
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefective S/N co-doped carbon cloth via a one-step process for effective electroreduction of nitrogen to ammonia†
Shaoan Cheng *,
Chaochao Li,
Zhen Yu,
Yi Sun,
Longxin Li and
Jiawei Yang
*,
Chaochao Li,
Zhen Yu,
Yi Sun,
Longxin Li and
Jiawei Yang
State Key Laboratory of Clean Energy Utilization, Department of Energy Engineering, Zhejiang University, Hangzhou 310027, PR China. E-mail: shaoancheng@zju.edu.cn
First published on 6th March 2020
Abstract
The electroreduction of nitrogen (N2) has gained increasing attention as a promising route to achieve green and sustainable ammonia (NH3) production. However, the construction of an active and durable electrocatalyst for N2 reduction reaction (NRR) remains a significant challenge. Herein, we, for the first time, report that S/N co-doped carbon cloth (CC) with abundant defects can serve as an efficient NRR electrocatalyst at ambient conditions. The S/N co-doped CC was prepared through a novel one-step method by using ammonium persulfate (APS) as the source of nitrogen and sulfur. The catalyst prepared at 800 °C (CC-APS 800) showed abundant defects and heteroatoms as the active and stable electrocatalytic sites for NH3 electrosynthesis. Based on this, a sizeable NH3 yield of 9.87 × 10−10 mol s−1 cm−2 and high faradaic efficiency of 8.11% were obtained in 0.05 M H2SO4 at −0.3 V (vs. reversible hydrogen electrode, RHE), respectively. Furthermore, the electrocatalytic mechanism on CC-APS 800 was elucidated using the electrochemical in situ Fourier transform infrared technique, and follows an associative reaction pathway. Our work would provide a new guideline for designing metal-free self-standing electrocatalysts for the NRR and other applications.
Introduction
As an active form of nitrogen (N2), ammonia (NH3) has become an indispensable ingredient of the modern agriculture and chemistry industry.1 However, the industrialized Haber–Bosch (H–B) method for modern ammonia synthesis requires high temperature (∼500 °C) and high pressure (20–40 MPa), and uses H2 as reductant (from steam reforming of natural gas).2 As a result, the H–B method contributes to 1–2% of the global energy consumption and over 300 million tons of CO2 emission per year. Therefore, given the serious environmental concerns and climate change, there is an urgent need to explore green and sustainable alternatives to the H–B method.The technique of nitrogen reduction reaction (NRR) has attracted increasing attention with less energy consumption, little climate change, and low cost. On the surface of the catalyst, the absorbed N2 molecules will break or dissociate on the active sites, further combine with the protons, and finally form the NH3 with the cooperation of electrons.  . Up to now, a large variety of electrocatalysts based on noble metals (such as Au,3 Pt,4 Ru,5 Ag,6 etc.) and transition metals (such as Co,7 Sn,8 Mn,9 Ti,10 Mo,11 V,12 Fe,13 etc.) has been developed for ammonia electrosynthesis. However, the practical utilization of these metal-based catalysts was often limited by the material cost and environmental impact. Therefore, developing an efficient and durable metal-free catalyst has been regarded as one of the most potential breakthroughs for the NRR technique.
. Up to now, a large variety of electrocatalysts based on noble metals (such as Au,3 Pt,4 Ru,5 Ag,6 etc.) and transition metals (such as Co,7 Sn,8 Mn,9 Ti,10 Mo,11 V,12 Fe,13 etc.) has been developed for ammonia electrosynthesis. However, the practical utilization of these metal-based catalysts was often limited by the material cost and environmental impact. Therefore, developing an efficient and durable metal-free catalyst has been regarded as one of the most potential breakthroughs for the NRR technique.
As an earth-abundant metal-free catalyst, carbon-based electrocatalysts are promising for NRR owing to their unique superiorities on the electric conductivity, chemical durability, surface area, and electronic structure.5 However, to better weaken the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bonding (∼945.6 kJ mol−1), the modulation of the electronic structure of carbon is very important, and the method of heteroatom doping has been proved as a reliable tool.14 For recent years, various heteroatoms owning similar electronic orbits to carbon (such as nitrogen,15 sulfur,16 fluorine,17 and boron18) have been investigated to regulate the electronic configuration of carbon, and thus promote the NRR performance. Furthermore, it is considered that co-doping carbon materials with two kinds of heteroatoms may co-activate the adjacent C atom and construct some unexpected electronic structures, which usually provides much more active sites than that of one heteroatom doping, and enhances the NRR performance eventually. However, dual-dopant pairs reported in co-doped carbon materials for NRR are merely restricted to N–P19,20 and N–B21,22 couples. Sulfur/nitrogen co-doped carbon has gained remarkable progress in multiple electrocatalysis fields, including oxygen reduction reaction (ORR),23 oxygen evolution reaction (OER),24 and hydrogen evolution reaction (HER).25 Recent literatures have reported the theoretical calculation results of the S/N-codoped graphene as the catalyst for NRR.26,27 Due to the higher electronegativity of N (3.04) and S (2.58) relative to C (2.55), the N/S dopants can induce the positive charge densities on the adjacent carbon atoms, which favor the adsorption of N2. Thus, the N/S-adjacent carbon atoms act as the active sites for N2 adsorption and subsequent activation. According to the DFT results, the N2 adsorption energy of S/N-codoped carbon is predicted to be 0.42 eV, which is much higher than those of N-doped carbon (0.31 eV) and S-doped carbon (0.19 eV). This suggests that S/N-codoping can co-activate the neighboring C atoms to enhance the N2 adsorption. Nevertheless, the performance and reaction mechanism of S/N co-doped carbon for N2 electroreduction remains unexplored.
N bonding (∼945.6 kJ mol−1), the modulation of the electronic structure of carbon is very important, and the method of heteroatom doping has been proved as a reliable tool.14 For recent years, various heteroatoms owning similar electronic orbits to carbon (such as nitrogen,15 sulfur,16 fluorine,17 and boron18) have been investigated to regulate the electronic configuration of carbon, and thus promote the NRR performance. Furthermore, it is considered that co-doping carbon materials with two kinds of heteroatoms may co-activate the adjacent C atom and construct some unexpected electronic structures, which usually provides much more active sites than that of one heteroatom doping, and enhances the NRR performance eventually. However, dual-dopant pairs reported in co-doped carbon materials for NRR are merely restricted to N–P19,20 and N–B21,22 couples. Sulfur/nitrogen co-doped carbon has gained remarkable progress in multiple electrocatalysis fields, including oxygen reduction reaction (ORR),23 oxygen evolution reaction (OER),24 and hydrogen evolution reaction (HER).25 Recent literatures have reported the theoretical calculation results of the S/N-codoped graphene as the catalyst for NRR.26,27 Due to the higher electronegativity of N (3.04) and S (2.58) relative to C (2.55), the N/S dopants can induce the positive charge densities on the adjacent carbon atoms, which favor the adsorption of N2. Thus, the N/S-adjacent carbon atoms act as the active sites for N2 adsorption and subsequent activation. According to the DFT results, the N2 adsorption energy of S/N-codoped carbon is predicted to be 0.42 eV, which is much higher than those of N-doped carbon (0.31 eV) and S-doped carbon (0.19 eV). This suggests that S/N-codoping can co-activate the neighboring C atoms to enhance the N2 adsorption. Nevertheless, the performance and reaction mechanism of S/N co-doped carbon for N2 electroreduction remains unexplored.
In this work, we present, for the first time, a defective S/N co-doped carbon cloth (CC) for electrocatalytic N2 fixation. The S/N co-doped CC with abundant defects was prepared via a facile one-step thermochemical treatment with ammonium persulfate (APS) as the dopant source of N and S. The NRR performance of the resulting catalyst was investigated at a low potential of −0.3 V vs. RHE. The reaction pathway of NRR was pursued through the electrochemical in situ Fourier transform infrared (FTIR) technique.
Results and discussion
Defective S/N co-doped CC was prepared via a facile thermochemical treatment (Fig. 1a, details in ESI†) on commercial CC. As a common supporting material for electrocatalysis, the self-standing CC is binder-free, chemical stable, and cost-effective. In this novel process, APS is reported as the nitrogen and sulfur source for the first time. As-prepared samples treated with APS under different temperatures were denoted as CC-APS X (X represents the temperature of 200, 400, 600, or 800 °C, respectively). The thermal interaction between APS and CC was determined through thermogravimetric analysis (TGA) measurements. As shown in Fig. S1,† the mass of APS decreases sharply to zero at about 455 °C, which implies that APS has been completely pyrolyzed into gases (including NH3, SO2, SO3, H2O, and O2).28 With the temperature further increasing, the mass of APS-CC blend presents another noticeable decrease at 625 °C, while that of CC showed no obvious change during the whole process. This result is attributed to the further reaction of CC with those pyrolysis gases, which probably leads to the introduction of heteroatoms. | ||
| Fig. 1 (a) Schematic illustration of the synthesis process of the defective S/N co-doped CC. Through a similar process with different treating temperatures (X °C), CC-APS X (X = 200, 400, 600, and 800, respectively) can be easily fabricated. SEM images of (b) pristine CC, (c) CC-APS 200, (d) CC-APS 400, (e) CC-APS 600, and (f) CC-APS 800. TEM images of (g) pristine CC, (h) CC-APS 200, (i) CC-APS 400, (j) CC-APS 600, and (k) CC-APS 800. | ||
The SEM images also show that more and more corrosion has been made on the CC surface, with the treating temperature increasing (illustrations in Fig. 1b–f). Particularly, complex 3D nanostructures composed of irregular holes and grooves have been dramatically formed on the surface of CC-APS 800. The transmission electron microscopy (TEM) images of the treated CCs (Fig. 1g–k) demonstrate clearly that the disorder of lattice fringes in the samples has been aggravated as the treating temperature increases. The distortion of the atomic arrangement possibly reflecting the abundant doping of heteroatoms or carbon vacancies.29,30 Also, some ultra-thin carbon layers have been formed on the edge of the carbon fiber after the heating process. Those irregular carbon nanosheets may provide rich active sites for nitrogen absorption and activation.31 We also investigated the treated CC obtained at 900 °C; however, the as-fabricated sample lost 76% of its original mass, and finally became too fragile to support itself as an electrode for electrocatalysis.
X-ray photoelectron spectroscopy (XPS) analysis was employed to explore the elementary configuration and chemical state of as-prepared CC catalysts. As shown in Fig. 2a, the XPS overall spectra of treated CCs depicts obvious S (∼164.8 eV), C (∼285.2 eV), N (∼400.0 eV), and O (∼531.5 eV) peaks,32 while that of the pristine CC only shows C, O, and weak N peaks. Moreover, the content of S and N increases with the temperature increasing (Table S1, Fig. S2†). As a result, CC-APS 800 shows the maximum atomic ratio of N/C and S/C of 8.23 at% and 2.51 at%, respectively. Accordingly, C–S (281.2 eV) and C–N (282.0 eV) peaks can be found in the C 1s spectra (Fig. 2b), confirming the resultful doping of S and N.33 Moreover, N 1s spectrum (Fig. 2c) of CC-APS 800 can be resolved into three peaks at 398.6, 399.8, and 401.0 eV, which are assigned to pyridinic N (24.97 at%), pyrrolic N (39.02 at%), and graphitic N (36.01 at%), respectively.29 Pyridinic and pyrrolic N occupies 63.99 at% of the total amount of N in CC-APS 800, producing massive catalytic sites for N2 electroreduction.15,34 As shown in Fig. 2d, the peaks of 164.0 eV and 168.4 eV in the S 2p spectrum are assigned to C–S–C and C–SOx–C (x = 2, 3, 4), respectively.35 The strong signal of thiophene-S indicates that the incorporated S atoms are mainly bonded with two C atoms at the edges or defects of disordered carbon networks, which are chemically active sites for electroreduction reaction.36 The surface elementary compositions were further analyzed using energy dispersive spectrum (EDS). The EDS elemental mapping for CC-APS 800 (Fig. S3†) indicates that both N and S atoms have been evenly introduced into the resulting carbon cloth with considerable quantities.
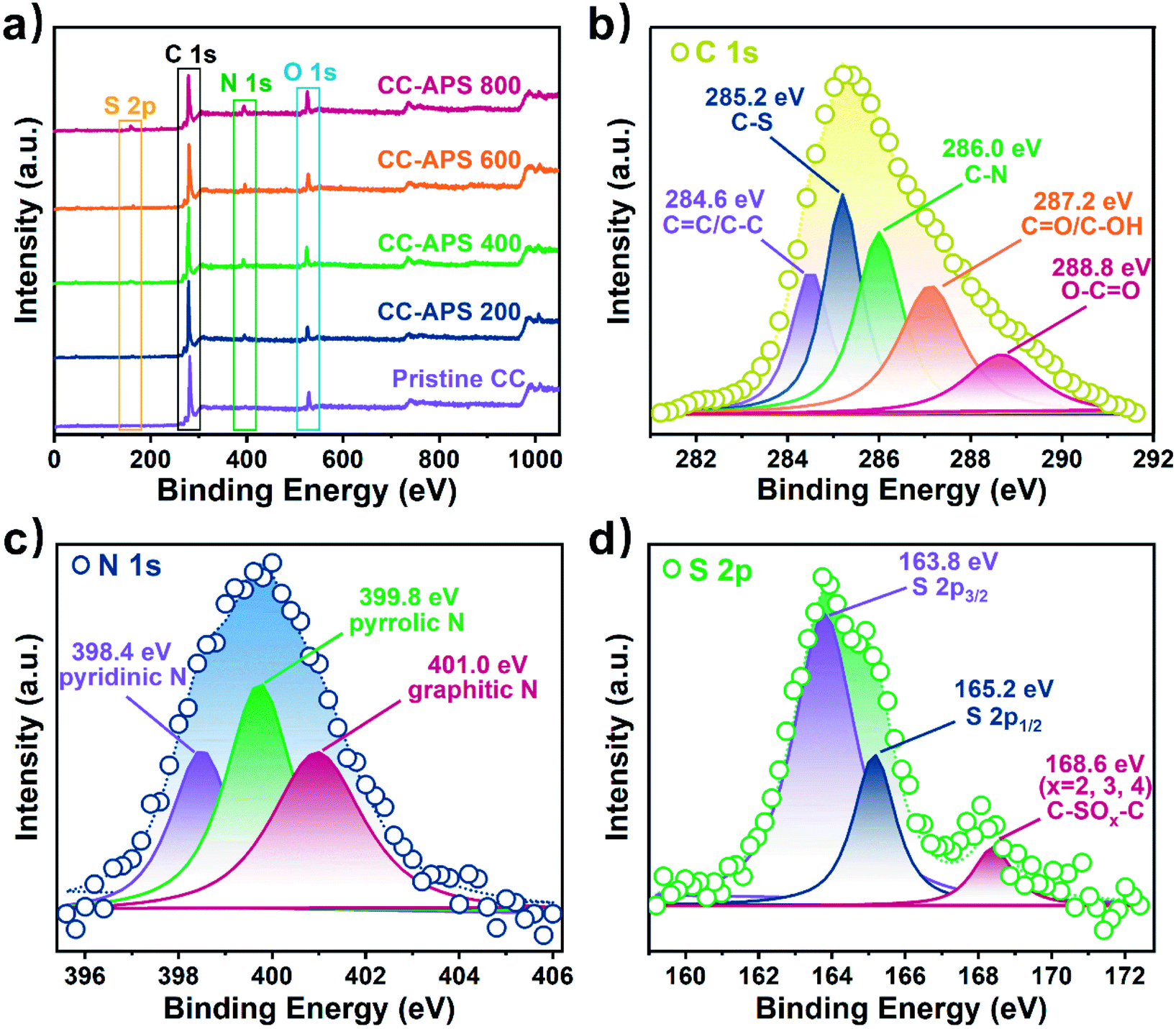 | ||
| Fig. 2 (a) XPS overall spectra of the pristine CC, CC-APS 200, CC-APS 400, CC-APS 600, and CC-APS 800. XPS spectra of (b) C 1s, (c) N 1s, and (d) S 2p of CC-APS 800. | ||
The isothermal N2 adsorption–desorption test was utilized to study the N2 adsorption ability and pore properties of the catalyst surface. CC-APS 800 presents a type-IV isotherm with an H2 shaped hysteresis loop in the p/p0 range of 0.4–0.8 (Fig. 3a), usually indicating massive mesoporous structures on the surface.37 As calculated by the BJH (Barrett–Joyner–Halenda) method, CC-APS 800 exhibits dominant mesopores centered at ∼3.5 nm and micropores concentrated at ∼1.7 nm, while nearly no pore structure is observed for the pristine CC (Fig. 3b). Correspondingly, CC-APS 800 presents a large surface area of 610.26 m2 g−1 as calculated through BET (Brunauer–Emmett–Teller) method, while the pristine CC only has a surface area of 1.19 m2 g−1. Furthermore, the electrochemical active surface area (ECSA) of the catalysts was measured by cyclic voltammetry (Fig. S4†).38 The ECSA of CC increases from 0.0042 to 1.7754 mF cm−2 after the calcination at 800 °C, demonstrating the generation of massive electrochemically active sites. Besides the large surface area, according to the contact-angle test results, the change of surface wettability also accounts for the improved ECSA (Fig. S5†). By treating CC with APS at high temperature (>400 °C), the surface contact angle increases form ∼127.4° to ∼180°. The surface hydrophilicity is significantly enhanced, which constructs the fast supply of hydrogen for hydrogenation reactions.39 The FTIR spectra (Fig. S6†) revealed the generation of abundant N- and S-containing functional groups on the surface of CC-APS 800. Those functional groups with strong polarity mainly endow CC-APS 800 with a super-hydrophilic surface.40 Temperature-programmed desorption (TPD) has been further carried out to investigate the capability of S/N-codoped carbon cloth for N2 adsorption (Fig. S9†), which is a prerequisite step of NRR. As the result, the pristine CC performed very weak physisorption (at about 100 °C) and chemisorption (at about 335 °C) of N2.41,42 However, for the S/N-codoped carbon cloth (CC-APS 800), the physisorption and chemisorption were significantly enhanced, suggesting that sulfur and nitrogen dopants induce the physical and chemical adsorption sites for N2 molecules. As a summary, the supplement of N2 gas (nitrogen source), water (hydrogen source), and electrons (energy source) are dramatically enhanced on the triple-phase boundary on CC-APS 800.
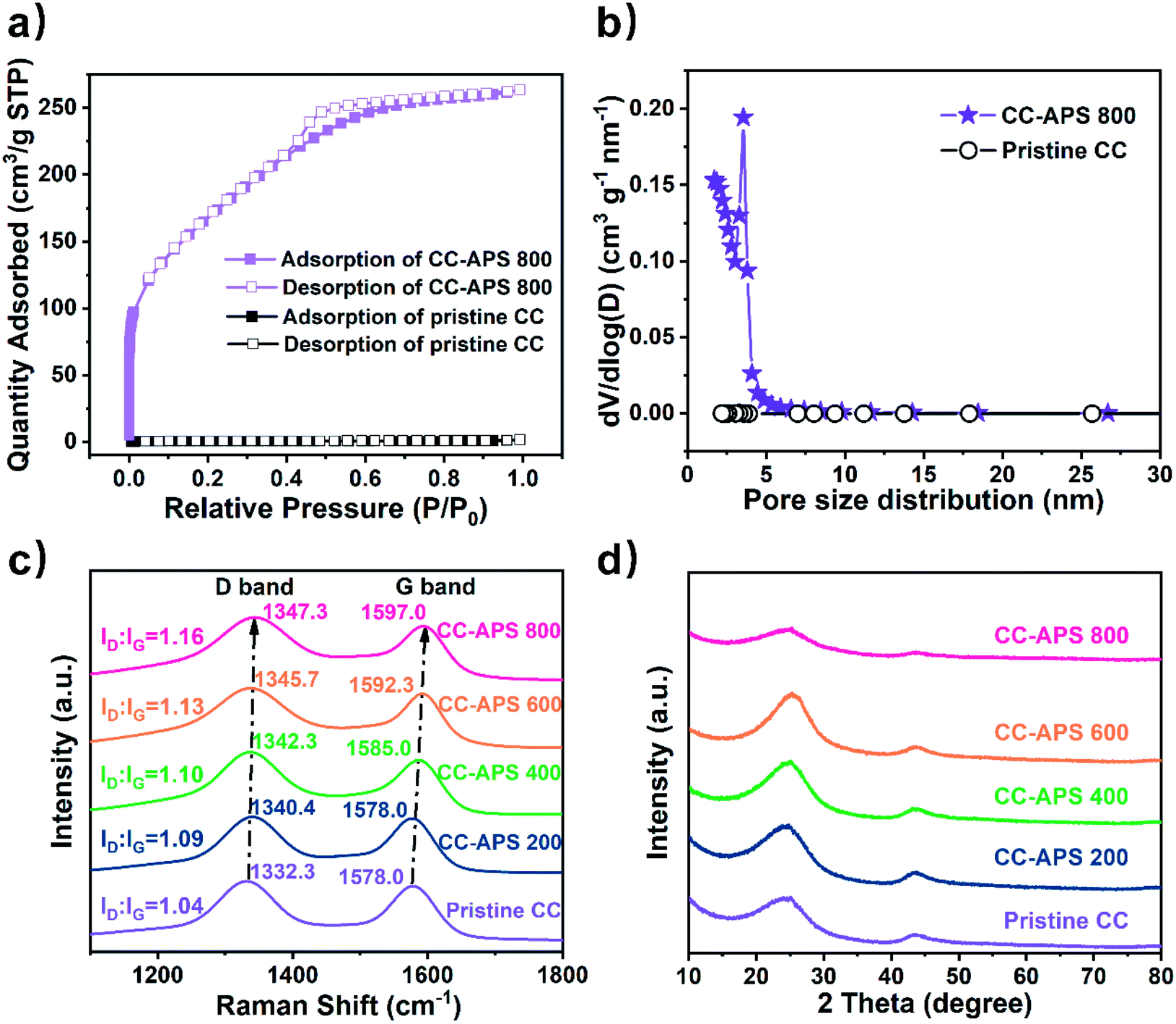 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms and (b) corresponding pore size distributions of the pristine CC and CC-APS 800, respectively. (c) Raman and (d) XRD spectra of the pristine CC, CC-APS 200, CC-APS 400, CC-APS 600, and CC-APS 800. | ||
The Raman spectra were investigated to characterize the defects of CC-APS X (Fig. 3c). The G band corresponding to 1530–1660 cm−1 was attributed to graphitic in-plane vibrations with E2g symmetry, whereas the D band indexed to 1270–1430 cm−1 generally was ascribed to the presence of defects and disorder in the carbon atomic array. These defects were probably caused by the in-plane substitutional heteroatoms.43 Notably, both the G band and D band shifted to higher wavenumbers after the thermochemical treatment, which probably resulted from the etching of graphite crystallites and the electron-cloud migration caused by doping heteroatoms.44 The intensity ratio of the D band and G band (ID/IG) is a credible indicator for the defect extent in carbon materials.45 CC-APS 800 showed the higher ID/IG of 1.15 than those of other samples, which implies the more abundant defects existing. These facts usually contribute to the superior NRR activity.29,30,46 In the XRD patterns of CC-APS X (Fig. 3d), the typical diffraction peaks at 25.2° and 43.6° could be well ascribed to the (002) and (101) planes of the hexagonal graphite (JCPDS no.75-1621), respectively. Notably, the diffraction peaks of CC-APS 800 are dramatically lower and broader than other CC-APS X, which demonstrated a sharp transformation from graphitic to amorphous carbon in CC-APS X. This transformation was probably caused by the etching and doping effects of 800 °C calcination, which was also consistent with the Raman results.
To investigate the performance of CC-APS X quantitatively, a typical three-electrode system established in a typical H-type reactor47 was used to measure the NH3 yield (details in ESI†). To avoid any trace amount of N-containing contaminant, the catalysts, the electrolyte solution, the bubbling gas, and the Nafion membrane have been well-purified or cleaned according to the protocol for NRR before each test (details in ESI†).48–50 0.05 M H2SO4 solution was used as the electrolyte to evaluate the stability and selectivity of the catalyst in the acidic condition so that to determine the industrial feasibility of the catalyst.51,52 The NH3 yield and faradaic efficiency (FE) were related to the cathode potential. To determine the optimal cathode potential, the chronoamperometry method was utilized to measure CC-APS 800 at a series of potentials (Fig. 4a). After a reaction time of 2 h, CC-APS 800 with a potential of −0.2 V performed a low NH3 yield of 5.32 × 10−11 mol s−1 cm−2 and an FE of 0.47%. Negatively shifting the potential to −0.3 V, CC-APS 800 dramatically demonstrated a maximum NH3 yield of 9.87 × 10−10 mol s−1 cm−2 and an FE of 8.11%. CC-APS 800 exhibited the better performance for NRR than those of many other carbon-based catalysts for NRR (Table S2†) as well as the lower preparation cost and less electricity consumption. The performance of CC-APS 800 for NRR declined apparently when the cathode potential further decreased from −0.3 V to −0.5 V. These results were probably attributed to the weaker competitiveness of the NRR process compared to HER process owing to the kinetic limits on the N2 adsorption and multielectron transfer.53 In a typical NRR electrolysis, the NH3–N accumulates almost linearly as the time increases (Fig. S10†).
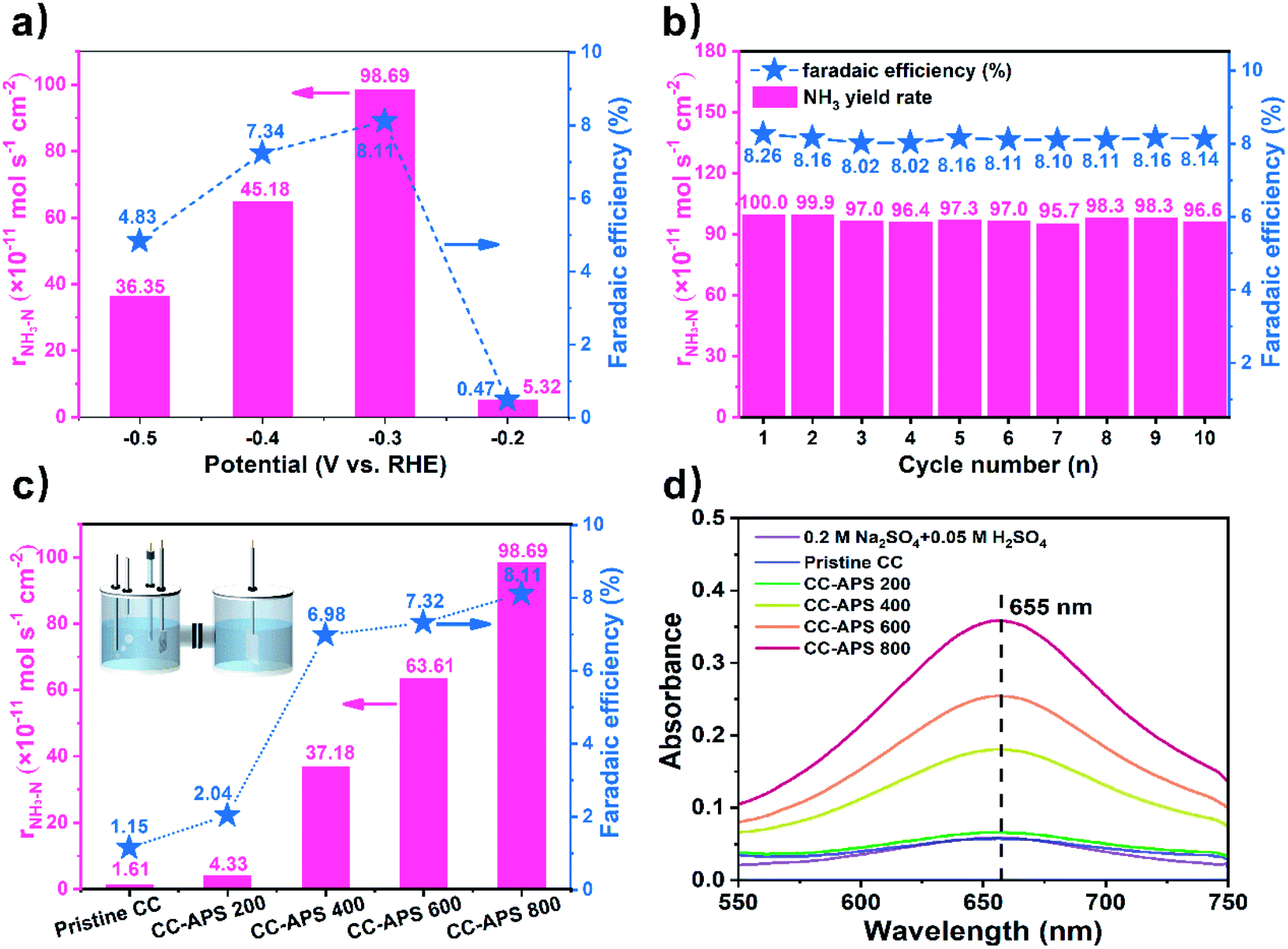 | ||
| Fig. 4 (a) NH3 yields and FEs of CC-APS 800 at different potentials. (b) Long-term NRR test of CC-APS 800 at −0.3 V vs. RHE for ten cycles (2 h for each cycle). (c) NH3 yield and FEs of the pristine CC, CC-APS 200, CC-APS 400, CC-APS 600, and CC-APS 800 at −0.3 V vs. RHE. (d) UV-vis spectra of N2-saturated electrolyte stained with indophenol indicator after 2 h NRR test at −0.3 V vs. RHE using the pristine CC and CC-APS X. | ||
Following protocols put forward by Andersen et al.54 and Suryanto et al.,55 the isotopic labeling experiment using 15N2 gas (15N enrichment of 99 atom%) has been carried out to ensure the genuine capability of the as-synthesized S/N-codoped carbon cloth for NRR (Fig. 5). According to the published literature,45,54,56 the nitrogen species produced during the NRR process could be distinguished by 1H NMR spectra due to the chemical shift of triplet coupling of 14N and doublet coupling of 15N. During the isotopic labeling experiment, the as-synthesized S/N-codoped carbon cloth (CC-APS 800) was applied with a potential of −0.3 V vs. RHE for 10 h in the electrolyte solution saturated with Ar, 15N2 or 14N2 gas as the feeding gas, respectively. To remove impurities of reducible, labile nitrogen-containing compounds such as NOx which is inevitable in the N2 gas,57 a Cu impurity trap was introduced, composed of 2 g Cu–Zn–Al oxide catalyst in a U-shaped stainless steel tubing.58,59 As a result, when using Ar gas as the feeding gas, no peaks of 15NH4+ or 14NH4+ can be observed. It proves that there is no ammonia or other labile N-containing species in the reactor, the electrolyte solution, or the catalysts. However, 1H NMR spectrum from the experiment with 15N2 depicted distinct doublet coupling of 15N in 15NH4+ and weak triplet peaks of 14N in 14NH4+. It revealed that the nitrogen atoms in the synthesized ammonia mainly contributed from the feeding gas of N2. The small quantity of 14N atoms may come from the background contamination of 14N2 in the feeding gas or the breaking of the doped nitrogen in the S/N-codoped carbon cloth.
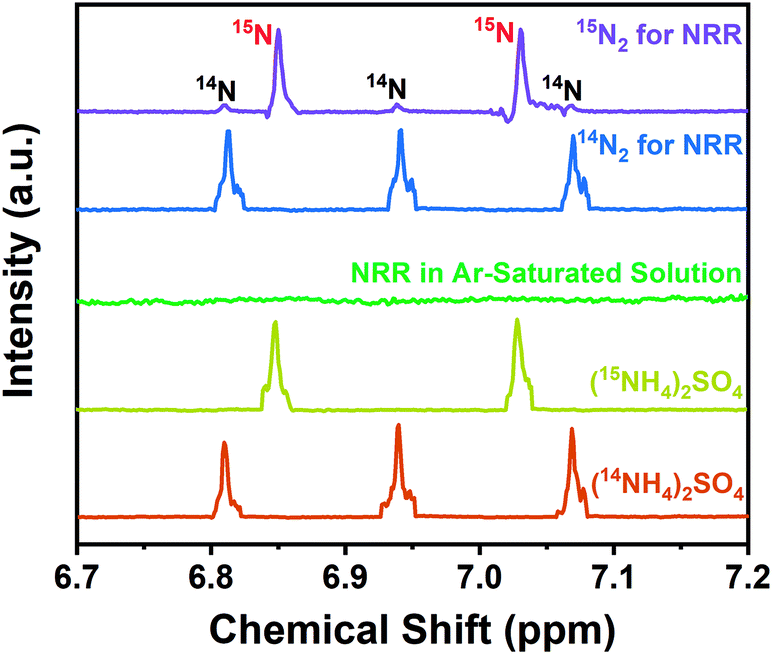 | ||
| Fig. 5 1H NMR spectra of both 14NH4+ and 15NH4+ produced from the NRR using 14N2, 15N2, and Ar as the feeding gas and using the CC-APS 800 as the electrocatalyst. | ||
The durability of CC-APS 800 was also tested by cyclic electrolysis at −0.3 V (details in ESI†). As shown in Fig. 4b, CC-APS 800 exhibited highly reproducible NH3 yield rates and FEs in 20 h cycles. After the 20 h cycles, the NH3 yield rate and FE of CC-APS 800 only decreased by 3.4% and 1.4%, respectively. Besides, an uninterrupted electrolysis test of CC-APS 800 for 20 h was also operated (Fig. S11a†). Illegible change in current density was observed after the first durability test, proving the long-time reliability of CC-APS 800. Repeating the NRR test for another 20 h, the current density gradually increased from 5.0 to 5.9 mA cm−2 (Fig. S11b†), but the yield rate and FE decreased to 8.36 × 10−10 mol cm−2 s−1 and 7.36%, respectively (Fig. S12†). The SEM images of the degenerated catalyst (Fig. S13†) revealed negligible change in the surface morphology. It indicates that the passivation of the S/N-codoped carbon cloth may lead from a chemical deactivation rather than the changes in physical structures. Besides, CC-APS 800 after electrolysis performed a very similar Raman spectrum with that of original catalyst (Fig. S14†). After electrolysis in acidic solutions for 40 h, the intensity ratio of the D band and G band (ID![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :IG) of CC-APS 800 decreased very slightly from 1.17 to 1.16, which demonstrated the defects were still abundant in the deactivated catalyst. Furthermore, the XPS spectra of CC-APS 800 after two times durability tests (Fig. S15 and Table S3†) revealed negligible changes in the total S and N content. Even the total content of pyridinic and pyrrolic N is maintained. However, after long-term electrolysis, the ratio of pyridinic N increased to 54.23% in the total N content, while the pyrrolic N decreased to 14.16%. It can be speculated that pyrrolic-N atoms exhibited better performance for NRR than those of pyridinic-N atoms in the N–S co-doped CC. This unique phenomenon has rarely been observed in previous N-doped carbon materials, which may result from the synergistic effect of co-doping of S atoms.
:IG) of CC-APS 800 decreased very slightly from 1.17 to 1.16, which demonstrated the defects were still abundant in the deactivated catalyst. Furthermore, the XPS spectra of CC-APS 800 after two times durability tests (Fig. S15 and Table S3†) revealed negligible changes in the total S and N content. Even the total content of pyridinic and pyrrolic N is maintained. However, after long-term electrolysis, the ratio of pyridinic N increased to 54.23% in the total N content, while the pyrrolic N decreased to 14.16%. It can be speculated that pyrrolic-N atoms exhibited better performance for NRR than those of pyridinic-N atoms in the N–S co-doped CC. This unique phenomenon has rarely been observed in previous N-doped carbon materials, which may result from the synergistic effect of co-doping of S atoms.
Under the same conditions, the NRR performance of the CC-APS X was evaluated at −0.3 V vs. RHE (Fig. 4c). Because of the lack of defects and active heteroatoms, both the pristine CC and CC-APS 200 exhibit low yield (1.61 and 4.33 × 10−11 mol s−1 cm−2, respectively) and FE (1.15% and 2.04%, respectively). CC-APS 400 performed an improved NH3 yield of 3.72 × 10−10 mol s−1 cm−2 and FE of 6.98%. As for CC-APS 600, the NH3 production and FE have been further enhanced to 6.36 × 10−10 mol s−1 cm−2 and 7.32%, respectively. The change of the NRR performance is consistent with that of S and N content in CC-APS X, confirming that the co-doping of S–N heteroatom pairs may enhance the NRR performance of the carbon through a synergistic effect.60–62 The doping quantity of N in CC-APS 800 is higher than many other N-doped carbon materials prepared through conventional thermochemical methods,63–65 suggesting that introducing S as a secondary dopant may benefit the co-doping of N in carbon materials.
Fig. 4d displays the UV-vis absorption spectra of the electrolyte incubated by the indophenol blue method after electrolysis at −0.3 V vs. RHE for 2 h using different catalysts. The result of the NRR test without any CC catalyst (Fig. 4d, 0.2 M Na2SO4 + 0.05 M H2SO4) proves that the Pt electrode holder lacks the ability of NRR in experimental conditions. Besides, there is no product in the Ar atmosphere for 2 h (Fig. S16†), which demonstrated that NH3 was produced exclusively from the electrochemical process with N2 gas bubbling on the CC-APS 800 electrode. For another comparison, CC treated in the N2 atmosphere at 800 °C without APS was fabricated, which is signed as CC-800. The XPS spectra of CC-800 (Fig. S17†) displays a weak peak of N 1s and no observable peak of S 2p. The ratio of N/C content is 2.07 at%, which is close to the value of the pristine CC. The result indicates the N2 gas flow is not an available source for the N doping in CC-APS 800. The low ratio of ID/IG (Fig. S18†) and the weak ability of NRR (r = 2.33 × 10−11 mol cm−2 s−1, FE = 2.05%) proves that N2 lacks the activity to form defects or introduce N atoms in the CC.
Comparatively speaking, CC-APS 800 displays an FE higher than the pristine CC (1.15%) and defect-rich commercial CC (6.92% (ref. 30)), which even exceeds the N-doped porous carbon (4.2% (ref. 16)), S-doped graphene (7.07% (ref. 66)), and N–P codoped porous carbon (0.075% (ref. 17) and 4.2% (ref. 16)). Some carbon-based catalysts with low dimensions for NRR (graphene, carbon nanotubes, porous carbon nanosheets, carbon nano spikes, etc.) may suffer from high preparation costs, weak stability, and high dependency on supporting or binding substances.67 In contrast, the self-standing CC-APS 800 catalyst displays high mechanical strength but low costs, which is applicable for scaling-up and further commercialization.
A series of electrochemical analysis has been applied to characterize the electrochemical performance of the resultant samples. Electrolysis tests of CC-APS X in Ar or N2 atmosphere were operated to measure the instant electrochemical response to N2 (Fig. 6a). The current density of CC-APS X in the acidic electrolyte solution reflects the mixture reaction of NRR and HER. Once switching the bubbling gas from Ar to N2, the current density of CC-APS 400, CC-APS 600, and CC-APS 800 ascended quickly by 1.4%, 5.5%, and 6.7%, respectively. CC-APS 800 performs a lower current density of HER than CC-APS 600 (under Ar bubbling), showing the side reaction of HER has been suppressed in CC-APS 800. These results are consistent with the NRR performance of CC-ASP X (Fig. 4c), indicating the abundant electrochemical cites for nitrogen reduction on the surface of CC-APS 800. What's more, the linear sweep voltammetry (LSV) curves of CC-APS X in N2 or Ar gas saturated electrolyte have been illustrated in Fig. S19.† A high current density (j = 4.8 mA cm−2) at −0.3 V vs. RHE in CC-APS 800 in N2 saturated condition indicates the robust electroreduction reaction. The LSV curves in the different atmospheres confirmed that CC-APS 800 is electrochemically active over N2 within a wide potential range.
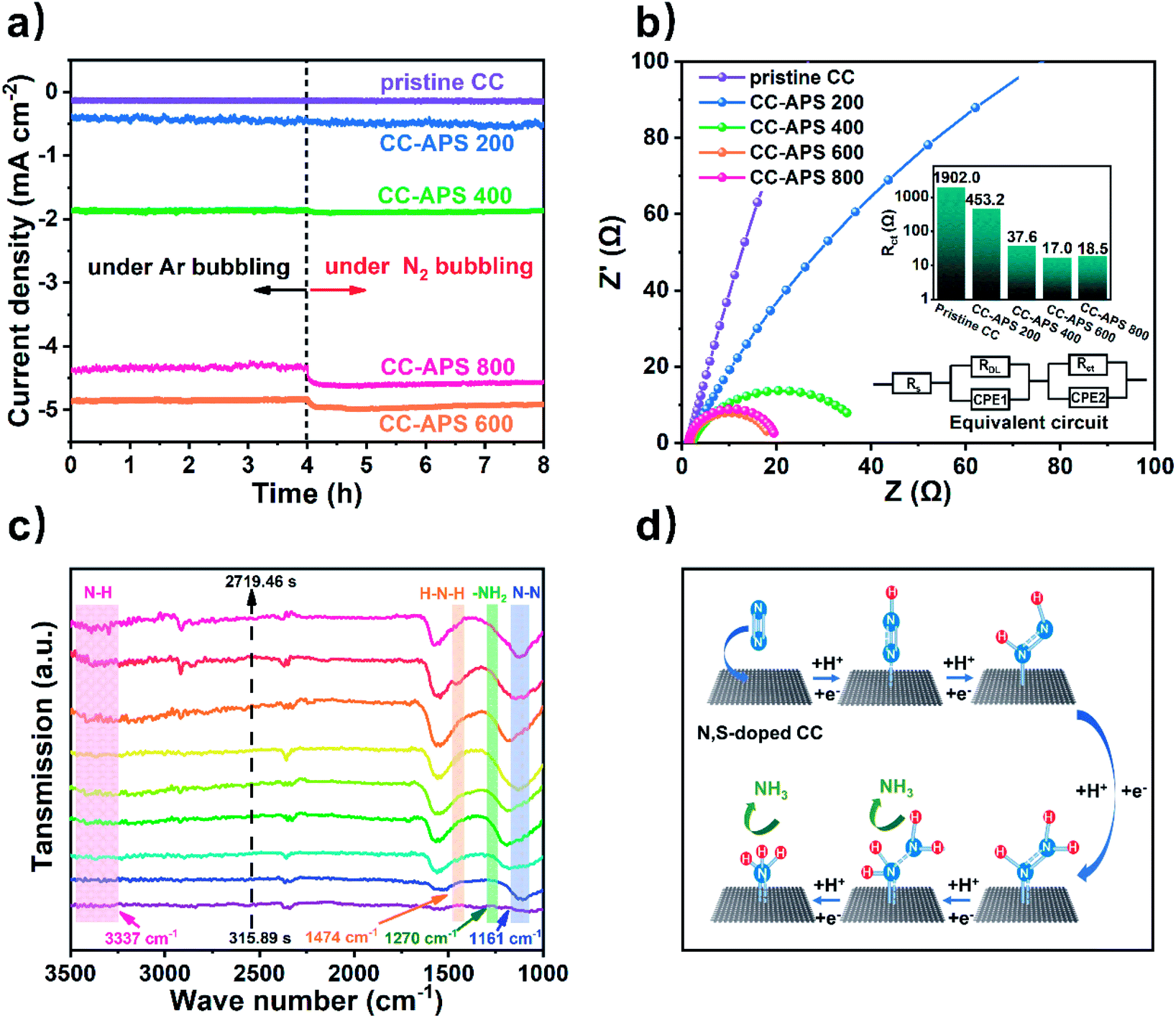 | ||
| Fig. 6 (a) I–t curves under −0.3 V vs. RHE of the prepared catalyst with Ar or N2 gas bubbling and (b) the electrochemical impedance spectra under −0.3 V vs. RHE in the N2 saturated electrolyte. (c) The electrochemical in situ FTIR spectra on CC-APS 800 in NRR. (d) Associative mechanism of the NRR process on CC-APS 800. | ||
To further analyze the electrochemical kinetics on CC-APS X, electrochemical impedance spectroscopy (EIS) tests have been operated to measure the electrochemical resistances of CC-APS X at −0.3 V vs. RHE (Fig. 6b). An arc on low and middle frequencies can be observed in the Nyquist plot, reflecting the hybrid electron transfer resistance (Rct) of HER and NRR in CC-APS X. Apparently, as the temperature of the heating process increases, the value of Rct inclines from 1902.0 to 17.0 Ω, indicating the electron transfer activity in the resultant sample has been dramatically enhanced. However, the value of Rct rises slightly to 18.5 Ω in CC-APS 800. It is because the abundant production of defects and heteroatoms stimulates the NRR but suppresses the dominant HER, and thus heightens the total hybrid resistance of Rct.
Note that N2H4 was not detected in the electrolytes after the electrocatalysis of CC-APS X (Fig. S20†), implying that CC-APS 800 has excellent selectivity for the NRR. The specific NRR pathway in CC-APS 800 is further investigated by electrochemical in situ Fourier transform infrared spectroscopy (in situ FTIR). Fig. 6c shows the electrochemical in situ FTIR spectra that have been observed on the surface of CC-APS 800 at −0.3 V vs. RHE for 2719.46 s in the N2 saturated electrolyte solution. The absorption at 1161 cm−1 is assigned to the N–N stretching, the intensity of which increased with increasing reaction time. Simultaneously, three weak absorptions at 1270, 1474, and 3337 cm−1 were observed, which are attributed to –NH2 wagging, H–N–H bending, and N–H stretching of H–N–H bending, respectively.17 These results verified that N2Hy (1 ≤ y ≤ 4) species were formed on the surface of CC-APS 800 in the NRR process, suggesting that the production of NH3 in CC-APS 800 follows an associative mechanism (Fig. 6d). Through this reaction pathway, the N2 molecules are initially adsorbed on the surface of CC-APS 800. Then, protons get attached to N2 to weaken the robust NN bond and form a –NN–H bond. In the third step, another proton further hydrogenates the weakened –NN–H bond and forms a –H–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–H bond. Thereafter, –H–N–NH2 occurred with the continuous electron transfer and hydrogenation of –H–NN–H intermediates. Eventually, NH3 is produced on the surface of CC-APS 800, and the nitrogen is electrochemically fixed under ambient conditions. The technique of in situ FTIR is powerful for detecting the transient states in the NRR process, through which the reaction pathway can be revealed directly.19,20,68 The NRR associated reaction path of S/N-codoped carbon cloth (NN → –NN–H → –HNNH → –HN–NH2 → NH–NH3) is consistent with many doped carbon materials in recent reports.19,20,69
N–H bond. Thereafter, –H–N–NH2 occurred with the continuous electron transfer and hydrogenation of –H–NN–H intermediates. Eventually, NH3 is produced on the surface of CC-APS 800, and the nitrogen is electrochemically fixed under ambient conditions. The technique of in situ FTIR is powerful for detecting the transient states in the NRR process, through which the reaction pathway can be revealed directly.19,20,68 The NRR associated reaction path of S/N-codoped carbon cloth (NN → –NN–H → –HNNH → –HN–NH2 → NH–NH3) is consistent with many doped carbon materials in recent reports.19,20,69
Conclusion
In this paper, we have firstly prepared an S/N dual-doped carbon cloth catalyst by a novel one-pot thermochemical method. Different from the conventional techniques, S/N elements were facilely introduced into carbon cloth via the co-calcination of APS and carbon cloth for the first time. The catalysts with different electrochemical performance can be obtained controllably by adjusting the calcination temperature. The carbon cloth treated with APS at 800 °C (CC-APS 800) exhibited the highest ammonia yield of 9.87 × 10−10 mol s−1 cm−2 with an excellent faradaic efficiency of 8.11% in 0.05 M H2SO4 at the applied potential of −0.3 V vs. RHE. As expected, there was no hydrazine formation during the NRR tests. Abundant S/N heteroatoms and the defects on CC-APS 800 were probably as the active and stable electrocatalytic sites for nitrogen reduction to ammonia. Moreover, the electrochemical in situ FTIR spectra of CC-APS 800 further revealed a very possible mechanism of the associated hydrogenation pathway for the electro-conversion of N2 to NH3. Our work may provide a new guideline for designing metal-free self-supporting electrocatalysts with high efficiency for the NRR and other applications.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFA0901300) and the National Natural Science Foundation of China (No. 51778562).References
- B. H. R. Suryanto, H. L. Du, D. B. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- C. X. Guo, J. R. Ran, A. Vasileff and S. Z. Qiao, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- J. Y. Zheng, Y. H. Lyu, M. Qiao, R. L. Wang, Y. Y. Zhou, H. Li, C. Chen, Y. F. Li, H. J. Zhou and S. P. Jiang, Chem, 2019, 5, 617–633 CAS.
- Y. Yao, S. q. Zhu, H. j. Wang, H. Li and M. h. Shao, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS PubMed.
- K. Kugler, M. Luhn, J. A. Schramm, K. Rahimi and M. Wessling, Phys. Chem. Chem. Phys., 2015, 17, 3768–3782 RSC.
- H. H. Huang, L. Xia, X. F. Shi, A. M. Asiri and X. P. Sun, Chem. Commun., 2018, 54, 11427–11430 RSC.
- K. Chu, Y. P. Liu, Y. B. Li, H. Zhang and Y. Tian, J. Mater. Chem. A, 2019, 9, 4389–4394 RSC.
- K. Chu, Y. P. Liu, Y. B. Li, J. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 31806–31815 CrossRef CAS PubMed.
- K. Chu, Y. P. Liu, Y. B. Li, Y. L. Guo, Y. Tian and H. Zhang, Appl. Catal., B, 2020, 264, 31806–31815 CrossRef.
- R. Zhang, X. Ren, X. F. Shi, F. Y. Xie, B. Z. Zheng, X. D. Guo and X. P. Sun, ACS Appl. Mater. Interfaces, 2018, 10, 28251–28255 CrossRef CAS PubMed.
- L. Zhang, X. Q. Ji, X. Ren, Y. J. Ma, X. F. Shi, Z. Q. Tian, A. M. Asiri, L. Chen, B. Tang and X. P. Sun, Adv. Mater., 2018, 30, 1800191 CrossRef PubMed.
- R. Zhang, Y. Zhang, X. Ren, Y. L. Luo and X. P. Sun, ACS Sustainable Chem. Eng., 2018, 6, 9545–9549 CrossRef CAS.
- X. J. Zhu, Z. C. Liu, Q. Liu, Y. L. Luo, X. F. Shi, A. M. Asiri, Y. P. Wu and X. P. Sun, Chem. Commun., 2018, 54, 11332–11335 RSC.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839–2855 RSC.
- Y. M. Liu, Y. Su, X. Quan, X. F. Fan, S. Chen, H. T. Yu, H. M. Zhao, Y. B. Zhang and J. J. Zhao, ACS Catal., 2018, 8, 1186–1191 CrossRef CAS.
- L. Xia, J. J. Yang, H. B. Wang, R. B. Zhao, H. Y. Chen, W. H. Fang, A. M. Asiri, F. Y. Xie, G. L. Cui and X. P. Sun, Chem. Commun., 2019, 55, 3371–3374 RSC.
- J. X. Zhao, J. J. Yang, L. Ji, H. B. Wang, H. Y. Chen, Z. G. Niu, Q. Liu, T. S. Li, G. L. Cui and X. P. Sun, Chem. Commun., 2019, 55, 4266–4269 RSC.
- X. M. Yu, P. Han, Z. X. Wei, L. S. Huang, Z. X. Gu, S. J. Peng, J. M. Ma and G. F. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS.
- P. F. Song, H. Wang, L. Kang, B. C. Ran, H. H. Song and R. M. Wang, Chem. Commun., 2019, 55, 687–690 RSC.
- P. F. Song, L. Kang, H. Wang, R. Guo and R. M. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 12408–12414 CrossRef CAS PubMed.
- C. Chen, D. F. Yan, Y. Wang, Y. Y. Zhou, Y. Q. Zou, Y. F. Li and S. Y. Wang, Small, 2019, 15, 1805029 CrossRef PubMed.
- Y. Kong, Y. Li, B. Yang, Z. J. Li, Y. Yao, J. G. Lu, L. C. Lei, Z. H. Wen, M. H. Shao and Y. Hou, J. Mater. Chem. A, 2019, 7, 26272–26278 RSC.
- J. J. Li, Y. M. Zhang, X. H. Zhang, J. Z. Huang, J. C. Han, Z. H. Zhang, X. J. Han, P. Xu and B. Song, ACS Appl. Mater. Interfaces, 2017, 9, 398–405 CrossRef CAS PubMed.
- K. G. Qu, Y. Zheng, S. Dai and S. Z. Qiao, Nano Energy, 2016, 19, 373–381 CrossRef CAS.
- K. Qu, Y. Zheng, X. Zhang, K. Davey, S. Dai and S. Z. Qiao, ACS Nano, 2017, 11, 7293–7300 CrossRef CAS PubMed.
- Y. Tian, D. Xu, K. Chu, Z. Wei and W. J. Liu, J. Mater. Sci., 2020, 54, 9088–9097 CrossRef.
- X. Chen, Y. Guo, X. Du, Y. Zeng, J. Chu, C. Gong, J. Huang, C. Fan, X. Wang and M. Xiong, Adv. Energy Mater., 2020, 10, 1903172 CrossRef CAS.
- I. A. Vorsina and Y. I. Mikhailov, Russ. Chem. Bull., 1996, 45, 539–542 CrossRef.
- S. Mukherjee, D. A. Cullen, S. Karakalos, K. Liu, H. Zhang, S. Zhao, H. Xu, K. L. More, G. Wang and G. Wu, Nano Energy, 2018, 48, 217–226 CrossRef CAS.
- W. Y. Li, T. X. Wu, S. B. Zhang, Y. Y. Liu, C. J. Zhao, G. Q. Liu, G. Z. Wang, H. M. Zhang and H. J. Zhao, Chem. Commun., 2018, 54, 11188–11191 RSC.
- D. F. Yan, H. Li, C. Chen, Y. Q. Zou and S. Y. Wang, Small Methods, 2019, 3, 1800331 CrossRef.
- B. B. Huang, X. Hu, Y. C. Liu, W. Qi and Z. L. Xie, J. Power Sources, 2019, 413, 408–417 CrossRef CAS.
- J. Geng, H. J. Zhang, S. W. Yao, C. L. Cai, Z. F. Ma, J. C. Wang, H. B. Wei and J. H. Yang, RSC Adv., 2019, 9, 24770–24776 RSC.
- C. J. Zhao, S. B. Zhang, M. M. Han, X. Zhang, Y. Y. Liu, W. Y. Li, C. Chen, G. Z. Wang, H. M. Zhang and H. J. Zhao, ACS Energy Lett., 2019, 4, 377–383 CrossRef CAS.
- A. M. El-Sawy, I. M. Mosa, D. Su, C. J. Guild, S. Khalid, R. Joesten, J. F. Rusling and S. L. Suib, Adv. Energy Mater., 2016, 6, 1501966 CrossRef.
- Y. Zheng, Y. Jiao, S. Qiao and A. Vasileff, Angew. Chem., Int. Ed., 2017, 130, 7568–7579 Search PubMed.
- N. Habibi, H. Arandiyan and M. Rezaei, RSC Adv., 2016, 6, 29576–29585 RSC.
- X. L. Zheng, Y. F. Ji, J. Tang, J. Y. Wang, B. F. Liu, H.-G. Steinrück, K. Lim, Y. Z. Li, M. F. Toney and K. R. Chan, Nat. Catal., 2019, 2, 55–61 CrossRef CAS.
- J. Y. Zheng, Y. H. Lyu, M. Qiao, R. L. Wang, Y. Y. Zhou, H. Li, C. Chen, Y. F. Li, H. J. Zhou, S. P. Jiang and S. Y. Wang, Chem, 2019, 5, 617–633 CAS.
- C. C. Li and S. A. Cheng, Crit. Rev. Biotechnol., 2019, 39, 1015–1030 CrossRef CAS PubMed.
- C. J. Zhao, S. B. Zhang, M. M. Han, X. Zhang, Y. Y. Liu, W. Y. Li, C. Chen, G. Z. Wang, H. M. Zhang and H. J. Zhao, ACS Energy Lett., 2019, 4, 377–383 CrossRef CAS.
- L. Xia, X. Wu, Y. Wang, Z. Niu, Q. Liu, T. Li, X. Shi, A. M. Asiri and X. Sun, Small Methods, 2019, 3, 1800251 CrossRef.
- K. Mamtani, D. Jain, D. Dogu, V. Gustin, S. Gunduz, A. C. Co and U. S. Ozkan, Appl. Catal., B, 2018, 220, 88–97 CrossRef CAS.
- C. A. Cooper, R. J. Young and M. Halsall, Composites, Part A, 2001, 32, 401–411 CrossRef.
- A. D. Tan, K. Wan, Y. F. Wang, Z. Y. Fu and Z. X. Liang, Catal. Sci. Technol., 2018, 8, 335–343 RSC.
- C. D. Lv, Y. M. Qian, C. S. Yan, Y. Ding, Y. Y. Liu, G. Chen and G. H. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246–10250 CrossRef CAS PubMed.
- X. H. Li, T. S. Li, Y. J. Ma, Q. Wei, W. B. Qiu, H. R. Guo, X. F. Shi, P. Zhang, A. M. Asiri and L. Chen, Adv. Energy Mater., 2018, 8, 1801357 CrossRef.
- L. Q. Li, C. Tang, D. Z. Yao, Y. Zheng and S. Z. Qiao, ACS Energy Lett., 2019, 4, 2111–2116 CrossRef CAS.
- C. Tang and S. Z. Qiao, Joule, 2019, 3, 1573–1575 CrossRef.
- C. Tang and S. Z. Qiao, Chem. Soc. Rev., 2019, 48, 3166–3180 RSC.
- C. J. Van der Ham, M. T. Koper and D. G. Hetterscheid, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- S. Zhao, D. W. Wang, R. Amal and L. J. Dai, Adv. Mater., 2019, 31, 1801526 CrossRef PubMed.
- W. S. Zhi, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 4998 CrossRef PubMed.
- S. Z. Andersen, V. Čolić, S. Yang, J. A. Schwalbe, A. C. Nielander, J. M. McEnaney, K. Enemark-Rasmussen, J. G. Baker, A. R. Singh and B. A. Rohr, Nature, 2019, 570, 504–508 CrossRef CAS PubMed.
- B. H. Suryanto, H. L. Du, D. B. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- C. Chen, D. F. Yan, Y. Wang, Y. Y. Zhou, Y. Q. Zou, Y. F. Li and S. Y. Wang, Small, 2019, 15, 1805029 CrossRef PubMed.
- R. Dabundo, M. F. Lehmann, L. Treibergs, C. R. Tobias, M. A. Altabet, P. H. Moisander and J. Granger, PLoS One, 2014, 9, 110335 CrossRef PubMed.
- L. Ma, Y. Cheng, G. Cavataio, R. W. McCabe, L. Fu and J. Li, Appl. Catal., B, 2014, 156, 428–437 CrossRef.
- L. Ma, Y. S. Cheng, G. Cavataio, R. W. McCabe, L. X. Fu and J. H. Li, Chem. Eng. J., 2013, 225, 323–330 CrossRef CAS.
- Y. Tian, D. Z. Xu, K. Chu, Z. Wei and W. M. Liu, J. Mater. Sci., 2019, 54, 9088–9097 CrossRef CAS.
- J. C. Li, X. P. Qin, P. X. Hou, M. Cheng, C. Shi, C. Liu, H. M. Cheng and M. H. Shao, Carbon, 2019, 147, 303–311 CrossRef CAS.
- X. R. Chen, Y. T. Guo, X. C. Du, Y. S. Zeng, J. W. Chu, C. H. Gong, J. W. Huang, C. Fan, X. F. Wang and F. Xiong, Adv. Energy Mater., 2019, 122, 1903172 Search PubMed.
- X. F. Sima, S. F. Jiang, X. C. Shen and H. Jiang, Ind. Eng. Chem. Res., 2019, 58, 13868–13878 CrossRef CAS.
- X. Xin, H. L. Qin, H. P. Cong and S. H. Yu, Langmuir, 2018, 34, 4952–4961 CrossRef CAS PubMed.
- J. J. Wu, C. Wen, X. L. Zou, J. Jimenez, J. Sun, Y. J. Xia, M.-T. Fonseca Rodrigues, S. Vinod, J. Zhong, N. Chopra, I. N. Odeh, G. Q. Ding, J. Lauterbach and P. M. Ajayan, ACS Catal., 2017, 7, 4497–4503 CrossRef CAS.
- H. Y. Chen, X. J. Zhu, H. Huang, H. B. Wang, T. Wang, R. B. Zhao, H. G. Zheng, A. M. Asiri, Y. L. Luo and X. P. Sun, Chem. Commun., 2019, 55, 3152–3155 RSC.
- S. L. Zhao, D. W. Wang, R. Amal and L. M. Dai, Adv. Mater., 2019, 31, 1801526 CrossRef PubMed.
- P. F. Song, H. Wang, X. M. Cao, N. Liu, Q. Wang and R. M. Wang, ChemElectroChem, 2020, 7, 212–216 CrossRef CAS.
- C. V. S. Kumar and V. Subramanian, Phys. Chem. Chem. Phys., 2017, 19, 15377–15387 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, the elemental composition of the catalysts based on the XPS spectra, the schematic illustration of the fabrication of CC-APS 800; TGA curves of CC, APS, and CC-APS composite; Raman and FTIR spectra, photos of contact angle tests, SEM and EDS images, CV and LSV curves of the samples, UV-vis absorption and calibration curves of NH3–N and N2H4·H2O, time-dependent current density curves and NH3 yield. See DOI: 10.1039/d0ra00155d |
| This journal is © The Royal Society of Chemistry 2020 |