
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and in vitro biological evaluation of isoxazol-4-carboxa piperidyl derivatives as new anti-influenza A agents targeting virus nucleoprotein†
Shuchen Pei ,
Shihao Xia,
Fating Yang,
Junlin Chen,
Mengdie Wang,
Wanlin Sun,
Ziqiang Li,
Kangyao Yuan and
Jun Chen*
,
Shihao Xia,
Fating Yang,
Junlin Chen,
Mengdie Wang,
Wanlin Sun,
Ziqiang Li,
Kangyao Yuan and
Jun Chen*
School of Chemistry and Chemical Engineering, Chongqing University of Science and Technology, Chongqing 401331, P. R. China. E-mail: wxchenjun@163.com; Tel: +86-023-65022211
First published on 27th January 2020
Abstract
Influenza infection is a major cause of morbidity and mortality during seasonal epidemics and sporadic pandemics. It is important and urgent to develop new anti-influenza agents with a new mechanism of action. Nucleozin has been reported as a potent antagonist of nucleoprotein accumulation in the nucleus. In this study, a new series of isoxazol-4-carboxa piperidyl derivatives 1a–j were synthesized and their chemical structures were confirmed by 1H, 13C NMR and mass spectral data. Furthermore, all the synthesized compounds were evaluated for in vitro anti-influenza virus activity against influenza virus (A/PR/8/34 H1N1). Among all the compounds, 1a, 1b, 1c, 1f and 1g exhibited more potent activity than the standard drug, and compound 1b has showed most promising anti-influenza virus activity. These results are also consistent with the docking study results in terms of the design of compounds targeting influenza A via viral nucleoprotein.
Introduction
Seasonal and pandemic influenza represent one of the major threats to public health. The annual influenza epidemic results in 250![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–500000 deaths worldwide.1 During the past century, the 1918 Spanish flu, 1957 Asian flu and 1968 Hongkong flu pandemics caused millions of fatalities.2 Influenza infection is a major cause of morbidity and mortality during seasonal epidemics and sporadic pandemics. Nucleoproteins (NP) that encapsulate viral RNA forming ribonucleoprotein (RNP) particles in influenza viruses are responsible for vital functions of transcription, assembly and packaging.3,4 More importantly, the viral genetic shift may eventually cause a breakthrough of interspecies and inter-human transmission barriers and raise the possibility of transmission from animals to human beings or vice versa (human beings to animals), which would be disastrous to public health.5–7 Two classes of anti-influenza drugs are available in the market, M2 inhibitors and neuraminidase inhibitors. However, the efficacies of these drugs are limited due to the rapid emergence of mutated viral strains and side effects.8,9 Thereby, it is important and urgent to develop new anti-influenza agents with a new mechanism of action.10,11
000–500000 deaths worldwide.1 During the past century, the 1918 Spanish flu, 1957 Asian flu and 1968 Hongkong flu pandemics caused millions of fatalities.2 Influenza infection is a major cause of morbidity and mortality during seasonal epidemics and sporadic pandemics. Nucleoproteins (NP) that encapsulate viral RNA forming ribonucleoprotein (RNP) particles in influenza viruses are responsible for vital functions of transcription, assembly and packaging.3,4 More importantly, the viral genetic shift may eventually cause a breakthrough of interspecies and inter-human transmission barriers and raise the possibility of transmission from animals to human beings or vice versa (human beings to animals), which would be disastrous to public health.5–7 Two classes of anti-influenza drugs are available in the market, M2 inhibitors and neuraminidase inhibitors. However, the efficacies of these drugs are limited due to the rapid emergence of mutated viral strains and side effects.8,9 Thereby, it is important and urgent to develop new anti-influenza agents with a new mechanism of action.10,11
Influenza virus is a single-stranded, negative-sense, segmented RNA virus of the Orthomyxovirus family. As such, it uses a heterotrimeric RNA-dependent RNA polymerase in complex with its RNA genome and associated nucleoprotein (NP) to synthesize the requisite viral RNAs (vRNAs), mRNAs, and cRNAs needed for replication.12,13 NP plays several roles during the viral life cycle, including ribonucleoprotein (RNP) formation, transport of vRNA-polymerase complex to the nucleus, viral replication, and virion assembly.14–17 Consequently, inhibitors of NP function may affect multiple stages of the life cycle.
Nucleozin has been reported as a “potent antagonist of NP accumulation in the nucleus”.18 Nucleozin induces the formation of NP aggregates and antagonizes its nuclear accumulation, leading to cessation of viral replication. It was reported that a series of nucleozin derivatives showed a good antiviral activity in vivo.19 In previous work, we designed and synthesized a series of nucleozin derivatives with prominent antiviral activities against influenza A H1N1.20 In the present study, target compounds of isoxazol-4-carboxa piperidyl derivatives were designed and synthesized. All the newly synthesized compounds were evaluated in cell toxicity assays and viral inhibition assays.
Results
Chemistry
Nucleozin was the lead compound of a series of derivatives selected by Kao and coworkers,18 and it was comprised of four components: aryl group, isoxazole, piperazinyl and 2-chloro-4-nitrophenyl. The authors studied the effect of the substitution on the aryl portion of phenyl with different groups such as Cl-, MeO-, both on anti-influenza virus activity and cytotoxicity. The work highlighted the importance of the substitution on the aromatic ring, in particular they found nucleozin impeded influenza A virus replication in vitro with a nanomolar median effective concentration (EC50) and protected mice challenged with lethal doses of avian influenza A H5N1.In order to assess the relevance of the piperazinyl group and its importance for anti-influenza virus activity, we planned to synthesize nucleozin piperidyl derivatives and test them against influenza virus. The aim was to evaluate modifications such as the introduction of substituents on the aromatic ring and the piperidyl group substituted, as depicted in Fig. 1.
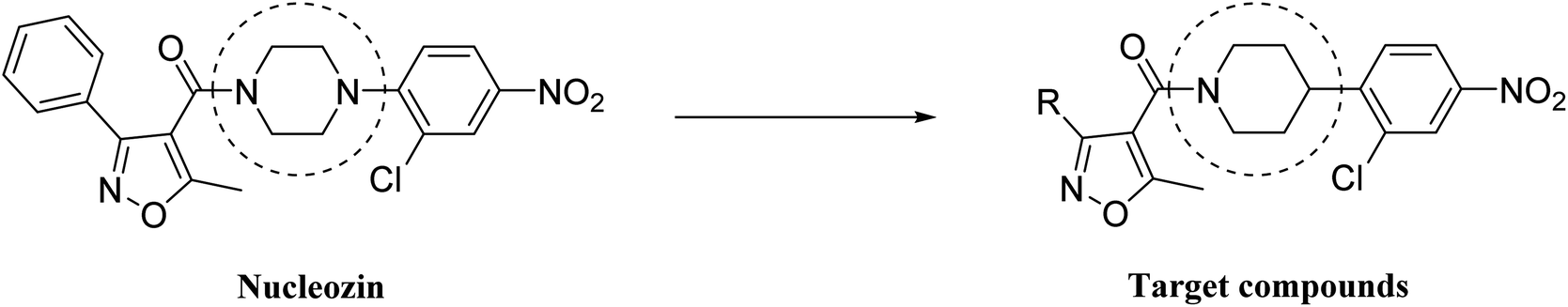 | ||
| Fig. 1 Design of isoxazol-4-carboxa piperidyl derivatives. | ||
The synthetic route to prepare all the isoxazol-4-carboxa piperidyl derivatives is depicted in Scheme 1. 5-Methyl-isoxazole-4-carboxylic acid 4a–j and 4-(2-chloro-4-nitrophenyl) piperidine 9 are key intermediates for the preparation of the desired compound 1a–j. Firstly, ethyl acetoacetate was treated with different substituted aromatic nitriles in toluene and TiCl4 was added. Then the mixture was refluxed for 2 h to afford compounds 2a–j.21,22 The intermediates 2a–j were reacted with hydroxylamine hydrochloride in ethanol and the reaction mixture was stirred at reflux for 2 h to obtain compounds 3a–j. Further, these compounds 3a–j were hydrolyzed under basic condition to afford pure compounds 4a–j.
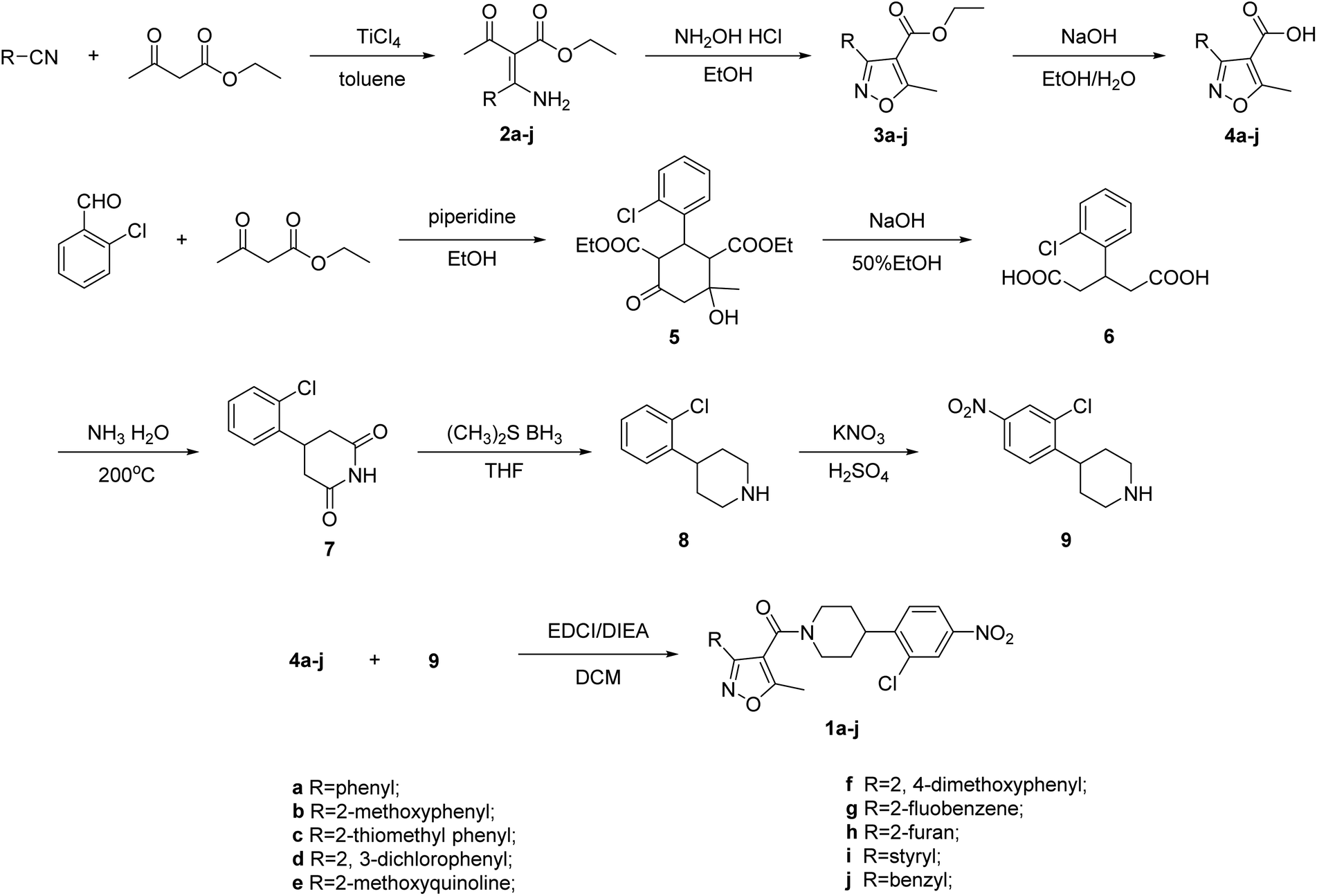 | ||
| Scheme 1 Synthesis of isoxazol-4-carboxa piperidyl derivative. | ||
On the other hand, the synthesis of 4-(2-chloro-4-nitrophenyl) piperidine 9 was carried out via a 5-step reaction. The formation of the intermediate 3-(2-chlorophenyl) pentanedioic acid 6 was obtained by the reaction of 2-chlorobenzaldehyde and ethyl acetoacetate with piperidine in ethanol at reflux temperature for 3 h. The subsequent hydrolyzation with NaOH in 50% ethanol at reflux temperature provided the compound 6. The intermediate 6 reacted with ammonia at 200 °C to obtain compound 7. The intermediate 9 was synthesized by the reduction and nitration of compound 7. Finally, these acid intermediates 4a–j were subjected to coupling reaction with intermediate 9 in the presence of EDCI, DIEA as coupling reagents and stirred at room temperature for 2 h in anhydrous DCM as the solvent to afford pure corresponding products 1a–j as shown in Scheme 1. The synthesized piperidyl derivatives 1a–j were characterized by 1H/13C NMR and mass spectral analysis.
Anti-influenza virus activity and cytotoxicity measurement
Discussion
Anti-influenza A virus activity
All the compounds prepared herein 1a–j were screened for their anti-influenza activity against influenza virus (A/PR/8/34 H1N1 strain) and the results acquired were incorporated in Table 1. Ribavirin was used as the standard reference drug and most of the tested compound displayed good to moderate activity with respect to all cell lines. The EC50 values of synthesized compounds ranged from 0.235 to 34.483 μM and the standard drug was at 9.891 μM. Among them, five compounds, 1a, 1b, 1c, 1f and 1g exhibited excellent activity than ribavirin. Further, compound 1b (R = 2-methoxyphenyl) showed the most promising activity (EC50 = 0.253 ± 0.001 μM, CC50 = 69.05 ± 0.11 μM, SI = 272.93). The replacement of (R = phenyl) with (R = 2-thiomethyl phenyl) resulted in compound 1c which exhibited lower activity (EC50 = 0.502 ± 0.002 μM, SI = 398.41) when compared with compound 1b. Compound 1a (R = phenyl) displayed good activity on the cell line (EC50 = 0.898 ± 0.004 μM, CC50 = 19.78 ± 0.07 μM, SI = 22.02) when compared with compound 1c. Whereas, compound 1f with (R = 2,4-dimethoxyphenyl) groups (EC50 = 2.036 ± 0.009 μM, CC50 = 27.85 ± 0.08 μM, SI = 13.68) and 1g with (R = 2-fluorobenzene) groups (EC5 = 1.356 ± 0.008 μM, CC50 = 16.64 ± 0.05 μM, SI = 12.27) exhibited comparable activity, respectively. The rest compounds 1d, 1e, 1h, 1i and 1j showed moderate activity on the cell line.| Entry | Compound | Cytotoxicity CC50b/(μM) | Anti-influenza virus activity | |
|---|---|---|---|---|
| EC50c/(μM) | SId | |||
| a All the data were the average values from three independent assays.b Compound concentration that reduces cell viability by 50% relative to uninfected MDCK cells.c Compound concentration that reduces viral replication by 50% relative to infected MDCK cells.d Selectivity index: CC50/EC50. | ||||
| 1 | 1a | 19.78 ± 0.07 | 0.898 ± 0.004 | 22.02 |
| 2 | 1b | 69.05 ± 0.11 | 0.253 ± 0.001 | 272.93 |
| 3 | 1c | >100 | 0.502 ± 0.002 | — |
| 4 | 1d | >100 | 21.572 ± 0.015 | — |
| 5 | 1e | 24.96 ± 0.09 | 9.905 ± 0.010 | 2.52 |
| 6 | 1f | 27.85 ± 0.08 | 2.036 ± 0.009 | 13.68 |
| 7 | 1g | 16.64 ± 0.05 | 1.356 ± 0.008 | 12.27 |
| 8 | 1h | >100 | 16.226 ± 0.012 | — |
| 9 | 1i | >100 | 34.483 ± 0.014 | — |
| 10 | 1j | >100 | 19.410 ± 0.012 | — |
| 11 | Ribavirin | 63.01 ± 0.05 | 9.891 ± 0.06 | 6.37 |
Cytotoxicity
Cytotoxic activities of the compounds against MDCK cells were also evaluated to monitor the potential cytotoxicity effects. Most of the derivatives show no obvious cellular growth inhibition against MDCK cells at concentrations below 500 μM (the highest test concentration); while ribavirin displays moderate toxicity against MDCK cells (CC50 = 201.80 μM). It is worth mentioning that all the compounds bear a great selectivity index.Molecular docking study
The X-ray crystal structure of influenza A virus nucleoprotein complex with inhibitors was disclosed. It was demonstrated that six inhibitors bridged two molecules of NP (NP_A and NP_B) protein to form a stable dimer complex. A computational study was performed using Schrodinger 2013 to investigate the differences of potential interactions between compounds 1b and nucleozin and virus nucleoprotein (PDB ID: 3RO5). The results indicate that compounds 1b and nucleozin have similar action modes. Although the 2-chlor-4-nitrophenyl fragments of three molecules overlapped well, there is a discrepancy on the other side of the molecule, which might be the reason why there is a disparity in the anti influenza viral activity (Fig. 2).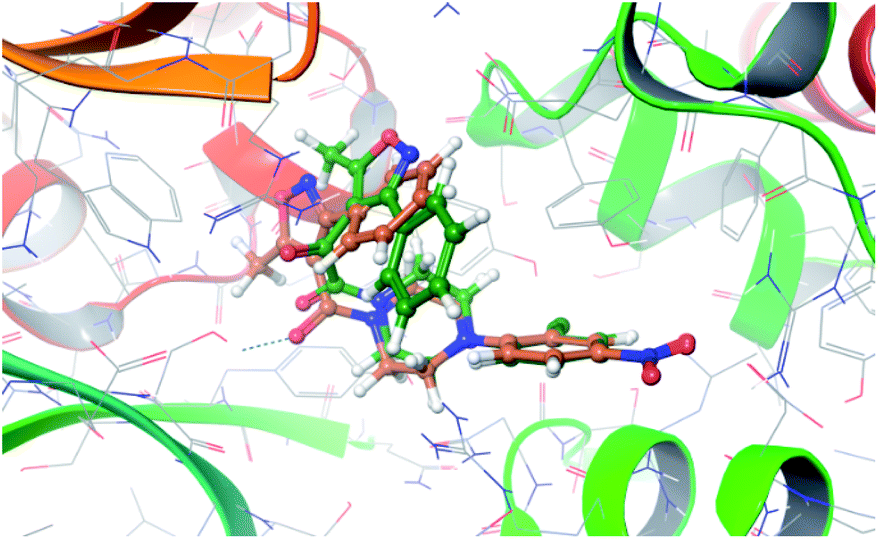 | ||
| Fig. 2 Orientation of the best docked poses of compound 1b and nucleozin in the active site of the viral nucleoprotein. | ||
As shown in Fig. 3, a strong π–π stacking interaction has been formed between 4-nitro-2-chloro-phenyl moiety and TYR289. The pyrazine ring of nucleozin also plays a crucial role in making the molecule into a more stable conformation so that nucleozin could link the two trimers more steadily. This gives a higher inhibitory activity. The diversity of the binding modes has aroused our great interests and encourages us to expansion additional study on this topic despite the fact that all the compounds we reported here have lower activity than nucleozin.
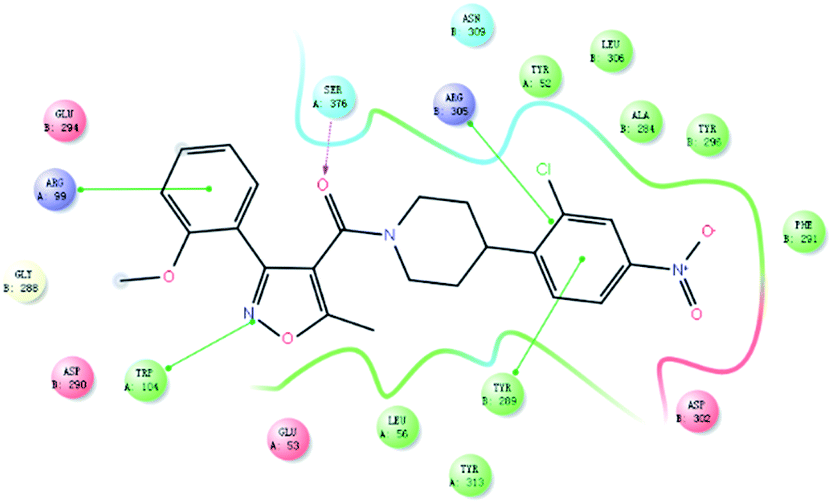 | ||
| Fig. 3 Interaction mode between viral nucleoprotein and compound 1b. | ||
Experimental
Generals
All the reactions were carried out under the protection of argon (Ar) atmosphere. Most chemicals and solvents were analytical grade and used without further purification. Thin layer chromatography (TLC) was performed using precoated silica gel GF254 (0.2 mm) while column chromatography was performed using silica gel (100–200 mesh). The melting point was measured on a YRT-3 melting point apparatus (Shantou Keyi Instrument & Equipment Co., Ltd, Shantou, China) without thermometer correction. 1H NMR spectra were collected on a Varian INOVA400 (Varian, Palo Alto, USA) using CDCl3, DMSO-d6 or D2O as solvent. Chemical shifts were expressed in δ, with tetramethylsilane (TMS) as the internal reference; coupling constants (J) were expressed in Hz. The high-resolution mass (HRMS) spectra were recorded on a Bruker maxis impact Q-TOF instrument (Bruker, Billerica, USA) coupled with a Dionex Ultimate 3000 spectrometer (Dionex, Sunnyvale, USA).General procedure for the preparation of compounds 2a–j
To a solution of nitriles (1 eq.), TiCl4 (1.2 eq.) and ethyl acetoacetate (1.2 eq.) were added at room temperature with stirring. The mixture was refluxed with stirring for 2 h. After cooling to room temperature, saturated sodium carbonate solution was added, and the mixture was extracted with ethyl acetate. The combined organic phases were washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The residue was purified by flash column chromatography on silica gel to afford pure compounds 2a–j.General procedure for the preparation of compounds 3a–j
To a solution of 2a–j (1 eq.) in ethanol was added hydroxylammonium chloride (1.5 eq.). the reaction mixture was refluxed with stirring for 2 h until TLC showed no starting materials. The suspension was cooled to room temperature and evaporated to dryness in vacuum followed by the addition of H2O and the extraction with ethyl acetate three times. The organic layer was combined, washed with brine three times and dried over Na2SO4. After the solvent was evaporated in vacuum to obtain a crude product, which was further purified by flash chromatography on silica gel to get 3a–j.General procedure for the preparation of compounds 4a–j
To a solution of 3a–j (1 eq.) in ethanol was added sodium hydroxide (5 eq.) in water. The reaction mixture was refluxed with stirring for 2 h until TLC showed no starting materials. The solution was evaporated in vacuum. The water layer was adjusted with 10% HCl until the pH was about 3. The mixture was filtered, the solid cake dried in a vacuum at 50 °C to give compounds 4a–j, which was used without further purification.Synthesis of 3-(2-chlorophenyl)pentanedioic acid (6)
To a solution of 2-chlorobenzaldehyde (20 g, 0.142 mol) in ethanol (100 ml) was added ethyl acetoacetate (36.2 ml, 0.284 mol) and piperidine (2 ml). The reaction mixture was stirred at room temperature for 4 h until TLC showed no starting materials. The reaction mixture was concentrated in vacuo and extracted with EtOAc (2 × 200 ml). The combined EtOAc layers were dried over Na2SO4 and concentrated to give the crude product 5, which was used without further purification.To a solution of crude product 5 in ethanol (220 ml) was added sodium hydroxide (55 g, 1.375 mol) in water (250 ml). The reaction mixture was refluxed with stirring for 3 h until TLC showed no starting materials. The reaction mixture was concentrated in vacuo. The pH value was adjusted to 4 with 1 mol L−1 HCl, and extracted with EtOAc (2 × 200 ml). The combined EtOAc layers were washed with brine and dried over Na2SO4. After the solvent was evaporated in vacuum, an off-white solid was obtained and further purified by recrystallization in acetone to obtain the product as a white crystal (19.5 g). Two steps yield 56.7%. mp 152–154 °C.
Synthesis of 4-(2-chlorophenyl)piperidine-2,6-dione (7)
To a solution of compound 6 (19.5 g) dissolved in ammonium hydroxide (25 ml), The reaction mixture was concentrated in vacuo and the residue was heated at 200 °C for 5 h. And then the residue was cooled to room temperature, and dissolved in ethyl acetate and washed with sat. NaHCO3, and brine. The combined EtOAc layers were dried over Na2SO4. After the solvent was evaporated in vacuum, an off-white solid was obtained and further purified by recrystallization in acetone to obtain the product as a white crystal (15.4 g). Yield 85.6% mp 155–157 °C; 1H NMR (CDCl3, 400 MHz, δ ppm): 7.43 (dd, 1H, J = 8.0 Hz), 7.30 (m, 2H), 7.21 (dd, 1H, J = 7.6 Hz), 3.91 (m, 1H), 2.98 (d, 2H, J = 8.0 Hz), 2.94 (d, 1H, J = 4.8 Hz), 2.76 (m, 2H). MS (ESI): 224.4 [M+ + H].Synthesis of 4-(2-chlorophenyl)piperidine (8)
A solution of compound 7 (10 g, 44.84 mol) in THF (100 ml) was cooled to 0 °C and borane dimethyl sulfide (25 ml, 89.68 mol) was added dropwise. The mixture was refluxed for 6 h. And then the residue was cooled to room temperature, and added 6 mol L−1 HCl (150 ml). The mixture was refluxed for 4 h until TLC showed no starting materials. The reaction mixture was concentrated in vacuo. The pH value was adjusted to 10 with NaOH, and extracted with EtOAc (2 × 200 ml). The combined EtOAc layers were washed with brine and dried over Na2SO4. After the solvent was evaporated in vacuum to obtain a crude product, which was further purified by flash chromatography on silica gel to get white solid (5.06 g). Yield 58.2% mp 210–215 °C; 1H NMR (CDCl3, 400 MHz, δ ppm): 7.33 (t, 2H, J = 6.4 Hz), 7.23 (m, 1H), 7.14 (t, 1H, J = 7.6 Hz), 3.64 (t, 1H, J = 4.8 Hz), 3.27 (d, 1H, J = 10.8 Hz), 3.12 (m, 1H), 2.59 (s, 1H), 2.31 (s, 1H), 1.93 (s, 2H), 1.75 (m, 2H), 1.26 (m, 1H). MS (ESI): 196.1 [M+ + H].Synthesis of 4-(2-chloro-4-nitrophenyl)piperidine (9)
A solution of compound 8 (5 g, 25.64 mmol) in con. H2SO4 (50 ml) was cooled to 0 °C and KNO3 (2.6 g, 25.64 mmol) was added. The mixture was stirred at room temperature overnight until TLC showed no starting materials. The solution was poured into ice. The pH value was adjusted to 10 with NaOH, and extracted with EtOAc (2 × 50 ml). The combined EtOAc layers were washed with brine and dried over Na2SO4. After the solvent was evaporated in vacuum to obtain a crude product, which was further purified by flash chromatography on silica gel to get yellow solid (4.94 g). Yield 80.3% 1H NMR (CDCl3, 400 MHz, δ ppm): 8.19 (s, 1H), 8.02 (dd, 1H, J = 8.0 Hz), 7.53 (d, 1H, J = 8.8 Hz), 3.74 (s, 1H), 3.32 (d, 1H, J = 12.4 Hz), 3.22 (t, 1H, J = 12.0 Hz), 2.86 (t, 1H, J = 12.0 Hz), 1.92 (d, 2H, J = 12.4 Hz), 1.72 (m, 2H). MS (ESI): 241.6 [M+ + H].General procedure for the preparation of compounds 1a–j
To a solution of 4a–j (1 eq.) in DCM was added compound 9 (2.5 eq.), HOBT (2.5 eq.), EDCI (4 eq.) and triethylamine (2.5 eq.). The reaction mixture was stirred at room temperature overnight until TLC showed no starting materials. The reaction mixture was washed with brine and the organic layers were dried over Na2SO4. After the solvent was evaporated in vacuum to obtain a crude product, which was further purified by flash chromatography on silica gel to get 1a–j.Conclusions
In conclusion, we have synthesized a new series of isoxazol-4-carboxa piperidyl derivatives 1a–j through a facile route and their anti-influenza virus activity was demonstrated. These compounds were tested for their preliminary anti-influenza virus activity against influenza virus (A/PR/8/34 H1N1) and ribavirin was used as the standard reference drug. All these molecules showed good to moderate activity. Among all the synthesized compounds, 1a, 1b, 1c, 1f and 1g exhibited more potent activity than standard drug, and compound 1b showed the most promising activity. Our results suggest that compound 1b has a significant potential for further development into a new class of antiviral therapeutics targeting of the influenza nucleoprotein.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Natural Science Foundation of Chongqing, grant number cstc2019jcyj-msxmX0083 and Scientific and Technological Research Program of Chongqing Municipal Education Commission, grant number KJQN201901501.Notes and references
- V. N. Petrova and C. A. Russell, The evolution of seasonal influenza viruses, Nat. Rev. Microbiol., 2017, 16, 47–60 CrossRef PubMed.
- M. Moriyama, T. Koshiba and T. Ichinohe, Influenza A virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses, Nature, 2019, 10, 1–14 CAS.
- F. Krammer, J. Gavin and D. Smith, et al. Influenza, Nat. Rev. Dis. Primers., 2018, 4, 1–21 CrossRef PubMed.
- T. J. Cheng and S. Y. Wang, et al. Chemical probes for drug-resistance assessment by binding competition (RABC): oseltamivir susceptibility evaluation, Angew. Chem., 2013, 52(1), 366–370 CrossRef CAS PubMed.
- R. Snacken, Pandemic planning, Vaccine, 2002, 20(16), 88–90 CrossRef.
- A. I. Karasin, M. M. Schutten and L. A. Cooper, et al. Genetic characterization of H3N2 influenza viruses isolated from pigs in North America, 1977-1999: Evidence for wholly human and reassortant virus genotypes, Virus Res., 2000, 68, 71–85 CrossRef CAS.
- K. S. Li, Y. Guan and J. Wang, et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia, Nature, 2004, 430, 209–213 CrossRef CAS.
- K. Das, J. M. Aramini and L. C. Ma, et al. Structures of influenza A proteins and insights into antiviral drug targets, Nat. Struct. Mol. Biol., 2010, 17, 530–553 CrossRef CAS PubMed.
- J. Gong, et al. Potential targets and their relevant inhibitors in anti-influenza fields, Curr. Med. Chem., 2009, 16, 3716–3739 CrossRef CAS PubMed.
- M. R. Krystal and N. A. Meanwell, Influenza-the case for combination therapy, Curr. Opin. Invest. Drugs, 2009, 10, 746–749 Search PubMed.
- E. A. Govorkova, A. Elena and R. G. Webster, Combination Chemotherapy for Influenza, Viruses, 2010, 2, 1510–1529 CrossRef CAS PubMed.
- A. Ishihama and K. Nagata, Viral RNA polymerases, Crit. Rev. Biochem., 1988, 23, 27–76 CrossRef CAS PubMed.
- P. Resa-Infante, N. Jorba, R. Coloma and J. Ortin, The influenza virus RNA synthesis machine: Advances in its structure and function, RNA Biol., 2011, 8, 207–215 CrossRef CAS PubMed.
- Y. Xu, Recent progress in human telomere RNA structure and function, Bioorg. Med. Chem. Lett., 2018, 28(15), 2577–2584 CrossRef CAS PubMed.
- T. Noda, H. Sagara and A. Yen, et al. Architecture of ribonucleoprotein complexes in influenza A virus particles, Nature, 2006, 439, 490–492 CrossRef CAS PubMed.
- J. F. Cros, A. García-Sastre and P. Palese, An Unconventional NLS is Critical for the Nuclear Import of the Influenza A Virus Nucleoprotein and Ribonucleoprotein, Traffic, 2005, 6, 205–213 CrossRef CAS PubMed.
- S. L. Noton, M. Simpson-Holley and E. Medcalf, et al. Studies of an Influenza A Virus Temperature-Sensitive Mutant Identify a Late Role for NP in the Formation of Infectious Virions, J. Virol., 2009, 83(2), 562–571 CrossRef CAS PubMed.
- R. Y. Kao, et al. Identification of influenza A nucleoprotein as an antiviral target, Nat. Biotechnol., 2010, 28, 600–605 CrossRef CAS PubMed.
- C. Y. Su, et al. High-throughput identification of compounds targeting influenza RNA-dependent RNA polymerase activity, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 19151–19156 CrossRef CAS PubMed.
- S. Zhang, B. Qu, H. Li and Y. Wu, Design, synthesis and antiviral activity evaluation of nucleozin derivatives of new inhibitors of influenza virus nucleoprotein, Chin. J. Med. Chem., 2016, 26, 1–9 CAS.
- S. Pei, C. Xue and L. Hai, et al. Synthesis of β-enaminodicarbonyl derivatives in the titanium chloride-promoted reactions of β-dicarbonyl compounds with nitriles, RSC Adv., 2014, 4, 38055–38058 RSC.
- Xu Cheng, S. Pei and C. Xue, et al. Reactions of b-diketone compounds with nitriles catalyzed by Lewis acids: a simple approach to β-enaminone synthesis, RSC Adv., 2014, 4, 63897–63900 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10828a |
| This journal is © The Royal Society of Chemistry 2020 |