
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis at room temperature of epoxides and linalool derivative pyrans in monolacunary Na7PW11O39-catalyzed oxidation reactions by hydrogen peroxide†
Castelo B. Vilanculo *a,
Márcio J. Da Silvab,
Milena Galdino Teixeirab and
Jesus Avendano Villarrealc
*a,
Márcio J. Da Silvab,
Milena Galdino Teixeirab and
Jesus Avendano Villarrealc
aChemistry Department, Pedagogic University of Mozambique, FCNM, Campus of Lhanguene, Av. de Moçambique, Km 1, Maputo, 4040, Mozambique. E-mail: castelovilanculo@gmail.com; Tel: +258 825573337
bChemistry Department, Federal University of Viçosa, Minas Gerais State 36590-000, Brazil
cChemistry Department Federal University of Minas Gerais, Minas Gerais State 31270-901, Brazil
First published on 21st February 2020
Abstract
In this work, we describe a new one-pot synthesis route of valuable linalool oxidation derivatives (i.e., 2-(5-methyl-5-vinyltetrahydrofuran-2-yl propan-2-ol) (1a)), 2,2,6-trimethyl-6-vinyltetrahydro-2H-pyran-3-ol (1b) and diepoxide (1c), using a green oxidant (i.e., hydrogen peroxide) under mild conditions (i.e., room temperature). Lacunar Keggin heteropolyacid salts were the catalysts investigated in this reaction. Among them, Na7PW11O39 was the most active and selective toward oxidation products. All the catalysts were characterized by FT-IR, TG/DSC, BET, XRD analyses and potentiometric titration. The main reaction parameters were assessed. Special attention was dedicated to correlating the composition and properties of the catalysts and their activity.
1. Introduction
The oxidation of terpenic alcohols, which are an abundant, renewable and attractive raw material, provide compounds useful for the synthesis of fine chemicals, fragrances, perfumes, and agrochemicals.1,2 Linalool is a tertiary alcohol whose cyclization derivatives and epoxides are highly valuable due to their chiral building blocks for the synthesis of drugs.3,4 The cyclization of linalool is an intramolecular process that results in the formation of tetrahydropyran and tetrahydrofuran derivatives, which are compounds with potential biological activity.5–7Several strategies have been developed to cyclize the linalool. Commonly, the first step of linalool cyclization requires a preliminary activation of the double bond through its transformation in epoxide or halogenate derivatives, which cyclizes to give the corresponding ether derivatives containing an additional hydroxyl or halogen functional group.8,9 Nonetheless, these multistep processes have low atomic efficiency and involve the use of environmentally hazardous stoichiometric oxidants, resulting in residual wastes.10
In this regard, to develop oxidative routes of linalool based on hydrogen peroxide, an inexpensive, atom-efficient and green oxidant that generates only water as a by-product is a challenge to overcome.11,12 Nonetheless, hydrogen peroxide requires the presence of a metal catalyst to be activated.13
Among the various catalysts used in oxidations with hydrogen peroxide, polyoxometalates should be highlighted due to their high compound versatility that has been effective in homogeneous and heterogeneous processes.14–16 Several heteropolyacids have redox properties that characterize them as an efficient catalyst in oxidation reactions with oxygen or hydrogen peroxide.17
Keggin heteropolyacids (HPAs) are the most used polyoxometalates in catalysis. However, they are strong Brønsted acids, a feature that compromises their use as a catalyst in oxidations of monoterpenes, which are substrates susceptible to acid-catalyzed concurrent reactions such as carbon skeletal rearrangement and nucleophilic addition.18,19 To circumvent this drawback, a rarely explored approach is to convert Keggin HPAs to neutral salts, exchanging their acidic protons by metal cations.20,21 Moreover, the removal of one MO unit of heteropolyanions (i.e., WO or MoO) results in a lacunar salt catalyst that may be more active than its precursor with saturated anion.22–24
The synthesis of linalool oxides and diepoxides, using lacunar polyoxometalates as catalysts, has not yet been reported in the literature. In this work, we present an innovative method of one-pot synthesis of linalool oxides with high conversion and selectivity, using a lacunar polyoxometalate as catalyst and hydrogen peroxide as an oxidant.
We synthesized three lacunar Keggin HPAs sodium salts and assessed their catalytic activity on the oxidation of linalool by hydrogen peroxide. All the lacunar salts were characterized by FT-IR, BET, XRD, TG/DSC techniques and their acidity strength were measured by n-butylamine titration. The effects of the main reaction variables were analyzed.
2. Experimental section
2.1. Chemicals
Linalool was purchased acquired by Sigma-Aldrich (99 wt%). Sodium hydrogen carbonate from Vetec (99 wt%). All the HPAs (i.e., H3PW12O40, H3PMo12O40, and H4SiW12O40; 99 wt%) were purchased from Sigma-Aldrich. Na2WO4·2H2O from Vetec (99 wt%), HCl(aq) (33 wt%) from Dinâmica and aqueous H3PO4(aq) (85 wt%) was from Sigma-Aldrich. CH3CN was acquired from Sigma (99 wt%) Aqueous hydrogen peroxide was from Alphatec (35 wt%). All reagents were used as received without further purification.2.2. Synthesis of lacunar Keggin heteropolyacid salts (Na7PW11O39)
The lacunar sodium salts preparation is very similar and they were prepared according to the reported procedure25. The only difference is the pH range used. Only the synthesis of lacunar phosphotungstate sodium salt was highlighted. Typically, 1 g of hydrate H3PW12O40 was dissolved in water (30 mL) and heated to 333 K with constant magnetic stirring. The solution's pH was adjusted to 4.8 using a NaHCO3 solution (Scheme SM1†). This results in the formation of the lacunar anion [PW11O39]7−. The solution with pH 4.8 was 3 h heated at 333 K. Finally, the Na7PW11O39 salt was obtained by solvent evaporation and recrystallization from water, followed by drying 5 h at 373 K. Once the synthesis of other lacunar salts (i.e., Na7PMo11O39, Na8SiW11O39) being very similar, it was omitted by simplification.2.3. Synthesis of saturated sodium phosphotungstate salt (Na3PW12O40)
For comparison, the sodium phosphotungstate catalyst was also synthesized.20 Na2WO4·2H2O (30 mmol, 10 g) was slowly added to 20 mL of distilled water and the mixture was magnetically stirred and warmed to 333 K. Then, after to add H3PO4(aq) (15 mmol, 1 mL) and HCl(aq) (100 mmol, 8 mL) the resulting mixture was stirred for 1 h. The white solid was washed with water and was recrystallized twice from hot water.2.4. Characterization of catalyst
Infrared spectra (FT-IR) were obtained on Varian 660-IR spectrometer at wavenumber range from 500 to 1700 cm−1, the fingerprint region of the main absorption bands of Keggin heteropolyanions. The bulk structure of the catalysts was examined by X-ray diffraction (XRD) technique on a Bruker D8-Discover diffractometer using Ni filtered Cu-kα radiation (λ = 1.5418 Å), working at 40 kV and 40 mA. The measurements were obtained with a count time of 1.0 s and in the 2θ range of 5–80 degrees.Thermogravimetric analysis (TGA) was performed using a PerkinElmer Simultaneous Thermal Analyzer (STA) 6000, to study the thermal decomposition and verify the hydration degree of the fresh catalyst. The masses of the samples used were between 10–50 mg, with a heating rate of 10 °C min−1 under nitrogen flow. Thermogram temperatures were recorded at each 0.1 °C range over a range of 30–600 °C.
The surface area and total pore volume of the catalysts were measured by N2 physisorption/desorption technique using NOVA 1200e High Speed, Automated Surface Area, and Pore Size Analyzer Quantachrome Instruments. The samples were outgassed by 1 h. The surface area was calculated by the Brunauer–Emmett–Teller equation applied to the isotherms.
Catalyst acidity was estimated by potentiometric titration, as described by Pizzio et al.26 The electrode potential variation was measured with a potentiometer (i.e. Bel, model W3B). Typically, 50 mg of HPA salt was dissolved in a CH3CN solution, magnetically stirred for 3 h and then titrated with a n-butylamine solution in toluene (ca. 0.05 mol L−1).
2.5. Identification of main reaction products
The major products were previously identified in a Shimadzu GC-2010 gas chromatographer coupled with a MS-QP 2010 mass spectrometer (i.e., electronic impact 70 eV, scanning range of m/z 50–450). Afterward, they were purified by column chromatography (silica 60G) using eluent hexane, containing 20% of ethyl acetate. Products (1a, 1b) were then characterized by 1H and 13C NMR and FT-IR (Varian FT-IR 660) spectroscopy analysis (see ESI†). The NMRs spectra were taken in CDCl3 solutions, using a Varian 600 spectrometer at 600 and 150 MHz, respectively. The chemical shifts were expressed as δ (ppm) relative to the tetramethylsilane (TMS) as an internal reference. Due to low yield, it was not possible to characterize diepoxide 1c; only their GC-MS data were obtained (see ESI†).2.6. Catalytic runs
The catalytic tests were carried out in acetonitrile under air (atmospheric pressure) in a closed three-necked glass flask (25 mL), equipped with a sampling system and a reflux condenser, and immersed in a thermostatic water bath. Typically, linalool (2.75 mmol) was solved in a CH3CN solution (ca. 10 mL), magnetically stirred and the reactions were carried out at room temperature. After the addition of the catalyst (ca. 0.33 mol%), the reaction was started and monitored during 4 h, periodically collecting aliquots and analyzing them in a GC equipment (Shimadzu 2010, FID).3. Results and discussion
3.1. Catalysts characterization
The FT-IR study provides information about the Keggin anion structure (i.e., primary structure); for instance, if it was retained or not after the removal of one WO unit. Fig. 1 shows infrared spectra of Keggin phosphotungstate salts (i.e., Na7PW11O39 and Na3PW12O40, respectively) and their parent heteropolyacid. | ||
| Fig. 1 FT-IR spectra of phosphotungstic acid, saturated sodium salt and lacunar sodium salt. | ||
The main absorption bands of H3PW12O40 were noticed at wavenumbers 1080, 980, 920, and 790 cm−1, which agree with data of literature.27 These bands were assigned to the stretching of P–Oa, W–Od, W–Ob–W and W–Oc–W bonds. The main absorption bands of Na3PW12O40 salt were like the pattern, meaning that the salt was successfully synthesized. The subscripts distinguish oxygen atoms; Ob are oxygen atoms belonging to the WO6 octahedral units sharing corners, Oc referrers to the oxygen atoms in edges, and Od are terminal oxygen atoms. All of them are connected to the tungsten atoms, while Oa are bonded to the phosphorous atom.28
The splitting of P–Oa bond absorption band at wavenumber 1080 cm−1, which resulted in two new bands (ca. 1020 and 1060 cm−1) is the main guarantee that the lacunar heteropolyanion was formed.29–31 This splitting is assigned to the decreasing symmetry of PO4 group, resultant from the removal of the WO unit.32 The same effect occurred when the Na7PMo11O39 lacunar salt was synthesized, nonetheless, the absorption bands appeared at wavenumbers 1078 and 1035 cm−1 (Fig. SM1†).33 Conversely, when the Na8SiW11O39 lacunar salt was synthesized, the stretching band of Si-Oa bond remained untouchable (Fig. SM2†).
While infrared spectra gave information about the primary structure of Keggin HPAs, XRD spectra provides data of the secondary structure. Fig. 2 shows clearly that the XRD spectrum of Na7PW11O39 lacunar salt displayed different peak patterns than those presented by parent H3PW12O40 and its saturated salt. It showed a lower level of crystallinity, with less intense peaks than the precursor heteropolyacid and the saturated phosphotungstate salt. This different crystallinity may be assigned to the exchange of protons by sodium cations and a changing on water hydration water molecules number (see Fig. 3). The other two lacunar sodium salts also presented a lower crystallinity level than acid precursors too (Fig. SM3 and SM4†).
 | ||
| Fig. 2 Powdered XRD patterns of Na7PW11O39 lacunar salt, parent H3PW12O40 and saturated Na3PW12O40. | ||
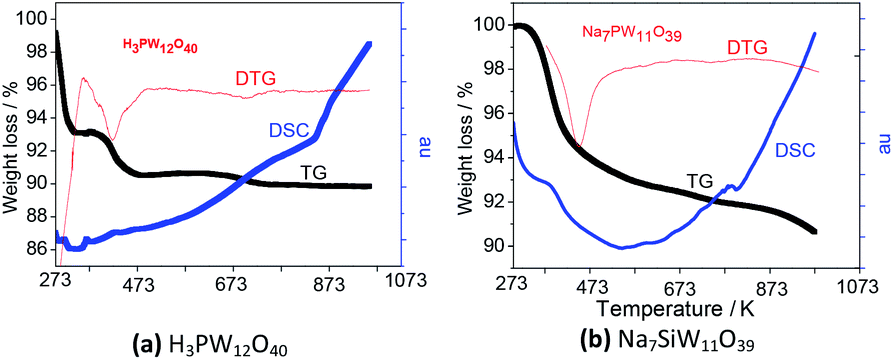 | ||
| Fig. 3 TG/TDTG and DSC curves: precursor H3PW12O40 (a) and lacunar salt Na7PW11O39 (b). | ||
TG/DTG curves obtained from lacunar Na7PW11O39 showed two regions of weight loss; the first one before 473 K assigned to loss of all water molecules. The second one, was assigned to the decomposition of P–Oa–W framework followed by the noticeable peak in DSC curves around 793 K. The final products are an oxides mixture (Fig. 3).
TG curves of the lacunar salt presented a weight loss of around 10%, likewise the precursor acid (Fig. 3a and b); nevertheless, it has occurred more quickly for the heteropolyacid sample, indicating that the lacunar salt was thermally more stable. The same behaviour was also observed on TG curves of silicotungstic and phosphomolybdic acids and their respective lacunar sodium salts (Fig. SM5 and SM6†).
The titration curves of the lacunar sodium salts and their precursor Keggin HPAs were obtained (Fig. 4). This procedure allows to classify the acidity strength of acid sites; Ei > 100 mV (very strong sites), 0 < Ei < 100 mV (strong sites), −100 < Ei < 0 (weak sites) and Ei < −100 mV (very weak sites).34
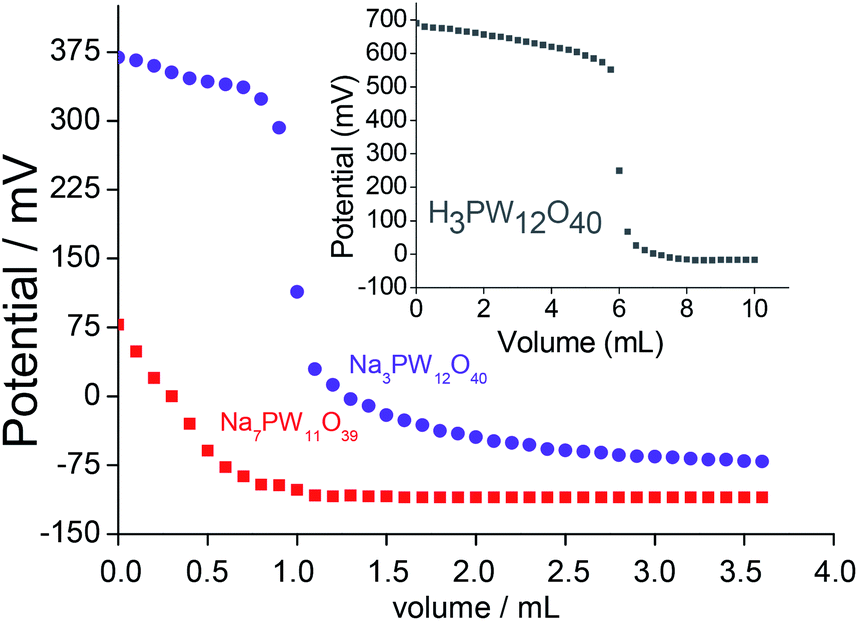 | ||
| Fig. 4 Potentiometric titration curves with n-butylamine of H3PW12O40, Na3PW12O40 and Na7PW11O39 catalysts. | ||
Regardless of the Keggin anion, the titration curves presented similar pattern; a quick decrease in the electrode potential after the addition of base minimum volume suggests that only a residual Brønsted acidity remained after the exchange of protons by the sodium cations, indicating that the protons were virtually removed.
After the initial period of the titration, the potential remained practically constant. This behaviour was completely distinct than the precursor heteropolyacids (Fig. 4, SM7 and SM8†).
The precursor HPA presented very strong acid sites (i.e., Ei = 700 mV, inset in Fig. 4); the exchange of protons by the sodium cations reduced this acidity (Ei = 375 mV), however, it was classified as very strong. Conversely, lacunar salt was less acid (Ei = 75 mV). Their strong acid sites may be assigned to the residual protons (Fig. 4).
The pore sizes presented by sodium salts were very similar to those displayed by the parent acids (ca. 1–4 m2 g−1), regardless of the Keggin anion. Aiming simplifies, only the isotherms and pores size distribution of the Na7PW11O39 salt was shown in Fig. 5. The quick initial increase corresponds to the formation of the first layer; therefore, an increase in pressure forms the second layer of adsorbed molecules, followed by another layer. The total reversibility of the adsorption–desorption isotherm was observed (i.e., absence of hysteresis cycle) for all catalysts, a condition noticed in this catalyst.
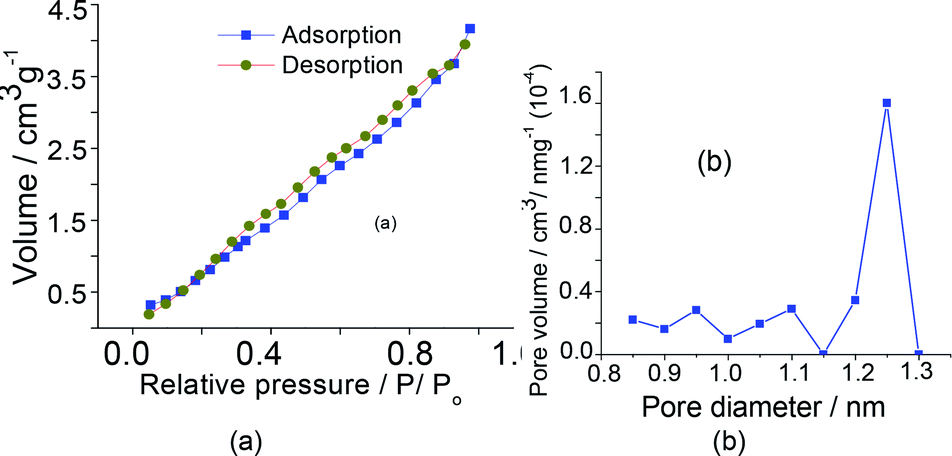 | ||
| Fig. 5 Isotherms of adsorption and desorption (a), volume and diameters porous (b) of the Na7PW11O39 catalyst. | ||
3.2. Catalytic tests
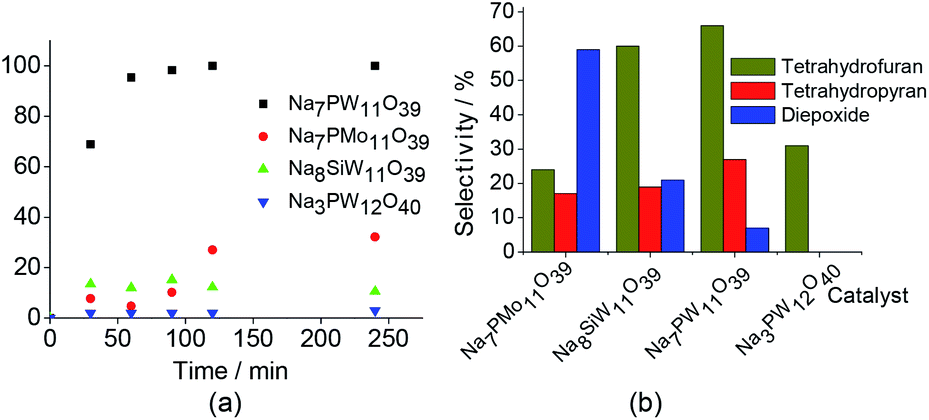 | ||
Fig. 6 Effect of catalyst nature on the conversion (a) and selectivity (b) of the linalool oxidation reaction with H2O2. aReaction conditions: linalool to H2O2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2); lacunar salt catalyst (0.33 mol%); temperature (298 K); CH3CN (10 mL). :2); lacunar salt catalyst (0.33 mol%); temperature (298 K); CH3CN (10 mL). | ||
Although not included in the Fig. 6, the H3PW11O39 -catalyzed oxidation reaction of linalool achieved only a poor conversion (ca. 9%) and no oxidation product was detected.
In addition to highest activity (conversion of ca. 100%), the lacunar Na7PW11O39 catalyst was also the most selective toward the goal-products; cis and trans 2-(5-methyl-5-vinyltetrahydrofuran-2-yl propan-2-ol) (1a), cis and trans 2,2,6-trimethyl-6-vinyltetrahydro-2H-pyran-3-ol (1b), which were formed at equimolar proportions. The diepoxide (1c) was also obtained in linalool (1) oxidation reaction (Scheme 1).6
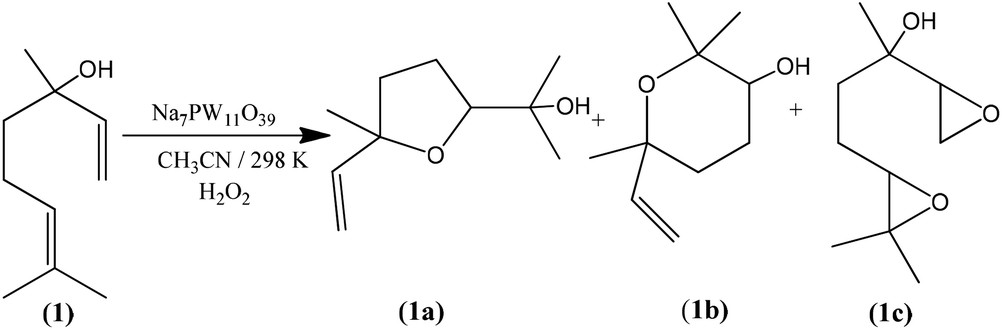 | ||
| Scheme 1 Linalool oxidation by H2O2 in presence of Na7PW11O39 catalyst. | ||
Note that the 2-(5-methyl-5-vinyltetrahydrofuran-2-yl propan-2-ol) (1a) and 2,2,6-trimethyl-6-vinyltetrahydro-2H-pyran-3-ol (1b) oxides were the major products, with selectivity of 66 and 27%, respectively (Scheme SM2†). On the other hand, the diepoxide (1c) had only a selectivity of 7% (Fig. 6b). It indicates that this catalyst system is highly selective for the synthesis of the linalool oxides. The formation of the tetrahydrofuran derivative (1a) with a five-members ring was more selective in all the reactions, excepted when the catalyst was Na7PMo11O39. Probably, (1a) is the kinetic product and thus is formed faster than (1b) product, which has a six-member ring.
Conversely, the diepoxide (1c) was the major product in Na7PMo11O39-catalyzed reaction and the secondary one in reaction with Na8SiW11O39. As we will demonstrate, the epoxide is directly involved in the formation of linalool oxides.
:1 ratio of H2O2 to linalool, we investigate what was the effect on conversion and selectivity using a lower load of oxidant (ca. 1:1.5 and 1:1, Fig. SM9†).As shown in Fig. SM9a,† the conversion of linalool to linalool oxides and diepoxide increased gradually with the increase of the amount of oxidant. The final conversions after 4 h of reaction were ca. 38%, 74% and 100%, when the proportions of H2O2 to linalool were 1:1, 1:1.5 and 1:2 respectively. Nonetheless, the final selectivity was approximately the same; 66%, 27% and 7%, for (1a), (1b) and (1c) products, respectively (Fig. SM9b†). When the reactions were carried out in the absence of the catalyst, no oxidation product was detected, regardless of the oxidant load.
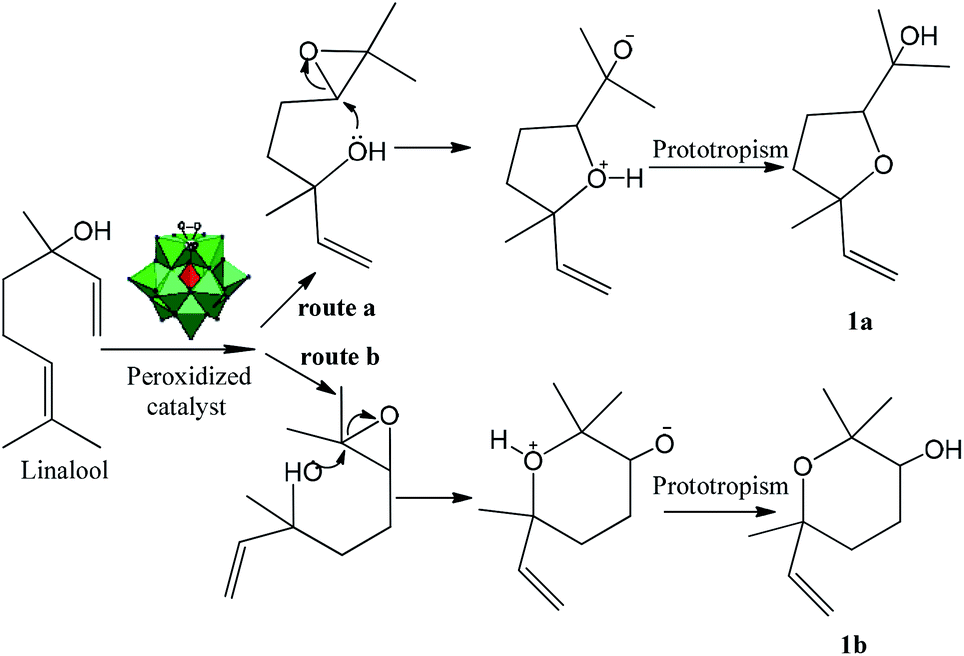 | ||
| Scheme 2 Possible reaction pathway for the formation of (1a) and (1b) linalool oxides. | ||
Thus, it is plausible that the intermediate 1 may be formed, which may transfer the oxygen atom to the double bond of the linalool. This transformation primarily results in an epoxide, which may be easily cyclized to give the corresponding ether derivatives (i.e., linalool oxides; 1a and 1b) bearing an additional hydroxyl group. Therefore, we believe that the formation of epoxide is essential for that the reaction continuance (Scheme 2). In Scheme 2, we will demonstrate how the epoxide is converted to product (1a) or (1b).
The intramolecular nucleophilic attack of the hydroxyl group belonging to linalool epoxide is the key-step that governs the reaction selectivity. After the formation of epoxide, its oxirane ring maybe then opened when the electron pairs of hydroxyl group attack the less hindered electrophilic carbon. This intramolecular cyclization step results in the tetrahydrofuran, after a proton transfer to the anionic intermediate (route a, Scheme 2).
Conversely, when the pair of electrons of the hydroxyl group attack the more hindered electrophilic carbon, the tetrahydropyran is the product obtained (route b, Scheme 2). Note that with the epoxides, the regioselectivity is not as simple, even with acid catalysts, the ANDN substitution at a primary carbon atom is very fast.35,36
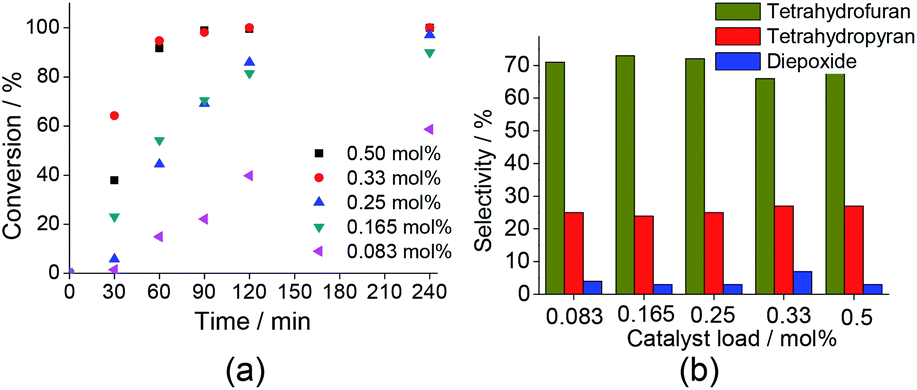 | ||
| Fig. 7 Impacts of catalyst load on the conversion (a) and products selectivity (b) of Na7PW11O39-catalayzed oxidation reaction of linalool with H2O2. aReaction conditions: linalool (2.75 mmol); H2O2 (5.5 mmol); temperature (298 K); CH3CN (10 mL). | ||
When we investigated the effect of H2O2 load, we have found that while the reaction conversions were strongly affected, the reaction selectivity remained almost unaltered. The variation of the catalyst load triggered the same effect. The reaction selectivity was practically constant, regardless of the catalyst load; conversely, an increase of the catalyst concentration of 0.083 to 0.33 mol%, raised the conversions of 59 to 100%.
Although at the studied concentration the initial rates of the reactions have been close, the exothermic character of the oxidation of linalool was confirmed by the results shown in Fig. 8a. Undoubtedly, it was verified that the linalool oxidation to oxides and diepoxide was more selective at room temperature. Nonetheless, from Fig. 8b we can also conclude that high temperatures increased the diepoxide selectivity and reduced the formation of the tetrahydrofuran derivative.
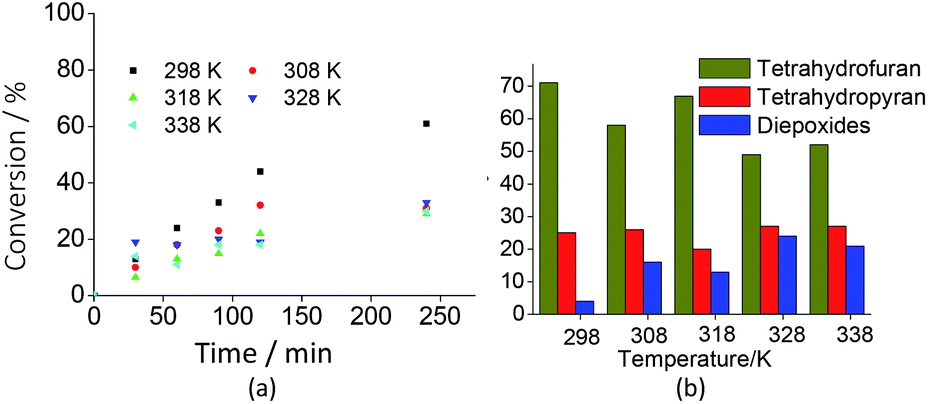 | ||
| Fig. 8 Effects of temperature on the conversion (a) and selectivity (b) of Na7PW11O39-catalyzed oxidation reactions of linalool with H2O2. aReaction conditions: linalool (2.75 mmol); H2O2 (5.5 mmol); Na7PW11O39 (0.083 mol%); CH3CN (10 mL). | ||
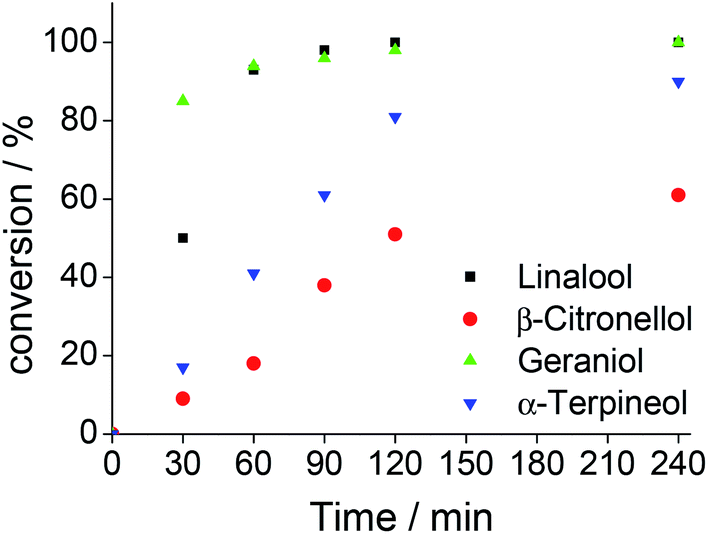 | ||
| Fig. 9 Oxidation of different terpenic alcohols with hydrogen peroxide in Na7PW11O39-catalyzed reactions. aReaction conditions: alcohol (2.75 mmol); H2O2 (5.5 mmol); Na7PW11O39 (0.33 mol%); temperature (298 K); CH3CN (10 mL). | ||
When an allylic alcohol (i.e., geraniol, Fig. SM10†) was the substrate, a complete conversion within 4 hours of reaction was achieved with a high selectivity for geraniol-epoxide (ca. 89%), meaning that only the allylic double bond of alcohol was reactive, while the more hindered double bond which remained intact.
The primary alcohol (i.e., β-citronellol, Fig. SM10†) was less active than the allylic alcohol; only a conversion of 61% was achieved, with a high epoxide selectivity. Solely traces of aldehydes were detected. The tertiary cyclic alcohol (α-terpineol, Fig. SM10†) was very reactive; a conversion close of ca. 90% was achieved with a selectivity of 39, 34 and 27% for keto-alcohol, epoxide and glycol, respectively. We suppose that the glycol was formed through the initial epoxidation of the double bond and subsequent opening of the oxirane ring of epoxide, triggered by nucleophilic attack water present in the oxidant.
4. Conclusions
In this work, we developed an efficient synthesis route of the linalool oxides using an environmentally benign oxidant (i.e., H2O2) in lacunar Na7PW11O39-catalyzed reactions at room temperature. Three valuable compounds were selectively obtained after an adequate adjust of reaction parameters; the 2-(5-methyl-5-vinyltetrahydrofuran-2-yl propan-2-ol) (cis/trans 1:1 proportion, 1a) and 2,2,6-trimethyl-6-vinyltetrahydro-2H-pyran-3-ol (cis/trans 1:1 proportion, 1b) products were the oxides obtained. A diepoxide product (1c) was also formed. The excellent performance of the catalyst was assigned to the removal of one WO unit on the saturated anion (i.e., PW12O407−), to form the catalytic active lacunar sodium Keggin phosphotungstate (Na7PW11O39). The maximum selectivity combined toward the main products was close to 100%, with a conversion virtually complete of the linalool. The process described herein has attractive aspects; the reactions were performed at room temperature, the catalyst was easily synthesized and achieved excellent catalytic performance, being also efficiently recovered and reused. Therefore, this route demonstrated to be very suitable for the conversion of linalool into linalool oxides, which are important intermediates in chiral synthesis. In addition, Na7PW11O39 catalyst efficiently converted the geraniol, β-citronellol and α-terpineol to their oxidation products (mainly epoxides) at room temperature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from CNPq and FAPEMIG (Brasil). This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001.References
- M. J. da Silva, P. H. S. Andrade, S. O. Ferreira, C. B. Vilanculo and C. M. Oliveira, Catal. Lett., 2018, 148, 2516 CrossRef CAS.
- L. A. S. Viana, G. R. N. da Silva and M. J. da Silva, Catal. Lett., 2018, 148, 374 CrossRef CAS.
- S. Serra and D. De Simeis, Catalysts, 2018, 8, 362 CrossRef.
- T. L. B Boivin, Tetrahedron, 1987, 43, 3309 CrossRef.
- B. Azaad and S. Lakshmipathi, Atmos. Environ., 2018, 189, 235 CrossRef.
- E. M. Elgendy and M. Y. Semeih, Arabian J. Chem., 2019, 12, 966 CrossRef CAS.
- M. G. Kim, S. M. Kim, J. H. Min, O. K. Kwon, M. H. Park, J. W. Park, H. I. Ahn, J. Y. Hwang, S. R. Oh, J. W. Lee and K. S. Ahn, Int. Immunopharmacol., 2019, 74, 105706 CrossRef CAS PubMed.
- K. C. Nicolaou, C. V. C. Prasad, P. K. Somers and C. K. Hwang, J. Am. Chem. Soc., 1989, 111, 5330 CrossRef CAS.
- N. M. Nasir, K. Ermanis and P. A. Clarke, Org. Biomol. Chem., 2014, 12, 3323 RSC.
- S. S. Wang and G. Y. Yang, Chem. Rev., 2015, 115(11), 4893 CrossRef CAS PubMed.
- Z. Guo, B. Liu, Q. Zhang, W. Deng and Y. Yang, Chem. Soc. Rev., 2014, 43(10), 3480 RSC.
- P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35(9), 686 CrossRef CAS PubMed.
- M. J. da Silva, L. C. de Andrade Leles, S. O. Ferreira, R. C. da Silva, K. de V. Viveiros, D. M. Chaves and P. F. Pinheiro, Chem. Select, 2019, 4, 7665 CAS.
- I. V. Kozhevnikov, J. Mol. Catal. A: Chem., 2007, 262(1–2), 86 CrossRef CAS.
- M. J. da Silva, P. H. S. Andrade, S. O. Ferreira, C. B. Vilanculo and C. M. Oliveira, Catal. Lett., 2018, 148, 2516 CrossRef CAS.
- F. Cavani, Catal. Today, 1998, 41(1–3), 73 CrossRef CAS.
- M. J. da Silva, Curr. Org. Chem., 2016, 20, 1 Search PubMed.
- A. L. P. Meireles, K. A. S. Rocha, I. V. Kozhevnikov and E. V. Gusevskaya, Appl. Catal., A, 2011, 82, 409 Search PubMed.
- R. Karcz, P. Niemiec, K. Pamin, J. Połtowicz, J. Kryściak-Czerwenka, B. D. Napruszewska, A. Michalik-Zym, M. Witko, R. Tokarz-Sobieraj and E. M. Serwicka, Appl. Catal., A, 2017, 542, 317 CrossRef CAS.
- M. J. da Silva and C. M. de Oliveira, Curr. Catal., 2018, 7, 26 CrossRef.
- L. Li, B. Liu, Z. Wu, X. Yuan and H. Luo, Chem. Eng. J., 2015, 280, 670 CrossRef CAS.
- K. Kamata, M. Kotani, K. Yamaguchi, S. Hikichi and N. Mizuno, Chem.–Eur. J., 2007, 13, 639 CrossRef CAS PubMed.
- N. C. Coronel and M. J. da Silva, J. Cluster Sci., 2018, 29, 195 CrossRef CAS.
- M. J. da Silva, L. C. de Andrade Leles, R. Natalino, S. O. Ferreira and N. C. Coronel, Catal. Lett., 2018, 148, 1202 CrossRef.
- S. Rana and K. M. Parida, Catal. Sci. Technol., 2012, 2, 979 RSC.
- R. Tayebee, Asian J. Chem., 2008, 20, 8 CAS.
- L. R. Pizzio, P. G. Vásquez, C. V. Cáceres and M. N. Blanco, Appl. Catal., A, 2003, 256, 125 CrossRef CAS.
- J. B. Moffat, Metal-Oxygen Clusters: The Surface and Catalytic Properties of Heteropoly Oxometalates, Plenum, New York, 2001 Search PubMed.
- G. P. Romanelli, J. C. Autino, M. N. Blanco and L. R. Pizzio, Appl. Catal., A, 2005, 295, 209 CrossRef CAS.
- L. R. Pizzio and M. R. Blanco, Microporous Mesoporous Mater., 2007, 103, 40 CrossRef CAS.
- D. Bajuk-Bogdanovi, I. Holclajtner-Antunonovi, M. Todorovi, U. B. Mio and J. Zakakrzewska, J. Serb. Chem. Soc., 2008, 73, 197 CrossRef.
- J. H. Choi, J. K. Kim, D. R. Park, T. H. Kang, J. H. Song and I. K. Song, J. Mol. Catal. A: Chem., 2013, 371, 111 CrossRef CAS.
- S. Das and T. Punniyamurthy, Tetrahedron Lett., 2003, 44, 6033 CrossRef CAS.
- J. Clayden, N. Greeves, S. Warren and P. Wothers. Organic Chemistry. Oxford, 2000 Search PubMed.
- T. M. khomenko, L. E. Tatarova, D. V. Korchagina and V. A. Barkhash, Russ. J. Org. Chem., 2002, 38, 498 CrossRef CAS.
- C. B. Vilanculo, M. J. da Silva, S. O. Ferreira and M. G. Teixeira, Mol. Catal., 2019, 478, 110589 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00047g |
| This journal is © The Royal Society of Chemistry 2020 |