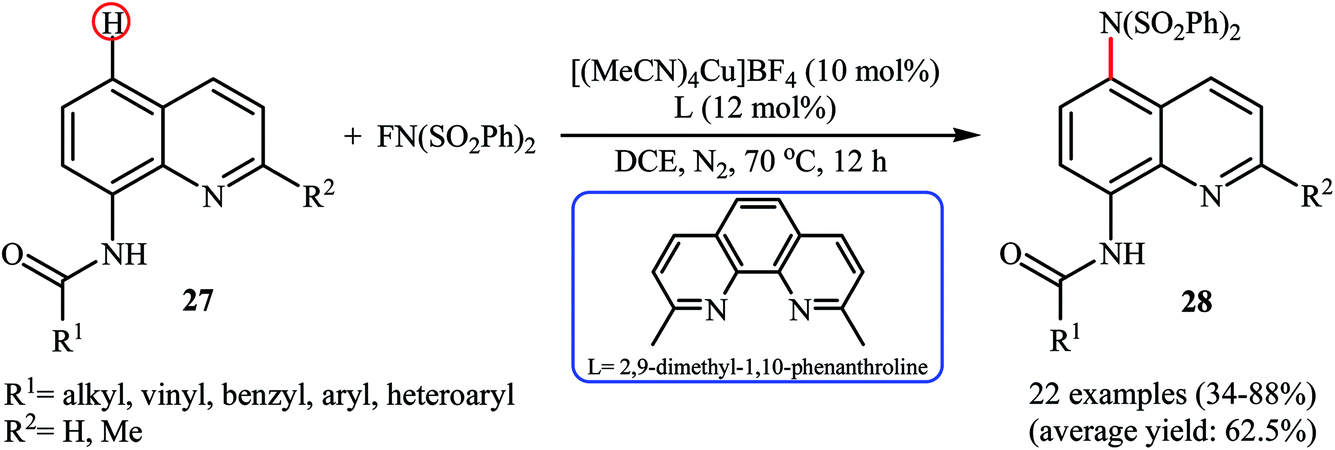
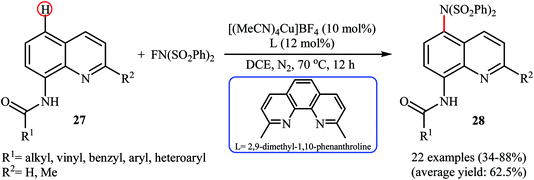
Subsequently, Zhang and colleagues broadened the applicability of Cu-catalyzed amination by developing a catalytic system ([(MeCN)4Cu]BF4, 2,9-dimethyl-1,10-phenanthroline) which allowed for the site-selective amination at the C5 position of 8-aminoquinoline derivatives 27 with NFSI under a nitrogen atmosphere (Scheme 16).38 Various N-(quinolin-8-yl) alkyl-, vinyl-, aryl-, and heteroaryl-amides were used to establish the general applicability of the protocol. It should be mentioned that the amination of quinolin-8-amine and N-methylquinolin-8-amine with NFSI failed to give any aminated quinoline products under the standard conditions. Moreover, the reaction did not occur when the quinoline ring was changed into naphthalene ring. Mechanistically, a free-radical process was likely involved in this reaction, which could be inhibited by 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) or 2,6-di-tert-butyl-4-methylphenol (BHT), radical scavengers. In a related investigation, Weng and Lu along with their co-workers disclosed that N-(naphthalen-1-yl)picolinamide can undergo selective amination with NFSI in the presence of Cu(OAc)2 + H2O/PhI(OAc)2 combination as a catalytic system under the open air to afford the C4-aminated product in 55% yield.39
 |
| | Scheme 16 Cu-catalyzed C5-amination of 8-aminoquinoline derivatives 27 with NFSI. | |
Recently, the group of Song explored the copper-mediated direct C3-amination of 2-aryl imidazopyridine derivatives 29 with NFSI using stoichiometric amounts of [(MeCN)4Cu]BF4 under the open air and without consuming any additive.40 This methodology efficiently provided the target C3-aminated imidazopyridines 30 in fair to excellent yields within 10 min (Scheme 17). Depending on the electronic and steric effects of substituents on the aryl ring, substrates bearing an electron-poor aryl ring gave higher yields compared to the substrates containing an electron-rich aryl ring, and para-substituted aryl ring containing substrates were more reactive than meta- and ortho-substituted ones. The results also proved that electron-donating groups (except OMe) in the pyridine ring periphery of the substrates led to higher yields than those with electron-withdrawing groups. The authors showed that the synthesized compounds could be easily converted to the corresponding monodesulfonylated products by heating (60 °C) in alkaline methanol through a reductive desulfonylation process.
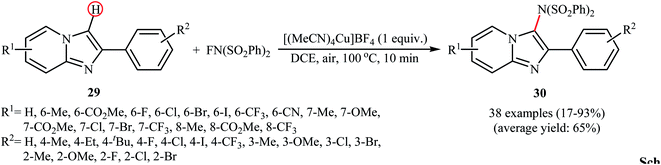 |
| | Scheme 17 Song's synthesis of C3-aminated imidazopyridines 30. | |
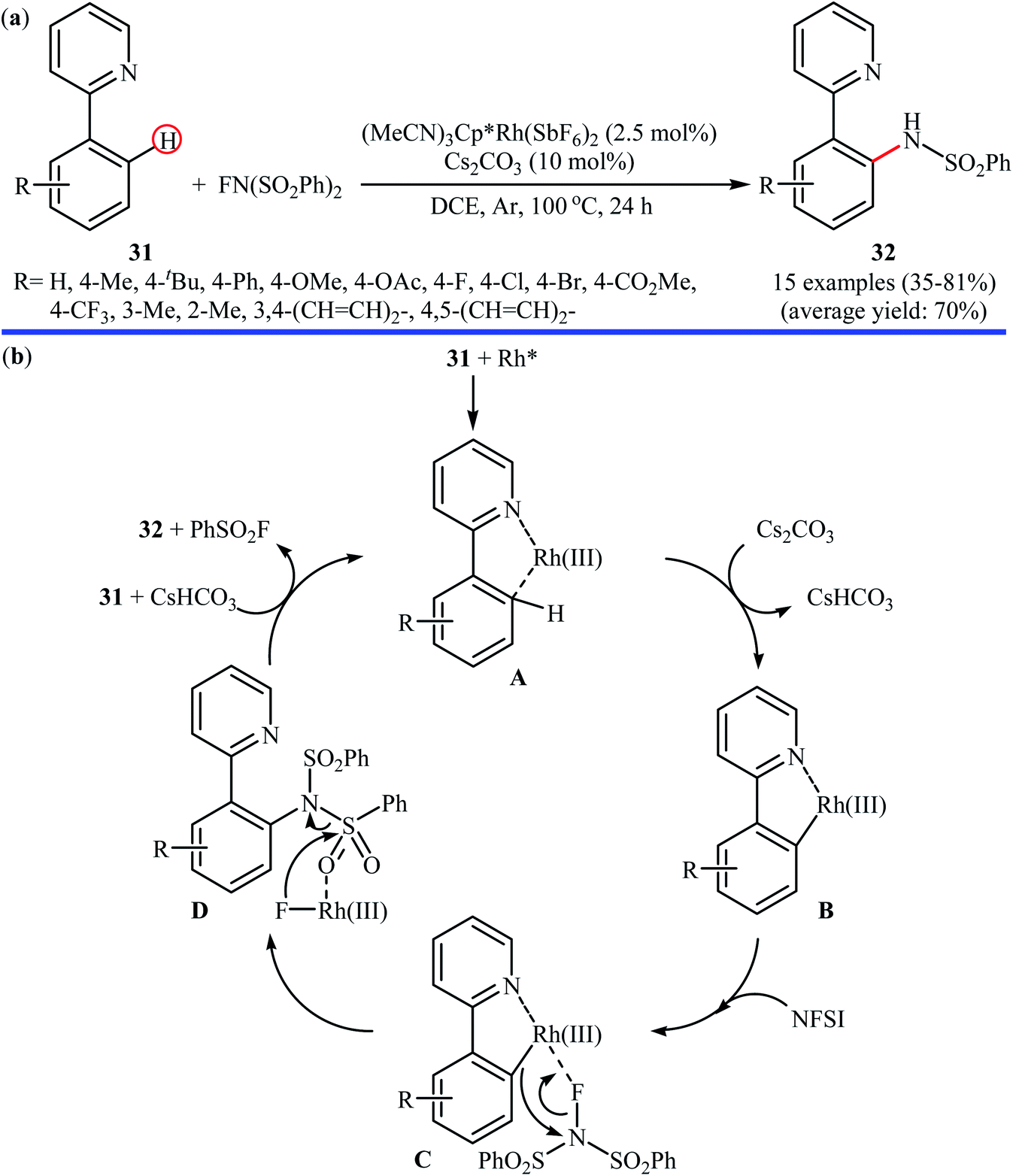
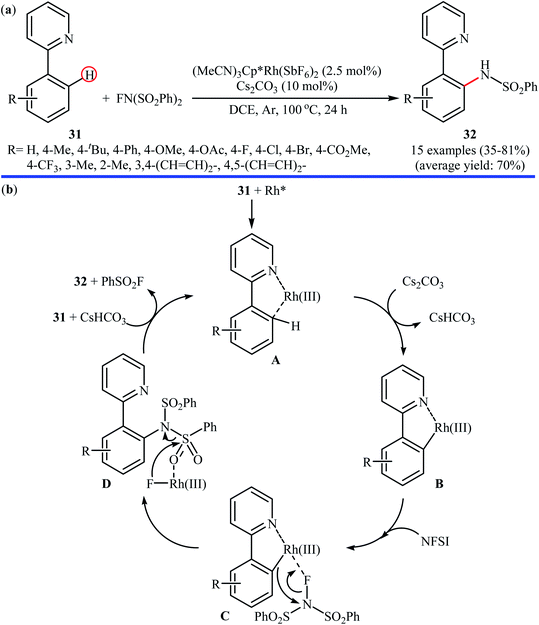
3.1.3 Rhodium. Rhodium-catalyzed direct amination of aromatic C–H bonds with NFSI has been scarcely explored; indeed, only one example of such a reaction was published in the literature till now. In 2013, Yang and Li along with their co-workers revealed that the treatment of arene substrates bearing pyridinyl directing group 31 with NFSI in the presence of only 2.5 mol% of (MeCN)3Cp*Rh(SbF6)2 as a catalyst and 10 mol% of Cs2CO3 as base in DCE afforded the corresponding ortho-sulfonamidated arenes 32 in good to high yields (Scheme 18a).41 The reaction tolerated various functional groups such as halides, ethers, and esters functionalities, and promised its potential applications in the further synthetic transformations. Other coordinating directing groups such as pyrimidinyl and pyrazolyl were also found to effectively mediate the direct and ortho-selective mono-sulfonamidation of the C(aryl)–H bonds under this condition, albeit the expected products were obtained in lower yields. The main drawback of the methodology is the requirement for an inert atmosphere (Ar) and an elevated reaction temperature (100 °C), which may limit its range of applications more or less. The authors suggested catalytic cycle for this transformation is displayed in Scheme 18b.
 |
| | Scheme 18 (a) Rh(III)-catalyzed direct amination of arenes 31 with NFSI reported by Yang and Li; (b) mechanism that accounts for the formation of 32. | |
3.1.4 Iron. The only example of utilizing an iron salt as a catalyst in direct amination of aromatic C–H bonds with NFSI was presented by Li et al. in 2017.42 To evaluate the activity of different metal catalysts and ligands, methyl 2-phenylthiazole-4-carboxylate was chosen as the model substrate. Among the various catalysts such as Pd(OAc)2, Cu(OAc)2, Cu(TFA)2, and FeCl2; FeCl2 was found to be more effective catalyst, which gave a better yield of aminated product. The ligands such as 1,10-phenanthroline, 2-(pyridin-2-yl)pyridine, benzil, and 1,3-diphenylpropane-1,3-dione were examined and a good yield of product was obtained when using 10 mol% of 1,3-diphenylpropane-1,3-dione (DPPD) as a ligand. Under optimized conditions, various 2,4-disubstituted azoles 33 reacted efficiently with NFSI to give the corresponding C5-aminated azoles 34 in moderate to excellent yields (Scheme 19). Competition experiments between electron-rich and electron-deficient azoles indicated that the electron-rich substrates reacted preferentially. Noteworthy, the reaction can be enlarged to gram scale (10 mmol) without a detrimental decrease in yield.
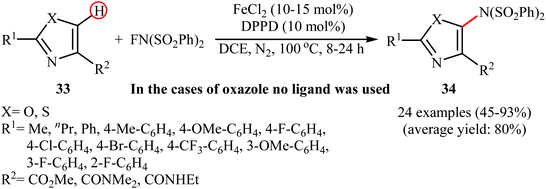 |
| | Scheme 19 Fe-catalyzed C5-amination of azoles 33 with NFSI. | |
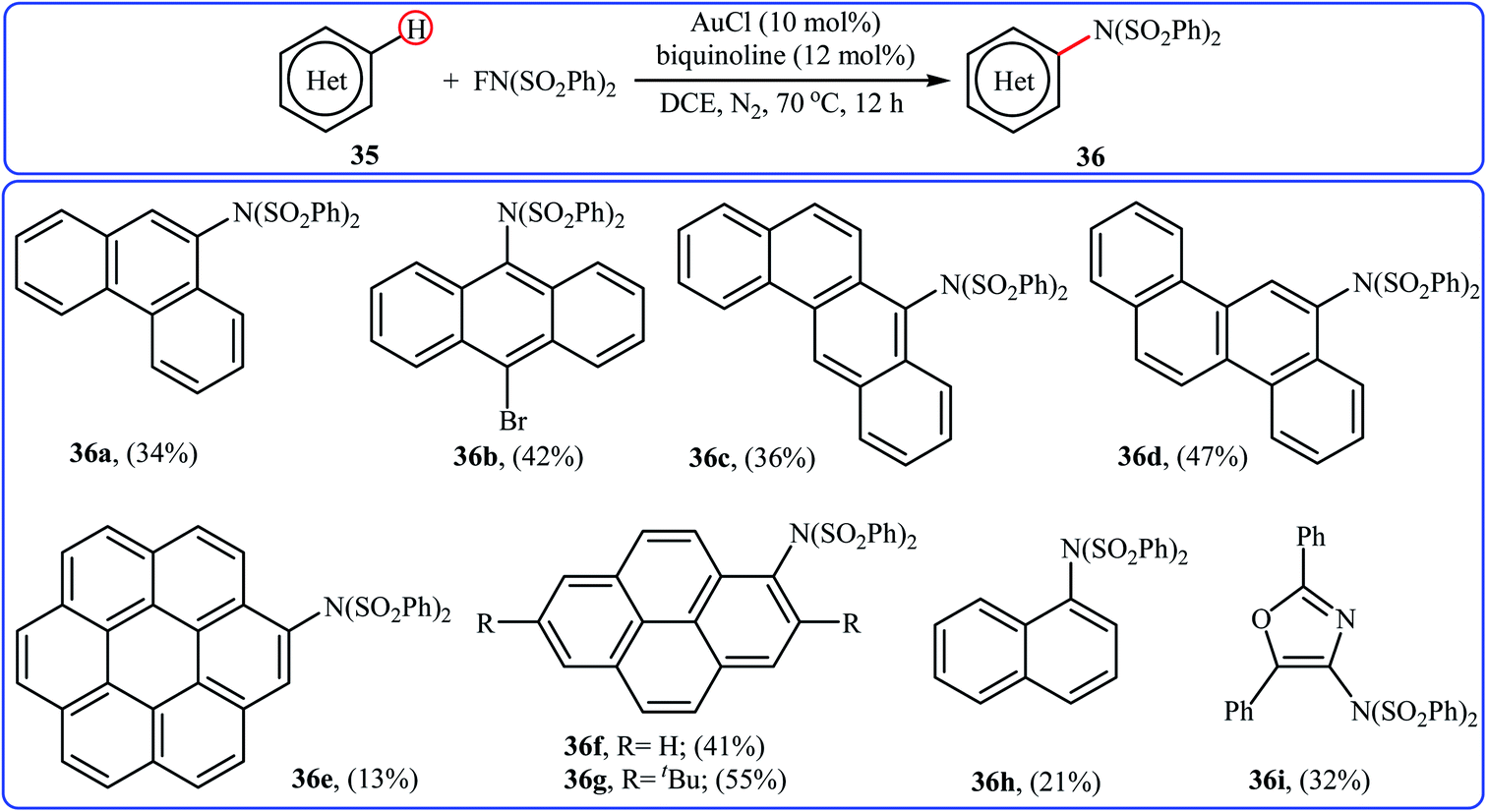
3.1.5 Gold. One of the latest metal catalysts to join the direct C–H amination of arenes with NFSI story is a gold salt. In 2018, Murakami and Itami have shown that the low-cost, commercially available AuCl effectively catalyzed the direct amination of polycyclic aromatic hydrocarbons 35 through C–H bond cleavage employing NFSI as the amination reagent.43 The reactions were occurred in the presence of bidentate 2,2′-biquinoline (BQ) as a ligand in DCE and afforded low to moderate yields of the synthetically challenging mono-aminated products 36, ranging from 13% to 55% (Scheme 20). Of note, the presence of a N,N-bidentate ligand is crucial for the success of the reaction. No product was found in the absence of a bidentate ligand.
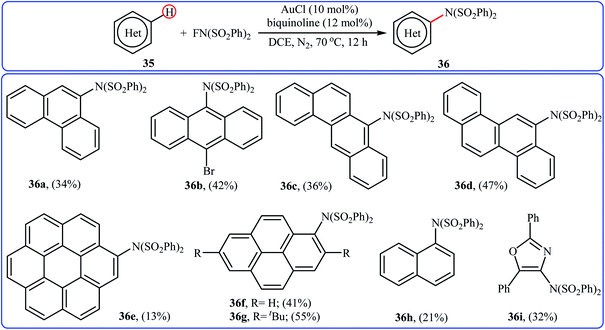 |
| | Scheme 20 Direct C–H amination of polycyclic aromatic hydrocarbons 35 catalyzed by AuCl. | |
3.2 Transition-metal-free reactions
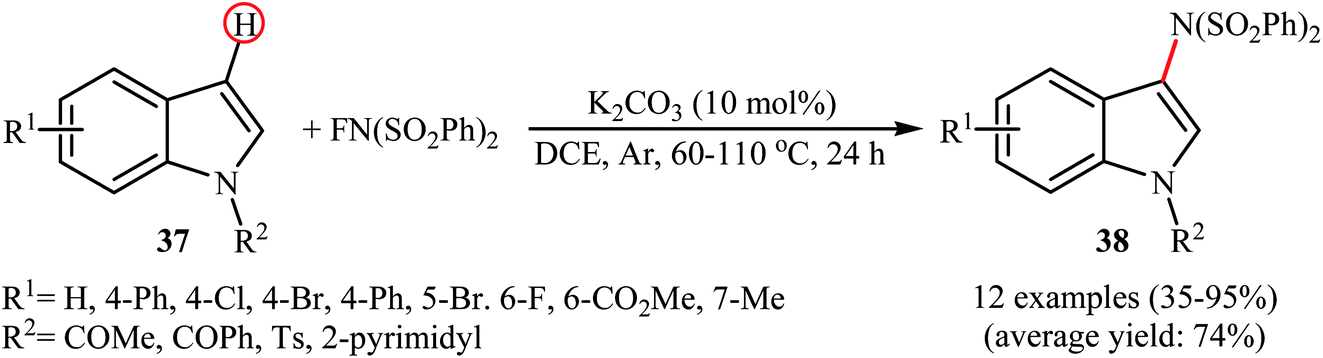
In 2013, Yang and colleagues reported the first example of transition metal-free amination of aromatic C–H bonds with NFSI.44 They showed that N-substituted indole derivatives 37 underwent regioselective C-3 amination with NFSI in the presence of a catalytic amount of K2CO3 under external oxidant-free conditions. The reactions were carried out under an inert atmosphere in DCE, tolerated a series of functional groups (e.g., halides, esters) and provided the target 3-aminated indoles 38 in moderate to excellent yields, ranging from 35% to 95% (Scheme 21). Remarkably, the corresponding 2-aminated products were not detected by in situ GC-MS and 1H NMR. Under similar conditions, the aminations of pyrrole substrates took place selectively on the C2-position and afforded the C2-aminated pyrroles in moderate to almost quantitative yields. Although the preliminary control experiments pointed toward a radical reaction mechanism, the authors do not propose a mechanistic scheme for the transformation.
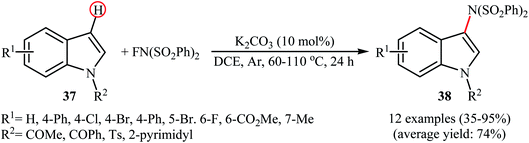 |
| | Scheme 21 Transition metal-free C3-amination of indoles 37 with NFSI. | |
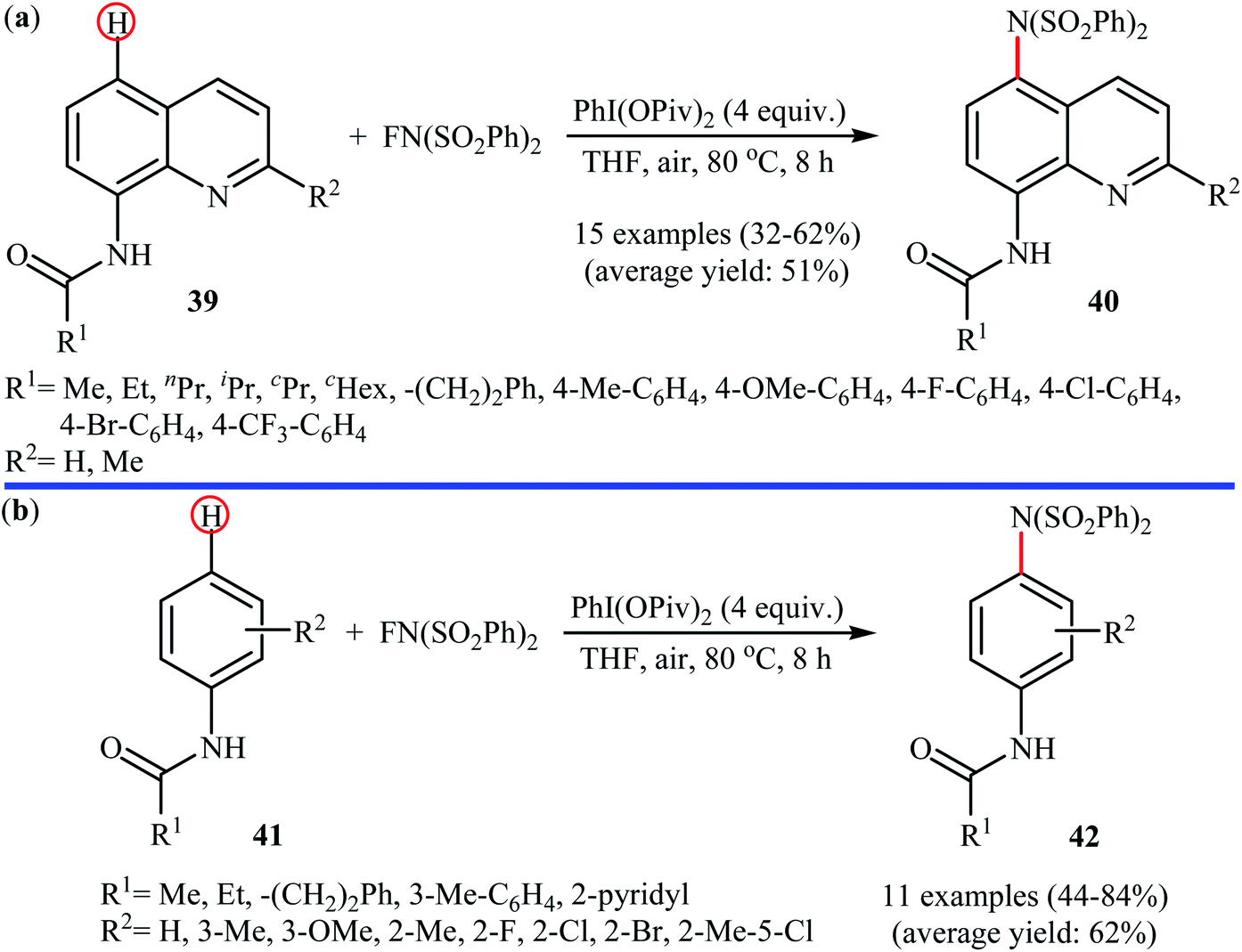
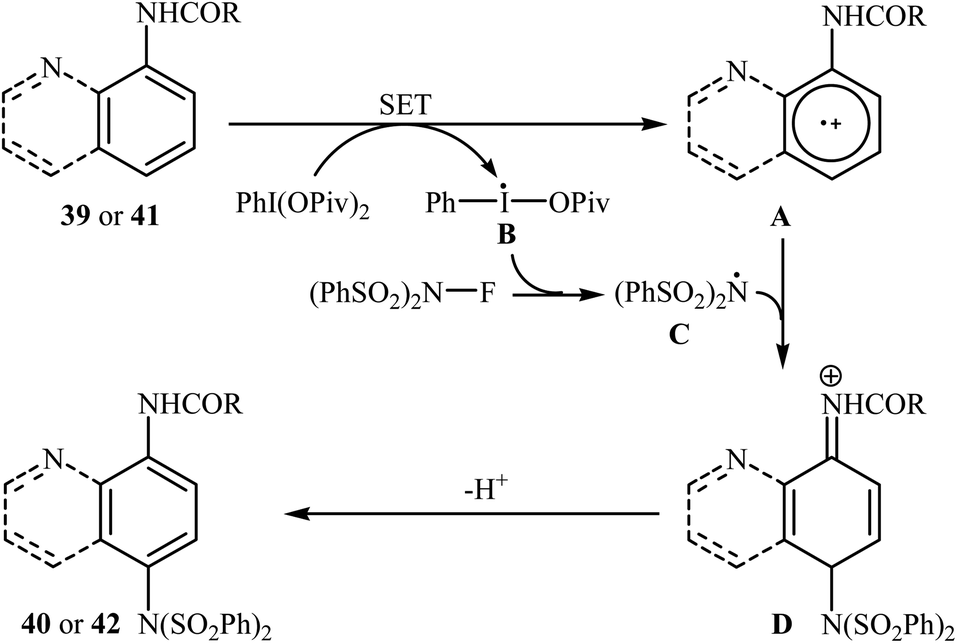
Shortly afterwards, Zhang and Li along with their co-workers extended the substrate scope of this chemistry to quinolines and obtained a library of C5-aminated 8-acylaminoquinolines 40 via the treatment of 8-acylaminoquinolines 39 with NFSI in the presence of a hypervalent iodine reagent under the open air (Scheme 22a).45 PhI(OPvi)2 was found to be more effective oxidant than PhI(OAc)2, and PhI(OCOCF3)2 proved to be completely ineffective. Among the various solvents such as toluene, chlorobenzene, 1,4-dioxane, THF, DCM, DCE; THF was clearly the most effective solvent and four equivalents of PhI(OPvi)2 are essential for this transformation. This methodology was also applicable for the highly selective synthesis of para-aminated anilides 42 from the corresponding anilides 41 (Scheme 22b). According to the authors, a plausible process for the formation of aminated products 40 and 42 should involve the formation of radical cation A and radical B intermediates through a single electron-transfer (SET) process from the 8-acylaminoquinolines 39 or anilides 41 to PhI(OPiv)2, followed by oxidation of the radical B by NFSI via one-electron F-atom transfer pathway to give the bis-sulfonylamidyl radical C, which after reaction with intermediate A generates the intermediate D. Finally, isomerization of intermediate D affords the expected product 40 or 42 (Scheme 23).
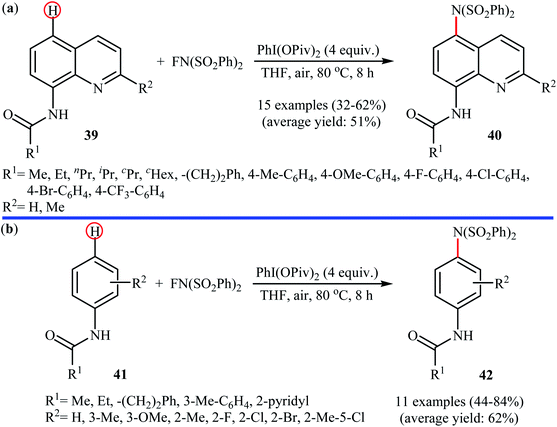 |
| | Scheme 22 PhI(OPiv)2-mediated C5-amination of 8-acylaminoquinolines 39 with NFSI; (b) regioselective para-amination of anilides 41 with NFSI mediated by a hypervalent iodine. | |
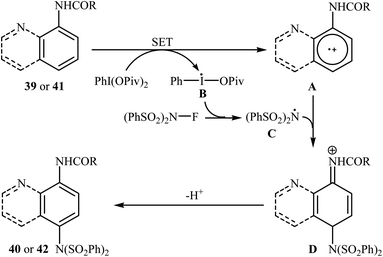 |
| | Scheme 23 Mechanistic proposal for the reaction in Scheme 22. | |
A similar example of the direct amination of aromatic C–H bonds by NFSI was disclosed by Hajra and his team in 2018, when they reported about the use of phenyliodine(III) diacetate (PIDA) to promote the regioselective amination of a series of C2-substituted imidazopyridines with NFSI in toluene at 60 °C.46 Under optimized conditions, both electron-deficient and electron-rich imidazopyridine substrates 43 were tolerated well and gave the C3-aminated products 44 in good to excellent yields (Scheme 24). 3-Phenylimidazo[1,2-a]pyridine did not take part in this amination reaction and therefore no other C3-substituted imidazopyridines were examined in the protocol. Applying this method, benzoimidazothiazoles, indoles and indolizines were also successfully aminated in good yields (up to 83%). They demonstrated that the N-(phenylsulfonyl)benzenesulfonamide group can be efficiently converted into the free amino (–NH2) group in a single-step procedure by treatment with TfOH in DCE. The control experiment in presence of various free-radical scavengers such as 2,2,6,6-tetramethylpiperidine-1-oxyl, 2,6-di-tert-butyl-4-methylphenol, and p-benzoquinone indicated that reaction proceeds through a radical mechanism. The authors proposed mechanistic pathway for this transformation is analogous to the one depicted in Scheme 23.
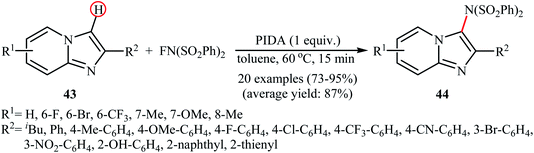 |
| | Scheme 24 PIDA-mediated C3-amination of imidazopyridines 43 with NFSI. | |
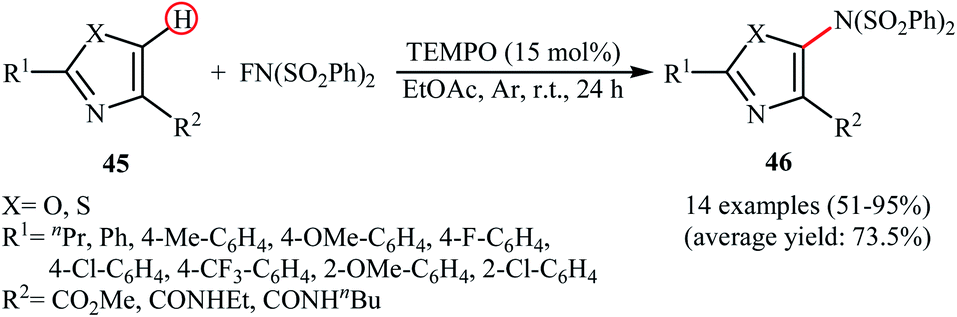
Very recently, Li and co-workers disclosed the direct and site-selective C–H amination of azoles 45 with NFSI under metal-free conditions, which allows for preparation of C5-aminated azoles 46 at room temperature (Scheme 25).47 The optimized reaction conditions were determined in the presence of TEMPO as the nitroxyl radical catalyst and EtOAc as the solvent. Various thiazole and oxazole derivatives underwent C–N bond formation successfully, affording moderate to excellent yields of the corresponding C5-aminated azoles. The results showed that electron-rich substrates afforded better yields compared to the electron-deficient ones, suggesting that this amination is probably an electrophilic substitution. Interestingly, steric hindrance had no significant impact on the facility of the reaction. The authors further demonstrated the applicability of their procedure for the direct amination of thiophene, furan, pyrrole, indole, benzofuran, benzothiophene, chromen-4-one, and mesitylene C(sp2)–H bonds. An intermolecular deuterium kinetic isotope effect (KIE) experiment of the amination of 2-d-benzofuran (kH/kD = 1.92) revealed that the cleavage of C–H bond was involved in the rate-determining step of the reaction.
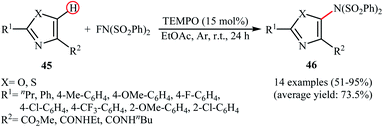 |
| | Scheme 25 TEMPO-catalyzed C5-amination of azoles 45 with NFSI. | |
4. Conclusion
This review summarized the available literature on the direct fluorination and amination of (hetero)aromatic C–H bonds utilizing commercially available N-fluorobenzenesulfonimide (NFSI) as both fluorine and nitrogen source. As illustrated, a variety of fluoro-(hetero)arenes and (hetero)aromatic amines could be easily achieved by this atom and step economic synthetic process indicating the versatility of the reagent NFSI. Interestingly, most of the reactions covered in this review showed a remarkable degree of regioselectivity, which is a great challenge in the direct functionalization of inert C–H bonds. Moreover, some of the reactions could be easily scaled up to provide gram quantities of the target products in satisfactory yields. Despite all these successes, most of the summarized reactions have been conducted under harsh conditions, which diminished their practicality, especially in the synthesis of complex molecules. Therefore, there is still further need for the exploration of novel efficient catalytic systems, which can allow these reactions under milder conditions.
Conflicts of interest
There are no conflicts to declare.
References
- T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16–29 CrossRef CAS.
-
(a) C. Isanbor and D. O'Hagan, J. Fluorine Chem., 2006, 127, 303–319 CrossRef CAS;
(b) S. Shafiei and S. Davaran, Chem. Rev. Lett., 2020, 3, 19–22 Search PubMed;
(c) S. Majedi and S. Majedi, J. Chem. Lett., 2020, 1, 2–8 Search PubMed.
-
(a) F. Babudri, G. M. Farinola, F. Naso and R. Ragni, Chem. Commun., 2007, 1003–1022 RSC;
(b) H. M. Hügel and N. Jackson, Appl. Sci., 2012, 2, 558–565 CrossRef.
-
(a) V. V. Grushin, Acc. Chem. Res., 2009, 43, 160–171 CrossRef PubMed;
(b) Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422–518 CrossRef CAS PubMed.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2013, 114, 2432–2506 CrossRef PubMed.
- M. G. Campbell and T. Ritter, Chem. Rev., 2014, 115, 612–633 CrossRef PubMed.
-
(a) S. Misaki, J. Fluorine Chem., 1981, 17, 159–171 CrossRef CAS;
(b) S. Misaki, J. Fluorine Chem., 1982, 21, 191–199 CrossRef CAS;
(c) M. Van Der Puy, Tetrahedron Lett., 1987, 28, 255–258 CrossRef CAS.
-
(a) I. Rozhkov and I. Y. Alyev, Tetrahedron, 1975, 31, 977–981 CrossRef CAS;
(b) J. H. Meurs, D. W. Sopher and W. Eilenberg, Angew. Chem., Int. Ed., 1989, 28, 927–928 CrossRef.
- M. J. Fifolt, R. T. Olczak, R. F. Mundhenke and J. F. Bieron, J. Org. Chem., 1985, 50, 4576–4582 CrossRef CAS.
- G. V. áDe Meio, J. Chem. Soc., Chem. Commun., 1990, 1065–1066 RSC.
- R. G. Plevey, R. W. Rendell and J. C. Tatlow, J. Fluorine Chem., 1982, 21, 159–169 CrossRef CAS.
-
(a) B. Šket and M. Zupan, Bull. Chem. Soc. Jpn., 1981, 54, 279–281 CrossRef;
(b) S. Stavber and M. Zupan, J. Org. Chem., 1983, 48, 2223–2226 CrossRef CAS;
(c) S. Anand, L. Quarterman, H. Hyman, K. Migliorese and R. Filler, J. Org. Chem., 1975, 40, 807–809 CrossRef CAS.
- T. Umemoto, K. Kawada and K. Tomita, Tetrahedron Lett., 1986, 27, 4465–4468 CrossRef CAS.
- P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C. H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS PubMed.
-
(a) G. S. Lal, G. P. Pez and R. G. Syvret, Chem. Rev., 1996, 96, 1737–1756 CrossRef CAS PubMed;
(b) A. S. Kiselyov, Chem. Soc. Rev., 2005, 34, 1031–1037 RSC;
(c) G. Borodkin, P. Zaikin, M. Shakirov and V. Shubin, Russ. J. Org. Chem., 2007, 43, 1451–1459 CrossRef CAS;
(d) J.-D. Yang, Y. Wang, X.-S. Xue and J.-P. Cheng, J. Org. Chem., 2017, 82, 4129–4135 CrossRef CAS PubMed.
- V. I. Sorokin, A. F. Pozharskii and V. A. Ozeryanskii, J. Fluorine Chem., 2013, 154, 67–72 CrossRef CAS.
- A. Rostami, Synlett, 2007, 2924–2925 CrossRef CAS.
- E. Differding and H. Ofner, Synlett, 1991, 187–189 CrossRef CAS.
- Y. Li and Q. Zhang, Synthesis, 2015, 47, 159–174 CAS.
-
(a) A. Monfared, S. Ebrahimiasl, M. Babazadeh, S. Arshadi and E. Vessally, J. Fluorine Chem., 2019, 220, 24–34 CrossRef CAS;
(b) M. Hamzeloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef;
(c) C. Yang, A. Hassanpour, K. Ghorbanpour, S. Abdolmohammadi and E. Vessally, RSC Adv., 2019, 9, 27625–27639 RSC;
(d) A. Hosseinian, Y. J. Sadeghi, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Sulfur Chem., 2019, 40, 565–585 CrossRef CAS.
-
(a) S. Sarhandi, M. Daghagheleh, M. Vali, R. Moghadami and E. Vessally, Chem. Rev. Lett., 2018, 1, 9–15 Search PubMed;
(b) M. Daghagheleh, M. Vali, Z. Rahmani, S. Sarhandi and E. Vessally, Chem. Rev. Lett., 2018, 1, 23–30 Search PubMed;
(c) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 23 CrossRef PubMed;
(d) S. Mohammadi, M. Musavi, F. Abdollahzadeh, S. Babadoust and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 49–55 Search PubMed;
(e) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 39 CrossRef PubMed;
(f) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 20 CrossRef PubMed;
(g) S. Farshbaf, L. Sreerama, T. Khodayari and E. Vessally, Chem. Rev. Lett., 2018, 1, 56–67 Search PubMed;
(h) S. Arshadi, A. Banaei, A. Monfared, S. Ebrahimiasl and A. Hosseinian, RSC Adv., 2019, 9, 17101–17118 RSC;
(i) Y. Liu, A. G. Ebadi, L. Youseftabar-Miri, A. Hassanpour and E. Vessally, RSC Adv., 2019, 9, 25199–25215 RSC;
(j) S. Shahidi, P. Farajzadeh, P. Ojaghloo, A. Karbakhshzadeh and A. Hosseinian, Chem. Rev. Lett., 2018, 1, 37–44 Search PubMed;
(k) X. Lu, Q. Yi, X. Pan, P. Wang and E. Vessally, J. Sulfur Chem., 2020, 41, 210–228 CrossRef CAS;
(l) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed;
(m) S. Majedi, S. Majedi and F. Behmagham, Chem. Rev. Lett., 2019, 2, 187–192 Search PubMed.
- S.-J. Lou, D.-Q. Xu, A.-B. Xia, Y.-F. Wang, Y.-K. Liu, X.-H. Du and Z.-Y. Xu, Chem. Commun., 2013, 49, 6218–6220 RSC.
- Q. Ding, C. Ye, S. Pu and B. Cao, Tetrahedron, 2014, 70, 409–416 CrossRef CAS.
- C. Testa, J. Roger, S. Scheib, P. Fleurat-Lessard and J. C. Hierso, Adv. Synth. Catal., 2015, 357, 2913–2923 CrossRef CAS.
- D. A. Gutierrez, W.-C. C. Lee, Y. Shen and J. J. Li, Tetrahedron Lett., 2016, 57, 5372–5376 CrossRef CAS.
- Y. Zhang, K. Shibatomi and H. Yamamoto, Synlett, 2005, 2837–2842 CAS.
- J. Ding, Y. Zhang and J. Li, Org. Chem. Front., 2017, 4, 1528–1532 RSC.
- Y. Hayakawa, M. Singh, N. Shibata, Y. Takeuchi and K. L. Kirk, J. Fluorine Chem., 1999, 97, 161–164 CrossRef CAS.
- P. M. O'Neill, R. C. Storr and B. K. Park, Tetrahedron, 1998, 54, 4615–4622 CrossRef.
- R. Andreev, G. Borodkin and V. Shubin, Russ. J. Org. Chem., 2009, 45, 1468 CrossRef CAS.
- J. M. Hatfield, C. K. Eidell and C. E. Stephens, Tetrahedron Lett., 2013, 54, 1025–1028 CrossRef CAS.
- V. Levchenko, Y. V. Dmytriv, A. V. Tymtsunik, K. Liubchak, A. Rudnichenko, A. V. Melnyk, S. Y. Veselovych, Y. V. Borodulin, O. M. Otsalyuk, A. A. Tolmachev and P. K. Mykhailiuk, J. Org. Chem., 2018, 83, 3265–3274 CrossRef CAS PubMed.
- G. I. Borodkin, I. R. Elanov, Y. V. Gatilov and V. G. Shubin, J. Fluorine Chem., 2019, 228, 109412 CrossRef.
- F. Sakurai, T. Yukawa and T. Taniguchi, Org. Lett., 2019, 21, 7254–7257 CrossRef CAS PubMed.
- K. Sun, Y. Li, T. Xiong, J. Zhang and Q. Zhang, J. Am. Chem. Soc., 2011, 133, 1694–1697 CrossRef CAS PubMed.
- G. B. Boursalian, M.-Y. Ngai, K. N. Hojczyk and T. Ritter, J. Am. Chem. Soc., 2013, 135, 13278–13281 CrossRef CAS PubMed.
- S. Wang, Z. Ni, X. Huang, J. Wang and Y. Pan, Org. Lett., 2014, 16, 5648–5651 CrossRef CAS PubMed.
- Y. Yin, J. Xie, F. Q. Huang, L. W. Qi and B. Zhang, Adv. Synth. Catal., 2017, 359, 1037–1042 CrossRef CAS.
- J.-M. Li, Y.-H. Wang, Y. Yu, R.-B. Wu, J. Weng and G. Lu, ACS Catal., 2017, 7, 2661–2667 CrossRef CAS.
- S. Lu, L.-L. Tian, T.-W. Cui, Y.-S. Zhu, X. Zhu, X.-Q. Hao and M.-P. Song, J. Org. Chem., 2018, 83, 13991–14000 CrossRef CAS PubMed.
- R. J. Tang, C. P. Luo, L. Yang and C. J. Li, Adv. Synth. Catal., 2013, 355, 869–873 CrossRef CAS.
- X. Wang, B. Lei, L. Ma, H. Jiao, W. Xing, J. Chen and Z. Li, Adv. Synth. Catal., 2017, 359, 4284–4288 CrossRef CAS.
- S. J. Yip, T. Kawakami, K. Murakami and K. Itami, Asian J. Org. Chem., 2018, 7, 1372–1375 CrossRef CAS.
- H. H. Liu, Y. Wang, G. Deng and L. Yang, Adv. Synth. Catal., 2013, 355, 3369–3374 CrossRef CAS.
- Y. Wang, Y. Wang, Z. Guo, Q. Zhang and D. Li, Asian J. Org. Chem., 2016, 5, 1438–1441 CrossRef CAS.
- M. Singsardar, S. Mondal, R. Sarkar and A. Hajra, ACS Omega, 2018, 3, 12505–12512 CrossRef CAS PubMed.
- Q. Miao, Z. Shao, C. Shi, L. Ma, F. Wang, R. Fu, H. Gao and Z. Li, Chem. Commun., 2019, 55, 7331–7334 RSC.
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b
*b