
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecularly engineered electroplex emission for an efficient near-infrared light-emitting electrochemical cell (NIR-LEC)†
Hashem Shahroosvand *a,
Leyla Heydaria,
Babak Nemati Bidehab and
Babak Pashaeia
*a,
Leyla Heydaria,
Babak Nemati Bidehab and
Babak Pashaeia
aGroup for Molecular Engineering of Advanced Functional Materials (GMA), Chemistry Department, University of Zanjan, Zanjan, Iran. E-mail: shahroos@znu.ac.ir; Fax: +98-24-33058202; Tel: +98-24-33052584
bFaculty of Chemistry, Bu-Ali Sina University, Hamedan, Iran
First published on 7th April 2020
Abstract
Electroplex emission is rarely seen in ruthenium polypyridyl complexes, and there have been no reports from light-emitting electrochemical cells (LECs) to date. Here, for the first time, near-infrared (NIR) emission via the electroplex mechanism in a LEC based on a new blend of ruthenium polypyridyl complexes is described. The key factor in the design of the new complexes is the 0.4 V decrease in the oxidation half-potential of Ru(II)/Ru(III) in [Ru(DPCO)(bpy)2]ClO4 (DPCO = diphenylcarbazone, bpy = 2,2 bipyridine), which is about one-third of the value for benchmark [Ru(bpy)3](ClO4)2, as well as the long lifetime of excited states of 350–450 ns. The LEC based on the new blend with a narrow band gap (≈1.0 eV) of a Ru(DPCO) complex and Ru(bpy)32+ can produce an electroluminescence spectrum centred at about 700 nm, which extends to the NIR region with a high external quantum efficiency (EQE) of 0.93% at a very low turn-on voltage of 2.6 V. In particular, the very simple LEC structure was constructed from indium tin oxide (anode)/Ru(DPCO):Ru(bpy)32+/Ga:In (cathode), avoiding any polymer or transporting materials, as well as replacing Al or Au by a molten alloy cathode. This system has promising applications in the production of LECs via microcontact or inkjet printing.
1. Introduction
For scientists who work on light-emitting devices (LEDs), organic light-emitting diodes (OLEDs) are of great interest because of their elevated brightness and maximum external quantum efficiency (EQE).1 OLEDs are now the first choice for use in smart phones and tablets as they are able to achieve brilliant and efficient flexible displays with the best image quality. The OLED market is currently estimated at around 15 billion dollars, and is set to grow rapidly in the coming years.2 OLEDs are generally constructed from several layers, including a hole transport layer (HTL), electron transport layer (ETL), hole injection layer (HIL), electron injection layer (EIL) and emitting layer (EL).3 Contact layers, including the anode and cathode, are costly and require vacuum thermal evaporation techniques, and need to be deposited onto other layers. In recent years, a new type of OLED, known as a light-emitting electrochemical cell (LEC), has been introduced to reduce the number of layers used.4,5 A typical LEC consists only of an emitter between two electrodes, which meets the requirement for simplicity. This simple configuration makes opposite charges in a LEC move rapidly between electrodes, leading to lower turn-on voltage of below 4 V. As a result, LECs are able to operate without an HTL or ETL, as well as without using electrodes that are sensitive to air requiring vacuum deposition.6 Therefore, the employed active layer has a key role in producing the required electroluminescence emission. The most active layer in an LEC consists of ionic transition metal complexes (iTMCs), which contain positive and negative charges that migrate between the two electrodes. The benchmark emitter in LEC is [Ru(bpy)3](ClO4)2, which has shown remarkable photophysical and photochemical properties, including long excited-state lifetimes (τ ≈ 1 μs), high molar absorption of metal-to-ligand charge transfer (3MLCT) and luminescence quantum yields (Φ ≈ 0.095).7 Bard et al. have devoted considerable effort to introduce ruthenium polypyridyl complexes as emitters in LECs with an elevated brightness and low turn-on voltage. Regardless of the above-stated advantages of [Ru(bpy)3](ClO4)2, the luminescence of its molecules is quenched over a long period, which can be attributed to the ligand being replaced with aqua molecules to form [Ru(bpy)2(H2O)2]2+ species.8 This problem can be fixed by changing the 2,2′-bipyridine (bpy) ligand with other derivatives of polypyridyl ligands. After this insight, many more ruthenium polypyridyl complexes have been reported and used to explore the influence of the ancillary ligand on enhancing the performance of LEC.9 Ruthenium polypyridyl complexes have also attracted considerable attention because of their potential ability to produce emissions in a near-infrared (NIR) region, as well as their exclusive applications such as bio-imaging,10–12 telecommunications,13–15 and wound healing.16,17 However, the intrinsic difficulty of the energy gap law limits emission in the NIR region, resulting in a lowering of EQE into the range of 0.1%. Therefore, there is a gap in the literature regarding electroluminescence in the NIR region.9,18,19 In this framework, two approaches have been proposed to overcome this limitation: first, substituting electron donor moieties20,21 as well as extending π-conjugation on the ancillary ligand; second, using one host polymer, which is generally a macromolecule.22 To solve this problem, we examined the application of a ruthenium complex to tune the electroluminescence wavelength to NIR electroluminescence. It was shown that a small amount of an ionic transition metal complex as a guest molecule in a thin film of [Ru(bpy)3]2+ (host) can significantly improve the electroluminescence features.18 In the following description, this concept was employed to introduce Ir-iTMC in which an orange emitter was used as guest in a matrix of the green emitter as host.23 These results were attributed to a decrease in self-quenching in the emitter layer. Recently, [Zn(bpy)3]2+ was reported as a promising additive to the iridium blue emitter to improve the electroluminescence properties of LEC. The significant enhancement of LEC performance was attributed to facilitated electron injection/transport.24 According to the achievements of our previous work, a zinc polypyridyl–diphenylcarbazone (DPCO) complex was reported as an efficient additive to the thin layer of [Ru(bpy)3]2+.25 Therefore, with the aim of enhancing the efficiency of NIR electroluminescence, we used a ruthenium polypyridyl–diphenylcarbazone complex as the doping material. Here, we used two unchanged bipyridine ligands similar to [Ru(bpy)3]2+, while the third ligand was altered by an N^O chelate, which is a homologue of aluminium tris-8-hydroxyquinoline (Alq3); the benchmark emitter in the OLED field.2. Results and discussion
This chapter includes four subsections. In Section 2.1, the characterisation of the molecularly engineered complexes is explained, as well as details of the synthesis procedure (also given in the ESI†). The photophysical properties of complexes including ultraviolet-visible (UV-vis) and photoluminescence studies plus density functional theory (DFT) calculations to assign the electronic transitions are investigated in Section 2.2. The electrochemical properties of complexes is investigated and used to determine the energy levels in Section 2.3. Finally, in Section 2.4, to tune the electroluminescence wavelength, a blend of synthesised complexes with [Ru(bpy)3]2+ is introduced. The performance and mechanism of electroluminescence is also reported.2.1 Characterisation of complexes
Fig. 1(a) shows the chemical structures of Ru(LH4), Ru(LH5) and Ru(LH6), [Ru(N^N)2(DPCO)](ClO4), in which the ancillary ligand (N^N) was changed, including 2,2 bipyridine (bpy), 1,10-phenanthroline (phen) and 4,4-dimethyl 2,2 bipyridin (dmbpy), respectively. The procedure for the synthesis and full characterisations of the complexes are incorporated into ESI (ESI S1–S4†). The equilibrium between enol-to-azo forms can be investigated by 1H nuclear magnetic resonance (NMR) spectroscopy. In the 1H NMR spectra of the complexes, the sharp single signal at about 4.5 ppm, which can be attributed to the N–H of the 1,5-diphenylcarbazide (DPC) ligand, was removed (ESI, Fig. S1, S3 and S5†).26 The Fourier-transform infrared (FT-IR) instrument is a useful tool to investigate the coordination of N^O ligands to metal. When the DPC converted to DPCO and was coordinated to ruthenium, the v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and v(NN) bands shifted from 1700 and 1570 cm−1 to 1630 and 1510 cm−1, respectively (ESI, Fig. S7†). This evidence clearly shows the coordination of the ligand to ruthenium through (C–O) and (N–N).27 Microanalysis, including CHN analysis and mass spectroscopy, also confirmed the formation of the suggested complexes (ESI, Section S1 and Fig. S2, S4 and S6†).
O) and v(NN) bands shifted from 1700 and 1570 cm−1 to 1630 and 1510 cm−1, respectively (ESI, Fig. S7†). This evidence clearly shows the coordination of the ligand to ruthenium through (C–O) and (N–N).27 Microanalysis, including CHN analysis and mass spectroscopy, also confirmed the formation of the suggested complexes (ESI, Section S1 and Fig. S2, S4 and S6†).
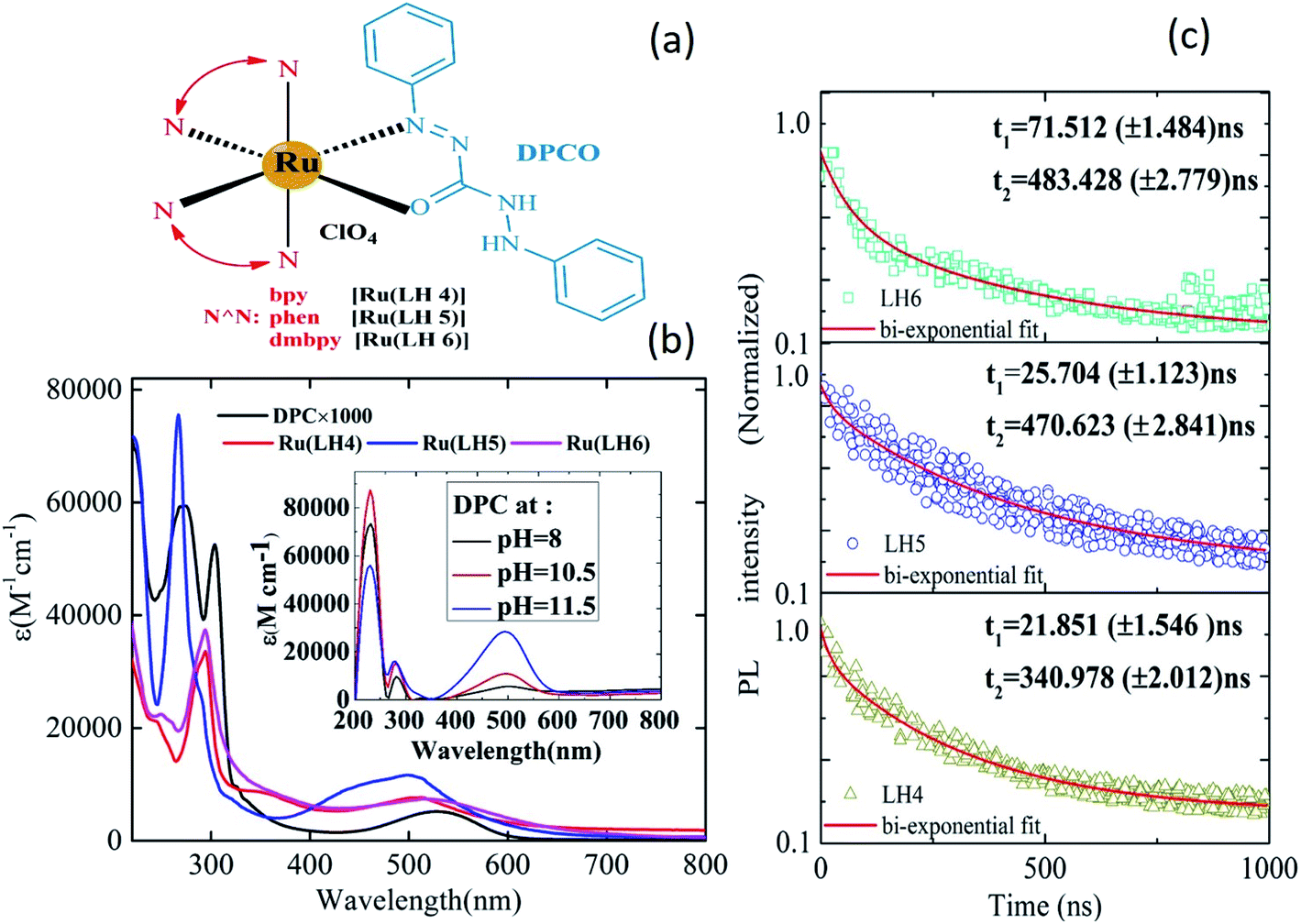 | ||
| Fig. 1 (a) The chemical structures of Ru(LH4–6). (b) UV-vis spectra of Ru(LH4–6) in methanol solvent. Inset: UV-vis spectra of DPC at varied pH values. (c) Time-resolved photoluminescence studies of Ru(LH4–6) complexes supported on a non-conducting glass substrate. Photoluminescence decay kinetics (PLDK) are measured at λmax upon excitation at 408 nm. For PLDK experiments, solid lines are the fits obtained after using a bi-exponential decay model. | ||
2.2 Photophysical properties of complexes
The ultraviolet-visible (UV-vis) spectrum of the DPC ligand has a unique profile because of the obvious broad band in the visible region at about 550 nm.28 Moreover, because of the presence of four acidic protons, DPC is a pH-sensitive ligand, and when the pH increases to 12, the molar absorptivity gradually increases (Fig. 1(b) inset). These interesting photochemical behaviours make DPC act as a promising ligand for use in electron-transfer systems. Note that DPC could be fully deprotonated in two steps: initially, two of the protons are deprotonated, to give DPCO and then, the other two remained protons are separated, to give DPDO (ESI, Scheme 1†).29The increasing intensity of the charge transfer band in the absorption spectra clearly indicated the deprotonation of DPC, as shown in the inset of Fig. 1(b). In addition, Fig. 1(b) shows the absorption spectra of the complexes Ru(LH4–6). Typically, ruthenium polypyridyl complexes showed intense and sharp bands in the UV region, which can be assigned to intra-ligand charge transfer (ILCT), as well as a weak and broad band in the visible region, which can be attributed to the MLCT.30 For all investigated complexes, there were two distinguished absorption regions. However, there was a blue shift in the MLCT band in the investigated complexes compared with the DPC ligand, confirming the coordination of DPCO to ruthenium metal. Moreover, the broadening of the MLCT band of novel ruthenium complexes has been increased compared with other polypyridine ruthenium complexes. As shown in the ESI (Fig. S8 and Table S1†), the maximum wavelengths of MLCT of Ru(LH4) and Ru(LH5) were also blue shifted compared with Ru(bpy)2Cl2 and Ru(phen)2Cl2, respectively, while that of Ru(LH6) was red-shifted compared with Ru(dmbpy)2Cl2. The photoluminescence spectra of Ru(LH4–6) were recorded in methanol solution with maximum wavelength of 628, 624 and 674 nm, respectively, which are in accordance with the homologue ruthenium complexes (Table 1 and ESI Fig. S9†).
| Complex | Absorbancea, λmax [nm](log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) ε) |
PLmax [nm] (PL quantum yield, %) | Ru(II/III)b oxi. | EHOM0c | ELUMOd | Egape | |
|---|---|---|---|---|---|---|---|
| ILCT | MLCT | E1/2(ΔE) (mV) | |||||
| a In methanol solution (1 × 10−5 M).b From CV measurements, E1/2 = 1/2(Epa + Epc); 0.1 M methanol/TBAP versus Ag/AgCl.c From the formula EHOMO = [−e(Eox − E1/2(Fc/Fc+))] − 4.8 eV.d From the formula ELUMO = [−e(Ered − E1/2(Fc/Fc+))] − 4.8 eV.e From the formula Egap = EHOMO − ELUMO (eV). | |||||||
| Ru(LH4) | 296 (4.52) | 516 (3.87) | 628(5.85) | 0.38 (73) | 4.78 | 3.69 | 1.09 |
| Ru(LH5) | 266 (4.68) | 500 (4.05) | 624(6.21) | 0.42 (81) | 4.82 | 3.61 | 1.21 |
| Ru(LH6) | 293 (4.56) | 518 (3.90) | 674 (7.55) | 0.40 (124) | 4.80 | 3.8 | 1.0 |
| Ru(bpy)32+ | 290 (4.91) | 451 (4.17) | 621 (9.54) | 1.29 (79) | −5.66 | −3.34 | 2.32 |
A suitable lifetime of excited states of an emitter molecule is another pre-condition for an efficient light-emitting device. To investigate the lifetime of excited states of complexes, the time-resolved photoluminescence decay was measured. The relatively long lifetime of all three complexes was estimated by fitting the photoluminescence (PL) decay traces with the bi-exponential decay model (Fig. 1(c)). The lifetime values were estimated to be in the range of 1–1000 ns for a large number of ruthenium polypyridyl complexes,31–35 with values of 340.97, 470.62 and 483 ns observed for the investigated Ru(LH4–6), respectively.
The bi-exponential decay results are presumably produced from two closely-lying levels of luminophors.36 In most Ru(II)-polypyridine complexes, the lowest excited state of luminescence is 3MLCT,37–40 which actually consists of several close-lying states including ligand charge (LC) with different degrees of singlet–triplet mixing and, thus, different intrinsic rates of decay to the ground state.41,42 Generally, 3MLCT emission is easily observable in fluid solution at room temperature, with lifetimes in the order of 100–1000 ns.43,44 However, in fluid solution at room temperature, 3LC emission can seldom be observed, because of the occurrence of faster (thermally activated) un-molecular decay processes and/or bimolecular quenching processes.45 Consequently, based on our lifetime results, the origin of two lifetime values can be attributed to 3LC (τ1 < 100 ns) and 3MLCT (τ2 > 100 ns) states, respectively.
These elongated lifetimes along with the reversible Ox/Red of the ruthenium centre indicated an exhibit-promising material for a light-emitting device. It should be noted the use of the N^O chelating ligand is a better electron donor than bpy (because it is deprotonated and a mono-anion), resulting in the decreasing of redox potential. Moreover, the dimethyl-bpy as an ancillary ligand in Ru(LH6) could also increase electron-donating properties compared with the bpy ligand. For clarify this point, we performed a DFT calculation on the complexes, and found that the methyl groups of dimethyl-bpy caused an increase in negative charge density distribution on the nitrogen atoms (−0.33), leading to a decrease in the positive charge on the ruthenium metal (+0.71) (ESI, Fig. S10†). These results caused a decrease in band gap to −1.31 eV in Ru(LH6) compared to Ru(LH4) with a band gap of −1.44 eV (ESI, Table S1†). The calculated highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies of the Ru(LH4) complex are −5.01 and −3.57 eV, while those of the Ru(LH6) complex equalled −5.06 and −3.75 eV, respectively. In fact, the HOMO energy level of the Ru(LH6) complex was stabilised due to the presence of methyl groups, which was also demonstrated by the cyclic voltammetry (CV) results (Table 1).
As shown in the ESI (Fig. S11†), electron density in the HOMO is delocalised on the diphenylcarbazone and bpy ligand for Ru(LH6) and Ru(LH4), respectively. However, the electron density in the LUMO is focused on the dimethyl-bpy and bpy moieties for Ru(LH6) and Ru(LH4), respectively. This means that electronic transition from HOMO to LUMO is more efficient for Ru(LH6) containing dimethyl-bpy than Ru(LH4).
2.3 Electrochemical behaviour of complexes
Electrochemical studies can determine the value of an emitter coordination complex for use in light emission diode. In particular, ruthenium polypyridyl complexes have singular electrochemical properties for two reasons. First, a reversible obvious half-wave of Ox/Red in the positive region for conversion of Ru(II)/Ru(III). Second, several irreversible half-waves of polypyridyl ligands in the negative region,46 which are ligand-based and occur in a stepwise manner for each π* system of polypyridyl ligands, and are analogous to the behaviour observed for [Ru(bpy)3]2+.47,48 In fact, precise assignment of the redox loci are not straightforward as in the family of [Ru(bpy)2(bpx)]2 (bpx = 2,2-bipyrimidine, 2,2-bipyrazine).49,50 Here, the complexes showed reversible Ox/Red of the Ru centre (Fig. 2(a)). Separation between the anodic and cathodic current peaks for this process was similar (∼0.08 V) to that of the Fc+/Fc wave, supporting the assignment of this feature to a one-electron redox process (Fig. 2b and ESI). Fig. S12† also shows the oxidation wave of Ru(LH6) at different scan rates at 125, 150 and 200 mV s−1. Moreover, as shown in Fig. 2(c), the linear correlation between υ½ and the anodic current can be attributed to mass-transport phenomenon that controls the kinetics of the overall process, and shows the reversibility of this process. Parenthetically, the peek current of the ruthenium polypyridyl complexes is in the order of microamperes.51–55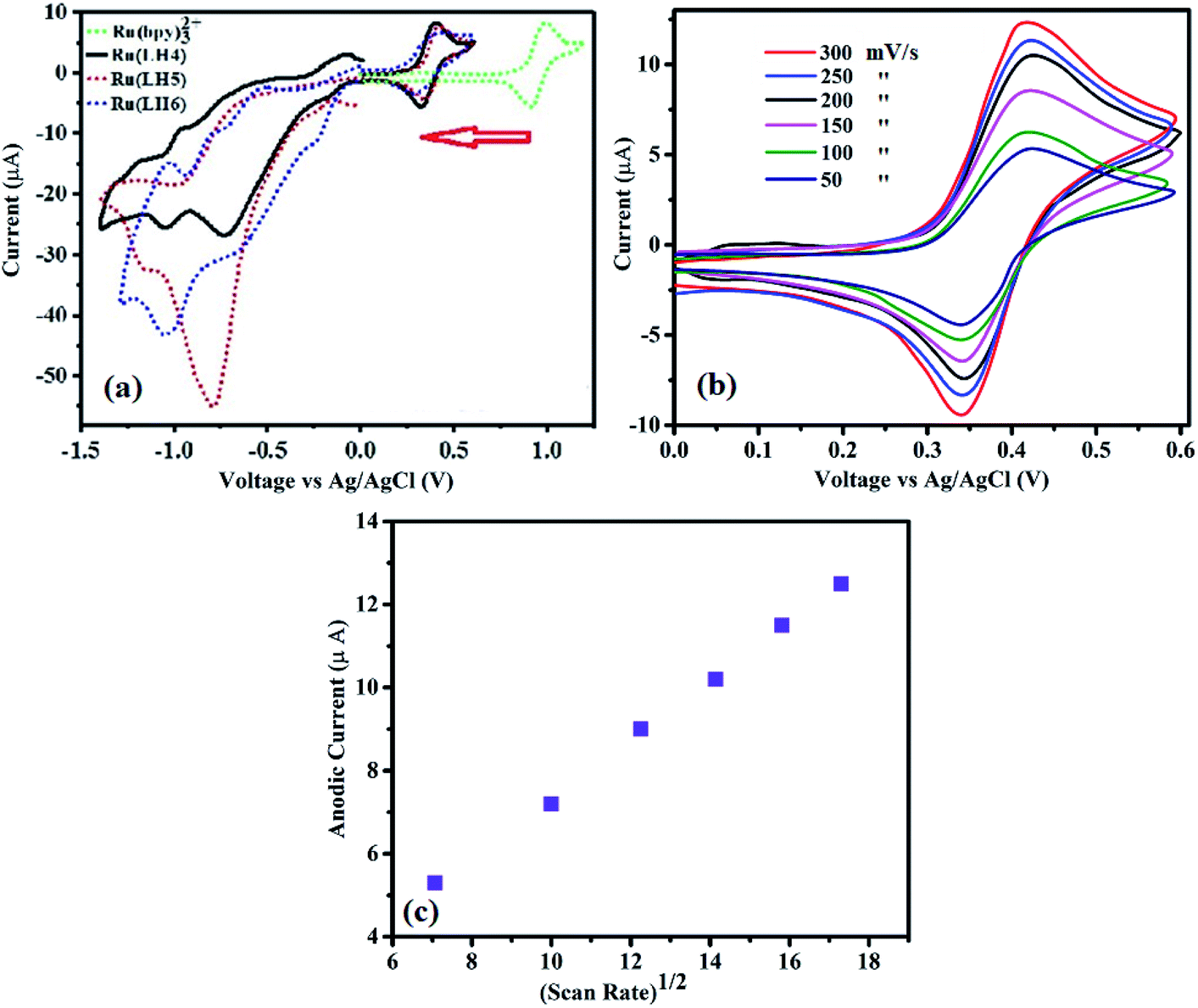 | ||
| Fig. 2 (a) Cyclic voltammetry studies of Ru(LH4–6) in acetonitrile solution. (b) CVs from Ru(LH6) at varied scan rates of 50, 100, 150, 200, 250 and 300 mV s−1. (c) Survey of the diffusion current of Ru(LH6). | ||
The electrochemical behaviour of the DPC ligand includes the oxidation of DPC to DPCO, and then the DPCO oxidises to DPCDO with a potential of around 0.83 (V) and 1.5 (V), respectively31 (ESI, Fig. S13†). In the reversed potential, the cathodic reduction of the oxidised products of DPC was formed, which was observed in the form of one peak of −0.3 vs. Ag/AgCl, corresponding to the DPCO reduction to DPC. Surprisingly, the value of EOx1/2 of the complex was about 0.4 V, which dramatically decreased compared with the ruthenium polypyridyl complexes that showed an EOx1/2 value in the range of 1.2–1.5 V.56–58 When DPCO was substituted with the bpy ligand in [Ru(bpy)3](ClO4)2 to form [Ru(bpy)2(DPCO)](ClO4), the EOx1/2 value dramatically decreased from 1.3 V to 0.5 V.59 This remarkable difference was also observed for [Ru(phen)3](ClO4)2, compared with [Ru(phen)2(DPCO)](ClO4)2, resulting in a decreased EOx1/2 value from 1.35 to 0.5 V.60 These low redox properties of ruthenium confirmed the importance of the DPCO ligand to enrich the electron-donating ability around the ruthenium centre. From our previous knowledge, this EOx1/2 value is the lowest among all the ruthenium polypyridyl families to date (ESI, Table S3†). Noticeably, this low EOx1/2 value for a ruthenium polypyridyl family could be useful for bio-sensors, displays and other optoelectronic applications, which fundamentally need low applied voltage.
2.4 LEC based on electroplex emission
According to the interesting results explained in previous sections, LECs based on the three investigated complexes with the configuration of indium tin oxide(ITO)/Ru(LH4–6):Ru(bpy)3(ClO4)2/Ga:In were fabricated. Two interesting novelties in this arrangement included: (1) avoiding any conductive polymers such as PEDOT:PSS or other transport layers; (2) the replacement of Al or other metal cathodes, which need the hard condition for deposition with Ga:In low molten alloy, resulting in the production of LEC by microcontact printing or inkjet. As a new strategy for increasing the mobility of ions in cells, we employed a blend of the investigated complexes with [Ru(bpy)3](ClO4)2. To seek the underlying electroluminescence mechanism in constructed LEC, the consistency and matching of HOMO and LUMO of Ru(LH4–6) with [Ru(bpy)3](ClO4)2 was investigated. Fig. 4(c) demonstrates the proposed schematic representation of charge transfer in the fabricated LEC. Under an applied bias, ClO4− ions in [Ru(bpy)3]2+(ClO4)2− complex drift into the counter electrode, leading to the accumulation of negative counter-ion and cationic Ru complexes in close proximity to holes and electrons, respectively.61 However, because a barrier energy of approximately 0.5 eV is produced at the interface of [Ru(bpy)3]2+/Ru(DPCO) complexes, it is difficult for the holes to be injected into Ru(DPCO) complexes. Therefore, holes (electrons) will be blocked by Ru complexes, and accumulate at the interface of the [Ru(bpy)3]2+/Ru(DPCO) complexes. In both sides near the [Ru(bpy)3]2+/Ru(DPCO) complex interfaces, the electric field in the bulk is redistributed and the electric field in the [Ru(bpy)3]2+/Ru(DPCO) layer moves higher than the one in the [Ru(bpy)3]2+ layer alone. This explanation is in good agreement with the observed red-shifted and broadening EL, which is routinely seen in electroplex emission.62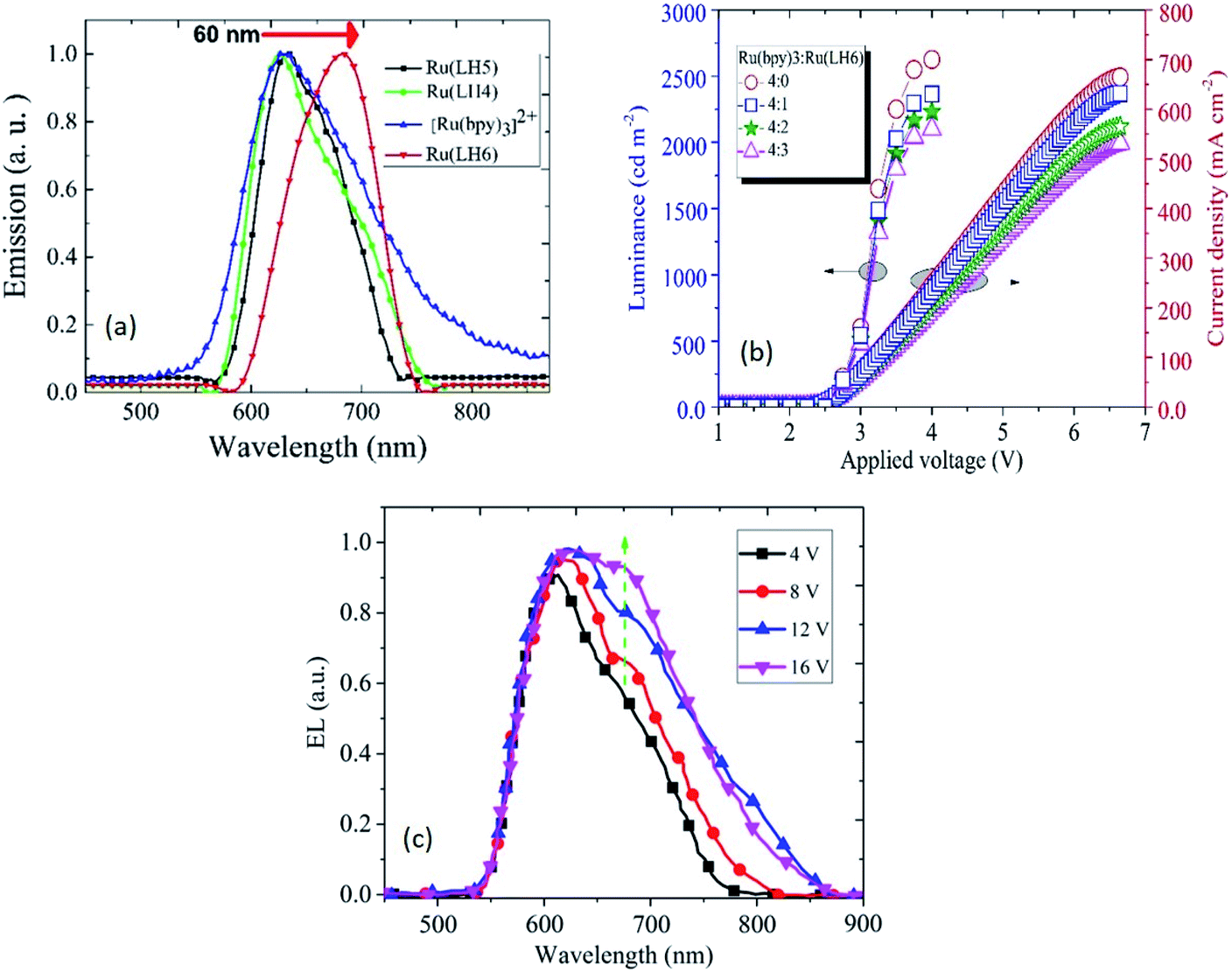 | ||
| Fig. 3 (a) Electroluminescence spectra of ITO/[Ru(bpy)3]2:Ru(LH4–6)(4:1)/Ga:In and ITO/[Ru(bpy)3]2/Ga:In. (b) Current density and luminance over the applied voltage for a LEC based on [Ru(bpy)3]2:Ru(LH6) at varied weight ratios of 4:0, 4:1, 4:2, and 4:3. (c) Electroluminescence spectra of Ru(bpy)32+/Ru(LH4) (4/1) at different applied voltages (4, 8, 12 and 16 V). | ||
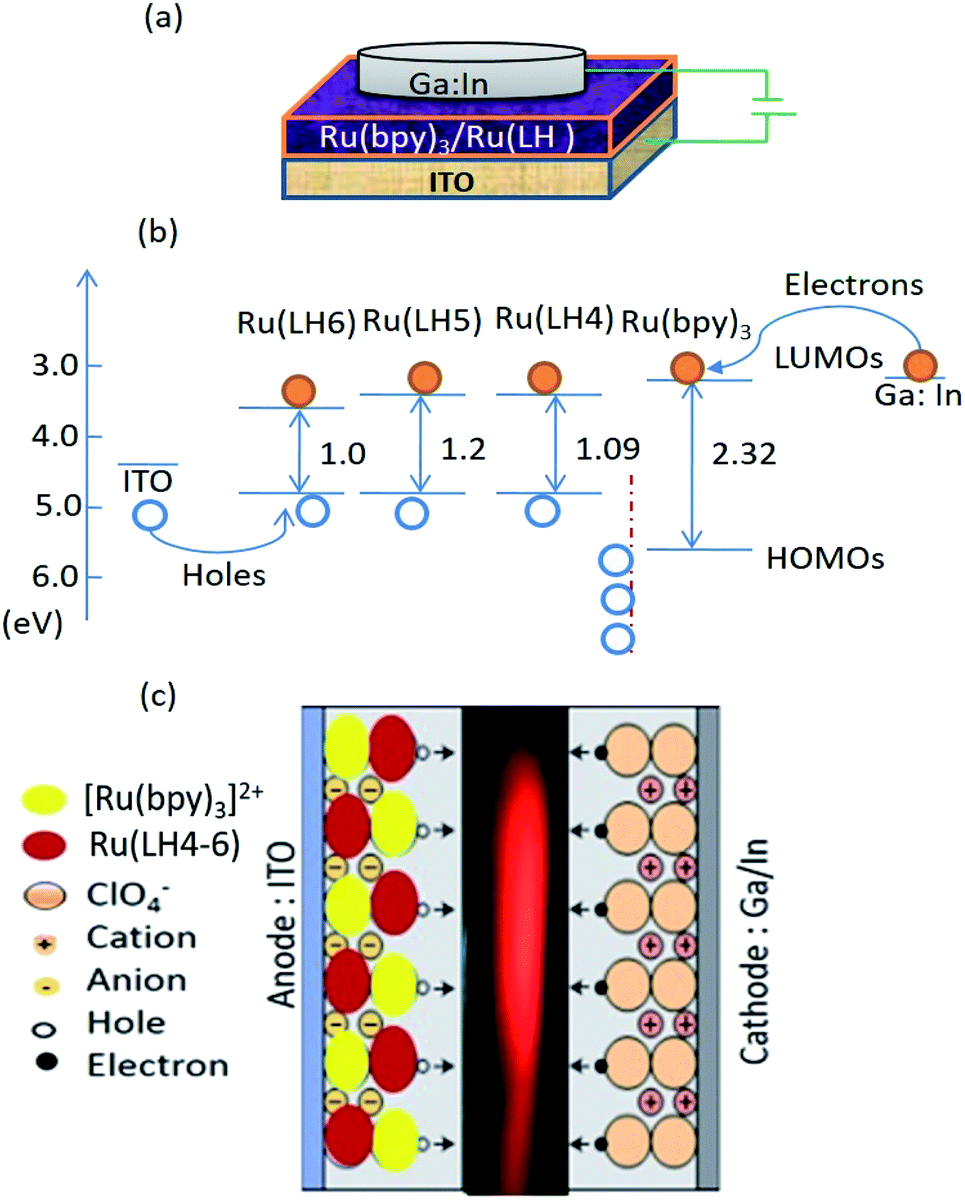 | ||
| Fig. 4 (a) Layer arrangement of a LEC based on the blend method. (b) Positions of the energy levels of components of the constructed LEC. (c) A schematic representation of a state-of-the-art LEC based on [Ru(bpy)3]2+–Ru(LH4–6). The movement of ions in the single layer under an applied voltage allows for efficient charge carrier injection from air-stable electrodes. | ||
As shown in Fig. 4(b), the HOMO of the Ru(LH4–6) complexes was higher than that of the [Ru(bpy)3]2+ ones, which means that the positive charge (holes) could efficiently drift from [Ru(bpy)3]2+ to Ru(LH4–6), confirming the promising hole-transporter behaviour of Ru(LH4–6). Fig. 4(b) shows a schematic of the layer arrangement along with the energy levels of the fabricated LEC.
In light of all of the data presented above, a low oxidation potential value for oxidising Ru(II) to Ru(III) in the investigated Ru(LH4–6) complexes is a promising candidate for use in a light-emitting device. These remarkable results suggest that the presence of shift-base ligands, such as the DPCO ligand in the ruthenium polypyridyl complex, can dramatically decrease the oxidation potential of the ruthenium centre, consequently leading to the achievement of hole transporting properties in solid state devices. Fig. 3(c) clearly shows that the electroluminescence spectrum of Ru(LH6) was shifted to a higher wavelength of about 60 nm compared with the other two complexes, Ru(LH4) and Ru(LH5) as well as [Ru(bpy)3]2+, extending the wavelength to the NIR region. This long red shift can be attributed to the presence of the electron-donating group of methyl moieties on the ancillary ligand of Ru(LH6).63 The colorimetric, densitometry and RGB data of LEC based on Ru(LH4–6) and Ru(bpy)32+ are given in the ESI (Table S4†). The optimisation of the weight ratio between the investigated complexes and [Ru(bpy)3]2+ was a crucial factor in achieving the best electroluminescence characteristics. As shown in Fig. 3(b), when the molar ratio of the novel complexes in the blend of [Ru(bpy)3]2+/Ru(LH6) increased from 4:1 to 4:3, the current density and luminance of LEC slowly decreased. Based on the aim of this study, which is to find an efficient NIR light electrochemical cell, we selected a ratio of 4:1 of [Ru(bpy)3]2+ to Ru(LH6) as a modified molar ratio. Considering the modified ratio, the EL spectrum of LEC based on the blend of [Ru(bpy)3]2+/Ru(LH6) showed an extending EL band around 700 nm, with a current density of 647 mA cm−2 at 6.5 V and 2430 cd m−2 for luminance at 4 V, as well as a very low turn-on voltage of 2.6 V. The electroluminescence metrics of the other complexes were modified in a 4:1 ratio of [Ru(bpy)3]2+:Ru(LH4)–Ru(LH6), as summarised in Table 2. The EQE value of 1.05 for Ru(LH5)/[Ru(bpy)3]2+ represents the current state-of-the-art for an NIR-LEC based on the ruthenium polypyridyl family with a configuration of anode(ITO)/Ru polypyridyl complex/cathode (Ga:In) (see ESI, Table S4†).64–67
:1)/Ga:In
| Complex | λmax [nm] | CIEa | FWHM [nm] | Jb | Vonc | Ld | CEe | EQEf |
|---|---|---|---|---|---|---|---|---|
| a CIE(x, y): Commission Internationale de L'Eclairage, with the D65 white reference CIE(x, y) = (0.31, 0.33).b Current density (mA m−2) at 4 V.c Turn-on voltage (V).d Luminance (cd m−2) at 4 V.e Efficacy, CE, (cd A−1) at 4 V.f External quantum efficiency at 4 V, EQE, (%).g LEC device based on pure [Ru(bpy)3]2+. | ||||||||
| Ru(LH4) | 633 | [0.711,0.288] | 112 | 192 | 3.2 | 1780 | 0.92 | 0.75 |
| Ru(LH5) | 627 | [0.703, 0.296] | 100 | 218 | 3.0 | 2270 | 1.04 | 1.05 |
| Ru(LH6) | 695 | [0.734, 0.265] | 95 | 239 | 2.6 | 2430 | 1.01 | 0.93 |
| [Ru(bpy)3]2+g | 632 | [0.710, 0.289] | 137 | 255 | 2.3 | 2790 | 1.10 | 1.31 |
3. Conclusions
In summary, we found that the DPCO ligand can effectively decrease the EOx1/2 value of Ru(II)/Ru(III) to one-third of the reported value. This significant decrease in the potential value leads to a dramatic reduction in the band gap of the investigated complexes to about 1.0 eV. This outstanding electrochemical behaviour of novel complexes, as well as the extremely long lifetime of the excited states of the Ru(DPCO) complexes in the range of 350–450 ns, provides promising hole-transporter materials for LEDs. In particular, a LEC based on the blend of [Ru(bpy)3]2+/Ru(LH4–6) showed efficient NIR electroluminescence with prominent electroluminescence metrics, including an EQE of 0.93% at 4 V and luminance of 2430 cd.m−2 along with a turn-on voltage of 2.6 V for Ru(LH6).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the University of Zanjan for financial support.References
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- OLED-Info: the OLED experts, https://www.oled-info.com/ Search PubMed.
- S. Welter, K. Brunner, J. Hofstraat and L. De Cola, Nat. Biotechnol., 2003, 421, 54–57 CAS.
- G. G. Malliaras, J. D. Slinker, J. A. DeFranco, M. J. Jaquith, W. R. Silveira, Y.-W. Zhong, J. M. Moran-Mirabal, H. G. Craighead, H. D. Abruña and J. A. Marohn, Nat. Mater., 2008, 7, 168–169 CrossRef CAS.
- Q. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086–1088 CrossRef CAS PubMed.
- G. Kalyuzhny, M. Buda, J. McNeill, P. Barbara and A. J. Bard, J. Am. Chem. Soc., 2003, 125, 6272–6283 CrossRef CAS.
- C.-Y. Liu and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 4190–4191 CrossRef CAS PubMed.
- F. G. Gao and A. J. Bard, Chem. Mater., 2002, 14, 3465–3470 CrossRef CAS.
- R. D. Costa, E. Orti, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178–8211 CrossRef CAS PubMed.
- S. A. Priola, A. Raines and W. S. Caughey, Science, 2000, 287, 1503–1506 CrossRef CAS PubMed.
- H. T. Whelan, R. L. Smits Jr, E. V. Buchman, N. T. Whelan, S. G. Turner, D. A. Margolis, V. Cevenini, H. Stinson, R. Ignatius and T. Martin, J. Clin. Laser Med. Surg., 2001, 19, 305–314 CrossRef CAS PubMed.
- E. Desurvire and M. N. Zervas, Phys. Today, 1995, 48, 53 CrossRef.
- N. Tessler, V. Medvedev, M. Kazes, S. Kan and U. Banin, Science, 2002, 295, 1506–1508 CrossRef PubMed.
- L. Slooff, A. Polman, F. Cacialli, R. Friend, G. Hebbink, F. Van Veggel and D. Reinhoudt, Appl. Phys. Lett., 2001, 78, 2122–2124 CrossRef CAS.
- R. Curry and W. Gillin, Appl. Phys. Lett., 1999, 75, 1380–1382 CrossRef CAS.
- J.-C. G. Bünzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824–842 CrossRef.
- A. F. Rausch, M. E. Thompson and H. Yersin, J. Phys. Chem. A, 2009, 113, 5927–5932 CrossRef CAS PubMed.
- A. R. Hosseini, C. Y. Koh, J. D. Slinker, S. Flores-Torres, H. D. Abruña and G. G. Malliaras, Chem. Mater., 2005, 17, 6114–6116 CrossRef CAS.
- H. J. Bolink, L. Cappelli, E. Coronado and P. Gavina, Inorg. Chem., 2005, 44, 5966–5968 CrossRef CAS PubMed.
- H. Shahroosvand, S. Abaspour, B. Pashaei, E. Radicchi, F. De Angelis and F. Bonaccorso, Chem. Commun., 2017, 53, 6211–6214 RSC.
- B. N. Bideh, C. Roldán-Carmona, H. Shahroosvand and M. K. Nazeeruddin, Dalton Trans., 2016, 45, 7195–7199 RSC.
- S. Tang and L. Edman, Electrochim. Acta, 2011, 56, 10473–10478 CrossRef CAS.
- H.-C. Su, C.-C. Wu, F.-C. Fang and K.-T. Wong, Appl. Phys. Lett., 2006, 89, 261118 CrossRef.
- L. He, X. Wang and L. Duan, ACS Appl. Mater. Interfaces, 2018, 10, 11801–11809 CrossRef CAS PubMed.
- H. Shahroosvand, L. Heydari, B. N. Bideh, B. Pashaei, S. Tarighi and B. Notash, ACS Omega, 2018, 3, 9981–9988 CrossRef CAS PubMed.
- S. Liu and J. Zubieta, Polyhedron, 1989, 8, 677–688 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, John Wiley & Sons, Hoboken, NJ, USA, 2009 Search PubMed.
- M. T. Ramírez, A. Morales-Pérez, A. Rojas-Hernández and I. González, J. Electroanal. Chem., 1996, 410, 203–212 CrossRef.
- K. Sivakumar, T. Stalin and N. Rajendiran, Spectrochim. Acta, Part A, 2005, 62, 991–999 CrossRef CAS PubMed.
- D. L. Ashford, C. R. Glasson, M. R. Norris, J. J. Concepcion, S. Keinan, M. K. Brennaman, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2014, 53, 5637–5646 CrossRef CAS PubMed.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Photochemistry and Photophysics of Coordination Compounds I, Springer, 2007, pp. 117–214 Search PubMed.
- M. Cavazzini, P. Pastorelli, S. Quici, F. Loiseau and S. Campagna, Chem. Commun., 2005, 5266–5268 RSC.
- F. Barigelletti, L. Flamigni, J.-P. Collin and J.-P. Sauvage, Chem. Commun., 1997, 333–338 RSC.
- J. P. Collin, P. Gaviña, V. Heitz and J. P. Sauvage, Eur. J. Inorg. Chem., 1998, 1998, 1–14 CrossRef.
- F. Barigelletti and L. Flamigni, Chem. Soc. Rev., 2000, 29, 1–12 RSC.
- C. D. Geddes and J. R. Lakowicz, Reviews in fluorescence 2006, Springer, 2005 Search PubMed.
- K. Kalyanasundaram, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 1992, 31, 5243–5253 CrossRef CAS.
- N. H. Damrauer and J. K. McCusker, J. Phys. Chem. A, 1999, 103, 8440–8446 CrossRef CAS.
- N. H. Damrauer, G. Cerullo, A. Yeh, T. R. Boussie, C. V. Shank and J. K. McCusker, Science, 1997, 275, 54–57 CrossRef CAS PubMed.
- A. T. Yeh, C. V. Shank and J. K. McCusker, Science, 2000, 289, 935–938 CrossRef CAS PubMed.
- G. Hager and G. Crosby, J. Am. Chem. Soc., 1975, 97, 7031–7037 CrossRef CAS.
- M. Abrahamsson, H. Wolpher, O. Johansson, J. Larsson, M. Kritikos, L. Eriksson, P.-O. Norrby, J. Bergquist, L. Sun and B. Åkermark, Inorg. Chem., 2005, 44, 3215–3225 CrossRef CAS PubMed.
- J. D. Henrich, H. Zhang, P. K. Dutta and B. Kohler, J. Phys. Chem. B, 2010, 114, 14679–14688 CrossRef CAS PubMed.
- S. Stagni, A. Palazzi, S. Zacchini, B. Ballarin, C. Bruno, M. Marcaccio, F. Paolucci, M. Monari, M. Carano and A. J. Bard, Inorg. Chem., 2006, 45, 695–709 CrossRef CAS PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. l. Belser and A. Von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- K. Brunner, A. van Dijken, H. Börner, J. J. Bastiaansen, N. M. Kiggen and B. M. Langeveld, J. Am. Chem. Soc., 2004, 126, 6035–6042 CrossRef CAS PubMed.
- D. Saha, S. Das, S. Mardanya and S. Baitalik, Dalton Trans., 2012, 41, 8886–8898 RSC.
- B. Sullivan, D. Salmon and T. Meyer, Inorg. Chem., 1978, 17, 3334–3341 CrossRef CAS.
- D. P. Rillema, G. Allen, T. Meyer and D. Conrad, Inorg. Chem., 1983, 22, 1617–1622 CrossRef CAS.
- K. M. Lancaster, J. B. Gerken, A. C. Durrell, J. H. Palmer and H. B. Gray, Coord. Chem. Rev., 2010, 254, 1803–1811 CrossRef CAS.
- B. Ghosh, P. Adak, S. Naskar, B. Pakhira, P. Mitra, R. Dinda and S. K. Chattopadhyay, Inorg. Chim. Acta, 2017, 459, 1–14 CrossRef CAS.
- K. Majumder and S. Bhattacharya, Polyhedron, 1999, 18, 3669–3673 CrossRef CAS.
- V. Aranyos, A. Hagfeldt, H. Grennberg and E. Figgemeier, Polyhedron, 2004, 23, 589–598 CrossRef CAS.
- F. Cheng, C. He, M. Ren, F. Wang and Y. Yang, Spectrochim. Acta, Part A, 2015, 136, 845–851 CrossRef CAS PubMed.
- S. E. Domínguez and F. Fagalde, Inorg. Chem. Commun., 2017, 77, 31–34 CrossRef.
- R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef CAS.
- V. W.-W. Yam, V. C.-Y. Lau and K.-K. Cheung, Organometallics, 1995, 14, 2749–2753 CrossRef CAS.
- F. Wu, E. Riesgo, A. Pavalova, R. A. Kipp, R. H. Schmehl and R. P. Thummel, Inorg. Chem., 1999, 38, 5620–5628 CrossRef CAS PubMed.
- S. D. Bergman, I. Goldberg, A. Barbieri, F. Barigelletti and M. Kol, Inorg. Chem., 2004, 43, 2355–2367 CrossRef CAS PubMed.
- A. Bouskila, B. Drahi, E. Amouyal, I. Sasaki and A. Gaudemer, J. Photochem. Photobiol., A, 2004, 163, 381–388 CrossRef CAS.
- H. Shahroosvand, L. Najafi, A. Sousaraei, E. Mohajerani, M. Janghouri and F. Bonaccorso, J. Phys. Chem. C, 2016, 120, 24965–24972 CrossRef CAS.
- J. Kalinowski, M. Cocchi, P. Di Marco, W. Stampor, G. Giro and V. Fattori, J. Phys. D: Appl. Phys., 2000, 33, 2379 CrossRef CAS.
- X. Gong, P. K. Ng and W. K. Chan, Adv. Mater., 1998, 10, 1337–1340 CrossRef CAS.
- S. Xun, J. Zhang, X. Li, D. Ma and Z. Y. Wang, Synth. Met., 2008, 158, 484–488 CrossRef CAS.
- A. Breivogel, M. Park, D. Lee, S. Klassen, A. Kühnle, C. Lee, K. Char and K. Heinze, Eur. J. Inorg. Chem., 2014, 2014, 288–295 CrossRef CAS.
- H. J. Bolink, E. Coronado, R. D. Costa, P. Gaviña, E. Ortí and S. Tatay, Inorg. Chem., 2009, 48, 3907–3909 CrossRef CAS PubMed.
- J.-H. Hsu and H.-C. Su, Phys. Chem. Chem. Phys., 2016, 18, 5034–5039 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full synthesis details and other characterizations. See DOI: 10.1039/c9ra10761d |
| This journal is © The Royal Society of Chemistry 2020 |