
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergetic catalysis of a cobalt-based coordination polymer for selective visible-light driven CO2-to-CO conversion†
Sheng-Jie Li,
Yong-Bin Chang,
Ming Li,
You-Xiang Feng and
Wen Zhang *
*
MOE International Joint Laboratory of Materials Microstructure, Institute for New Energy Materials and Low Carbon Technologies, School of Material Science and Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: zhangwen@email.tjut.edu.cn
First published on 11th May 2020
Abstract
Herein, based on the strategy of synergetic catalysis, we report a cobalt-based coordination polymer PEI6-Co. As a heterogeneous catalyst, PEI6-Co shows a selectivity of 95% and a yield of 1170 mmol g−1 for visible-light-driven CO2-to-CO conversion in a water containing system, which is almost 2.8 times that of the mononuclear cobalt catalyst CoL1 and is comparable to that of the dinuclear cobalt catalyst Co2L.
Energy shortages and greenhouse gas emissions caused by the consumption of fossil fuels have become an indisputable obstacle to the sustainable development of human beings. Solar-driven conversion of CO2 to carbon fuels is regarded as an ideal strategy to deal with these energy and environmental issues.1–7 In this context, many researchers have devoted themselves to promote the activity and efficiency of photocatalytic CO2 conversion.8–11 Unlike the reduction of H2O to H2, the CO2 reduction reactions are more complicated. Firstly, CO2 is an inert molecule, it requires high energy to be activated. Secondly, as the carbon atom in CO2 is the highest valence, various reduction products including CO,12 formic acid,13 formaldehyde,14 methane15 and some multi-carbon products16 can be obtained. Besides, due to the competitive reduction of protons, the selectivity of CO2 reduction is usually low in water containing medium.17 In this context, the development of a catalyst for the efficient and highly selective reduction of CO2 to a single valuable product is still challenging, especially in water containing system.
In recent decades, many molecular catalysts for the reduction of CO2 have been designed elaborately. For example, the catalysts based on Ru,18,19 Re,20,21 Ir,22 Fe,23,24 Co,25,26 Ni,27,28 Mn,29 etc., have been developed to reduce CO2 and the mechanisms for the activation and conversion of CO2 have also been investigated reasonably.30,31 Notable among them is dinuclear metal synergistic catalysis (DMSC) in which two active centers are involved into the catalytic reaction concurrently and used to decrease the reaction barriers.32–34 In our previous works,25 dinuclear cobalt cryptate Co2L (Scheme 1) which was composed of aza-cryptand ligand and two adjacent cobalt ions displayed excellent performance in the conversion of CO2 to CO. Due to the immobilization and synergistic catalysis of two adjacent cobalt active sites to CO2 molecules, the selectivity and the turnover numbers (TON) of Co2L reached as high as 98%, and 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 896, respectively. Although molecular catalysts are significant in the study of catalytic mechanism and optimizing catalyst structure, the synthesis operations of such molecular catalyst are tedious and not conducive to its large-scale practical applications.
896, respectively. Although molecular catalysts are significant in the study of catalytic mechanism and optimizing catalyst structure, the synthesis operations of such molecular catalyst are tedious and not conducive to its large-scale practical applications.
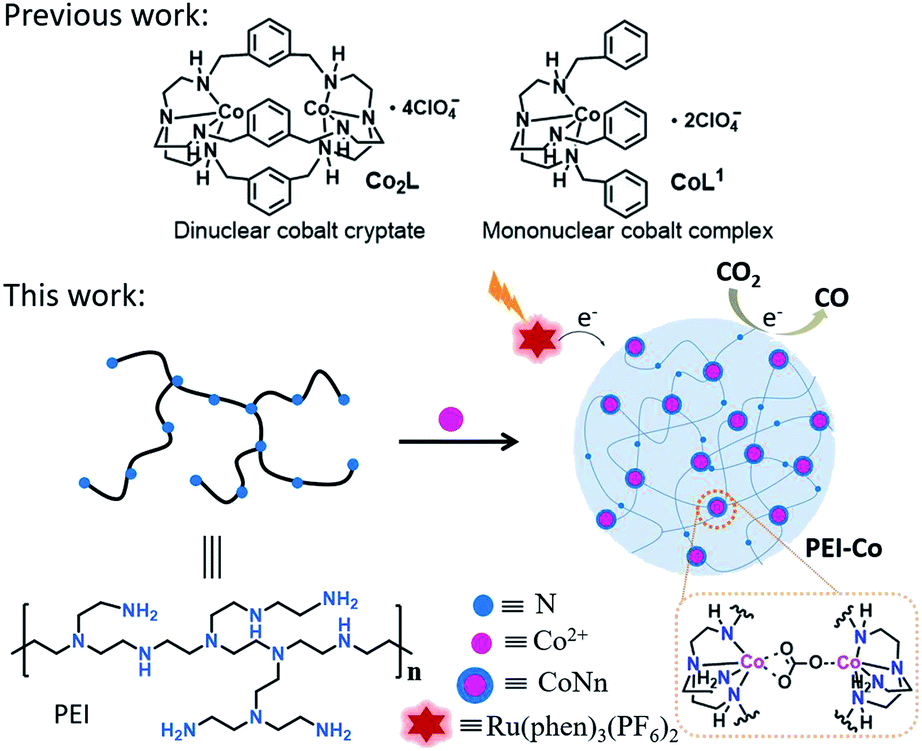 | ||
| Scheme 1 The structures of Co2L, CoL1, PEI-Co and the proposed catalysis process of PEI-Co for CO2-to-CO conversion. | ||
Herein, we choose polyethyleneimine (PEI) as the analogue of aza-cryptand and synthesize a cobalt-based coordination polymer (PEI6-Co) as a heterogeneous catalyst for the photocatalytic reduction of CO2 (Scheme 1). The reason for choosing PEI as a ligand is that both the structures of aza-cryptand and PEI have secondary amine and tertiary amine groups which can coordinate with Co2+. The formation of the coordination polymer can shorten the distance between cobalt active sites, which is similar to that of Co2L, so that they may function synergistically in the photocatalytic reactions. Besides, PEI has been commonly used as a CO2 absorbent in the past research of electrocatalytic reduction of CO2.35 As a result, when PEI6-Co is used as a heterogeneous catalyst for the reduction of CO2 in water containing system, the selectivity for CO reaches up to 95%, and the yield for CO2-to-CO conversion reaches 1170 mmol g−1, which is almost 2.8 times of molecular catalyst mononuclear cobalt CoL1 and comparable with that of dinuclear cobalt catalyst Co2L.
Co2L and CoL1 were prepared according to the literature.25 PEIx-Co (x represents the N/Co ratio) was synthesized through the coordination between the amine groups of PEI and cobalt ions and the exact contents of cobalt in the catalyst are listed in Table S1 (see ESI†). At the beginning, the formation of PEI6-Co was characterized by TEM, DLS, element mapping, XPS and cyclic voltammetry measurements. As shown in Fig. 1a, PEI6-Co was formed as a nanoparticle which resulted from the coordination crosslinking between the amino groups of PEI and Co ions. DLS measurement in Fig. 1b shows that the average diameter of this nanocomposite is about 50 nm. The elemental mapping images confirm that the Co, N and C elements uniformly distributed over the nanostructures of PEI6-Co (Fig. 1c). The XPS measurement shows that there are only Co, N, C, O and Cl element in PEI6-Co (Fig. S1†). These results indicate that amine groups in the PEI chain can coordinates with the cobalt ions, causing the long chain of the PEI to be twisted and crosslinked to form nanoparticles and giving the chance for cobalt centers to get closer. Besides, the redox property of PEI6-Co was investigated using cyclic voltammogram (CV). The results in Fig. 1d show that compared with the CV curve under argon condition, the current has an obvious enhancement under CO2 atmosphere and the onset potential (Eonset = −0.62 V vs. NHE) is more negative than that of Co2L (Eonset = −0.68 V vs. NHE, Fig. S2†) and CoL1 (Eonset = −0.76 V vs. NHE, Fig. S3†), indicating that PEI6-Co can reduce CO2 more easily than Co2L and CoL.1 Besides, the redox potential of photosensitizer [Ru(phen)3]3+/2+* was reported as −0.84 V vs. NHE,26 which is more negative than the onset potential of PEI6-Co to CO2 reduction, thus, it is feasible for [Ru(phen)3](PF6)2 to donate electrons to PEI6-Co under light irradiation, which is one of the foundations for driving the reduction of CO2.
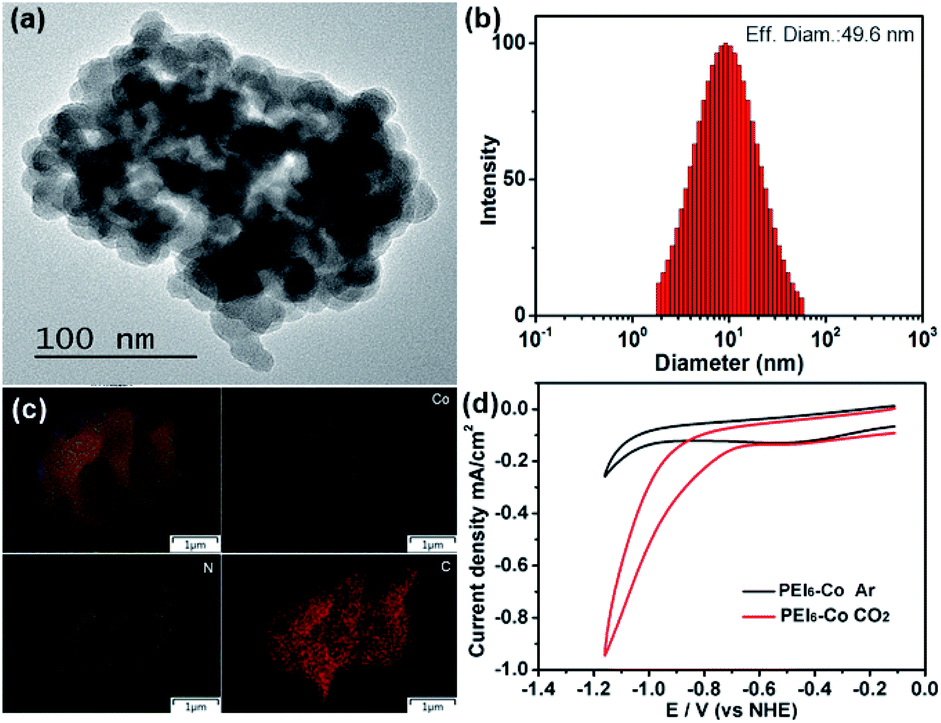 | ||
| Fig. 1 (a) TEM image of PEI6-Co. (b) DLS data of PEI6-Co. (c) Mapping images of PEI6-Co. (d) CV curves of PEI6-Co in CH3CN/H2O (4:1) solution under an Ar (black) and CO2 atmosphere (red) at 25 °C. | ||
In addition, to examine the gathering ability of PEI6-Co to CO2, CO2 adsorption experiment was carried out. As shown in Fig. 2a, PEI6-Co has an adsorption capacity of 2.63 cm3 g−1 towards CO2. The IR spectra in Fig. 2b shows that after the treatment of CO2 atmosphere, both PEI6-Co and Co2L have two peaks at 1628 cm−1 and 1473 cm−1, which belong to the stretching vibrations of the carbonate group. All these results indicate that PEI6-Co, just like the molecular catalyst binuclear cobalt Co2L, has the ability of adsorbing CO2, which is favorable for the reduction of CO2 in the heterogeneous catalysis process.
 | ||
| Fig. 2 (a) The adsorption curve for carbon dioxide of PEI6-Co. (b) IR spectra of PEI6-CO (red) and Co2L (black) after adsorption of CO2. | ||
Encouraged by the positive results above, we optimized the atom ratio of N and Co in the catalysts for the reduction of CO2. In the photoreaction system, the reaction solutions were prepared by adding photocatalyst PEIx-Co and photosensitizer [Ru(phen)3](PF6)2 to CH3CN/H2O (v/v = 4:1) solutions with triethanolamine (TEOA) as a reducing agent. The quartz reaction bottle was sealed by the rubber tube and filled with CO2 for 30 min, then, followed by the irradiation of a 450 nm LED light. With the increase of N/Co ratio in the catalyst, the yield of CO product shows a volcano-type trend (Table 1). The highest yield of 1170 mmol g−1 for CO is achieved when the ratio between N and Co reaches 6:1. This result indicates that the appropriate coordination structure is feasible for the reduction of CO2. When the ratio of N/Co is low (entries 1 and 2), the uncoordinated N atoms is insufficient to adsorb CO2. While too much N atoms would saturate the cobalt center and prevent it from acting as an active center to catalyze CO2 reduction (entries 4 and 5). Besides, in the liquid phase, only trace amount of formate (∼0.4 μmol) was detected by ion chromatograph (Fig. S4†). In addition, photoreactions with different catalyst dosage show that the production of CO is a first order dependence on the concentration of PEI6-Co (Fig. S5†).
| Entry | Catalyst | nH2 (μmol) | nCO (μmol) | Selectivity of CO | Activity (mmol g−1) |
|---|---|---|---|---|---|
| a Reaction conditions: PEIx-Co (5 μg), [Ru(phen)3] (PF6)2 (0.4 mM), TEOA (0.3 M), 5 mL CH3CN/H2O (v/v = 4:1) solution, 25 °C, 12 h. 450 nm LED lamp (100 mW cm−2, irradiation area 0.8 cm2). |
|||||
| 1 | PEI4-Co | 0.35 | 4.35 | 93% | 870 |
| 2 | PEI5-Co | 0.36 | 4.71 | 93% | 942 |
| 3 | PEI6-Co | 0.34 | 5.84 | 95% | 1170 |
| 4 | PEI7-Co | 0.30 | 5.09 | 96% | 1020 |
| 5 | PEI8-Co | 0.26 | 4.90 | 95% | 981 |
To investigate the synergistic catalysis effect of PEI6-Co, the reported dinuclear cobalt cryptate Co2L and mononuclear cobalt CoL1 complex were chosen as the counterpoints and the photoreactions were carried out under similar conditions (the amount of Co ion was kept same in these catalysts). As shown in Fig. 3a, for PEI6-Co, the CO product increased nonlinearly under the visible light irradiation, giving an activity of 1170 mmol g−1 for CO2-to-CO conversion (Fig. 3a, black line). This value is almost 2.8 times of mononuclear cobalt catalyst CoL1 (416 mmol g−1, Fig. 3a, blue line) and comparable with dinuclear cobalt catalyst Co2L (884 mmol g−1, Fig. 3a, red line), indicating that the easy-to-synthesized coordination polymer can also achieve the efficient reduction of CO2 as elaborate molecular catalyst. It is worth noting that when the reaction time reaches 6 h, the evolution rate of CO slows down and even stop. We speculate that the instability of photosensitizer may be a cause of the reaction stopping. Thus we investigate the stability of [Ru(phen)3](PF6)2 before and after the photoreaction using mass spectrometry. The results in Fig. S6 and S7† show that one phenanthroline ligand is substituted by two H2O molecule after 10 h light irradiation and form [Ru(phen)2(H2O)2](PF6)2, which has no effect for CO2 reduction as photosensitizer. In addition, the tracking of UV-visible absorption spectrum of [Ru(phen)3](PF6)2 shows that the absorbance of [Ru(phen)3](PF6)2 decreases with the light irradiation (Fig. S8†). These results suggest that [Ru(phen)3](PF6)2 decomposed with the light irradiation. In addition, we performed a cyclic stability test on the reaction system by re-adding the photosensitizer [Ru(phen)3](PF6)2 to the stopped photoreactor. The results in Fig. 3b show that with the addition of extra [Ru(phen)3](PF6)2, the photoreaction could restart and the evolution rate of CO is almost maintained, which suggests that the decomposition of [Ru(phen)3](PF6)2 is the main fact for the reduced rate of CO evolution while PEI6-Co is relatively stable as a photocatalyst for CO2 conversion in water containing system.
 | ||
| Fig. 3 (a) Comparison of photocatalytic carbon dioxide reduction yield of PEI6-Co Co2L and CoL1. (b) Stability test of PEI6-CO. | ||
To illustrate the role of each component in the photoreaction, a sequence of control experiments were carried out (Table 2). In the absence of PEI6-Co, a little amount of CO generated (Table 2, entry 1), indicating that PEI6-Co was significant to drive the reduction of CO2 and [Ru(phen)3](PF6)2 could catalyze the conversion of CO2-to-CO weakly. Further control experiments suggested that the existence of [Ru(phen)3](PF6)2, TEOA, and light was indispensable for the generation of CO (Table 2, entries 2–4). Additionally, when Ar was used as the substitution of CO2 (Table 2, entries 5), no CO was detected in the photocatalytic system, indicating that the producing of CO was originated from the reduction of CO2 instead of other organic components.
Conclusions
In summary, based on the synergic catalytic strategy of dinuclear cobalt, we designed and synthesized a transition metal coordination polymer PEI6-Co and investigated its catalytic activity towards CO2 reduction. The results show that because of the effective adsorption of CO2 by PEI and the synergic catalytic effect of the adjacent cobalt active sites in PEI6-Co, the conversion of CO2-to-CO with high yield of 1170 mmol g−1 and selectivity of 95% were achieved in water containing medium. Such performance is almost 2.8 times of mononuclear cobalt catalyst CoL1 and comparable with dinuclear cobalt catalyst Co2L. This work provides a prospective strategy for the transformation of molecular catalyst to heterogeneous catalyst in a convenient and cost-effective way.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by NSFC (21702146) and Natural Science Foundation of Tianjin (19JCQNJC05500).Notes and references
- S. Chen, Y. Qi, C. Li, K. Domen and F. Zhang, Joule, 2018, 2, 2260–2288 CrossRef CAS.
- J. Jian, G. Jiang, R. van de Krol, B. Wei and H. Wang, Nano Energy, 2018, 51, 457–480 CrossRef CAS.
- D. Kim, K. K. Sakimoto, D. Hong and P. Yang, Angew. Chem., Int. Ed., 2015, 54, 3259–3266 CrossRef CAS PubMed.
- A. K. Singh, J. H. Montoya, J. M. Gregoire and K. A. Persson, Nat. Commun., 2019, 10, 443 CrossRef CAS PubMed.
- H. Tong, S. Ouyang, Y. Bi, N. Umezawa, M. Oshikiri and J. Ye, Adv. Mater., 2012, 24, 229–251 CrossRef CAS PubMed.
- C. Gao, Q. Meng, K. Zhao, H. Yin, D. Wang, J. Guo, S. Zhao, L. Chang, M. He, Q. Li, H. Zhao, X. Huang, Y. Gao and Z. Tang, Adv. Mater., 2016, 28, 6485–6490 CrossRef CAS PubMed.
- T. Arai, S. Sato and T. Morikawa, Energy Environ. Sci., 2015, 8, 1998–2002 RSC.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- D. C. Grills and E. Fujita, J. Phys. Chem. Lett., 2010, 1, 2709–2718 CrossRef CAS.
- K. Li, B. Peng and T. Peng, ACS Catal., 2016, 6, 7485–7527 CrossRef CAS.
- X. Li, J. Yu, M. Jaroniec and X. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- Q.-Q. Bi, J.-W. Wang, J.-X. Lv, J. Wang, W. Zhang and T.-B. Lu, ACS Catal., 2018, 8, 11815–11821 CrossRef CAS.
- S. Gao, Y. Lin, X. Jiao, Y. Sun, Q. Luo, W. Zhang, D. Li, J. Yang and Y. Xie, Nature, 2016, 529, 68 CrossRef CAS PubMed.
- P. S. Nabavi Zadeh, M. Zezzi do Valle Gomes, B. Åkerman and A. E. C. Palmqvist, ACS Catal., 2018, 8, 7251–7260 CrossRef CAS.
- L.-Y. Wu, Y.-F. Mu, X.-X. Guo, W. Zhang, Z.-M. Zhang, M. Zhang and T.-B. Lu, Angew. Chem., Int. Ed., 2019, 58, 9491–9495 CrossRef CAS PubMed.
- H. Jung, S. Y. Lee, C. W. Lee, M. K. Cho, D. H. Won, C. Kim, H.-S. Oh, B. K. Min and Y. J. Hwang, J. Am. Chem. Soc., 2019, 141, 4624–4633 CrossRef CAS PubMed.
- J. L. White, M. F. Baruch, J. E. Pander, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- R. Kuriki, M. Yamamoto, K. Higuchi, Y. Yamamoto, M. Akatsuka, D. Lu, S. Yagi, T. Yoshida, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2017, 56, 4867–4871 CrossRef CAS PubMed.
- T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 16825–16828 CrossRef CAS PubMed.
- T. Morimoto, C. Nishiura, M. Tanaka, J. Rohacova, Y. Nakagawa, Y. Funada, K. Koike, Y. Yamamoto, S. Shishido, T. Kojima, T. Saeki, T. Ozeki and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 13266–13269 CrossRef CAS PubMed.
- S. Sato, T. Morika, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- C. Cometto, R. Kuriki, L. Chen, K. Maeda, T.-C. Lau, O. Ishitani and M. Robert, J. Am. Chem. Soc., 2018, 140, 7437–7440 CrossRef CAS PubMed.
- P. G. Alsabeh, A. Rosas-Hernandez, E. Barsch, H. Junge, R. Ludwig and M. Beller, Catal. Sci. Technol., 2016, 6, 3623–3630 RSC.
- T. Ouyang, H.-H. Huang, J.-W. Wang, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2017, 56, 738–743 CrossRef CAS PubMed.
- T. Ouyang, H.-J. Wang, H.-H. Huang, J.-W. Wang, S. Guo, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Angew. Chem., Int. Ed., 2018, 57, 16480–16485 CrossRef CAS PubMed.
- V. S. Thoi, N. Kornienko, C. G. Margarit, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 14413–14424 CrossRef CAS PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- E. Torralba-Peñalver, Y. Luo, J.-D. Compain, S. Chardon-Noblat and B. Fabre, ACS Catal., 2015, 5, 6138–6147 CrossRef.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
- W. Kubo and T. Tatsuma, J. Am. Chem. Soc., 2006, 128, 16034–16035 CrossRef CAS PubMed.
- L. Zong, C. Wang, A. M. P. Moeljadi, X. Ye, R. Ganguly, Y. Li, H. Hirao and C.-H. Tan, Nat. Commun., 2016, 7, 13455 CrossRef PubMed.
- N. P. Mankad, Chem. – Eur. J., 2016, 22, 5822–5829 CrossRef CAS PubMed.
- A. Magnuson, M. Anderlund, O. Johansson, P. Lindblad, R. Lomoth, T. Polivka, S. Ott, K. Stensjo, S. Styring, V. Sundstrom and L. Hammarstrom, Acc. Chem. Res., 2009, 42, 1899–1909 CrossRef CAS PubMed.
- S. Zhang, P. Kang, S. Ubnoske, M. K. Brennaman, N. Song, R. L. House, J. T. Glass and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 7845–7848 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Including XPS, CV, HRMS (ESI), UV absorption spectrum. See DOI: 10.1039/c9ra10962e |
| This journal is © The Royal Society of Chemistry 2020 |