
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLithium chloride (LiCl)-modified polyethersulfone (PES) substrate surface pore architectures on thin poly(dimethylsiloxane) (PDMS) dense layer formation and the composite membrane's performance in gas separation†
Zulfida Mohamad Hafis Mohd Shafie ab,
Abdul Latif Ahmad*a,
Siew Chun Lowa,
Sabine Rodeb and
Bouchra Belaissaouib
ab,
Abdul Latif Ahmad*a,
Siew Chun Lowa,
Sabine Rodeb and
Bouchra Belaissaouib
aSchool of Chemical Engineering, Engineering Campus, Universiti Sains Malaysia, 14300 Nibong Tebal, Penang, Malaysia. E-mail: chlatif@usm.my
bLaboratoire Réactions & Génie des Procédés (LRGP) (UMR 7274) ENSIC, Université de Lorraine, 1 Rue Grandville, 54001 Nancy, France
First published on 5th March 2020
Abstract
The use of pore forming agents has been notable for improving the water flux in a water-based separation membrane but are rarely being studied as a methodology to influence the substrate's surface architectures for composite membrane fabrication in gas separation. In this study, the influence of lithium chloride (LiCl) on the surface pore architectures and hence, the gas permeance, has been studied in both bare and composite forms with poly(dimethylsiloxane) (PDMS). 1–4 wt% of LiCl was mixed with the dope solution of PES/NMP in the ratio 0.19 and was casted via the dry–wet phase inversion method. Bare substrates were noted to possess increasingly larger surface pore sizes but at a diminishing surface pore density with maximum surface porosity at 2 wt% LiCl. The permeances were, however, significantly reduced with the increase in the LiCl content from 105![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 to 4300 GPU for N2 gas, presumably due to the thicker skin layer. Nevertheless, the porous surface morphology was confirmed and exhibited Knudsen selectivity with a CO2/N2 selectivity of about 0.8, signifying minimal gas flow resistance by the substrates. Upon coating with a similar amount of thin PDMS layer, the composite permeances retain the same trend with values from 361.9 GPU for 0 wt% LiCl substrates to 68.8 GPU for 4 wt% LiCl substrates for CO2 gas at a consistent selectivity of about 14. As the PDMS layer of the same volumes were used and no significant difference in the coating thickness was noted, the mixed influence of pore intrusion and lateral diffusion is hypothesised at the substrate–coating interface owing to the different surface pore architectures of the substrates.
300 to 4300 GPU for N2 gas, presumably due to the thicker skin layer. Nevertheless, the porous surface morphology was confirmed and exhibited Knudsen selectivity with a CO2/N2 selectivity of about 0.8, signifying minimal gas flow resistance by the substrates. Upon coating with a similar amount of thin PDMS layer, the composite permeances retain the same trend with values from 361.9 GPU for 0 wt% LiCl substrates to 68.8 GPU for 4 wt% LiCl substrates for CO2 gas at a consistent selectivity of about 14. As the PDMS layer of the same volumes were used and no significant difference in the coating thickness was noted, the mixed influence of pore intrusion and lateral diffusion is hypothesised at the substrate–coating interface owing to the different surface pore architectures of the substrates.
1. Introduction
A thin film composite (TFC) membrane, first developed by Peter S. Francis to be used as a reverse osmosis membrane, can be defined as a multilayer membrane structure made up of at least two different materials: (i) substrate support, which is usually a thick, porous, and mechanically sound structure and (ii) a selective layer with a thin and dense structure. With the advent of TFC in 1966, it became the new contender to the original Loeb–Sourirajan's anisotropic (non-composite) polymeric membranes, which significantly propelled the research and commercial interest in polymer based synthetic membranes a few years earlier.1,2 Ever since, the idea has been translated to gas separation application, with Jay M. S. Henis and Mary K. Tripodi as the pioneers in making TFC membrane economically feasible in industrial application in 1979.3As in the anisotropic membrane, gas separation is normally provided by the dense selective layer while the mechanical backbone is provided by the porous substrate. Notwithstanding the similarity, layers of TFC are fabricated separately, allowing the fabricators to have better control of the layers suited to their specific functions.4 On the other hand, separated layer fabrication methodology can allow the use of more expensive materials in the separating layer in a localized structure instead of being dispersed throughout the whole membrane. With a thinner selective layer, higher permeance value can also be achieved for highly selective polymers, which are normally permeability limited by their intrinsic physical characteristics (reduction in diffusion coefficient with tighter molecular spacing for highly selective polymer), as explained in the well-known Robeson upper bound limits.5,6
Nonetheless, with a better permeating selective layer, the impact of the substrate's mass transfer resistance would become much more prominent.7 As the intrinsic selective layer resistance decreases, the contribution by the substrate starts to become a problem. TFC also deviates from its ideal performance, which is reduced in the relative efficiency when the thinner selective layer is introduced. Geometric constrictions near to the substrate's surface pores can cause lateral diffusion through the thin film, limiting the actual permeability improvements of the whole membrane system.8,9 Depending on the layer's intrinsic properties, it has been recently reported that up to 30% of the total resistance can be contributed by lateral diffusion in the selective layer and another 5% by Knudsen diffusion in the porous substrate.7 The use of gutter layer has previously been suggested as a methodology for mitigating lateral diffusion in the dense upper layer. Nevertheless, the gutter layer can also reduce the apparent selectivity of the composite membrane, especially if the substrate is low in surface porosity.9 On the other hand, the nature of TFC fabrication requires the formation of normally non-viscous solution on top of the porous substrate. Pore intrusion of the selective layer polymer coating solution into the underlying substrate can hence significantly increase the effective separating layer thickness and significantly contribute to an increase in the gas transfer resistance at the interlayer.10,11 This also holds true for dense gutter layer formation, although to a lesser consequence, due to its higher permeability than the selective layer.
Being the gas entry point from the selective layer, the substrate surface structures are a subject of interest for composite membranes as both lateral diffusion and pore intrusion can be affected by the substrate's surface characteristics. Not to mention, the intrinsic compatibility between the layers, as an easily peeled skin coating, would surely be a poor combination for an industrially attractive membrane. Hence, the importance of the substrate layer should not be underestimated. With this enlightenment, in recent years, more and more attention has been given towards understanding and optimizing porous substrates to minimize the overall gas transport resistance. J. Wang et al. (2018) prepared polyamide selective layers through interfacial polymerization on polysulfone substrate layers with different surface pore size,12 whereby the interfacial characteristics of the bottom surface of the thin film formed were affected by the substrate's surface microstructures, although not linearly. From the theoretical point of view, Wijmans and Hao (2015) proved, through CFD, the influence of the substrate's surface pore architecture on the composite membrane with a very thin selective layer.8 In the study, the authors proposed a correlation to quantify the gas flow restriction imposed by the substrate by introducing a new dimensionless parameter named ‘Restriction Number’, NR. It is also worth noting that the pore restriction reduced the permeances but not the selectivity, hence providing an interesting area to dwell into, especially for the polymeric membranes, which are prone to the permeability-selectivity trade off. This theoretical correlation has been proven and modified by A. Ghadimi et al. (2018) to further improve the estimation's precision by incorporating pore density into the equation and was later verified with PES based substrate of different pore sizes and porosities.4
Based on the literature evaluation of the influence of porous substrate's surface pore architectures on the composite membrane formation and performance, minimal lateral diffusion can be achieved by substrates with high surface porosity and pore density but low surface pore sizes.4,8,13 On the other hand, similar characteristics can increase the effect of pore penetration by capillary action and surface wetting, depending on the chemistry of the coating solution and its underlying substrate.7,14 Together with the increased gas flow resistance of smaller pore-sized membranes,15 choosing substrates for composite membrane gas separation with optimum surface morphologies can become complicated. With selective layer thickness in the range of nanometers9 to a few micrometers,4 asymmetric ultrafiltration and microfiltration membranes could be the porous substrate candidates for this purpose as the surface pores are usually in this range (1 nm to 10 μm).16 Nevertheless, membrane fabrication methodology for water-based application has rarely studied the gas permeability of these membranes,17–19 while composite membrane gas research that uses commercial samples does not provide an insight into the fabrication methodology of these substrates in terms of materials and modifications involved, which can make it a good substrate.13 Hence, the influence of pore forming agents, usually used as hydrophilic additives for porous membrane formation in water-based application, is relatively unknown when used as porous substrates for composite membrane gas separation. D. Wu et al. (2018) tried to elucidate this idea by blending PES with hydrophilic additives to manipulate its interfacial properties and surface morphology.7 It was noted that enhanced substrate surface hydrophilicity through hydrophilic material incorporation such as PVP allows for thinner selective layer formation and adhesion with improved gas permeance for hydrophilic surface layer but too much increase in the hydrophilicity can induce excessive pore penetration. On the other hand, A. Ghadimi et al. (2018) also fabricated PES membranes using different solvents and concentrations to create substrates of varying surface architectures to validate their model on lateral diffusion.4 While their work did not incorporate any pore forming agents, the differences in the surface pore architectures were noted to influence the PEBAX coating formation and the composite's performance.
In this work, the influence of surface pore architectures of LiCl modified PES substrate layer on the PDMS coating formation and its performance for gas separation will be investigated. Unlike in water-based application research work, the substrate's bulk and surface structures, together with its influence on the coating formation and the resulting bare and composite performance, will be the main interest. LiCl was chosen as it has been reported to improve the permeability in water-based application with reduced mean pore sizes but increased the porosity of PES–DMF20 and PSf–NMP21 systems. On the contrary, LiCl can also generate a denser surface structure due to formation of complexes with NMP solvent, which significantly increases the dope solution's viscosity22,23 and hence, the kinetic hindrance during the phase inversion process. Increased viscosity of the PES–NMP system by increased PES content has been noted to exhibit larger pore size despite the surface densification. Hence, for this work, it is hypothesized that an initial increase in the surface porosity would be seen before being reduced as the substrate's surface layer became denser (Fig. 1). A thin layer of PDMS of equal volume will be introduced as a high permeance dense layer to further justify the substrate's performance in composite configuration. A higher substrate surface energy than the coating solution's surface tension would ideally suggest good wetting properties and hence, good formation of the PDMS layer with similar coating layer thickness, regardless of the substrate's surface structures. Nevertheless, pore penetration by capillary action of the coating solution on different surface pore sizes and porosities may influence the results. The results from this work would elucidate not only the effect of PES–LiCl–NMP system on substrate fabrication for gas separation but also an insight into the influence of kinetic hindrance by high viscous substrate polymer solution during phase inversion on the surface architectures.
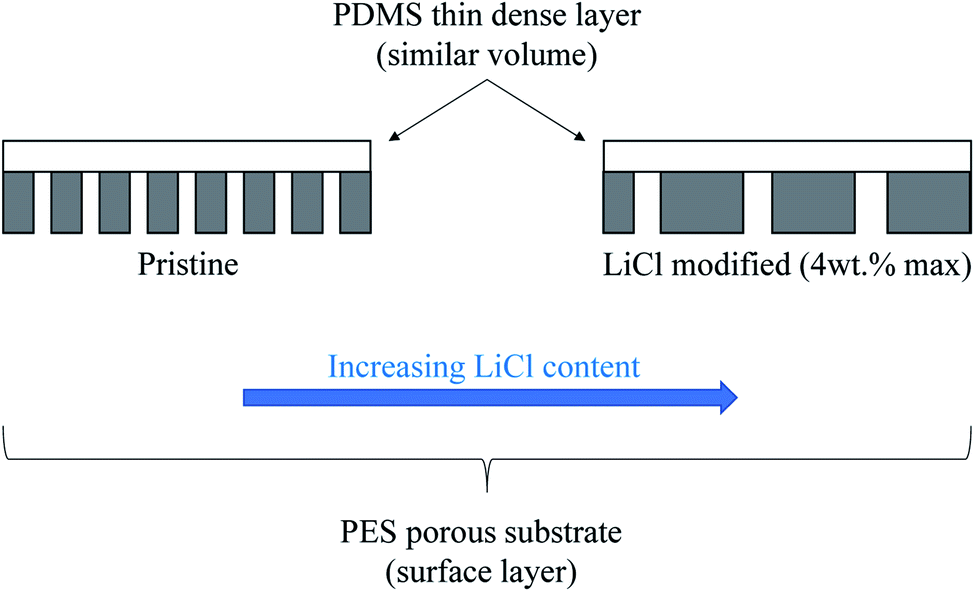 | ||
| Fig. 1 Formation of PDMS thin film on LiCl modified PES substrate. | ||
2. Experimental methodology
2.1. Materials
For substrate layer fabrication, polyethersulfone, PES (Ultrason E6020P) was purchased from BASF, while N-methyl-2-pyrrolidone, NMP (EMPLURA) and lithium chloride, LiCl (ACS Reag., Ph Eur.) were purchased from Merck. As for the PDMS surface layer, heptane (anhydrous, 99%), tetraethyl orthosilicate, TEOS (reagent grade, 98%), dibutyltin dilaurate, DBD (95%), and poly(dimethylsiloxane), PDMS (hydroxy terminated, viscosity 2550–3570 cSt) were all purchased from Sigma.2.2. Substrate layer fabrication
PES flakes were dried in a vacuum oven under the temperature of 110 °C at −0.6 bar (gauge) for about 5 h. LiCl was premixed with NMP using a magnetic stirrer for 1 h at 60 °C and 300 rpm. All the solutions were then mixed with PES flakes that were dried earlier for 18 h at 60 °C and 400 rpm. Once fully mixed, the dope solutions were placed in an ultrasonic bath for degassing purpose for another 1 h. The composition of the dope solution is summarized in Table 1. The PES/NMP mass ratios were maintained at 0.19 for all the samples, based on the results of preliminary studies, which can be found in the ESI.†| Samples | Compositions (wt%) | ||
|---|---|---|---|
| PES | NMP | LiCl | |
| Pristine | 16 | 84 | 0 |
| LiCl_1% | 16 | 83 | 1 |
| LiCl_2% | 16 | 82 | 2 |
| LiCl_3% | 16 | 81 | 3 |
| LiCl_4% | 15 | 81 | 4 |
Porous substrate layers were then fabricated using the synthesised dope solutions. Flat sheet membranes were casted using the dry–wet phase inversion method with filtered water as the non-solvent. Coagulation bath temperature was maintained at room temperature and casted with a blade at 200 μm thickness. The dry phase was maintained for 45 s before the samples were immersed in the coagulation bath for 24 h. The prepared samples were then dried at room temperature.
2.3. Dense coating fabrication
In order to test the performance of the substrate layer in the composite form, the thin film of PDMS layer was fabricated on top of the substrate. The PDMS solution was prepared using the formulation adapted from A. A. M. Salih et al. (2014).11 PDMS, DBD, and TEOS was mixed in the ratio of 3:1:1 by weight in heptane with PDMS concentration of 3 wt%. The mixed solution was sonicated for a few minutes in an ultrasonic bath before use. Prior to coating, the substrate of interest was sandwiched between a custom-made holder (Fig. 2), thoroughly cleaned using deionized water, and dried in an oven at 70 °C overnight. The prepared mould was then left to cool to room temperature. About 0.5 g of the prepared PDMS solution was then poured onto the assembled substrate and left to dry at room temperature for 3 h. The samples were then further dried overnight in an oven at 70 °C to fully cure the coating and remove any traces of the solvent. The composite membranes were then cut out from the mould and were ready to be tested.
 | ||
| Fig. 2 Custom holder assembled with an PES substrate layer for PDMS coating. | ||
2.4. Cross sectional and surface morphologies
The surface image and cross-sectional image for each sample was observed under a scanning electron microscope, SEM (Hitachi TM 3000 Tabletop) at 15 kV. The samples were coated with a thin layer of gold/palladium using a sputter coater (Quorum SC7620) for 90 seconds. As for the cross-sectional morphology, the clean cut of the samples was obtained by immersing the samples in liquid nitrogen and was gently fractured. The surface and cross section of both the bare substrates and coated membranes were taken.2.5. Surface pore architecture
The surface pores were characterized using the SEM images of the fabricated membranes at 3000× magnification (equivalent to the substrate's surface projected area of 60 μm × 45 μm). The images were analysed using ImageJ software by applying a bandpass filter in the range of 3–100 pixels and suppressing the horizontal stripes to obtain much clearer and sharper images. Next, the micrograph's threshold was set automatically using ‘Yen’ method for better consistency. The number of pores and the area of ‘black’ pores on the cleaned micrographs were then calculated. The size distribution of the pores was calculated by assuming each ‘black spot’ as a surface pore opening, finding its equivalent diameters, and then segregating them into groups according to their diameter size in increments of 0.1 μm. The process was repeated at least thrice on different images of the samples and the average statistics were taken.2.6. Bulk and surface porosity
The bulk porosity, εb of the substrate layer was determined through its dry–wet weight. The samples were immersed in deionized water for 24 h. Then, the weight of the wet samples was measured after wiping off excess water on the outer surfaces using a filter paper. Afterwards, the wet samples were dried in an oven for another 24 h before they were weighted again. The bulk porosity can hence be calculated using the following equation:
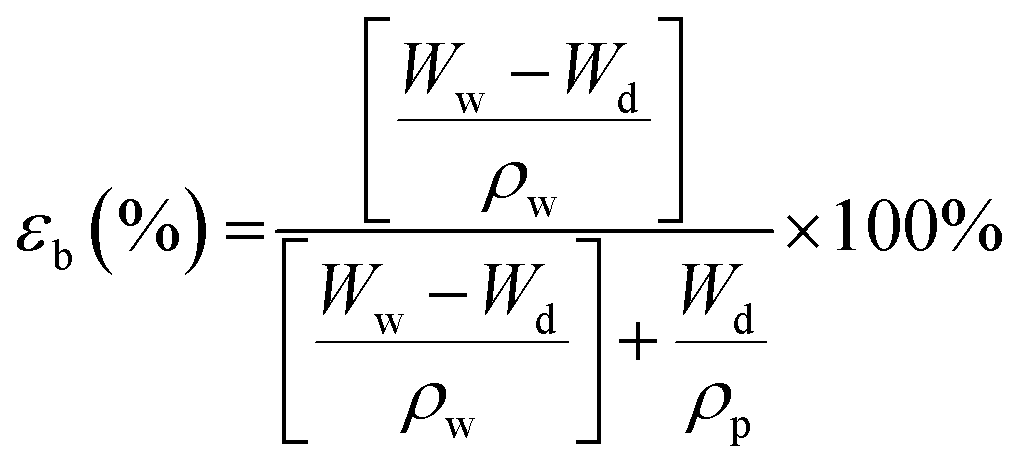 | (1) |
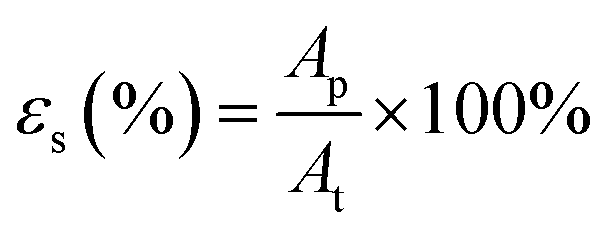 | (2) |
2.7. Substrate layer/composite membrane performance
Gas flow permeation performance of the substrate layer was tested in the dead-end mode using the setup in Fig. 3. Pieces of the substrate samples were cut in circles with an effective diameter of 1.6 cm and fitted into a suitable module with metal porous support underneath. N2 and CO2 gas was used as the model gas, which was introduced across the samples with a feed pressure between 1 and 6 bar gauge with 1 bar increment. Permeated gas was released to atmospheric pressure. The pressure was allowed to stabilize for a certain amount of time before the permeate flow rate was registered using a bubble soap flowmeter. The time taken for the bubble to travel by 50 mL was noted. The experiment was conducted at room temperature and repeated at least thrice. The gas permeance,![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) i was calculated using the equation:
i was calculated using the equation:
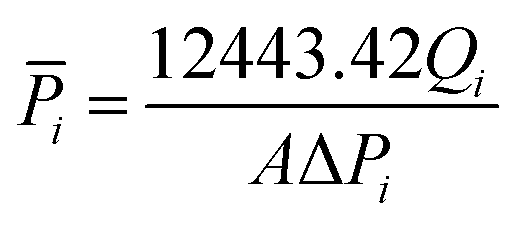 | (3) |
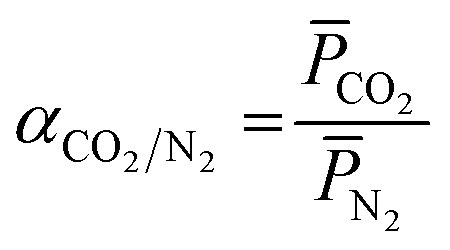 | (4) |
CO2 and N2 is the gas permeance of CO2 and N2, respectively. The performance tests were conducted on at least 3 different samples to obtain an average.
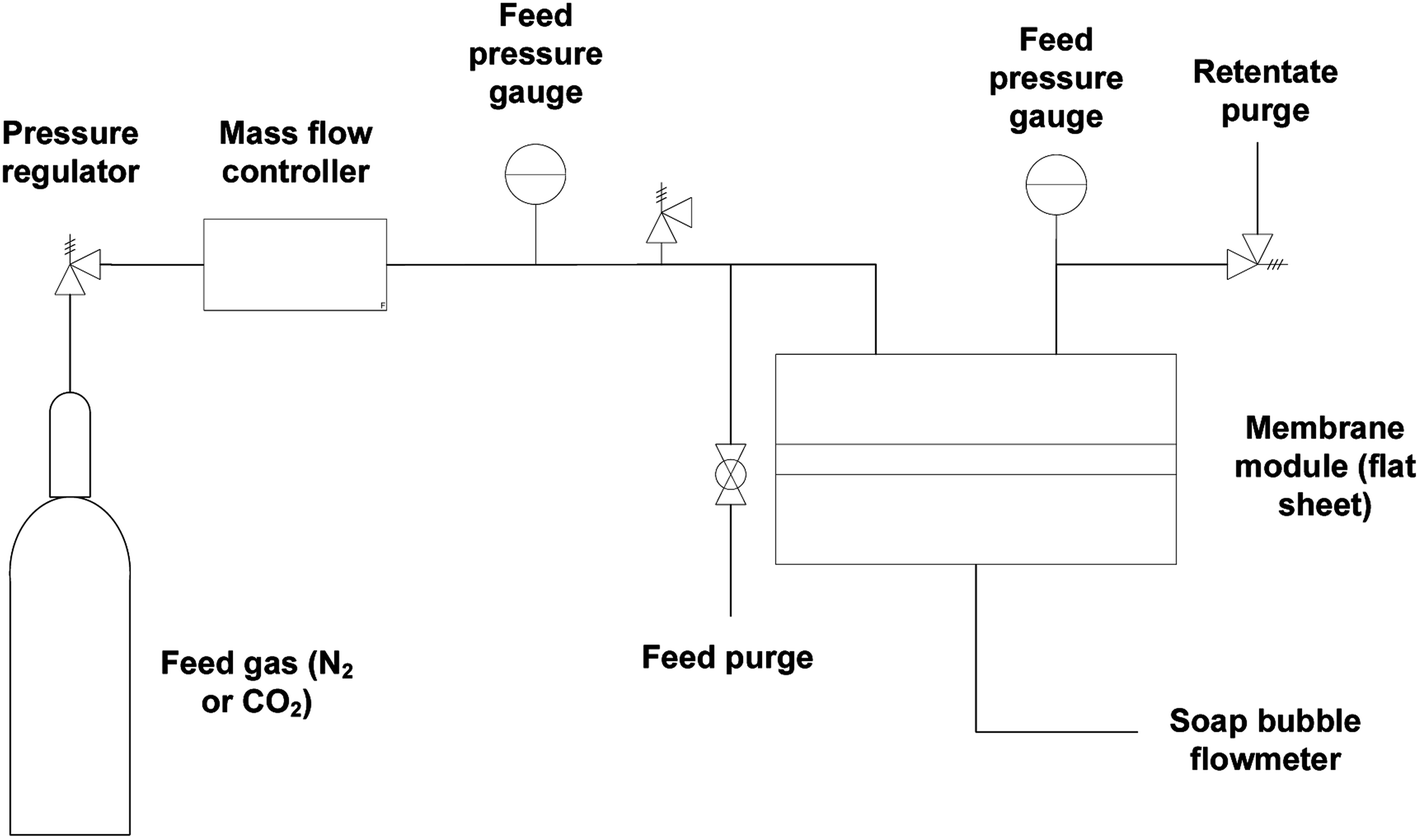 | ||
| Fig. 3 Membrane gas permeation test rig configuration. | ||
3. Results and discussion
3.1. Substrate's surface and bulk structure analysis
Fig. 4 represents the surface and cross-sectional micrograph of the substrate samples at ×3000 and ×1000 magnification, respectively, with the pore structures highlighted on each photo. Significant differences can be seen between the samples with increasing concentration of LiCl. The surface pore size seems to be increase with increasing LiCl concentration but at the same time, significant reduction in the surface pore density can be noted visually. Nevertheless, the cross-sectional micrograph suggested a reduction in the macro-void size with LiCl content with longer finger-like pore structure extruding from the surface. This suggested better mechanical strength of the substrate but at the probable cost of the substrate's gas flow resistance.24 A similar trend could be seen in the literature for LiCl incorporated PVDF hollow fiber membranes where the morphology transformed from finger-like to sponge-like with the increase in LiCl content,22 although the use of different base polymer may show different interactions between the mixture's components. Similarly, the work by L. Zheng et al. (2016) on PVDF/CTFE/PEG/LiCl/DMAc membranes noted an increase in the surface pore interconnectivity and cross-sectional void size reduction with increasing LiCl content, although the surface pore density seems to be increased in the mentioned work.25 This is, however, arguable for comparison as the presence of PEG (also a pore forming agent) may contribute to differences in the trend. Another work by H. J. Lee et al. (2002) for poly(amic acid)/LiCl/NMP membrane also reported the evolution of the samples' cross-sectional micrograph from finger-like to sponge-like with the increase in LiCl concentration.23 A higher magnified cross-sectional micrograph near the surface can be found in Fig. 7 later.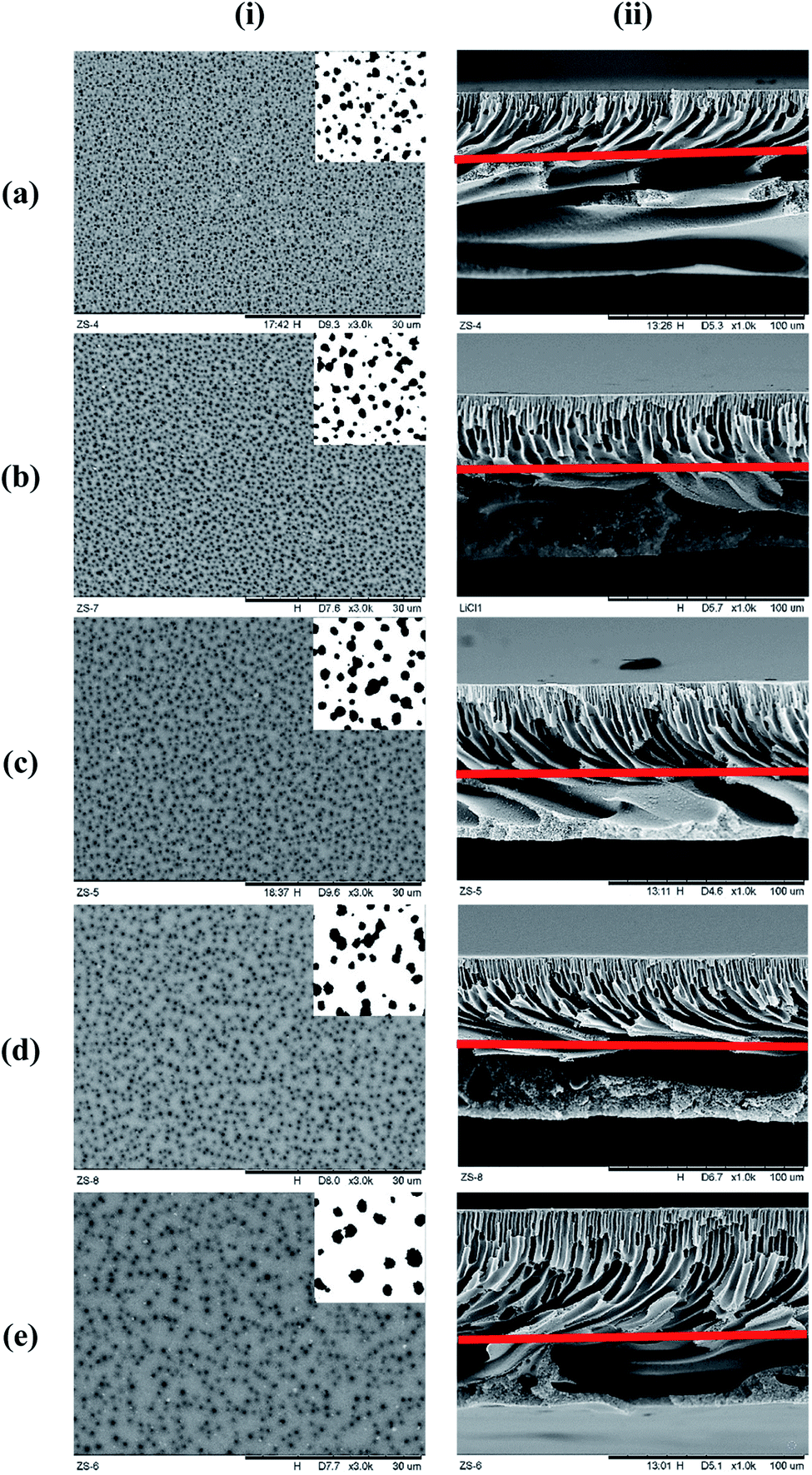 | ||
| Fig. 4 Substrate layer surface and cross-sectional micrograph for all the samples. (i) Surface at ×3000 magnification, (ii) cross-section at ×1000 magnification, (a) pristine, (b) LiCl_1%, (c) LiCl_2%, (d) LiCl_3%, (e) LiCl_4%. Subpicture for the surface images are the cleaned surface pore images using methodology noted in Section 2.5 while the line in the cross-sectional images are the separation line between the regularly aligned finger-like pores from the surface and the underneath macro void structures. | ||
A significant increase in the viscosity of LiCl containing the dope solution has been known to be caused by the formation of LiCl complex with a polar, aprotic solvent such as NMP due to their strong ion–dipole interactions.23,26 The increment in viscosity can decrease the solvent–nonsolvent kinetics, causing slower rate of demixing during the coagulation period. Possible consequences include the suppression of macro-void formation and tendency towards sponge-like morphologies due to slower propagation of the polymer-poor nucleus to form macro void finger-like structures.24,27 Based on these observations, the visual morphology for the cross-section corresponds well with the hypothesis proposed earlier on the size of the macropores, thanks to a thicker and denser skin formation slowing down the precipitation of the layer underneath.22 Nevertheless, there has been a lack of literature data on the influence of pore forming agents on surface pore distribution and architecture, which is important in the context of thin composite membrane for gas separation. The kinetic hindrance and pore forming properties of LiCl would then have a competing effect between the surface pore improvements and denser skin structure. Whether the increase in pore size will compensate the reduction in surface pore density visually seen here, in terms of the surface porosity, is hence unknown. This will be discussed later.
The SEM micrographs presented above suggest a minimal difference in the substrate thickness with increasing LiCl content. Table 2 represents the measured substrate thickness using a micrometer screw gauge at over 5 different points. Thickness variation was noted in ranges between 78–100 μm between the lower and upper limit (22 μm differences). The final membrane thickness is to be determined by the influence of kinetics and thermodynamics during phase inversion. Lower thickness are to be expected when kinetic effects outweigh the thermodynamic instability of the system.28 In this condition, the diffusion of non-solvent into the coagulating membranes would be minimized, resulting in lower final thickness when LiCl content increases. This is indeed the case for this work where high kinetic resistance of the LiCl samples exhibits lower thickness, except for LiCl_1%. Nevertheless, with minimal impact of thickness in few micrometer ranges for porous structures, these fluctuations in the overall thickness should not significantly influence the resulting flux and contribute to its performance.
| Substrate samples | Thickness (μm) |
|---|---|
| Pristine | 90 ± 2 |
| LiCl_1% | 98 ± 2 |
| LiCl_2% | 82 ± 4 |
| LiCl_3% | 86 ± 1 |
| LiCl_4% | 89 ± 7 |
3.2. Surface and bulk pore parameters
Quantitative analysis on the mean surface pore diameter, surface pore density, bulk porosity, and surface porosity is presented in Table 3. Mean surface pore diameter was noted to increase in the LiCl incorporated samples with an increase of 98% for the 4 wt% sample as compared to the pristine membrane. The quantitative values are in line with the visuals from Fig. 4. Nevertheless, this is contrary to the literature where mean pore size was noted to decrease to values far smaller than the one in this work with increasing LiCl content,17,20,22 although in all cases the mean pore size was determined from the solute transport data and gas permeation test. These methodologies represent the bulk mean pore size along the thickness, which can be influenced by the gas flow chokepoint inside the substrate, if any. While this chokepoint will affect the substrate's gas permeation performance, it will not contribute to pore intrusion and lateral diffusion on the surface unless it is located at the surface.| Substrate samples | Mean surface pore diameter (nm) | Surface pore density (×1000 mm−2) | Bulk porosity (%) | Surface porosity (%) |
|---|---|---|---|---|
| Pristine | 456 ± 1 | 972 ± 4 | 78.6 ± 0.2 | 15.9 ± 0.1 |
| LiCl_1% | 550 ± 10 | 815 ± 7 | 78.9 ± 0.2 | 19.4 ± 0.6 |
| LiCl_2% | 733 ± 14 | 472 ± 13 | 79.6 ± 0.2 | 19.9 ± 0.2 |
| LiCl_3% | 742 ± 23 | 363 ± 15 | 79.6 ± 0.1 | 15.6 ± 0.3 |
| LiCl_4% | 901 ± 13 | 200 ± 6 | 80.6 ± 0.4 | 12.8 ± 0.1 |
On the other hand, bulk porosity was noted to increase by 1.99% for 4 wt% LiCl sample as compared to the pristine membrane. Again, the results are contrary to the reported literature for the PVDF–LiCl sample where the bulk porosity was noted to be lower than that of the pristine membrane.22 Nevertheless, it is possible for the bulk porosity to increase upon increasing the LiCl concentration despite the reduction in micro-void size due to the formation of fine spongy pores,23 which might be the case for this work. The use of PVDF, which are interacting with the LiCl, might also contribute to the different influence. Interestingly, surface pore density was consistently reduced with LiCl concentration due to larger surface pores and increased pore distance at higher LiCl concentration, as qualitatively noted from the SEM images. While changes in the bulk porosity are minimal, it is also noted to be much higher than the surface porosity in all the cases due to its anisotropic design; significant increase was noted from pristine to 2 wt% LiCl. Nevertheless, the relative trend between the increase in the surface pore size and the decrease in the surface pore density caused the surface porosity to maximize at 2 wt% but starts to decrease beyond 3 wt% LiCl. Thermodynamic instability is known to influence the bulk porosity more while demixing kinetics influence the surface pore size and macro-void density more. This is also supported by the work of M. Sadrzadeh et al. (2013) where the bulk porosity increased significantly as the thermodynamic influence increases.29 As LiCl–NMP solution is prone to high viscosity increase with increasing LiCl concentration, it is suggested that the pore forming ability of LiCl (due to thermodynamic instability) starts to be overwhelmed by its kinetic hindrance (due to higher viscosity) at about 2–3 wt% LiCl. This trend is also in line with that in the literature.22
To further elucidate the surface characteristics of the substrate, surface pore size distribution was plotted. Fig. 5 represents the surface pore size distribution for the substrate samples. The distribution fits well into Weibull distribution with R2 between 0.94–0.99, as noted by the fitted line's graph. The mode of distribution is noted to be in between 0.3–0.9 μm with 90% of the distribution below the pore diameter of 0.8 μm (for pristine) and 1.2 μm (for LiCl_4%), thus placing the substrates at the interface between ultrafiltration and microfiltration regime.
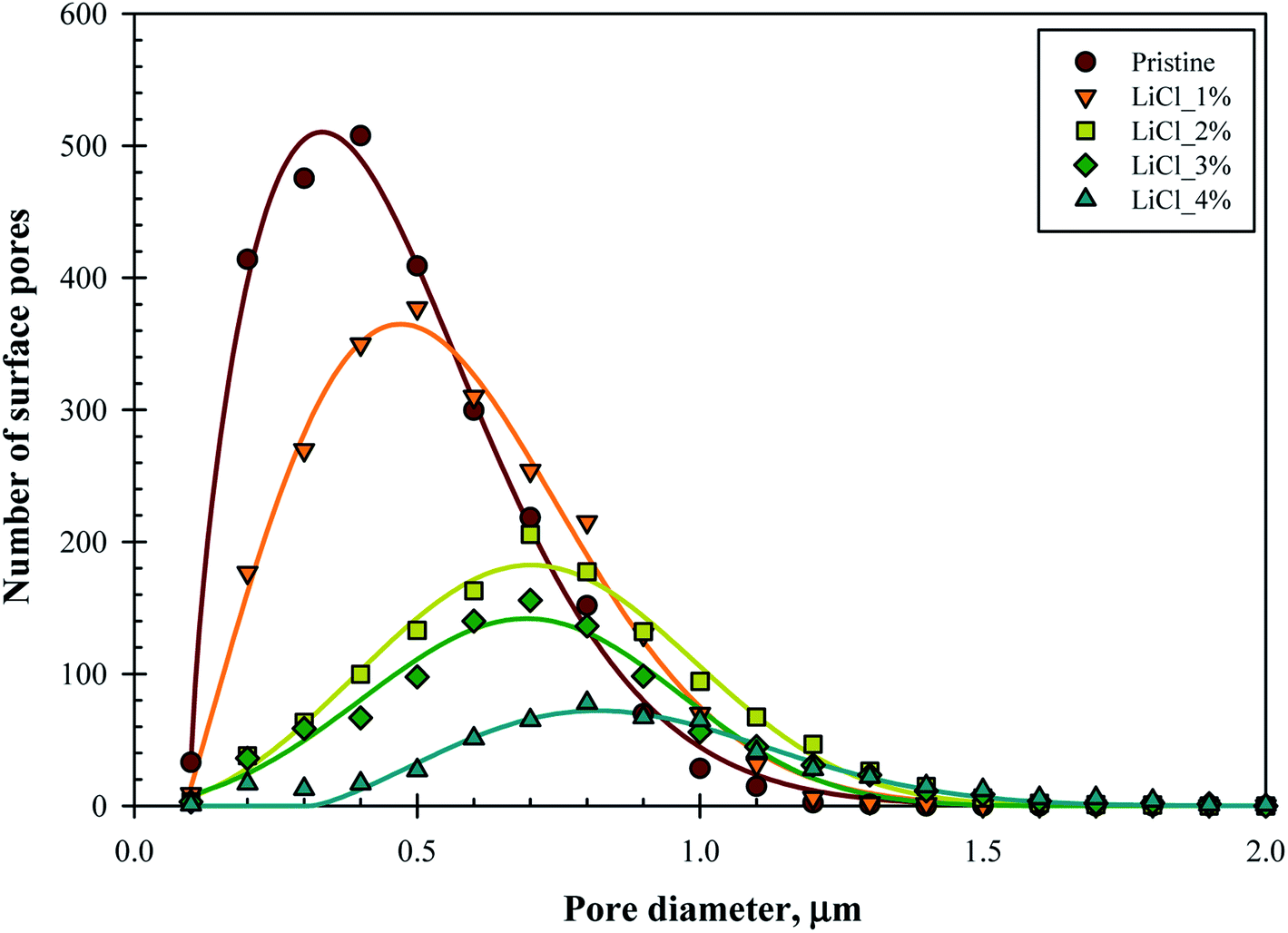 | ||
| Fig. 5 Number of surface pores and pore size distribution of the membrane substrates over the substrate's surface projected area (60 μm × 45 μm). | ||
While the mode value of the distribution (surface pore diameter) significantly increased with LiCl concentration, it came with reduced frequency (lower number of surface pores). The frequency of the modal pore diameter decreased rapidly from 508 for pristine samples down to only 78 for LiCl_4% over the same surface area. This finding supports the hypothesis earlier where the increased pore size came at the cost of reduced pore density, which could be important for the design of the composite thin film membrane. On the other hand, the pore distribution tends to become symmetrical at higher LiCl content, which suggested that the skewed distribution at lower LiCl concentration might be due to limited and poor pore detection at lower ranges below 0.1 μm. Hence, the decrease in surface pore density might be more severe in this case.
The use of NMP as the solvent has been suggested to create the highest number of surface pores among common solvents for PES polymer.4 This shows that the decrease in surface pore is contributed to only by the pore forming agents. It was also noted that permeance efficiency decreased with higher PES concentration due to reduced surface pore count but increase in the average pore size, characteristics similar to in this work. While the PES concentration was maintained in this work, an indirect correlation can be made with the possible structures of the substrate as the increased PES concentration will also affect its viscosity, hence affecting the phase inversion pathways through increase in the kinetic resistance for solvent exchange. Hence, it is hypothesized that the pore forming agents will also produce the same results, especially for highly viscous solutions. Nevertheless, the use of pore forming agents will also affect the thermodynamics of phase inversion, so the use of other pore forming agents may show significantly different results.
Careful judgement needs to be used in interpreting the results in this Section. SEM imaging requires metal coating to increase the conductivity and to subject the samples to the electron beam, which may alter the surface pore structures.30 On the other hand, as SEM in this work was conducted at 15 kV, it would be high enough to penetrate beyond the immediate substrate surface and the registered pores located just underneath it.31 Hence, this exaggerates the surface pore size. If the choke point is really at the apparent surface and as small as in the literature, this suggests that there is a steep change in the pore depth between the surface pores and the pores underneath. This information can be important as continuously increasing the pore circumference into the depth of the substrate can give an important insight into the coating solution's penetration, which can be beneficial (or not) for composite formation. Further discussions will be made in the next section. Nevertheless, similar protocol used for all the samples would suffice for direct comparison between the substrate samples.
3.3. Substrate gas flow permeation
Fig. 6 represents the N2 gas flux for all the substrate samples with increasing pressures; all the data points were taken after a pressure holding time of at least 5 minutes. The substrates were able to withstand up to a maximum of 6 bars of pressure without bursting, except for the pristine membrane due to its high gas flux and limitation of the set-up used. Nevertheless, subsequent tests on an alternative set-up verified that all the substrates were able to withstand up to 10 bars of transmembrane pressure even for the pristine membrane, although the pressure holding time for this equipment was less than 1 minute. An almost linear increment in the gas flux over 6 bar transmembrane pressure suggested that no significant compaction nor pore collapse occurred during the testing period.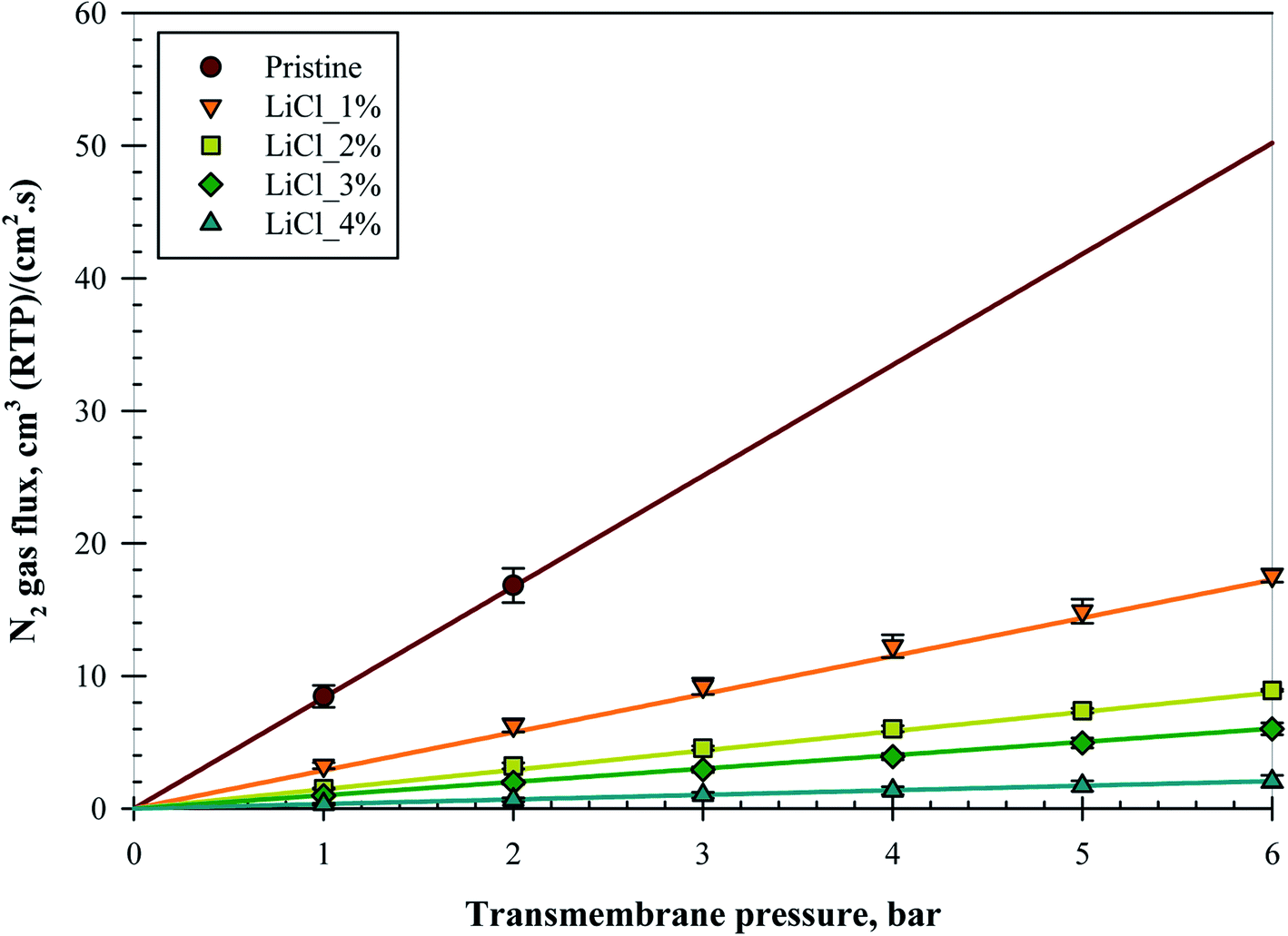 | ||
| Fig. 6 Membrane substrate layer's N2 gas flux over different transmembrane pressures. | ||
Despite the increase in surface pore size and surface porosity at 1 and 2 wt% LiCl, as noted in the previous section, surprisingly, the gas flux was reduced. Reduction was noted in the slope of the linear fitting in the form of decrease in the exponential where the flux ratio between the samples was 0.64, 0.50, 0.34, and 0.66 for pristine/LiCl_1%, LiCl_1%/LiCl_2%, LiCl_2%/LiCl_3%, and LiCl_3%/LiCl_4%, respectively. This trend suggested that increased LiCl concentration reduced the substrate's gas flux but at a diminishing rate up to 2 wt% before the reduction rate started to increase again. It is unclear whether this trend has anything to do with the increase in surface porosity but it might partially explain the phenomena.
So far, the literature data has suggested otherwise. In contrast, the highest gas permeance was noted when high LiCl content was incorporated for the sample of PAA based membrane due to the formation of fine porous sponge-like structure by LiCl.23 Nevertheless, the extend of macro-void reduction is far less in this work as compared to the mentioned work, where the macro-void was able to be removed completely at 5 wt% of LiCl. Interestingly, the incorporation of LiCl in PPTA/PVDF was also shown to possess the same pore evolution trend (long finger-like pores with increased porosity and pore diameter) as in this work but with increased water flux and declined PEG rejection.19 The increase in water flux was also noted in several other literatures.17,20 It is highly likely that the use of LiCl creates more hydrophilic structures, which would help to channel the water feed. In this case, it would help the membrane in water-based application but not as in gas separation. Due to this reason, it is hard to quantify and compare the results with literature. The best comparison found so far the work of A. Mansourizadeh et al. (2010) where the N2 permeance decreased with LiCl content.22 However, the mean pore size was also noted to decrease in this case, although it was calculated using the gas permeation test.
As mentioned previously, the sensitivity of the substrate's gas flow resistance was expected to be minimal. In the range of ±22 μm from the pristine membrane thickness, negligible thickness effect can be concluded from flux reduction this high. Insignificant differences in the bulk porosity also suggested that it should not be the reason for this flux reduction. One explanation is the skewed pore size distribution at lower LiCl concentration, as mentioned in the previous section. Sub 0.1 μm surface pores might significantly be abundant for the pristine sample, which would contribute to the substrate's flux results. If the sub 0.1 μm hypothesis is true, it would reduce the value of average surface pore size and increase the surface porosity presented in Table 3, especially for the pristine substrate. However, calculations using the Knudsen diffusion equation with the surface pore size and porosity suggested that there should be an increase in the flux for all LiCl modified substrates (will be presented later in Table 5). Another possible explanation is the formation of chokepoint in the substrate, as mentioned in the previous section. Literature has so far suggested that the skin layer is the one responsible for the separation performance,16,32 which means that the smallest pores should be located on the surface. In this sense, the apparent surface would be filled with sub 100 nm pores, which went a few nanometers deeper before reaching the pores noted from the SEM image. Some literatures have also noted that pore collapse could contribute to the decline in permeability as the collapsed porous structures became dense a polymer film.33–35 In this work particularly, the substrates were air-dried directly after the end of the coagulation process, which increases the possibility of pore collapse. Notwithstanding the concern, no sign of pore collapse can be seen for all the samples, based on the SEM images in Fig. 4 and later on in Fig. 7, while the gas flux for the pristine membrane was already higher than the others even at low pressure difference, suggesting that pore collapse is minimal, occurred only at a higher pressure, or did not happen at all in this experiment.
 | ||
| Fig. 7 PES–PDMS interface cross-sectional micrograph at ×5000 magnification, (a) pristine, (b) LiCl_1%, (c) LiCl_2%, (d) LiCl_3%, (e) LiCl_4%. | ||
Before further conclusion is made, the permeance of the substrate layer was calculated. The performance of the substrate layer can sometimes significantly impact the overall composite performance if the substrate possesses the correct morphology to selectively separate the permeating gas (e.g., dense surface layer). Hence, the influence of the layer on the gas permeation performance should be elucidated. Table 4 represents the CO2/N2 permeance of the substrates and their ideal selectivity. Indeed, a similar trend was noted for CO2 where the gas flux (hence, the permeance) decreased with LiCl content. However, this confirmed that the substrate is governed mainly by the Knudsen regime, with an average selectivity of about 0.8 for CO2/N2, which is in line with the Knudsen flow selectivity for the gas pairs. All the permeances had a standard deviation of 2 from the sample average. A large permeance range can be noted (100000 GPU difference between the pristine and LiCl_4% substrate for N2).
| Substrate samples | CO2 permeance, ×1000 (GPU) | N2 permeance, ×1000 (GPU) | Selectivity (CO2/N2) |
|---|---|---|---|
| a All permeance and selectivity values were taken at 6 bars transmembrane pressure value except for pristine due to equipment limitation (1 bar). | |||
| Pristine | 86.4 ± 8.9 | 105.3 ± 10.2 | 0.82 ± 0.03 |
| LiCl_1% | 27.8 ± 2.4 | 36.5 ± 1.0 | 0.76 ± 0.06 |
| LiCl_2% | 15.3 ± 0.4 | 18.5 ± 0.2 | 0.83 ± 0.01 |
| LiCl_3% | 10.3 ± 0.4 | 12.5 ± 0.9 | 0.83 ± 0.03 |
| LiCl_4% | 3.6 ± 0.9 | 4.3 ± 1.0 | 0.84 ± 0.03 |
Substrate samples of at least 2900 GPU in this work should possess permeance high enough to be used as the substrate layer. It is recommended that the substrate should be at least 5–10 times more permeable than the selective layer to ensure minimal flow resistance by the substrate.9,16 Hence, the maximum selective layer intrinsic permeance suitable for the fabrication of composite membrane with this substrate should be 290 GPU, which is high enough for a lot of known materials. Any resistance contribution by the substrate layer can also be said to be originated by Knudsen flow.
In search of the reason for discrepancy between the characterization and the experiment, theoretical Knudsen and Poiseuille flow for the porous substrate was calculated using the equation:16
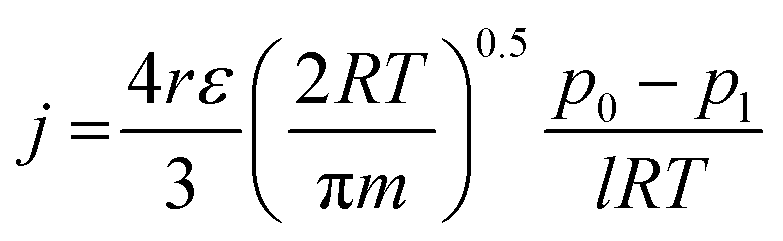 | (5) |
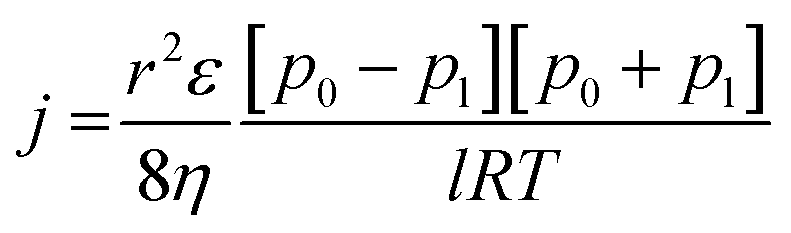 | (6) |
Table 5 represents the comparison between the permeance of the experimental results with the Knudsen and Poiseuille flow. All the theoretical values are far below the one registered by the experiments. For the model to reach experimental permeance, 3 parameters can be modified; (i) pore radius, (ii) porosity, and (iii) thickness. As experimental results have already shown that the gas selectivity follows Knudsen diffusion, gas permeation is highly unlikely to be mainly governed by Poiseuille flow.
| Substrate samples | Experimental permeance, ×1000 (GPU) | Poiseuille permeance, ×1000 (GPU) | Knudsen permeance, ×1000 (GPU) |
|---|---|---|---|
| Pristine | 105.3 | 0.5 | 0.3 |
| LiCl_1% | 36.5 | 2.0 | 0.4 |
| LiCl_2% | 18.5 | 4.4 | 0.7 |
| LiCl_3% | 12.5 | 3.4 | 0.5 |
| LiCl_4% | 4.3 | 3.9 | 0.5 |
On the other hand, back calculation using Knudsen equation suggested strong evidence that the permeance behaviour is due to a thicker skin layer formation. Both the results from pore radius and porosity are unlikely to be true due to their excessive values. In fact, results greater than the characterization done in Table 3 are highly unlikely. While it is hard to measure the skin thickness accurately, these values are logical even after comparison with the SEM images (Fig. 7). In fact, skin layer formation has been noted to become thicker and denser with increased pore forming agent concentration such as LiCl.22 Nevertheless, theoretical skin thickness was excessively overestimated for LiCl_4%, suggesting that both the actual surface pore size and surface porosity might be slightly different than the measured values (Table 6).
| Substrate samples | Theoretical pore radius (nm) | Theoretical porosity (%) | Theoretical thickness (μm) |
|---|---|---|---|
| Pristine | 79433 |
5537 | 0.3 |
| LiCl_1% | 24281 |
1710 | 1.1 |
| LiCl_2% | 10027 |
545 | 3.0 |
| LiCl_3% | 9006 | 380 | 3.5 |
| LiCl_4% | 3973 | 113 | 10.1 |
3.4. Support layer formation and performance with PDMS layer
In order to test the performance of the substrates in the composite form, a thin layer of PDMS was administered. Fig. 7 represents the substrate–PDMS interface cross-section for all the substrates at ×5000 magnification. The thin PDMS layer was confirmed to be deposited successfully. Significant differences were noted in the pore design between the samples, where the surface pores became more orderly at higher LiCl concentration. In fact, small finger-like pores from the surface start to disappear at 2 wt% LiCl. On the other hand, a thicker skin layer could be noted at high LiCl concentration. Nevertheless, no significant differences in the PDMS thickness were noted visually between the samples. Proper quantification of 6 micrographs for each sample is presented in Table 7, which supports this claim, although pristine samples showed relatively lower thickness than the others. On the other hand, higher substrate surface energy than the coating solution's surface tension would ideally suggest good wetting properties and hence, good formation of the PDMS layer with similar coating layer thickness, regardless of the substrate's surface structures. This is confirmed by the good coating structures of all the samples. Nevertheless, although no confirmation can be made, pore penetration by capillary action of the coating solution on different surface pore sizes and porosities, particularly on the nano porous skin layer,36 may influence the composite permeation results and needs to be elucidated.| Substrate samples | Measured selective thickness (μm) |
|---|---|
| Pristine | 1.5 ± 0.2 |
| LiCl_1% | 2.1 ± 0.3 |
| LiCl_2% | 2.1 ± 0.4 |
| LiCl_3% | 2.6 ± 0.4 |
| LiCl_4% | 2.1 ± 0.2 |
Despite the reduction in the gas permeance of the substrates by LiCl incorporation, these values are in the range of thousands. Hence, in the ideal composite design, it should not contribute to the overall selectivity nor resistance. However, Table 8, which represents the CO2 and N2 permeances and selectivity for the PDMS coated substrates, shows some reduction in permeance. Meanwhile, CO2/N2 selectivity was noted to increase as compared to that of the substrate, with the average selectivity over all the samples about 14.6 ± 0.6, thus confirming the formation of the PDMS layers. This value is, however, a bit higher than the reported selectivity of about 9.5–9.8.37,38 In comparison, reduction in the composite permeances from its substrate layer is of the order of 150–260 for CO2 and 2500–4000 for N2, suggesting minimal flux resistance contribution by the substrate. This is interesting as although the substrate by itself did not contribute to the selectivity and is far permeable than the dense layer, it indirectly affects the PDMS permeance when made into a thin film. A typical explanation would be the pore intrusion but, as noted before and by back calculation from the PDMS permeability, this could not be the only explanation for the reduction, as PDMS thickness needs to be of the order of 9.6–12.9 μm for the pristine substrate and 50.7–66.6 μm for LiCl_4%. While lateral diffusion could become important based on the thickness/pore radius ratio,8 more experiments need to be done to confirm this claim.
| Substrate samples | CO2 permeance (GPU) | N2 permeance (GPU) | CO2/N2 selectivity |
|---|---|---|---|
| a All permeance and selectivity was taken at 4 bars value. | |||
| Pristine | 361.9 ± 31.8 | 27.3 ± 2.9 | 13.5 ± 0.6 |
| LiCl_1% | 109.8 ± 9.2 | 6.9 ± 1.0 | 16.1 ± 0.8 |
| LiCl_2% | 96.4 ± 9.9 | 7.3 ± 0.8 | 13.9 ± 1.5 |
| LiCl_3% | 55.2 ± 6.3 | 3.7 ± 0.8 | 16.0 ± 1.8 |
| LiCl_4% | 68.8 ± 6.5 | 5.3 ± 0.7 | 13.5 ± 1.2 |
4. Conclusion
PES based porous substrates have been fabricated with different LiCl concentrations. The surface pore architectures have been noted to be starkly different with increasing pore sizes and reducing pore density but with negligible bulk porosity differences. Gas permeation test confirms the porous nature and Knudsen diffusivity of the substrate but also suggested the formation of thicker skin layer with higher LiCl content. This in turn reduces the permeance significantly, although it should still be high for a substrate. Nevertheless, the formation of PDMS surface coating does not suggest significant differences in the thickness when fabricated at a similar volume but still possess reduced permeance even for the coated pristine substrates. This signifies the complex interaction between the composite layers when subjected to gas permeation. Nevertheless, more experiments need to be conducted to elucidate the culprit behind this phenomenon.Conflicts of interest
The authors have no other competing interests.Acknowledgements
The authors would like to acknowledge the financial support by Universiti Sains Malaysia USM-RUI grant (Grant no: 1001/PJKIMIA/8014063). Zulfida Mohamad Hafis Mohd Shafie would like to express his thanks to USM Fellowship and the Government of France through the French Embassy in Malaysia/Campus France for financing his study and research mobility.References
- J. E. Cadotte and R. J. Petersen, in Synthetic Membranes: Volume I Desalination, American Chemical Society, 1981, ch. 21, vol. 153, pp. 305–326 Search PubMed.
- P. S. Francis, F. C. D. Luzio, W. S. Gilla and A. Kotch, Fabrication and Evaluation of New Ultrathin Reverse Osmosis Membranes, Office of Saline Water, United States Department of the Interior, 1966 Search PubMed.
- C&EN, Chemical & Engineering News Archive, 1980, 58, 57–60 Search PubMed.
- A. Ghadimi, S. Norouzbahari, H. Lin, H. Rabiee and B. Sadatnia, J. Membr. Sci., 2018, 563, 643–654 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- D. Wu, Y. Han, W. Salim, K. K. Chen, J. Li and W. S. W. Ho, J. Membr. Sci., 2018, 565, 439–449 CrossRef CAS.
- J. G. Wijmans and P. Hao, J. Membr. Sci., 2015, 494, 78–85 CrossRef CAS.
- M. Kattula, K. Ponnuru, L. Zhu, W. Jia, H. Lin and E. P. Furlani, Sci. Rep., 2015, 5, 15016 CrossRef PubMed.
- J. M. S. Henis and M. K. Tripodi, J. Membr. Sci., 1981, 8, 233–246 CrossRef CAS.
- A. A. M. Salih, C. Yi, H. Peng, B. Yang, L. Yin and W. Wang, J. Membr. Sci., 2014, 472, 110–118 CrossRef CAS.
- J. Wang, R. Xu, F. Yang, J. Kang, Y. Cao and M. Xiang, J. Membr. Sci., 2018, 556, 374–383 CrossRef CAS.
- L. Zhu, M. Yavari, W. Jia, E. P. Furlani and H. Lin, Ind. Eng. Chem. Res., 2016, 56, 351–358 CrossRef.
- K. G. Kornev and A. V. Neimark, J. Colloid Interface Sci., 2001, 235, 101–113 CrossRef CAS PubMed.
- U. Unije, R. Mücke, P. Niehoff, S. Baumann, R. Vaßen and O. Guillon, J. Membr. Sci., 2017, 524, 334–343 CrossRef CAS.
- R. W. Baker, Membrane technology and applications, John Wiley & Sons, England, 2nd edn, 2004 Search PubMed.
- A. Idris, I. Ahmed and M. A. Limin, Desalination, 2010, 250, 805–809 CrossRef CAS.
- A. Urkiaga, D. Iturbe and J. Etxebarria, Desalin. Water Treat., 2015, 56, 3415–3426 CrossRef CAS.
- H.-B. Li, W.-Y. Shi, Y.-F. Zhang, D.-Q. Liu and X.-F. Liu, Polymers, 2014, 6, 1846–1861 CrossRef.
- I. Ahmed, A. Idris and N. F. C. Pa, J. Appl. Polym. Sci., 2010, 115, 1428–1437 CrossRef CAS.
- S. H. Elahi and I. C. Escobar, in Modern Applications in Membrane Science and Technology, American Chemical Society, 2011, vol. 1078, ch. 16, pp. 271–283 Search PubMed.
- A. Mansourizadeh and A. F. Ismail, Chem. Eng. J., 2010, 165, 980–988 CrossRef CAS.
- H. J. Lee, J. Won, H. Lee and Y. S. Kang, J. Membr. Sci., 2002, 196, 267–277 CrossRef CAS.
- G. R. Guillen, Y. Pan, M. Li and E. M. V. Hoek, Ind. Eng. Chem. Res., 2011, 50, 3798–3817 CrossRef CAS.
- L. Zheng, Z. Wu, Y. Wei, Y. Zhang, Y. Yuan and J. Wang, J. Membr. Sci., 2016, 506, 71–85 CrossRef CAS.
- A. El-Kafrawy, J. Appl. Polym. Sci., 1982, 27, 2435–2443 CrossRef CAS.
- S. Mohsenpour, A. Safekordi, M. Tavakolmoghadam, F. Rekabdar and M. Hemmati, Polym, 2016, 97, 559–568 CrossRef CAS.
- C. Zhou, Z. Hou, X. Lu, Z. Liu, X. Bian, L. Shi and L. Li, Ind. Eng. Chem. Res., 2010, 49, 9988–9997 CrossRef CAS.
- M. Sadrzadeh and S. Bhattacharjee, J. Membr. Sci., 2013, 441, 31–44 CrossRef CAS.
- K. C. Khulbe, C. Y. Feng and T. Matsuura, in Synthetic Polymeric Membranes, ed. H. Pasch, Springer, 2008 Search PubMed.
- W. Zhou, R. Apkarian, Z. L. Wang and D. Joy, in Scanning Microscopy for Nanotechnology: Techniques and Applications, eds. W. Zhou and Z. L. Wang, Springer New York, New York, NY, 2007, pp. 1–40, DOI:10.1007/978-0-387-39620-0_1.
- H. C. Shih, Y. S. Yeh and H. Yasuda, J. Membr. Sci., 1990, 50, 299–317 CrossRef CAS.
- I. Pinnau and B. D. Freeman, in Membrane Formation and Modification, American Chemical Society, 1999, vol. 744, ch. 1, pp. 1–22 Search PubMed.
- V. P. Khare, A. R. Greenberg, S. S. Kelley, H. Pilath, I. Juhn Roh and J. Tyber, J. Appl. Polym. Sci., 2007, 105, 1228–1236 CrossRef CAS.
- J. Gao and T.-S. Chung, J. Membr. Sci., 2019, 572, 223–229 CrossRef CAS.
- D. R. Ceratti, M. Faustini, C. Sinturel, M. Vayer, V. Dahirel, M. Jardat and D. Grosso, Nanoscale, 2015, 7, 5371–5382 RSC.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, Wiley, 1999 Search PubMed.
- T. C. Merkel, V. I. Bondar, K. Nagai, B. D. Freeman and I. Pinnau, J. Polym. Sci., Part B: Polym. Phys., 1999, 38, 415–434 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra00045k |
| This journal is © The Royal Society of Chemistry 2020 |