
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeted delivery of thermoresponsive polymeric nanoparticle-encapsulated lycopene: in vitro anticancer activity and chemopreventive effect on murine skin inflammation and tumorigenesis
Sameena Bano *a,
Faheem Ahmedb,
Farha Khanc,
Sandeep Chand Chaudharyd and
M. Samim*a
*a,
Faheem Ahmedb,
Farha Khanc,
Sandeep Chand Chaudharyd and
M. Samim*a
aDepartment of Chemistry, School of Chemical and Life Sciences, Jamia Hamdard (Deemed to be University), New Delhi, India. E-mail: sameenabano01@gmail.com; shamim_chem@yahoo.co.in; Tel: +91 9210707636 Tel: +91 11 26054685 ext. 5557
bDepartment of Community Medicine, Hamdard Institute of Medical Sciences and Research, Jamia Hamdard (Deemed to be University), New Delhi, India
cDepartment of Biochemistry, School of Chemical and Life Sciences, Jamia Hamdard (Deemed to be University), New Delhi, India
dDepartment of Dermatology, University of Alabama, Birmingham, USA
First published on 27th April 2020
Abstract
Naturally occurring lycopene has been reported for its chemopreventive and chemotherapeutic efficiency in various cancers, but its exceptional lipophilicity, poor aqueous solubility, instability, and consequently poor bioavailability limit its usage as a chemopreventive and chemotherapeutic agent. The present study aimed to synthesize co-polymeric nanoparticle-encapsulated formulations of commercial lycopene (NLY) and extracted lycopene (NLX) and evaluate their in vitro anticancer activity and inhibitory effect on 12-O-tetradecanoylphorbol-13-acetate (TPA)-promoted skin inflammation and tumorigenesis in Swiss albino mice. To prepare the nanoparticle-encapsulated formulations of lycopene, thermosensitive PNIPAAM–PEG-based co-polymeric nanoparticles were synthesized and characterized by FTIR spectroscopy, NMR spectroscopy, DLS, and TEM. Nanolycopene, unlike free lycopene, could be readily dispersed in aqueous media. Nanolycopene demonstrated stronger antioxidant activity and comparable in vitro anticancer efficacy to free lycopene against the melanoma cell line B16. Furthermore, nanolycopene showed comparable reduction of TPA-induced skin edema, expression of COX-2, and oxidative stress response. Additionally, it showed significant inhibition of tumor promotion. It also altered Bax and Bcl2 expressions, which led to the induction of apoptosis. The results also supported that the extracted lycopene-encapsulated nanoparticles may be a good alternative to the expensive commercial lycopene for cancer treatment.
Introduction
Cancer is the second leading cause of deaths in the United States of America and many other nations in the world. Despite modern advances in medical therapeutics worldwide, cancer continues to account for more than eighteen million new cases and more than nine million deaths each year.1 Conventional therapeutic approaches have not been able to control the incidence of most of the cancer types. Therefore, there is an urgent need to develop mechanism-based approaches for the management of cancer. Chemoprevention via non-toxic agents can be one such approach. Cancer chemoprevention involves the use of different natural or biological agents to inhibit or reverse tumor growth. Epidemiological and pre-clinical data suggest that various phytochemicals and nutraceuticals possess chemopreventive properties, and in vitro and animal studies support that these compounds may modulate the signalling pathways involved in cell proliferation and apoptosis in transformed cells, enhance the immune system, and sensitize malignant cells to cytotoxic agents.2–4Oxidative stress is recognized as one of the major contributors of an increased risk of cancer. Lycopene is one of the most potent antioxidants5 and has been suggested to prevent carcinogenesis and atherogenesis by protecting critical biomolecules, including lipids, low-density lipoproteins (LDLs), proteins, and DNA.6–8 Several studies have indicated that lycopene is an effective antioxidant and free radical scavenger. Lycopene, because of its high number of conjugated double bonds, exhibits higher singlet oxygen quenching ability compared to β-carotene or α-tocopherol.9 These results indicate that lycopene may, therefore, play an important role in the prevention of cancer. Although lycopene has shown remarkable promise as a potent chemopreventive agent in several bioassay systems,10–13 there is a long way to go for it to be developed as an agent for the chemoprevention/treatment of cancer due to its insolubility in water and poor bioavailability.14,15
Continued efforts are needed to enhance its bioavailability and targeting to desirable sites together with well-designed pre-clinical studies in animal models that closely mimic human diseases, with an aim to establish the usefulness of lycopene as a cancer chemopreventive agent. Its health benefits can be increased by preventing its degradation and improving its dissolvability in the aqueous phase through its incorporation in nanoformulations.16,17 Nano-drug-delivery systems can be used to overcome the several limitations of conventional drug-delivery systems, such as nonspecific biodistribution, targeting, lack of water solubility, poor oral bioavailability, and low therapeutic indices. A few nanocarriers for lycopene have already been reported in the literature, like vesicular nanocarriers, solid lipid nanocarriers, and lipid nanocarriers, which can ensure the improved properties of lycopene when they are administrated in nanoform.18,19 In the present study, we report the synthesis, characterization, and cancer-related application of nanoparticle-encapsulated formulations of lycopene. Pure lycopene (LY) and extracted lycopene (LX) were used for the nanoformulations. Already reported, this simple, fast, economic, and environmentally friendly process for the extraction of lycopene from tomato has been used with some modifications.18
PNIPAAM is a thermosensitive nanopolymer that is widely used as a successful drug-delivery system against various diseases.20,21 Pegylated PNIPAAM nanoparticles were found to be safe, biocompatible, and more suitable for cancer targeting in conjunction with hyperthermia due to their higher, lower critical solution temperature (LCST) than PNIPAAM.22 Polymeric nanoparticles have also proved their high potential for selective localized drug delivery, particularly at the site of inflamed skin.23–25 It has also been demonstrated that the increased selective penetration of polymeric nanoparticles in inflamed skin would potentially reduce the associated side effects resulting from an unintentional penetration of the healthy skin from the use of conventional treatment systems.26 PNIPAAM–PEG-based cross-linked polymeric nanoparticles with a hydrophobic core and a hydrophilic shell were used for the encapsulation of lycopene. The results of the present study support the idea that the lycopene-rich extract may be a good alternative to the expensive commercial lycopene for incorporation into advanced drug-delivery systems.
Experimental
Materials
The chemicals and biochemicals used in this study were either of analytical grade or of the highest purity available commercially. N-Vinyl-2-pyrrolidone (VP) and N-isopropylacrylamide (NIPAAM) were from Acros. Ammonium per sulfate (APS), glutathione reductase (GR), 1-chloro-2,4-dinitrobenzene (CDNB), reduced glutathione (GSH), oxidized glutathione (GSSG), 1,2-dithio-bis-nitrobenzoic acid (DTNB), copper sulfate (CuSO4), trichloroacetic acid (TCA), sodium hydroxide (NaOH), reduced nicotinamide adenine dinucleotide phosphate (NADPH), acrylic acid (AA), ethylenediaminetetraacetic acid (EDTA), disodium orthophosphate (Na2HPO4), sodium potassium tartarate, sodium dihydrogen phosphate (NaH2PO4), hydrogen peroxide (H2O2), sodium carbonate (Na2CO3), and N,N,N,N-tetramethylethylene diamine (TEMED) were from Sisco Research Laboratory (SRL). Ferrous ammonium sulfate (FAS) and sodium azide (NaN3) were from Thomas Baker. Lycopene, bovine serum albumin (BSA), N-nitrosodiethylamine, dithiothreitol, thiobarbituric acid, coomassie brilliant blue (CBB), ethylenediamine tetra acetic acid (EDTA), EGTA, phenylmethylsulfonyl fluoride (PMSF), 2-mercaptoethanol, Tween 80, Brij 35, ethanolamine, methoxyethanol, citric acid, DMSO, DMBA, and TPA were purchased from Sigma Aldrich. [14C] ornithine and [3H] thymidine were purchased from Amersham Biosciences. Mouse melanoma cancer cell line (B16) was purchased from National Centre for Cell Sciences (NCCS), Pune, India, and it was maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The Annexin V-FITC/propidium iodide (PI) apoptosis detection kit was purchased from BD Biosciences. All other chemical and solvents used were obtained commercially at the highest purity available.Extraction and identification of lycopene
An already-reported, simple, fast, economic, and environmental friendly process for the extraction of lycopene from tomato was used with some modifications.18 The whole procedure was performed in subdued lighting to prevent damaging the compound. Fresh tomatoes, purchased from a grocery store, were sliced and dried using a freeze dryer for 18 h. Powdered dried tomato was extracted for 1 h with ethyl acetate at room temperature, in the dark. The filtered extract was then concentrated using a rotary evaporator below 40 °C. The resulting extract was stored at −20 °C until it was used. Extracted lycopene was identified and compared to a commercial standard using UV-visible spectroscopy and mass spectrometry.27 A concentrated solution of lycopene from tomato was prepared by dissolving 15–20 mg in 10 ml dichloromethane and diluting to 100 ml with n-hexane. The sample was vortexed for 2 min to dissolve; from this, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 dilution in n-hexane was prepared and used for the spectrophotometric measurements at 472 nm. Generally, the concentration of carotenoids is calculated from their extinction coefficients in hexane. The percentage of lycopene in the extract was calculated using the following relation:
:100 dilution in n-hexane was prepared and used for the spectrophotometric measurements at 472 nm. Generally, the concentration of carotenoids is calculated from their extinction coefficients in hexane. The percentage of lycopene in the extract was calculated using the following relation:| % lycopene = (A × 10000)/(3450 × P) |
Lycopene fragmentation was studied by tandem mass spectrometry with an electrospray ionization source (ESIMS/MS). Mass spectra were obtained in “full scan” mode from m/z 250 to 800. MS/MS experiments gave the characteristic fragmentation of the molecular ion, using different collision energies (10–30 eV). Argon was used as the collision gas.
Preparation of the polymeric nanoparticles and determination of the LCST
A co-polymer of N-isopropylacrylamide (NIPAAM) with N-vinyl-2-pyrrolidone (VP) poly(ethyleneglycol)monoacrylate (PEG-A) was synthesized through a previously reported free radical polymerization with some modifications.20 The monomers were dissolved in water and the polymerization was carried out under a nitrogen (N2) atmosphere. To carry out the experiment, recrystallized NIPAAM (90 mg), freshly distilled VP (10 μl), and properly washed PEG-A (500 μl, 1% w/v) were dissolved in water. To cross-link the polymer chains, N,N′-methylene bis acrylamide (MBA, 30 μl, 0.049 g ml−1) was added to the aqueous solution of monomers. The dissolved oxygen was removed by passing N2 gas for 45 min. Then, FAS (20 μl 0.5% w/v), saturated APS (30 μl), and TEMED (20 μl) were added to initiate the polymerization reaction. The polymerization was carried out at 30 °C under a N2 atmosphere for 18 h. Upon completion of the polymerization, the polymeric aqueous solution was dialyzed for 48 h using a spectropore membrane dialysis bag (12 kDa cutoff) with a continuous change of distilled water every 4 h. Thereafter, the aqueous solution was lyophilized to obtain a dry powder for subsequent use.The LCST was determined by measuring the optical transmittance of the aqueous polymeric samples (2 mg ml−1) at 500 nm on a UV-vis spectrophotometer fitted with a temperature controller. The heating rate was 1.0 °C min−1 and the sample was allowed to equilibrate for 15 min at each temperature. The LCST was the temperature of the inflection point in the curve.
Drug loading and release kinetics
Commercial lycopene (LY) and extracted lycopene (LX) were separately physically entrapped inside the hydrophobic core of the polymeric micelles after the complete polymerization reaction. Thus, the process of loading is described as a post-polymerization loading. Briefly, 50 mg lyophilized powder was dispersed in 10 ml of double distilled water and stirred to re-constitute the micelles. Then, the drug solution in DMSO (5 mg ml−1) was gradually added in the co-polymeric solution and stirred at room temperature till no more settling of the drug occurred. The drug-loaded polymeric nanoparticles were lyophilized to obtain a dried powder product for further use. The encapsulation efficiency and drug-loading content% were calculated as follows:| Entrapment efficiency (%) = drug present in nanoparticles/initial amount of drug × 100 |
| Drug loading content (%) = drug present in nanoparticles/weight of nanoparticles × 100 |
| Release (%) = [lycopene]release/[lycopene]total × 100 |
Characterization
Free radical scavenging activity
The DPPH scavenging activities of lycopene (LY and LX) and nanolycopene (NLY and NLX) were measured by the reported method (Sreejayan and Rao, 1996)28 with minor modifications. Briefly, 0.5 ml of test samples at different concentrations was mixed with 0.5 ml of 100 mM solution of DPPH. After 30 min incubation in the darkness and at ambient temperature, the resultant absorbance was recorded at 517 nm. The lycopene concentration that reduced 50% of the absorbance of free DPPH radicals (IC50) was measured. Ascorbic acid was used as the standard. The percentage inhibition was calculated using the following formula:| Percentage inhibition = [abs(control) − abs(sample)]/abs(control) × 100 |
Cell culture and MTT assay
The mouse melanoma cell line B16 was purchased from the National Centre of Cell Sciences (Pune, India). B16 mouse melanoma cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% FBS, 100 units per ml penicillin, and 100 μg per ml streptomycin in a 37 °C incubator containing 5% CO2. The growth of B16 cells was tested by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a tetrazole) assay.29 Cells were treated with LY (100 μg ml−1), LX (100 μg ml−1), NLY1 (5 μg ml−1), NLY2 (10 μg ml−1), NLX1 (5 μg ml−1), and NLX2 (10 μg ml−1).Flow cytometric analysis
The Annexin V-FITC/propidium iodide (PI) apoptosis detection kit was purchased from BD Biosciences to measure the apoptotic cell levels. Cells were treated with LY (100 μg ml−1), LX (100 μg ml−1), NLY1 (5 μg ml−1), NLY2 (10 μg ml−1), NLX1 (5 μg ml−1), and NLX2 (10 μg ml−1). All the cells after the different treatments as described were harvested and washed with ice-cold PBS twice, and then incubated in a darkroom with Annexin V-FITC and PI for 15 min. Subsequently, the cells were analyzed by flow cytometry (BD LSR ll, BD Biosciences). The percentage of cells undergoing apoptosis was quantified.30Animals
Seven-to-eight-weeks-old Swiss albino mice (20–25 g) were obtained from the Central Animal House Facility of Jamia Hamdard (Hamdard University), New Delhi, India. The mice were housed in a well-ventilated room at 25 ± 2 °C under a light–dark cycle and provided with standard laboratory feed (Hindustan Lever Ltd., Bombay, India) and drinking water ad libitum. During all the animal procedures, the animals received Humane Care in accordance with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India, and prior permission was sought from the Institutional Animal Ethics Committee (regn. no. 173/GO/Re/2000/CPCSEA) of Jamia Hamdard, New Delhi, India. The dorsal skins of the mice were shaved with electric clippers followed by the application of hair removing cream (Anne French, Geoffrey Manners, Bombay, India) at least 2 days prior to the treatments. All the compounds were applied to the dorsal shaved area of the mice.Experimental protocol for the biochemical estimations
The effects of the pre-treatment of LX and NLX on TPA-induced skin inflammatory and oxidative stress response were studied by a random allocation of mice into five groups of six mice each. The mice in group 1 received a topical application of 200 μl vehicle (acetone alone) and served as the vehicle control, while the mice in group 2 were treated with TPA alone and served as the positive control. The mice in group 3 received topical applications of 10 μg of LX in 200 μl acetone, the mice in groups 4 and 5 received topical applications of 0.5 and 1 μg (NLX1 and NLX2) in 200 μl acetone, respectively. Then, 30 min after these treatments, all the groups except group 1 received topical applications of TPA (2 μg/200 μl acetone/animal). The treatments were carried out for 3 days. The mice were sacrificed by cervical dislocation 12 h after the last treatment of TPA and the skin of each mouse was excised and processed for the evaluation of edema, histopathology, immunohistochemistry, and biochemical estimations. To assay the activity of ODC in cutaneous cytosol, the allocation and treatment schedule of the mice was exactly the same as in the case of the oxidative stress study, except that the mice in all groups were sacrificed 6 h after the last TPA treatment and were processed for cytosolic preparation. For the cutaneous [3H] thymidine-incorporation studies, the treatment protocol was exactly the same as describe above. The only difference was that the mice in all groups were injected intraperitoneally with [3H] thymidine (20 μCi/animal/0.2 ml saline) after 18 h of TPA/acetone treatment and were sacrificed by cervical dislocation 2 h after the [3H] thymidine treatment.Biochemical assays
Skin tissue from the mice in each group was excised and washed with ice cold 0.85% NaCl. The tissue was homogenized in cold phosphate buffer (0.1 M, pH 7.4). A homogenate of 10% (w/v) was prepared from the skin tissue and processed for the preparation of post mitochondrial supernatant (PMS) and microsomes for the biochemical estimations. Reduced glutathione (GSH) was determined by the method of Jollow et al. (1974).31 Glutathione reductase (GR) and glutathione peroxidase (GPx) activities were assayed by the method of Mohandas et al. (1984).32 Glutathione-S-transferase (GST) activity was estimated by the method of Habig et al. (1974).33 Catalase (CAT) activity was measured by the method of Claiborne (1985).34The assay of lipid peroxidation (LPO) was done by following the method of Wright et al. (1981).35 The amount of malonaldehyde (MDA) formed in each of the samples was assessed by measuring the absorbance at 535 nm against a blank. The results are expressed as nmol MDA formed/h per g tissue at 37 °C taking a molar extinction coefficient of 1.56 × 105 M−1 cm−1.
Ornithine decarboxylase (ODC) activity and [3H] thymidine incorporation
ODC activity was assayed according to the method of Verma et al. (1979).36 For the estimation of ODC activity, the skin was minced into small pieces as describe above and then homogenized in ice-cold 50 mM Tris–HCl buffer, pH 7.5 containing 0.4 mM EDTA, 0.32 mM pyridoxal phosphate, 0.1 mM PMSF, 1.0 mM 2-mercaptoethanol, 4.0 mM dithiothreitol, and 0.1% Tween-80. The epidermal homogenate was centrifuged at 10000 × g for 30 min and a portion of this supernatant was centrifuged at 105000 × g for 60 min at 4 °C to obtain the microsomal pellet and cytosol.
The isolation of skin DNA and assessment of the incorporation of [3H] thymidine into DNA were done by the method of Smart et al. (1986).37 Briefly, to the homogenate, an equal volume of ice-cold 10% TCA (trichloroacetic acid) was added and centrifuged at 2500 × g for 10 min. The precipitate was washed with cold 5% TCA and then centrifuged. The precipitate thus obtained was incubated with cold 10% PCA (perchloric acid) at 4 °C overnight. After incubation, the mixture was centrifuged and the precipitate was washed with cold 5% PCA and dissolved in warm 10% PCA, followed by incubation in a boiling water bath for 30 min. The solution was then filtered through Whatman 50 filter paper to get a clear nucleotide solution, which was taken for DNA estimation by the diphenylamine method. The amount of [3H] thymidine incorporated was expressed as dpm per μg DNA.
Evaluation of skin edema and histological appearance of the skin
The effect of LX and NLX on TPA-induced skin edema was assessed by using the method of Afaq et al. (2005).38 To observe the histological changes, the skin was removed, fixed in 10% buffered formalin, and embedded in paraffin. Next, 5 mg sections were cut and stained with H&E and observed under the microscope.Immunohistochemistry
The dorsal skin of each mouse was prepared for the immunohistochemical analysis of COX-2 expression. Immunohistochemistry was done according to the method described in Chaudhary et al. (2009)39 with some modifications. The expression of BcI2 and Bax proteins in the skin tumors was also investigated using immunohistochemical staining with appropriate antibodies.Skin tumor studies
Tumor induction were initiated by a single topical application of DMBA (50 μg/200 μl in acetone) to the shaved dorsal skin of each mouse, and one week later, tumor growth was promoted with a topical application of TPA (2 μg/200 μl in acetone) using the standard protocol as described in Chaudhary et al. (2009).39 The first group of mice received the vehicle only and served as the vehicle control. In group 2, the mice were treated with TPA alone and served as the positive control. However, the other groups received treatments with different doses of drugs (NLX1 = 0.5 μg and NLX2 = 1.0 μg), 30 min before the application of TPA, three times weekly up to the termination of the experiments (i.e., at 18 weeks) and these groups served as the experimental groups. The treatments were continued until the termination of the experiment. The animals were observed for the appearance of tumors, and the number of tumors was counted weekly for 18 weeks. The data are expressed as the percentage of mice with tumors and the number of tumors/mouse as a function of weeks of the test.Statistical analysis
The level of significance between different groups was based on the analysis of variance test followed by Dunnett's test. A p-value of less than 0.05 was considered statistically significant. All the statistical analyses were carried out using SPSS v 20 software.Results and discussion
Extraction and identification of lycopene
In the present study, we used a simple, less expensive, and rapid method of extraction to obtain lycopene from tomatoes and compared it with the commercially available form. The yield of lycopene was found to be 20 mg of extracted lycopene per 100 g of dry tomato. Lycopene has been analyzed in food or biological samples by many analytical methods, such as UV-vis spectrophotometry and mass spectrometry. UV-vis spectroscopy remains a technique of first choice for the analysis of carotenoids because it is a rapid, reproducible, unambiguous analysis. In fact, using this technique it is possible to distinguish different carotenoids in accordance with the literature.27,40,41 For this reason, the high purity of lycopene was checked using the specific extinction coefficient 3450 using hexane as a solvent, and recording the UV spectrum at 472 nm, which is the maximum absorption for lycopene. The purity of this lycopene was found to be over 94.5%.When analyzed by mass spectrometry, both (commercial and extracted) lycopene samples presented similar mass spectra with a molecular ion at m/z 537. A fragment identified at m/z 522 corresponded to the loss of a terminal methyl group. The fragments detected at m/z 467 and 444 corresponded to the loss of the isoprene and toluene units, respectively.
Synthesis and characterization of the nanoparticles
The lower critical solution temperature (LCST) can be altered by co-polymerization with hydrophilic or hydrophobic monomers. The LCST increases with the increase in hydrophilic minomers.42 It has been previously reported that if the LCST of a thermally responsive polymer is between the physiological temperature and hyperthermia, there will be an increase in drug delivery to solid tumors.43,44 So in the present study we synthesised pegylated thermosensitive polymeric nanoparticles, which have an amphiphilic character with a hydrophobic core inside the micelles, and a hydrophilic outer shell composed of hydrated amides, pyrrolidone, and PEG moieties that project from the monomeric units.45,46The absence of strong peaks in the range of 800–1000 cm−1 corresponding to the stretching mode of the vinyl double bond in the spectrum of the polymer (Fig. 1) confirmed the formation of polymeric nanoparticles. The absorption band at 3309 cm−1 (–OH stretching) and a sharp peak at 1129 cm−1 (–C–O–C– stretching) showed the presence of PEGMA in the polymer structure. The presence of NIPAAM in the sample could be noted by an absorption band split at 3440 cm−1 due to –NH stretching and a doublet at 1366 and 1386 cm−1 due to –CH bending of the isopropyl group. The –CH– stretching vibration of the polymer backbone was manifested through peaks at 2971–2932 cm−1, while peaks at 1633 and 1536 cm−1 correspond to the amide carbonyl group and the bending frequency of the amide N–H group, respectively. Intense peaks at 1457 cm−1 could be attributed to –CH2 bending vibrations of the co-polymer.
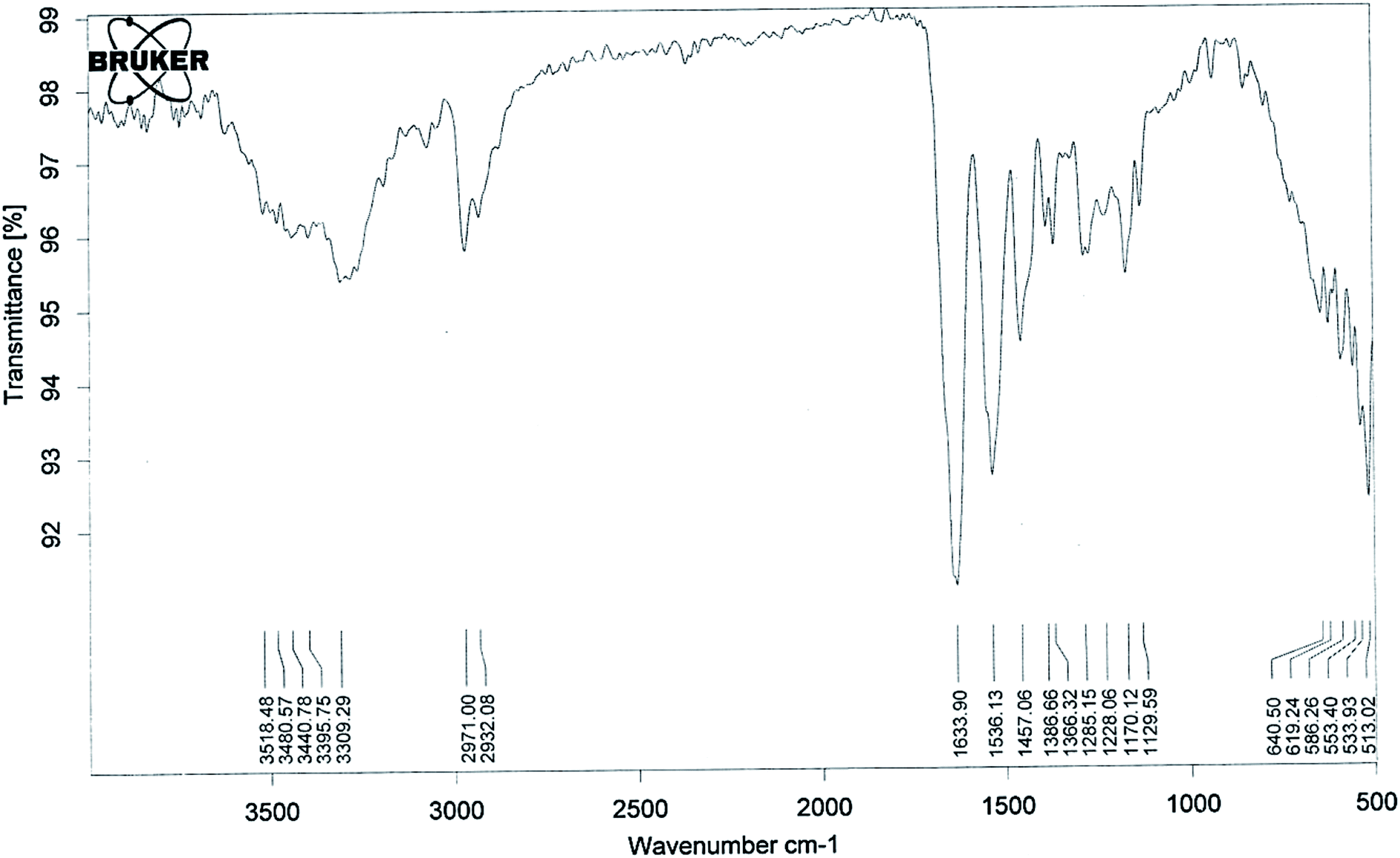 | ||
| Fig. 1 Fourier transform infrared (FTIR) spectra of the co-polymeric nanoparticles. | ||
Fig. 2 shows the 1H-NMR spectrum of the copolymer formed. Resonance observed in the upfield region (δ = 1.44–1.99 ppm) was attributed to the saturated protons of the polymeric network. The broad resonance peak at δ = 1.05 ppm was due to methyl protons of the isopropyl group. The signal peaks for the methyne proton (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CH–) of the N-isopropyl acrylamide group and methylene protons (–CH2–) of polyethylene oxide could be observed at 3.85 and 3.31 ppm, respectively.
CH–) of the N-isopropyl acrylamide group and methylene protons (–CH2–) of polyethylene oxide could be observed at 3.85 and 3.31 ppm, respectively.
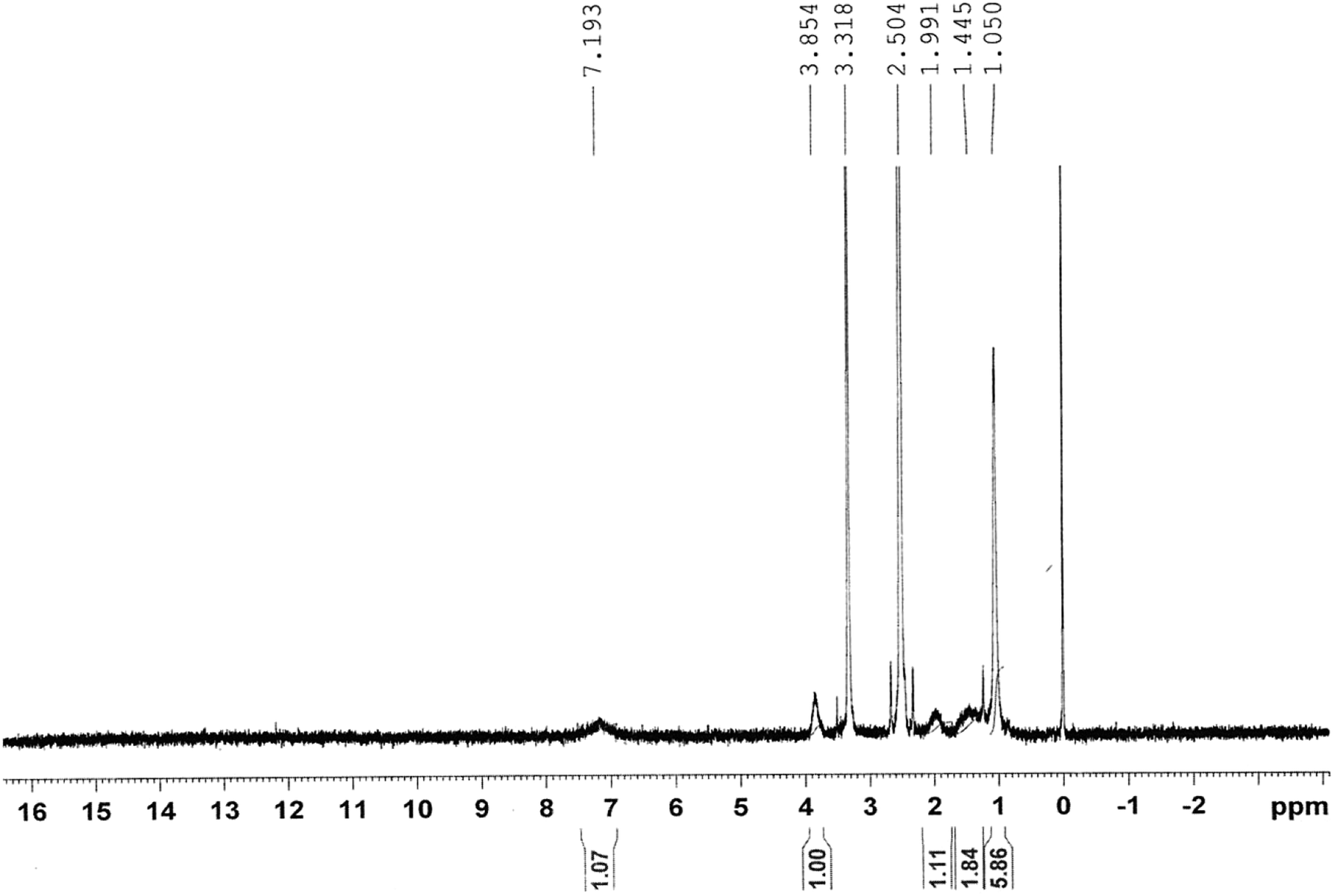 | ||
| Fig. 2 Nuclear magnetic resonance (NMR) spectrum of the co-polymeric nanoparticles. | ||
According to the dynamic light scattering (DLS) method and transmission electron microscopy (TEM) images, the particle size was found to be less than 100 nm with a spherical morphology and average polydispersity index (PDI) of 0.594 (Fig. 3).
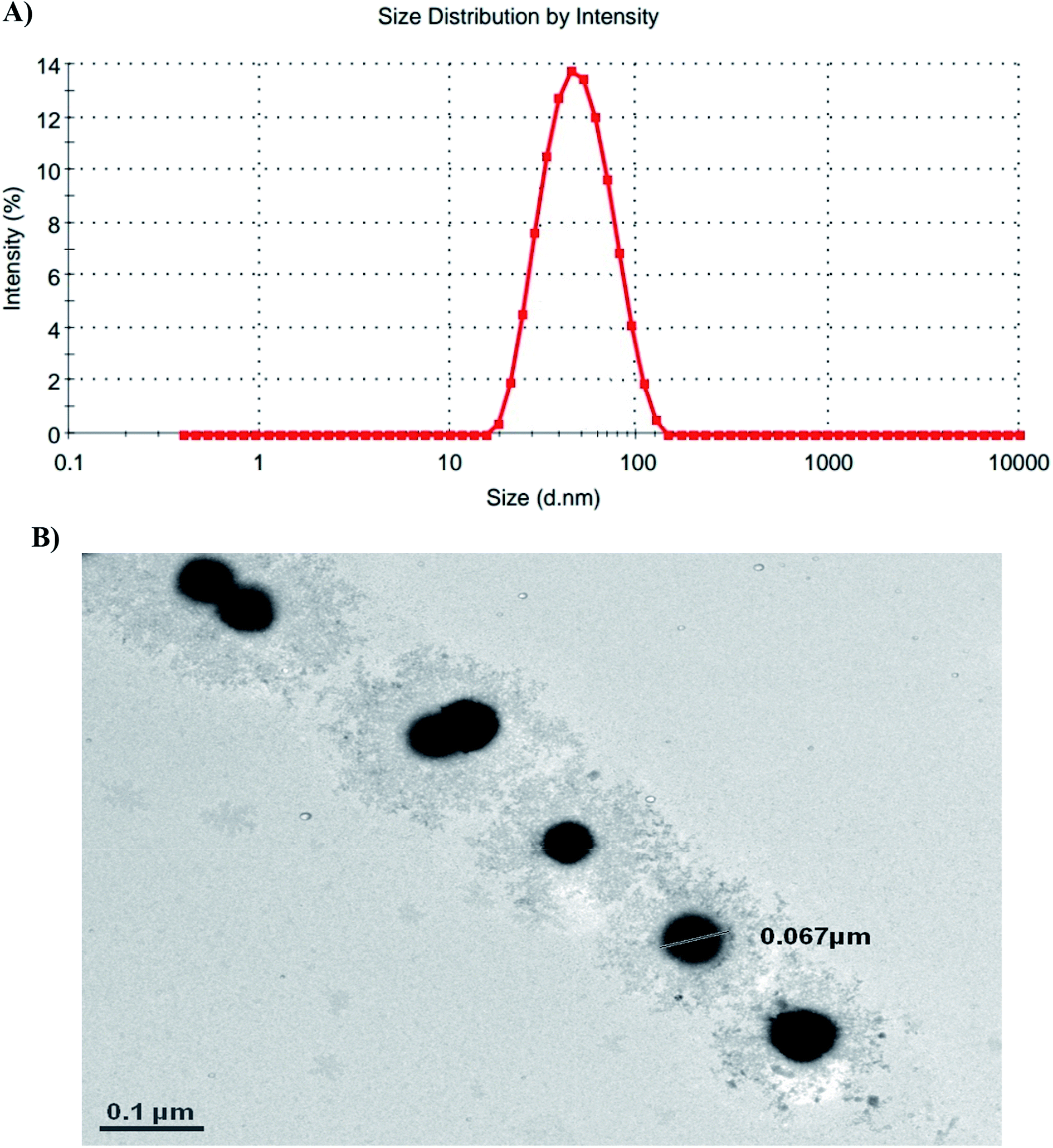 | ||
| Fig. 3 Size characterization of the polymeric nanoparticles. (A) DLS of the polymeric nanoparticles confirming a narrow size distribution in the 65 nm range with an average PDI of 0.594. (B) TEM picture demonstrates particles with a spherical morphology, with a size comparable to that observed in the DLS studies. (*0.1 μm = 100 nm). | ||
The thermoresponsive property of the nanoparticles was determined by measuring the optical transmittance as a function of temperature. The value obtained for the LCST of the synthesized nanoparticles was ∼38 ± 1 °C.
Entrapment efficiency and in vitro release kinetics
The entrapment efficiency of lycopene in the nanoparticles was found to be >85%. The lycopene-loaded nanoparticles showed a sustained release of lycopene from the nanopolymer at a physiological pH of 7.4 in phosphate buffer (Fig. 4).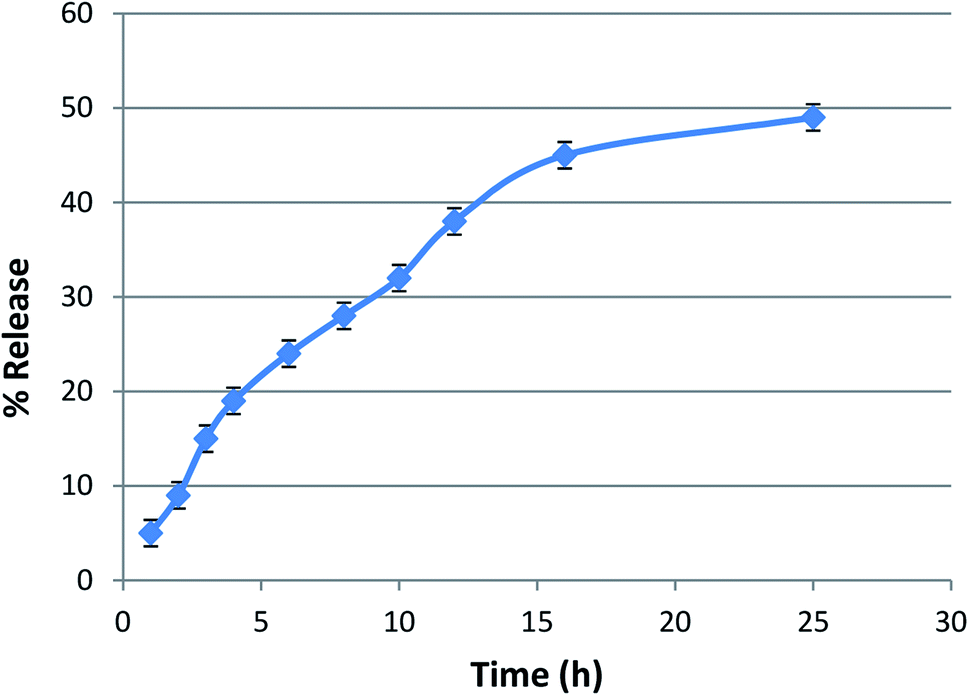 | ||
| Fig. 4 In vitro release kinetics of lycopene from polymeric nanoparticles. | ||
DPPH radical scavenging assay
The 1,1-diphenyl-2-picrylhydrazyl (DPPH) radical scavenging assay is the most commonly used method for screening antioxidant activity. Here, the change in absorbance occurred due to the acceptance of hydrogen atoms from the antioxidant by DPPH radicals. This could be visually noticed by a color change from purple to yellow. All the samples showed DPPH radical scavenging activity in a concentration-dependent manner. The concentration of the tested samples at which it scavenges DPPH radicals by 50% in the solution is defined as IC50. The IC50 values of LY, LX, NLY, and NLX were found to be 59.91, 53.82, 30.30, and 25.15 μg ml−1, respectively. The IC50 value for ascorbic acid was 53.08 μg ml−1. The radical scavenging ability of the lycopene-loaded nanoparticles was found to be significantly higher (p ≤ 0.001) than the free lycopene, which can be described as a sustained release of the drug, while the lycopene molecules inside the nanoparticles are strongly protected against oxidation. The extracted lycopene in both forms, free as well as encapsulated, showed better activity than commercial lycopene (Fig. 5).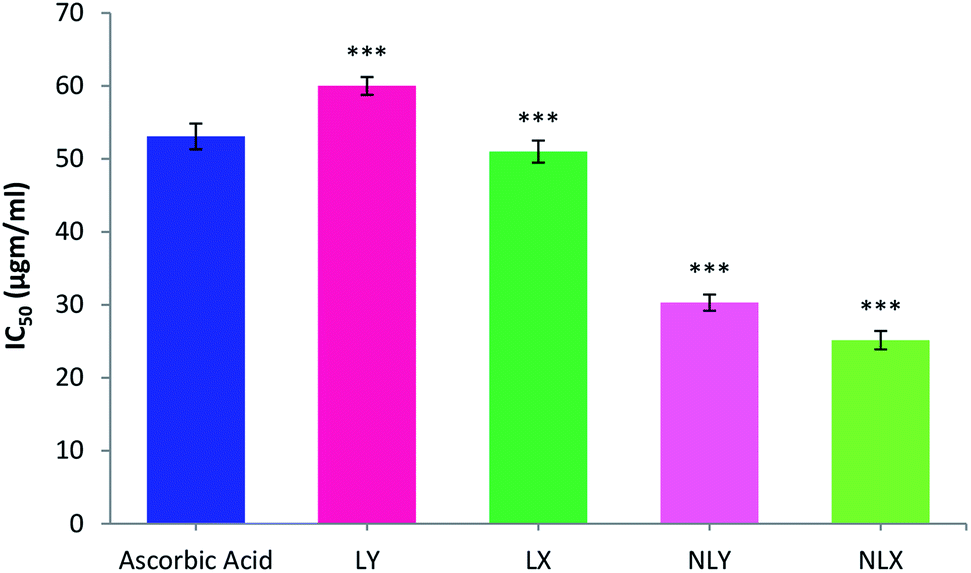 | ||
| Fig. 5 DPPH free radical scavenging activity of nanolycopene and free lycopene. All data are the mean ± S.D. (n = 6) with statistical difference at (***) p ≤ 0.001. | ||
In vitro anticancer activity
Several in vitro studies with various cancer cell lines have shown the cancer-preventive ability of lycopene by inducing apoptosis. Apoptosis helps maintain health by eliminating unhealthy, excess, or abnormal cells. When damaged cells fail to undergo apoptosis, they become immortal and can become cancer cells. Lycopene can promote apoptosis in these cells and cancer cells, and therefore might have potential as a chemopreventive and chemotherapeutic agent.47,48 The inhibiting effects of lycopene and nanolycopene on the melanoma cell line B16 was evaluated in vitro by using the MTT assay (Fig. 6A) and the apoptosis of B16 cells induced by treatment was measured by FACS (Fig. 6B). The results clearly demonstrated that the lycopene-encapsulated nanoparticles had enhanced cytotoxicity compared to the free lycopene at lower doses. The nanoparticles could deliver the free drug inside the cells at the site of action, so there will be a greater therapeutic concentration of lycopene inside the cells. The cell death with the lycopene-loaded nanoparticles was higher when compared with free lycopene even at significantly lower doses. The percentage of apoptotic cells with nanolycopene was also significantly higher than free lycopene. Better results with the extracted lycopene than the commercial form further encourage the use of this raw material.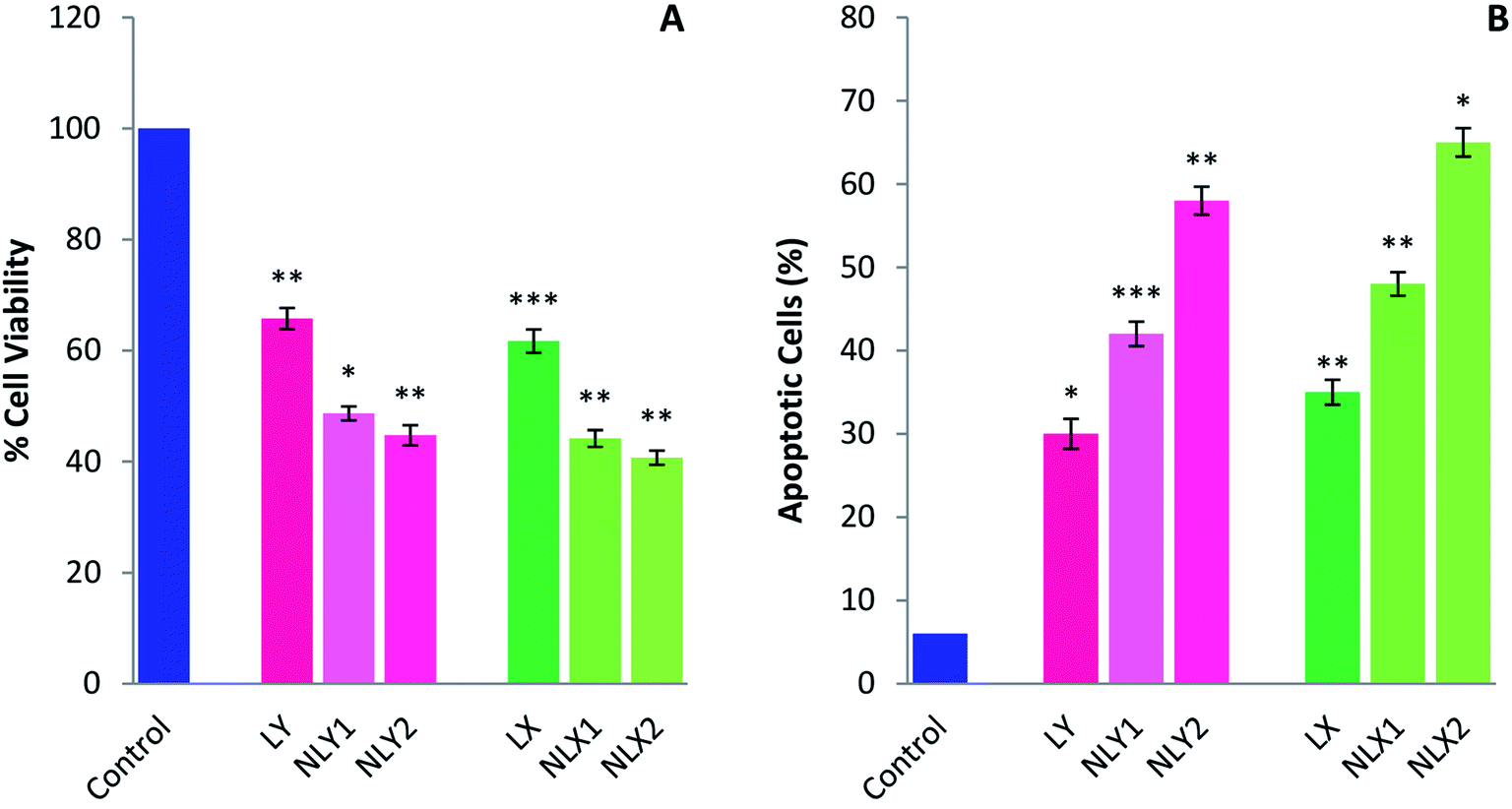 | ||
| Fig. 6 Nanolycopene and free lycopene inhibited cell growth and induced the apoptosis of melanoma B16 cells. (A) MTT assay with B16 cells. (B) Annexin-V analysis of apoptotic cells of B16 cells. Cells were treated with LY (100 μg ml−1), LX (100 μg ml−1), NLY1 (5 μg ml−1), NLY2 (10 μg ml−1), NLX1 (5 μg ml−1), and NLX2 (10 μg ml−1). Data are presented as the mean ± SEM from three separate experiments and statistically different from the control) (*p < 0.05, **p < 0.01, ***p < 0.001). | ||
Chemopreventive effect of lycopene
We investigated the chemopreventive effect of LX and NLX against TPA-induced skin edema, hyperplasia, COX-2 expression, and oxidative stress and DMBA/TPA-mediated murine skin tumorigenesis in a Swiss albino mice model. The role of lycopene in the induction of the apoptotic pathway by altering the expression of pro-apoptotic Box and antiapoptotic Bcl-2 protein was also demonstrated in the present study. Earlier studies revealed that the topical application of TPA to mouse skin and the treatment of certain epidermal cells represent excellent models to study the edema and inflammatory responses, oxidative responses, induction of ODC expression, thymidine incorporation into DNA, and cell proliferation leading to skin tumor promotion in DMBA-initiated mouse skin tumorigenesis.49,50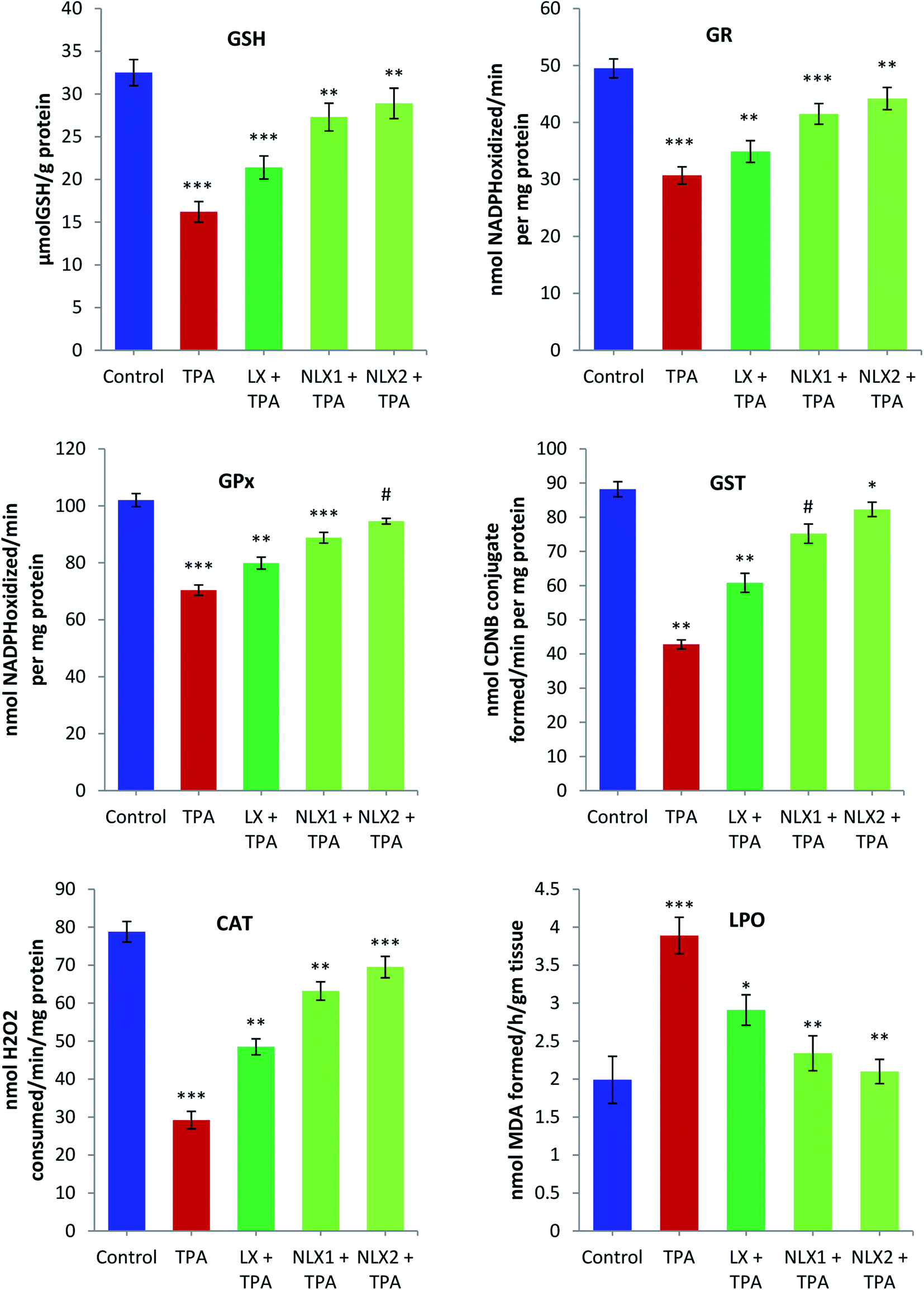 | ||
| Fig. 7 Effect of pre-treatment of LX and NLX on TPA-induced alterations in the levels of reduced glutathione (GSH) and malondialdehyde (MDA) and the activities of antioxidant enzymes (GR, GPx, GST, and CAT). Each value represents the mean ± SE of six animals. The group 2 (TPA-treated) is compared to the group 1 (acetone-treated) control. Group 3 (LX, 10 μg/0.2 ml acetone + TPA), group 4 (NLX1, 0.5 μg/0.2 ml acetone + TPA), and group 5 (NLX2 1 μg/0.2 ml acetone + TPA) are compared to group 2 (TPA-treated) (*p < 0.05, **p < 0.01, ***p < 0.001, # = not significant). | ||
An elevated level of ODC activity and unscheduled DNA synthesis play a significant role in tumor promotion.54,55 Our data indicated that the pre-treatment of LX and NLX significantly inhibited the induction of ODC and [3H] thymidine incorporation caused by TPA treatment in mouse skin (Fig. 8). The ODC activity and thymidine incorporation in DNA showed 5.9- and 2.7-fold increases, respectively, upon the treatment of mice skins with a single topical application of TPA. Pre-treatment with LX, NLX1, and NLX2 restored ODC activity by 144–416% and unscheduled DNA synthesis by 58–173%. Therefore, these findings confirmed that NLX at a very low dose more significantly inhibited cellular proliferation during the tumor promotion stage than for LX.
 | ||
| Fig. 8 Effect of pre-treatment of LX and NLX on TPA-induced epidermal ornithine decarboxylase (ODC) activity and [3H] thymidine incorporation in Swiss albino mice. Results represent the mean ± S.E. of six mice per group. Group 2 (TPA-treated) is compared to the group 1 (acetone-treated) control. Group 3 (LX, 10 μg/0.2 ml acetone + TPA), group 4 (NLX1, 0.5 μg/0.2 ml acetone + TPA), and group 5 (NLX2 1 μg/0.2 ml acetone + TPA) are compared to group 2 (TPA-treated) (*p < 0.05, **p < 0.01, ***p < 0.001, # = not significant). | ||
Topical application of LX (10 μg) 30 min prior to TPA application showed a significant inhibition (64.6%; p < 0.01) against TPA-induced skin edema, but the nanoformulation application NLX1 (0.5 μg) showed 77.8% (p < 0.01) and NLX2 (1 μg) showed 92% (p < 0.001) protective effects against TPA-induced skin edema (Table 1).
| Treatment groups | Weight of skin puncha (mg) | % Inhibition of edema |
|---|---|---|
| a Values represent the mean ± SE of three mice in each group. Group 2 (TPA-treated) is compared to the group 1 (acetone-treated) control. Group 3 (LX, 10 μg/0.2 ml acetone + TPA), group 4 (NLX1, 0.5 μg/0.2 ml acetone + TPA), and group 5 (NLX2 1 μg/0.2 ml acetone + TPA) are compared to group 2 (TPA-treated) (**p < 0.01, ***p < 0.001). | ||
| Control | 20.20 ± 0.32 | |
| TPA | 31.50 ± 0.72** | |
| LX + TPA | 24.20 ± 1.30** | 64.0 |
| NLX1 + TPA | 22.20 ± 1.10** | 77.8 |
| NLX2 + TPA | 21.10 ± 1.40*** | 92.0 |
Since skin tumor promotion is directly related to inflammation,49 so, the effect of the pre-treatment of LX and NLX on TPA-induced skin inflammation was observed (Fig. 9A). Marked changes were found in the histology of the skin in the TPA-treated mice as compared to the acetone-treated mice (Fig. 9A(a)), while the topical application of TPA alone (Fig. 9A(b)) showed obvious inflammation response with clear evidences of skin edema and inflammatory cells infiltration. Moreover, with the pre-treatment of LX, NLX1 and NLX2 (Fig. 9A(c–e)) significantly decreased inflammatory cell infiltration, while nanolycopene caused the most marked morphological alterations, compared to the TPA treatment alone. COX-2 plays a crucial role in inflammation, cellular proliferation, and tumor promotion.56 In the present study, the topical treatment of LX and NLX at the two doses, 30 min prior to TPA application, significantly inhibited the number of epidermal cells with the expression of COX-2 as compared to TPA treatment alone (Fig. 9B(a–e)). The results obtained confirmed that the pre-treatment of NLX at low doses more significantly inhibited the TPA-induced induction of COX-2 in epidermal cells as well as TPA-induced skin edema and the infiltration of inflammatory cells than LX.
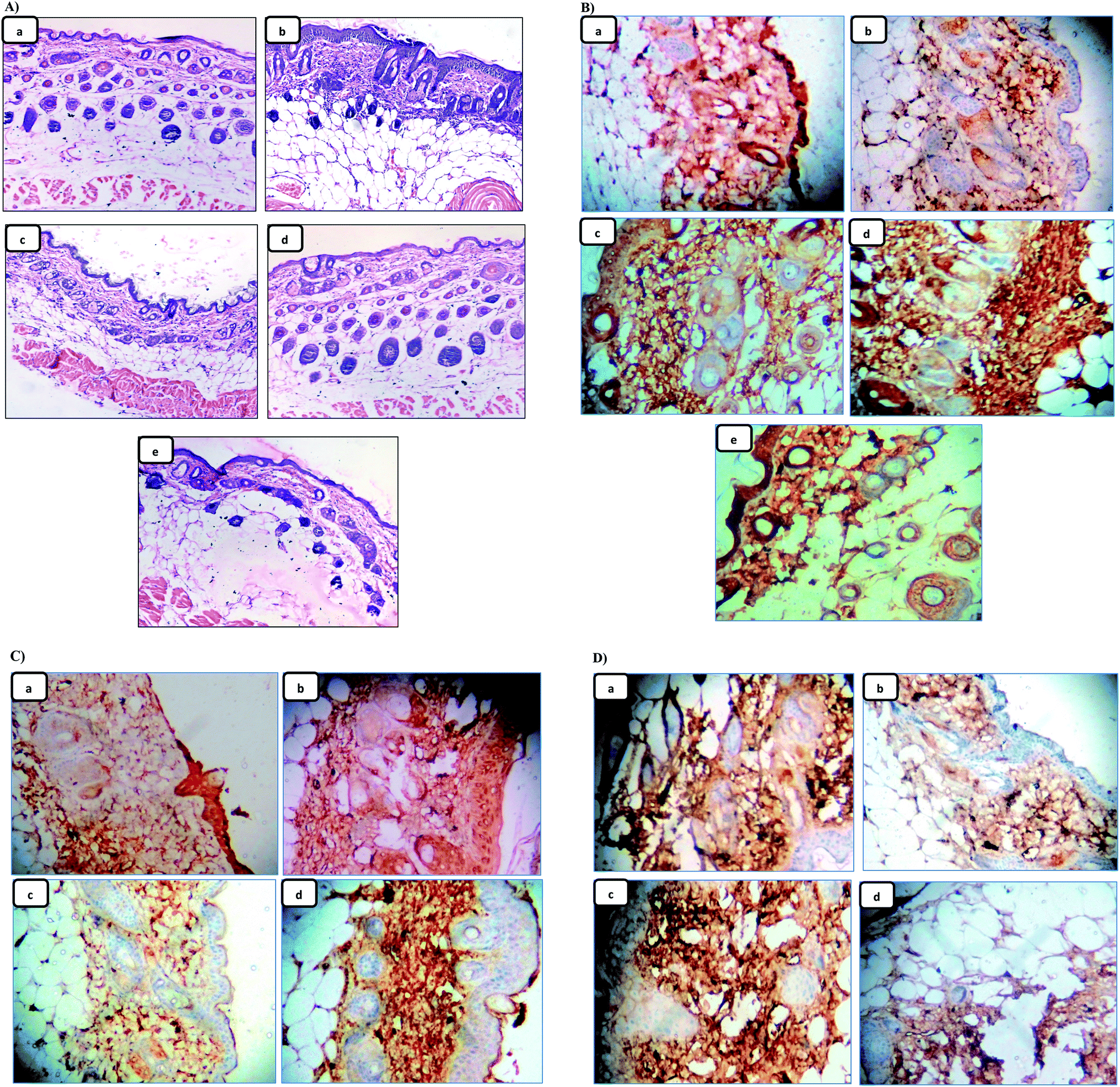 | ||
| Fig. 9 Histopathological sections of skin showing the effect of the pre-treatment of LX and NLX on TPA-induced histological alteration in skin (A) (a–e), and on the immunohistochemical expression of COX-2 in skin (B) (a–e). Effect of the pre-treatment of NLX on the DMBA/TPA-induced immunohistochemical expression of Bax and Bcl-2 in skin/tumors (C) (a–d) and (D) (a–d), respectively. | ||
| Treatment group | Percentage of mice with tumors | Tumors/mouseb | ||||
|---|---|---|---|---|---|---|
| 6 wk | 12 wk | 18 wk | 6 wk | 12 wk | 18 wk | |
| a Mice skin was initiated by a single topical application of DMBA (50 μg/200 μl) and promoted with TPA (2 μg/200 μl) by thrice weekly applications for 18 weeks.b The tumors per mouse are expressed as the mean ± SE from 15 mice. Group 2 (TPA-treated) is compared to the control, while group 3 (NLX1, 0.5 μg/0.2 ml acetone + TPA), and group 4 (NLX2 1 μg/0.2 ml acetone + TPA) are compared to group 2 (TPA-treated) (*p < 0.05, **p < 0.01). | ||||||
| Control | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| TPA | 44.8 | 84.7 | 97.3 | 3.3 ± 1.2** | 7.4 ± 1.0** | 12.7 ± 1.31** |
| NLX1 + TPA | 19.9 | 30.4 | 51.2 | 1.7 ± 0.32* | 3.4 ± 0.10** | 5.2 ± 1.10* |
| NLX2 + TPA | 15.5 | 22.9 | 41.4 | 0.91 ± 0.21* | 2.3 ± 0.09* | 4.1 ± 0.91* |
Thus, the improved anti-tumor promotional activity by NLX in murine skin can be explained by its effects on mechanisms involving inflammation, oxidative stress, ODC, and thymidine uptake and the induction of apoptosis.
Conclusions
Thermosensitive co-polymeric nanoparticles, used for lycopene encapsulation, were synthesized and characterized by IR, NMR, DLS, and TEM. In the present study, nanolycopene clearly demonstrated a comparable efficacy to free lycopene. The polymeric nanoparticles were able to deliver large amounts of drug at a targeted site. The enhanced chemopreventive activity demonstrated by NLX in murine skin was attributed to its effects on mechanisms involving inflammation, oxidative stress, ODC activity, thymidine uptake, and the induction of apoptosis. In addition, the topical drug delivery using polymeric nanoparticles is a promising strategy for the treatment of skin inflammatory disorders. This formulation increases aqueous solubility and the bioavailability of lycopene, and at the same time it reduces the required dose of the drug, and hence paves the way for future application as it improves the potential of the existing drug. Furthermore, the results reinforced that extracted lycopene is a better alternative to the expensive commercial lycopene for incorporation into advanced delivery systems.Conflicts of interest
The authors declare there are no conflicts of interest.Acknowledgements
Authors are thankful to the Hon'ble Vice-Chancellor of Jamia Hamdard for extending all the facilities to carry out this research work. We acknowledge the financial support of Science and Engineering Research Board (SERB), Department of Science and Technology, Government of India to perform this study.References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, CA-Cancer J. Clin., 2018, 68, 394–424 CrossRef.
- K. Aleksandrova, T. Pischon, M. Jenab, H. B. Bueno-de-Mesquita, V. Fedirko, T. Norat, D. Romaguera, S. Knüppel, M. C. Boutron-Ruault, L. Dossus, L. Dartois, R. Kaaks and K. Li, BMC Med., 2014, 12, 168 CrossRef.
- D. O. Garcia and C. A. Thomson, Nutr. Clin. Pract., 2014, 29, 768–779 CrossRef.
- M. Mehta and M. Shike, J. Natl. Compr. Cancer Network, 2014, 12, 1721–1726 CrossRef.
- N. J. Miller, J. Sampson, L. P. Candeias, P. M. Bramley and C. A. Rice-Evans, FEBS Lett., 1996, 384, 240–246 CrossRef CAS.
- S. Agarwal and A. V. Rao, Lipids, 1998, 33, 981–984 CrossRef CAS.
- A. V. Rao and S. Agarwal, Nutr. Cancer, 1998, 31, 199–203 CrossRef CAS.
- B. L. Pool-Zobel, A. Bub, H. Muller, I. Wollowski and G. Rechkemmer, Carcinogenesis, 1997, 18, 1847–1850 CrossRef CAS.
- P. D. Mascio, S. Kaiser and H. Sies, Arch. Biochem. Biophys., 1989, 274, 532–538 CrossRef.
- D. Aune, D. S. Chan, A. R. Vieira, D. A. Rosenblatt, R. Vieira, D. C. Greenwood and T. Norat, Breast Cancer Res. Treat., 2012, 134, 479–493 CrossRef CAS.
- J. D. Ribayo-Mercado, M. Garmyn, B. A. Gilchrest and R. M. Russell, J. Nutr., 1995, 125, 1854–1859 CrossRef.
- K. Steven and M. D. Clinton, Nutr. Rev., 1998, 56, 35–51 Search PubMed.
- M. Takeshima, M. Ono, T. Higuchi, C. Chen, T. Hara and S. Nakano, Cancer Sci., 2014, 105, 252–257 CrossRef CAS.
- W. Faisal, C. M. O'Driscoll and B. T. Griffin, J. Pharm. Pharmacol., 2010, 62, 323–331 CrossRef CAS.
- M. Vertzoni, G. Valsami and C. Reppas, J. Pharm. Pharmacol., 2006, 58, 1211–1217 CrossRef CAS.
- J. M. Gutierrez, C. Gonzalez, A. Maestro, I. Sole, C. M. Pey and J. Nolla, Curr. Opin. Colloid Interface Sci., 2008, 13, 245–251 CrossRef CAS.
- Q. Huang, H. Yu and Q. Ru, J. Food Sci., 2010, 75, 50–57 CrossRef.
- A. Ascenso, S. Pinho, C. Eleutério, F. C. Praça, M. V. L. B. Bentley, H. Oliveira, C. Santos, O. Silva and S. Simoes, J. Agric. Food Chem., 2013, 61, 7284–7293 CrossRef CAS.
- A. Singh, Y. R. Neupane, B. P. Panda and K. Kohli, J. Microencapsulation, 2017, 34, 416–429 CrossRef CAS.
- S. Bisht, G. Feldmann, S. Soni, R. Ravi, C. Karikar, A. Maitra and A. Maitra, J. Nanobiotechnol., 2007, 5, 3 CrossRef.
- M. Samim, S. Naqvi, I. Arora, F. J. Ahmad and A. Maitra, Ther. Delivery, 2011, 2, 223–230 CrossRef CAS.
- N. Gulatia, R. Rastogia, A. K. Dindac, R. Saxenad and V. Koula, Colloids Surf., B, 2010, 79, 164–173 CrossRef.
- M. M. Abdel-Mottaleb, B. Moulari, A. Beduneau, Y. Pellequer and A. Lamprecht, Eur. J. Pharm. Biopharm., 2012, 82, 151–157 CrossRef CAS.
- M. M. Abdel-Mottaleb, D. Neumann and A. Lamprecht, Eur. J. Pharm. Biopharm., 2011, 7936–7942 Search PubMed.
- J. Yokota and S. Kyotani, J. Chin. Med. Assoc., 2018, 81, 511–519 CrossRef.
- C. Try, B. Moulari, A. Béduneau, O. Fantini, D. Pin, Y. Pellequer and A. Lamprecht, Eur. J. Pharm. Biopharm., 2016, 100, 101–108 CrossRef CAS.
- D. Montesano, F. Fallarino, L. Cossignani, A. Bosi, M. S. Simonetti, P. Puccetti and P. Damiani, Eur. Food Res. Technol., 2008, 226, 327–335 CrossRef CAS.
- N. Sreejayan and M. N. A. Rao, Drug Res., 1996, 46, 169–173 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- J. Du, X. Lu, Z. Long, Z. Zhang, X. Zhu, Y. Yang and J. Xu, Molecules, 2013, 18, 757–767 CrossRef.
- D. J. Jollow, J. R. Metchell, N. Zampaglione and J. R. Gillete, Pharmacology, 1974, 11, 151–169 CrossRef CAS.
- J. Mohandas, J. J. Marshall, G. G. Dugin and J. S. Horvath, Cancer Res., 1984, 44, 5086–5091 CAS.
- W. H. Habig, M. J. Pabst and W. B. Jokoby, J. Biol. Chem., 1974, 249, 7130–7139 CAS.
- A. Claiborne, Catalase activity, in Handbook of Methods for Oxygen Radical Research, ed. R. A. Greenwald, CRC Press, Boca Raton, FL, 1985, pp. 283–284 Search PubMed.
- J. R. Wright, H. D. Colby and P. R. Miles, Arch. Biochem. Biophys., 1981, 206, 296–304 CrossRef CAS.
- A. K. Verma, B. G. Shapas, H. M. Rice and R. K. Boutwell, Cancer Res., 1979, 39, 419–425 CAS.
- R. C. Smart, M. T. Huang and A. H. Conny, Carcinogenesis, 1986, 7, 1865–1870 CrossRef CAS.
- F. Afaq, M. Saleem, C. G. Krueger, J. D. Reed and H. Mukhtar, Int. J. Cancer, 2005, 113, 423–433 CrossRef CAS.
- S. C. Chaudhary, M. S. Alam, M. S. Siddiqui and M. Athar, Chem.-Biol. Interact., 2009, 179, 145–153 CrossRef CAS.
- G. Britton, UV/visible spectroscopy, in Carotenoids: spectroscopy, ed. G. Britton, S. Liaaen-Jensen and H. Pfandser, Birkäuser, Basel, 1995, vol. 1B, pp. 13–62 Search PubMed.
- G. Britton, Carotenoids, in methods in plant biochemistry, Academic, London, 1991, vol. 7. pp. 473–518 Search PubMed.
- D. Schmaljohann, J. Oswald, B. Jørgensen, M. Nitschke, D. Beyerlein and C. Werneret, Biomacromolecules, 2003, 4, 1733–1739 CrossRef CAS.
- A. Chilkoti, D. E. Meyer, M. R. Dreher and D. Raucher, Adv. Drug Delivery Rev., 2002, 54, 613–630 CrossRef CAS.
- D. E. Meyer, B. C. Shin, G. A. Kong, M. W. Dewhirst and A. Chilkoti, J. Controlled Release, 2001, 74, 213–224 CrossRef CAS.
- S. Soni, A. K. Babbar, R. K. Sharma and A. Maitra, J. Drug Targeting, 2006, 14, 87–95 CrossRef CAS.
- R. Tyagi, S. Lala, A. K. Verma, A. K. Nandy, S. B. Mahato, A. Maitra and M. K. Basu, J. Drug Targeting, 2005, 13, 161–171 CrossRef CAS.
- B. Richard van Breemen and N. Pajkovic, Cancer Lett., 2008, 2(269), 339–351 CrossRef.
- N. F. Gloria, N. Soares, C. Brand, F. L. Oliveira, R. Borojevic and A. J. Teodoro, Anticancer Res., 2014, 34, 1377–1386 CAS.
- S. K. Katiyar and H. Mukhtar, Carcinogenesis, 1997, 18, 1911–1916 CrossRef CAS.
- M. Athar, Indian J. Exp. Biol., 2002, 40, 656–667 CAS.
- J. J. Reiners Jr, G. Thai, A. Pavone, T. Rupp and E. Kodari, Carcinogenesis, 1990, 11, 957–963 CrossRef CAS.
- D. R. Bicker and M. Athar, J. Invest. Dermatol., 2006, 126, 2565–2575 CrossRef.
- U. Giri, S. D. Sharma, M. Abdulla and M. Athar, Biochem. Biophys. Res. Commun., 1995, 209, 698–705 CrossRef CAS.
- M. Auvinen, A. Paasinen, L. C. Andersson and E. Holtta, Nature, 1992, 360, 355–358 CrossRef CAS.
- T. G. O'Brien, L. C. Megosh, G. Gilliard and A. P. Soler, Cancer Res., 1997, 57, 2630–2637 Search PubMed.
- S. M. Prescott and F. A. Fitzpatrick, Biochim. Biophys. Acta, 2000, 1470, 69–78 Search PubMed.
- J. Di Giovanni, Prog. Exp. Tumor Res., 1991, 33, 192–229 CAS.
- S. Cory, D. C. Huang and J. M. Adams, Oncogene, 2003, 22, 8590–8607 CrossRef CAS.
- M. Nihal, N. Ahmad, H. Mukhtar and G. S. Wood, Int. J. Cancer, 2005, 114, 513–521 CrossRef CAS.
- N. Kalra, K. Bhui, P. Roy, S. Srivastava, J. George, S. Prasad and Y. Shukla, Toxicol. Appl. Pharmacol., 2008, 226, 30–37 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |