
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of dopants on aW-phase antimonene: theoretical investigations
Qingxiao Zhou *ab,
Qian Zhanga,
Weiwei Jua,
Yanling Liua and
Jiahui Lia
*ab,
Qian Zhanga,
Weiwei Jua,
Yanling Liua and
Jiahui Lia
aCollege of Physics and Engineering, Henan University of Science and Technology, Luoyang 471023, People's Republic of China. E-mail: zhouqingxiao1989@163.com; Tel: +86 18567628290
bHenan Key Laboratory of Photoelectric Energy Storage Materials and Applications, Henan University of Science and Technology, Luoyang 471023, People's Republic of China
First published on 14th February 2020
Abstract
We systemically investigate the effect of dopants on the geometrics, electronic and magnetic properties of asymmetric washboard structure of antimonene (aW-Sb) by using density functional theory (DFT) calculations. The large binding energies and short bond lengths indicate the doped systems still maintain high stability. Pristine aW-Sb is a nonmagnetic semiconductor with a narrow band gap, while the doped aW-Sb exhibit metallic by doping. Furthermore, the Ti, V, Cr, Mn and Fe doping induced magnetic states, and the result of spin density indicates that the magnetic moments are mainly localized at dopant and the adjacent Sb atoms.
1. Introduction
Two-dimensional (2D) materials, such as graphene, transition metal dichalcogenides (TMDs), phosphorene, and silicene, have attracted considerable attention because of their intriguing mechanical, electronic, optical and thermal properties,1–6 which has led to these 2D materials being promising for new optoelectronic and nanoelectronic applications.7–10 Recently, antimonene, a monolayer of antimony, was predicted to exhibit a stable geometry and be a semiconductor by density functional theory (DFT).11 Subsequently, monolayer antimonene has been experimentally achieved on Bi2Te3 (111), Sb2Te3 (111),12 or PdTe2 (ref. 13) substrates. Furthermore, an asymmetric washboard structure of antimonene (aW-Sb) was found to maintain its stability at a high temperature of 1000 K, and its armchair and zigzag nanoribbons exhibit excellent electronic properties.Currently, for the exploration of 2D materials, doping is an effective method to achieve anticipant properties.14–20 Dai et al.21 investigated the effect of Fe-dopants on β-phase antimonene and found that the Fe-doped system exhibited stable room temperature ferromagnetism (RTFM). He et al.22 and Zhou et al.23 reported 3d transition metal (TM) doped β-phase antimonene, and the results suggested that the doped systems were magnetic semiconductors. Li et al.24 found that electric field and dopants improved the sensitivity to CO molecules of β-antimonene. Yang et al.25,26 investigated the effect of vacancies and 3d-transition-metal doped β-antimonene, and the results indicated that different vacancies and 3d-dopants resulted in different electronic structures and magnetic properties in antimonene. However, few studies explored the effect of dopants on the electronic and magnetic properties of aW-phase antimonene.
Therefore, in this work, we aim to systemically explore the effect of dopants (Ge, Se, Sn, Te, Sc, Ti, V, Cr, Mn, Fe, Co, and Ni) on the properties of aW-phase antimonene using DFT calculations. Firstly, we examined the stabilities of doped systems by calculating the binding energies, charge transfer, and bond lengths. Secondly, we investigated the change of electronic structures. Finally, the magnetic behaviors of doped systems were explored. We hope our results could provide useful information for designing new antimonene-based magnetic and electronic devices.
2. Calculation details and models
All calculations were done with the Vienna ab initio Simulation Package (VASP) (VASP).27–29 The generalized gradient approximation (GGA) using Perdew–Burke–Ernzerhof (PBE) parameterization was used to approximate the exchange-correlation potential,30 and the Projector-Augmented Wave (PAW) pseudopotentials were employed.31 A plane-wave basis set with the kinetic energy cutoff is 500 eV and the van ser Waals (vdW) was carried out by D3-Grimme correction (DFT-D3).32 Considering the localization of 3d TM-dopants, GGA+U is adopted to describe the strong on-site Coulomb interaction. Moreover, the spin–oribit couplings (SOC) act important role in the band structures33,34 and is also included to calculated the band structures in our study. The convergence criterion between two consecutive steps was set as 10−6 eV and a maximum force of 0.001 eV Å−1 was allowed on each atom. A 8 × 8 × 1 k-point sampling was used for 4 × 4 × 1 antimonene supercells. A vacuum space of 15 Å was set along the out-of-plane direction to avoid the interactions between the neighboring layers. To explore the stability of doped systems, the binding energy (Eb) for the doped systems, as follows:| Eb = Edoped-Sb − EV-Sb − Edopant | (1) |
3. Results and discussion
3.1 Geometry structures
Our optimized model of aW-phase antimonene (aW-Sb) is performed in Fig. 1(a) and (b). The lattice constants of a1 and a2 are calculated to be 4.28 Å and 4.77 Å, and the nearest-neighbor distances b1 and b2 are 2.93 Å and 2.87 Å, respectively. The calculated parameters of geometry structure of aW-Sb are well agreement with previous literatures.35,36 According to previous reports, aW-Sb is a semiconductor with narrow bandgaps. The band structure of aW-Sb is described in Fig. 1(c). It can be found that it exhibits a indirect band gap of 0.12 eV, which is consistent with previous PBE results.37,38 We also calculated the bandgap by HSE correction (PBE + HSE) and spin–orbit couplings method (PBE + SOC), and the values are 0.33 eV and 0.03 eV, and the HSE results is larger than PBE result. The SOC result is smaller than PBE result, which is due to the valence band near Y-point splits into two bands. Furthermore, the spin-up (majority) and spin-down (minority) channel are represented by black and blue color lines, which are completely symmetrical and the PBE band result is blue color. It suggests the aW-Sb is nonmagnetic. To examine the stability of aW-Sb, the phonon dispersion curves are shown in Fig. 1(d). It can be observed that no imaginary vibrating modes exist, and that illustrates the stability as monolayer antimonene.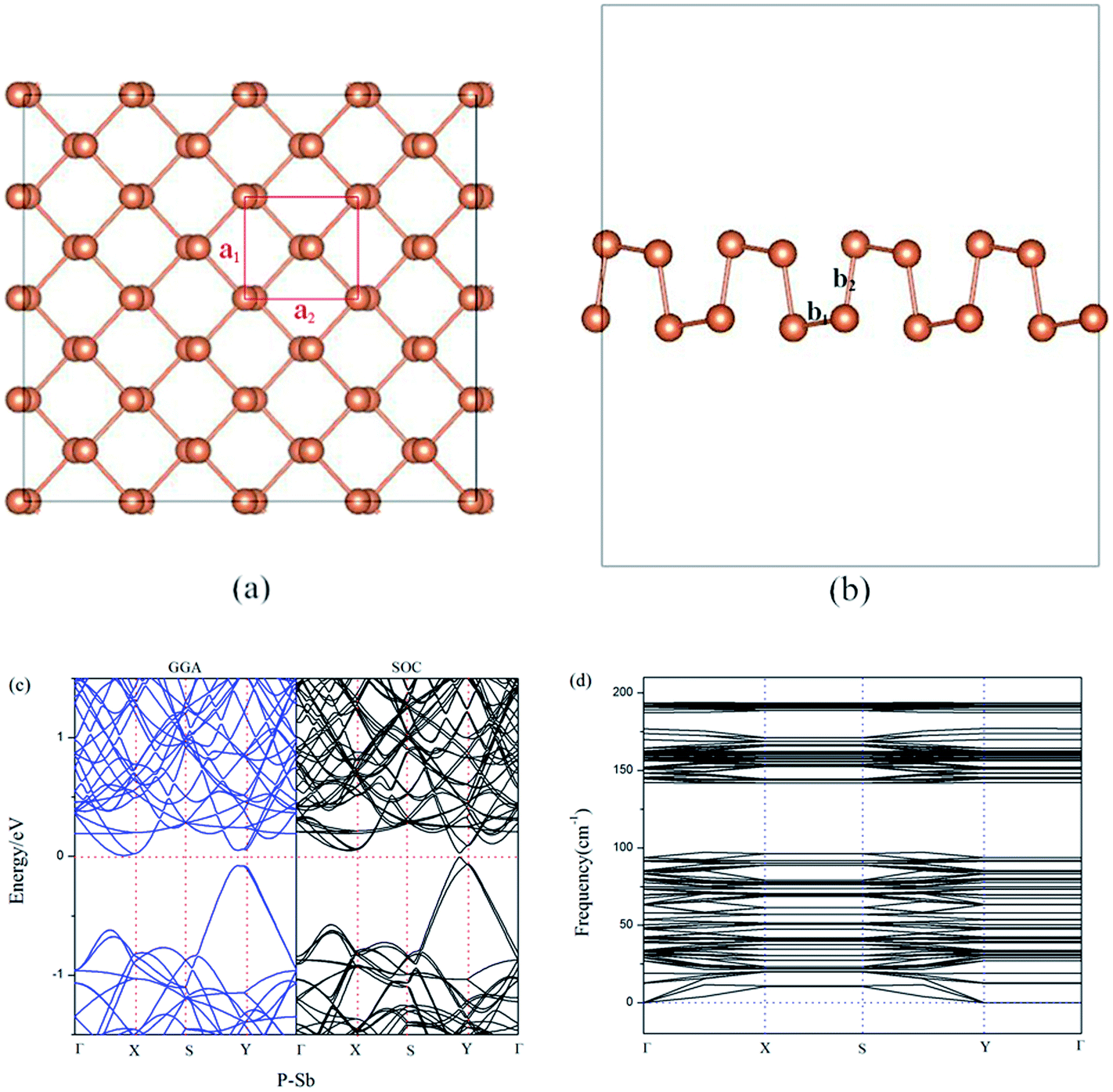 | ||
| Fig. 1 The top (a) and side (b) view of the optimized structure of pristine antimonene. (c) Band structure and (d) phonon dispersion curves structure of pristine antimonene. | ||
To systemically explore the influence of dopant on aW-Sb, we consider 10 types of dopants, including Ge, Se, Sn, Te, Sc, Ti, V, Cr, Mn, Fe, Co, and Ni. The doped aW-Sb structure was created by replacing a Sb atom by a dopant atom, and the doping concentration is 1.56%. Furthermore, the stability of doped systems is examined by the binding energy, and the nearest-neighbor distances between the Sb and dopants (d1 and d2) are also summarized in Table 1. The negative binding energies of doped systems indicate the stable geometry structures, and the Ti–Sb is the most stable with a binding energy of −6.40 eV. Moreover, we illustrate one type of optimized doped geometry structures in Fig. 2, namely, Ge–Sb. As shown in Fig. 2 and Table 1, after the doping of Ge, there is no obvious distortion appears and the value of b1 (2.93 Å) and b2 (2.87 Å) in pristine aW-Sb change to be 2.72 Å and 2.76 Å. The change of bond lengths of b1 varied in 0.01–0.44 Å, when that of b2 is 0.00–0.40 Å. Among the doped structures, the decrease of bond length in Co–Sb was the most obvious. Furthermore, the charge transfer of dopant is calculated by Bader charge analysis.39 The result indicates that Ge, Se, and Te act as acceptors, while the other dopants are donors. The large charge transfer between the dopants and aW-Sb substrate indicated the strong interaction present, and the Ti-dopant exhibits 0.89e, which is the largest value of Q and is agreement with its largest binding energy. Therefore, the small change of geometry structures, large binding energies and charge transfer are all imply the stability of doped systems.
| Adatom | d1 (Å) | d2 (Å) | Eb (eV) | Mtotal (μB) | Q (e) |
|---|---|---|---|---|---|
| Ge | 2.72 | 2.76 | −3.51 | 0 | −0.30 |
| Se | 2.76 | 2.67 | −3.51 | 0 | −0.53 |
| Sn | 2.91 | 2.98 | −3.24 | 0 | 0.18 |
| Te | 2.94 | 2.86 | −2.83 | 0 | −0.27 |
| Sc | 3.03 | 2.87 | −5.80 | 0 | 0.37 |
| Ti | 2.87 | 2.72 | −6.40 | 1.0 | 0.89 |
| V | 2.75 | 2.67 | −5.08 | 2.7 | 0.68 |
| Cr | 2.73 | 2.67 | −3.75 | 3.2 | 0.45 |
| Mn | 2.73 | 2.71 | −3.96 | 3.7 | 0.42 |
| Fe | 2.57 | 2.50 | −4.82 | 1.3 | −0.13 |
| Co | 2.49 | 2.47 | −5.88 | 0 | −0.36 |
| Ni | 2.55 | 2.50 | −5.70 | 0 | −0.31 |
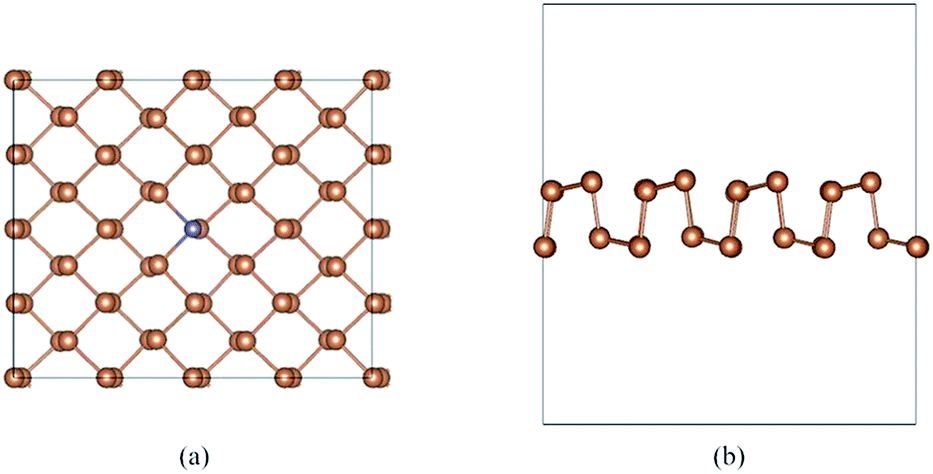 | ||
| Fig. 2 The top (a) and side (b) view of the optimized structure of Ge-doped antimonene. | ||
3.2 Electronic and magnetic properties
To further understand the effect of dopants on the electronic and magnetic structures, we further illustrate the band structure and projected density of states (PDOS) of all doped-Sb systems in Fig. 3 and 4. As performed in Fig. 3, the black and blue color lines represent spin-up and spin-down channels for the PBE results. The PBE results exhibit blue color, when the spin-up and spin-down bands are coincident. We first discuss the GGA (GGA+U) results. As shown in Fig. 3, the doped aW-phase antimonene exhibit metallic, due to the appearance of impurity-band around the Fermi level contributed by the dopant. We can classify the systems into two types, including weak conductivity and strong conductivity structures. It can be found that only a few channels of valence or conductive bands go through the Fermi level in Fig. 3(e)–(l) and then the Sc-, Ti-, V-, Cr-, Mn-, Fe-, Co-, and Ni-doped antimonene exhibit weak conductive character. Although, there are more bands through the Fermi level in the Ge-, Se-, Sn-, and Te-doped systems, indicating a character of relative strong conductivity. The pristine aW-phase antimonene is nonmagnetic structure, while the doping of Ti, V, Cr, Mn, and Fe induced magnetic states. As performed Fig. 3(f)–(j), the spin-up and spin-down band channels are unsymmetrical, and we summary the total magnetic moment of systems in Table 1. For the SOC results, it can be found that the splitting of electronic bands is not obvious, and the shapes of band structures of the doped systems with and without SOC are similar, which is agreement with previous report.22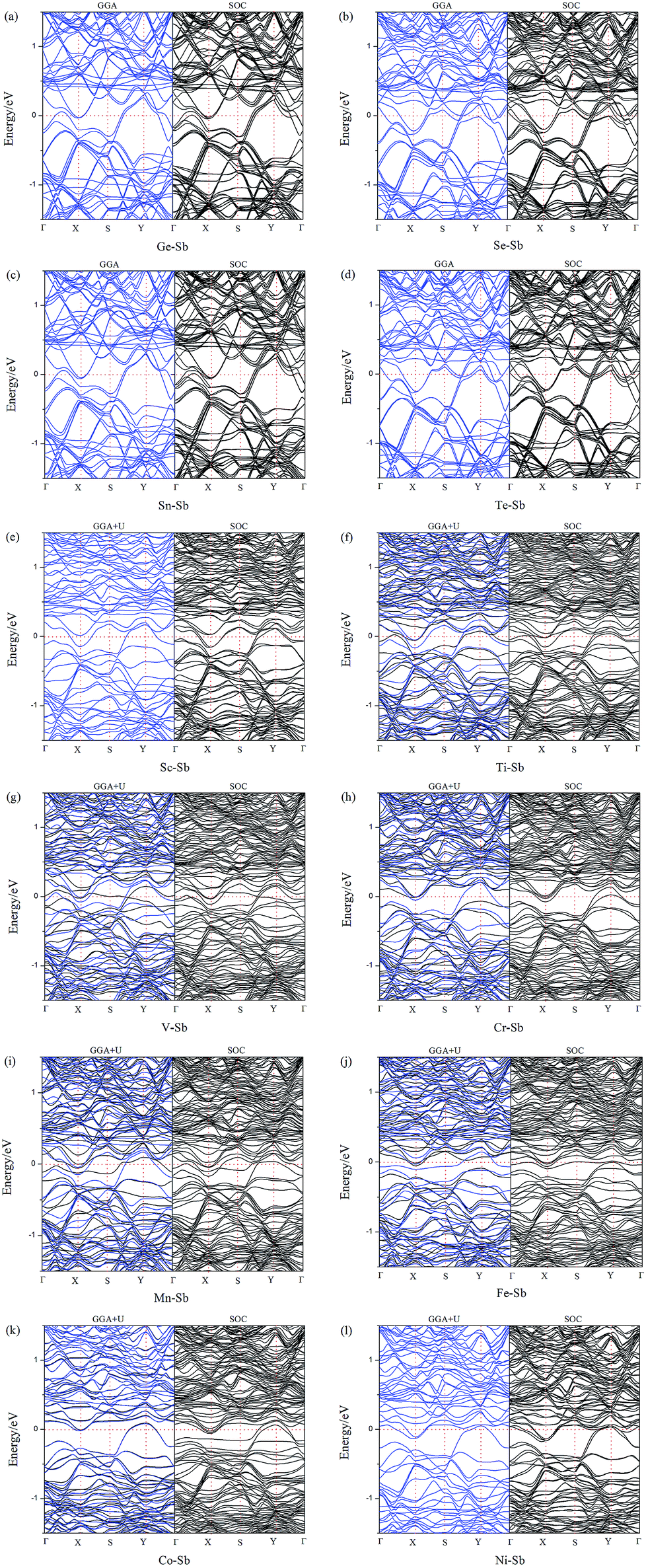 | ||
| Fig. 3 Band structures of (a) Ge–Sb, (b) Se–Sb, (c) Sn–Sb, (d) Te–Sb, (e) Sc–Sb, (f) Ti–Sb, (g) V–Sb, (h) Cr–Sb, (i) Mn–Sb, (j) Fe–Sb, (k) Co–Sb, and (l) Ni–Sb. For the GGA+U or GGA band structure results, the black and blue colors represent spin-up and spin-down channels, respectively. | ||
Aiming to deeply explore the contribution of magnetic states, we illustrate the PDOS of dopants and the adjacent Sb atoms in Fig. 4. Firstly, there are obvious hybridization between the dopant and adjacent Sb atoms above and below the Fermi level, which is agreement with the large binding energies and charge transfers, indicating that chemical bonds form and the excellent stability of doped systems. Transition-metal (TM) is well known as magnetic metal. After doping of TM-dopants, the Sc-, Co-, Ni-doped antimonene still maintain nonmagnetic state. It may be resulted from the coupling of unpaired d-orbital electrons of TM-dopants and the p-orbital of adjacent Sb atoms, and that is consistent with the strong hybridization performed in Fig. 4. For the five types of magnetic structures, the d-orbital of dopants dominate the magnetic properties of structures. For example, the unsymmetrical sharp peaks of PDOS in Ti–Sb is mainly contributed by dxy and dz2 orbital, and the magnetic property of Mn–Sb system is dominated by dx2−y2 and dyz orbital.
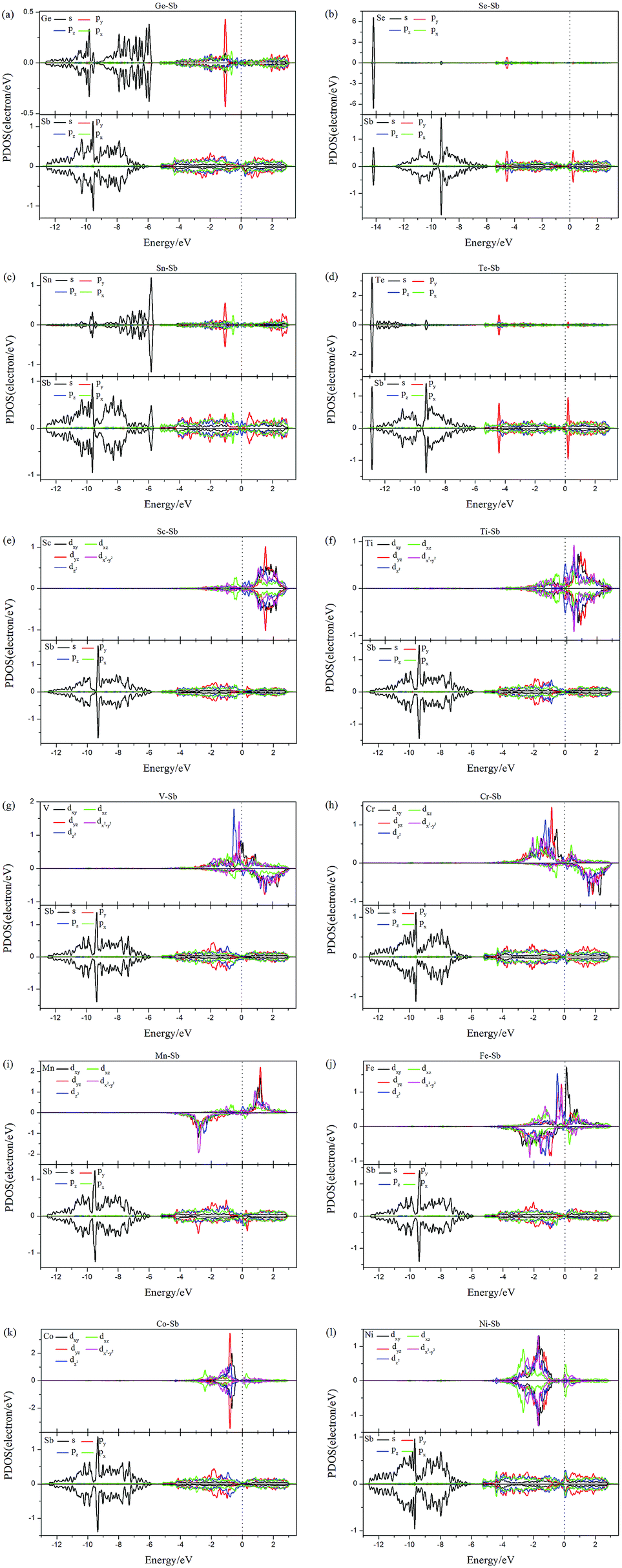 | ||
| Fig. 4 The PDOS of the dopants and the adjacent Sb atoms around the dopants: (a) Ge–Sb, (b) Se–Sb, (c) Sn–Sb, (d) Te–Sb, (e) Sc–Sb, (f) Ti–Sb, (g) V–Sb, (h) Cr–Sb, (i) Mn–Sb, (j) Fe–Sb, (k) Co–Sb, and (l) Ni–Sb. | ||
To deeply explore the magnetic properties, we also perform the spin density distributions of Ti–Sb, V–Sb, Cr–Sb, Mn–Sb, and Fe–Sb systems in Fig. 5. Firstly, it is obvious that spin density is mainly localized at dopant and adjacent Sb atoms, and that is agreement with the PDOS result (Fig. 4). Secondly, the magnetic moments of dopant are contrary to the adjacent Sb atoms. According to our calculated results, the magnetic moments of Ti-, V-, Cr-, Fe-, and Mn-dopant are 1.1 μB, 2.8 μB, 3.4 μB, 4.1 μB, and 2.3 μB, while the total magnetic moments of doped structures are 1.0 μB, 2.7 μB, 3.2 μB, 3.7 μB, and 1.3 μB (Table 1). Therefore, the doping could produce interesting magnetic properties in nonmagnetic aW-phase antimonene.
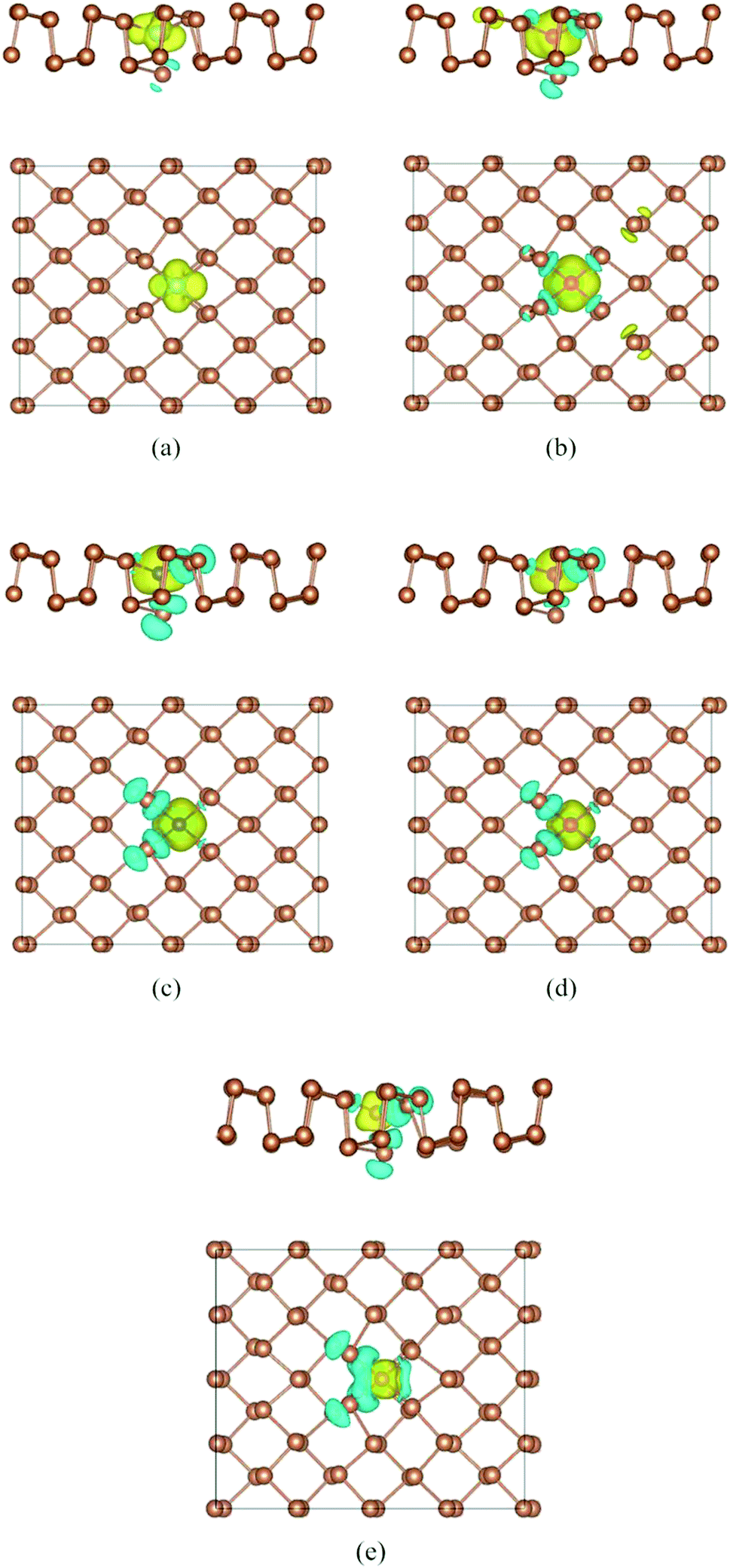 | ||
| Fig. 5 Top view of spin density of doped antimonene: (a) Ti–Sb, (b) V–Sb, (c) Cr–Sb, (d) Mn–Sb, and (e) Fe–Sb. The isosurface value is set to be 0.0015e Å−1. | ||
4. Conclusions
In summary, the geometry stability, electronic and magnetic properties of doped aW-phase antimonene (aW-Sb) have been systemically explored by using density functional theory (DFT) method. The large biding energies and charge transfer between the dopant and antimonene substrate indicate the stable geometry structures of doped systems. The pristine aW-Sb is nonmagnetic semiconductor with a narrow bandgap. After doping, the doped aW-Sb structures all exhibit metallic, and the V-, Cr-, Mn-, Fe-doped systems exhibited different magnetic properties. According to the results of PDOS and spin density distribution, it can be found that the dopant and adjacent Sb atoms dominate the magnetic behavior. Our result is expected to promote the application of antimonene in nanoelectronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by National Natural Science Foundation of China (NSFC, Grant No. 11604080 and 61874160), and the Student Research Training Program of School of Physics and Engineering (Grant No. WLSRTP201908).References
- D. Xiao, G. B. Liu, W. Feng, X. Xu and W. Yao, Coupled Spin and Valley Physics in Monolayers of MoS2 and Other Group-VI Dichalcogenides, Phys. Rev. Lett., 2012, 108, 196802 CrossRef PubMed.
- Z. Y. Zhu, Y. C. Cheng and U. Schwingenschlögl, Giant spin-orbit-induced spin splitting in two-dimensional transition-metal dichalcogenide semiconductors, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 153402 CrossRef.
- W. Qing Hua, K. Z. Kourosh, K. Andras, J. N. Coleman and M. S. Strano, Electronics and optoelectronics of two-dimensional transition metal dichalcogenides, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef PubMed.
- H. Shu, P. Luo, P. Liang, D. Cao and X. Chen, Layer-Dependent Dopant Stability and Magnetic Exchange Coupling of Iron-Doped MoS2 Nanosheets, ACS Appl. Mater. Interfaces, 2015, 7, 7534–7541 CrossRef CAS PubMed.
- D. Cao, T. Shen, P. Liang, X. Chen and H. Shu, Role of Chemical Potential in Flake Shape and Edge Properties of Monolayer MoS2, J. Phys. Chem. C, 2015, 119, 4294–4301 CrossRef CAS.
- A. A. Kistanov, A. R. Davletshin, S. V. Ustiuzhanina, I. Evazzade, D. Saadatmand, S. V. Dmitriev and E. A. Korznikova, Effects of substrate and environmental adsorbates on the electronic properties and structural stability of antimonene, J. Mater. Sci., 2018, 53, 15559–15568 CrossRef CAS.
- S. Zhang, S. Guo, Z. Chen, Y. Wang, H. Gao, J. Gómez-Herrero, P. Ares, F. Zamora, Z. Zhu and H. Zeng, Recent progress in 2D group-VA semiconductors: from theory to experiment, Chem. Soc. Rev., 2018, 47, 982–1021 RSC.
- T. P. Kaloni, G. Schreckenbach and M. S. Freund, Large Enhancement and Tunable Band Gap in Silicene by Small Organic Molecule Adsorption, J. Phys. Chem. C, 2014, 118, 23361–23367 CrossRef CAS.
- E. C. Anota, M. S. Villanueva, A. E. Morales and M. Castro, Stability, electronic and magnetic properties of the Octagraphene-like boron nitride Nanosheets: in silico studies, Fullerenes, Nanotubes, Carbon Nanostruct., 2018, 26, 93–99 CrossRef.
- R. G. Amorim and R. H. Scheicher, Silicene as a new potential DNA sequencing device, Nanotechnology, 2015, 26, 154002 CrossRef PubMed.
- S. Zhang, Z. Yan, Y. Li, Z. Chen and H. Zeng, Atomically Thin Arsenene and Antimonene: Semimetal–Semiconductor and Indirect–Direct Band-Gap Transitions, Angew. Chem., Int. Ed., 2015, 54, 3112–3115 CrossRef CAS PubMed.
- T. Lei, C. Liu, J.-L. Zhao, J.-M. Li, Y.-P. Li, J.-O. Wang, R. Wu, H.-J. Qian, H.-Q. Wang and K. Ibrahim, Electronic structure of antimonene grown on Sb2Te3 (111) and Bi2Te3 substrates, J. Appl. Phys., 2016, 119, 015302 CrossRef.
- P. Ares, F. Aguilar-Galindo, D. Rodríguez-San-Miguel, D. A. Aldave, S. Díaz-Tendero, M. Alcamí, F. Martín, J. Gómez-Herrero and F. Zamora, Mechanical Isolation of Highly Stable Antimonene under Ambient Conditions, Adv. Mater., 2016, 28, 6332–6336 CrossRef CAS PubMed.
- A. V. Krasheninnikov, P. O. Lehtinen, A. S. Foster, P. Pyykko and R. M. Nieminen, Embedding transition-metal atoms in graphene: structure, bonding, and magnetism, Phys. Rev. Lett., 2009, 102(12), 126807 CrossRef CAS PubMed.
- H. Bing, X. Hongjun, Y. Jaejun and W. Su-Huai, Effective control of the charge and magnetic states of transition-metal atoms on single-layer boron nitride, Phys. Rev. Lett., 2012, 108, 206802 CrossRef PubMed.
- D. Ma, W. Ju, T. Li, X. Zhang, C. He, B. Ma, Y. Tang, Z. Lu and Z. Yang, Modulating electronic, magnetic and chemical properties of MoS2 monolayer sheets by substitutional doping with transition metals, Appl. Surf. Sci., 2016, 364, 181–189 CrossRef CAS.
- L. Hu, X. Hu, X. Wu, C. Du, Y. Dai and J. Deng, Density functional calculation of transition metal adatom adsorption on graphene, J. Phys.: Condens. Matter, 2008, 405, 3337–3341 Search PubMed.
- M. Dongwei, L. Zhansheng, J. Weiwei and T. Yanan, First-principles studies of BN sheets with absorbed transition metal single atoms or dimers: stabilities, electronic structures, and magnetic properties, J. Phys.: Condens. Matter, 2012, 24, 145501 CrossRef PubMed.
- X. Q. Wang, W. G. Chen, Z. L. Zhu and Y. Jia, Electronic and Magnetic Properties Modulated by Adsorption of 3 d Transition Metal Atoms in Monolayer and Bilayer MoS2 Sheets, Acta Metall. Sin. (Engl. Lett.), 2015, 28, 793–798 CrossRef CAS.
- M. Y. Liu, Q. Y. Chen, Y. Huang, Z. Y. Li, C. Cao and Y. He, Electronic and magnetic properties of 3D transition-metal atom adsorbed arsenene, Nanotechnology, 2018, 29, 095203 CrossRef PubMed.
- S. Dai, W. Zhou, Y. Liu, Y.-L. Lu, L. Sun and P. Wu, Tunable electronic and magnetic properties of antimonene system via Fe doping and defect complex: a first-principles perspective, Appl. Surf. Sci., 2018, 448, 281–287 CrossRef CAS.
- C. He, M. Cheng and W. Zhang, Tunable electronic and magnetic properties of transition metals doped antimonene: a first-principles study, Mater. Res. Express, 2018, 5, 065059 CrossRef.
- Z. Yungang and L. Xudong, Effects of interstitial dopings of 3 d transition metal atoms on antimonene: a first-principles study, Appl. Surf. Sci., 2018, 458, 572–579 CrossRef.
- T. T. Li, C. He and W. X. Zhang, Electric field improved the sensitivity of CO on substitutionally doped antimonene, Appl. Surf. Sci., 2018, 427, 388–395 CrossRef.
- L. F. Yang, Y. Song, W. B. Mi and X. C. Wang, Prediction of spin-dependent electronic structure in 3d-transition-metal doped antimonene, Appl. Phys. Lett., 2016, 109, 022103 CrossRef.
- L. F. Yang, Y. Song, W. B. Mi and X. C. Wang, The electronic structure and spin–orbit-induced spin splitting in antimonene with vacancy defects, RSC Adv., 2016, 6, 66140–66146 RSC.
- G. Kresse, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Stefan, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2010, 27, 1787–1799 Search PubMed.
- Y. Song, X. C. Wang and W. B. Mi, Spin splitting and electric field modulated electron-hole pockets in antimonene nanoribbons, npj Quantum Mater., 2017, 2, 15 CrossRef.
- W. W. Ju, D. H. Wang, T. W. Li and S. J. Gong, Electric field control of Rashba spin splitting in 2D NIIIXVI(N = Ga, In; X = S, Se, Te) monolayer, J. Phys.: Condens. Matter, 2020, 32, 175503 CrossRef PubMed.
- D. Singh, S. K. Gupta, Y. Sonvane and I. Lukačević, Antimonene: a monolayer material for ultraviolet optical nanodevices, J. Mater. Chem. C, 2016, 4, 6386–6390 RSC.
- G. Wang, R. Pandey and S. P. Karna, Atomically Thin Group V Elemental Films: Theoretical Investigations of Antimonene Allotropes, ACS Appl. Mater. Interfaces, 2015, 7, 11490–11496 CrossRef CAS PubMed.
- O. Üzengi Aktürk, E. Aktürk and S. Ciraci, Effects of adatoms and physisorbed molecules on the physical properties of antimonene, Phys. Rev. B, 2016, 93, 035450 CrossRef.
- O. Ü. Aktürk, V. O. Özçelik and S. Ciraci, Single-layer crystalline phases of antimony: Antimonenes, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 235446 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, A grid-based Bader analysis algorithm without lattice bias, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2020 |