
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyester functional graphenic materials as a mechanically enhanced scaffold for tissue regeneration†
Stephen J. Schmidt ,
Brian D. Holt,
Anne M. Arnold and
Stefanie A. Sydlik*
,
Brian D. Holt,
Anne M. Arnold and
Stefanie A. Sydlik*
Carnegie Mellon University, 4400 Fifth Ave, Pittsburgh, PA 15213, USA. E-mail: ssydlik@andrew.cmu.edu
First published on 28th February 2020
Abstract
Traditional metal implants such as titanium, cobalt, and chromium have found wide utility in medicine; however, these come with a risk of toxicity. To overcome metal-related toxicity and enable degradability, polyesters including polycaprolactone (PCL), polylactic acid (PLA), and polyglycolic acid (PGA) show promise for the replacement of various biomedical applications of metals due to their accepted biocompatibility and FDA approval. However, polyesters are less stiff than their metallic counterparts, limiting their application to non-load bearing injury sites, such as fixation hardware for fingers. To improve mechanical properties, graphene oxide (GO)-polyester composites are a promising class of biodegradable scaffolds. Initial reports of these composites are encouraging, but mechanical properties still fall short. Traditional composites rely on non-covalent association between GO and the polyesters, which often leads to failure at the interface and weakens the overall strength of the material. Herein, we present a strategy for attachment of these FDA-approved polyesters onto a derivative of GO using a robust covalent bond. By covalently functionalizing the graphenic backbone with polyesters and without metal catalysts, we create functional graphenic materials (FGMs) to not only simultaneously retain biodegradability and compatibility, but also mechanically strengthen PCL, PLA, and PGA; we observed an average increase in the Young's modulus of over 140% compared to the graphenic backbone. These polyester-functionalized FGMs are a promising platform technology for tissue implants.
Introduction
Polycaprolactone (PCL), polylactic acid (PLA), and polyglycolic acid (PGA) have the approval of the Food and Drug Administration (FDA) for use in implants in humans1–3 and thus have been the subject of a wide range of research efforts in medicine. Decades of work in drug delivery and regenerative medicine have demonstrated the safety of these polyesters.4–11 Further, these polyesters are eliminated through natural pathways, with no reported side effects, suggesting that they are bioresorbable.6,12 However, their mechanical properties are inferior to those of metallic scaffolds, which limits their utility in tissue regenerative therapies.One application of the replacement of metal scaffolds with polyesters is in bone regeneration. Polyester-only screws have been FDA approved and can be used as fixation devices for bone injuries, but weak mechanical properties restrict implantation sites. The weaker properties are amplified when the rate of polymer degradation does not match that of bone regrowth, resulting in formation of a void. To address the limitations of polyesters, additives are a desirable option to enhance mechanical properties and tune degradation profiles to match those of tissue regeneration. One approach to overcome this mismatch is to include compounds found in bone naturally to stimulate bone growth, such as hydroxyapatite (HA).13 However, without appropriate pore size and extent of porosity, bone growth via HA may not be optimized in each implant.14 Furthermore, bone growth is favored in a porous structure of HA with limited interconnections, but porosity comes at the expense of mechanical strength.15
Inclusion of graphene oxide (GO) and functional graphenic material (FGM) additives into traditional scaffolds has been considered due the osteoinductive and mechanical properties of GO.16–19 These recent efforts include non-covalent composites of GO and polyesters, and these biomaterials have demonstrated utility as osteogenic scaffolds. However, improvements to the mechanical properties have been modest,20 and tunable degradation remains a challenge.21
Non-covalent interactions in these composites can limit mechanical properties since the polyester matrix cannot efficiently transfer stress to the reinforcing graphenic backbone.22 Furthermore, non-covalent composites may experience interfacial slip between the matrix and fiber, reducing the observed bulk mechanical properties.23 Non-covalent composites rely on van der Waals forces as well as hydrogen bonding between the polyesters and the carboxylic acid and hydroxyl functional groups on the GO surface to create adhesion, which are significantly weaker than covalent bonds.23,24 Consequently, the non-covalent addition of GO does not result in substantial improvements in mechanical properties and may even weaken the mechanical properties by as much as half.20
Rather than rely on non-covalent bonds, we use organic reactions and our graphene expertise to covalently attach clinically relevant polyesters to stiff, osteoinductive GO. We accomplish this by functionalizing and reducing GO to form Claisen graphene (CG).25 Alcohols on the basal plane of GO undergo acid-catalyzed conversion to methylene-spaced ethers, which are subsequently transformed into the carboxylic acids on CG via the Johnson–Claisen rearrangement.26,27 Covalent functionalization with polyesters is achieved by converting the basal-plane carboxylic acids of CG to acyl-chloride groups to create acyl chloride graphene (ACG). We then use ACG to end-cap controlled polyester polymerizations to give polyester-functionalized CG (Fig. 1).
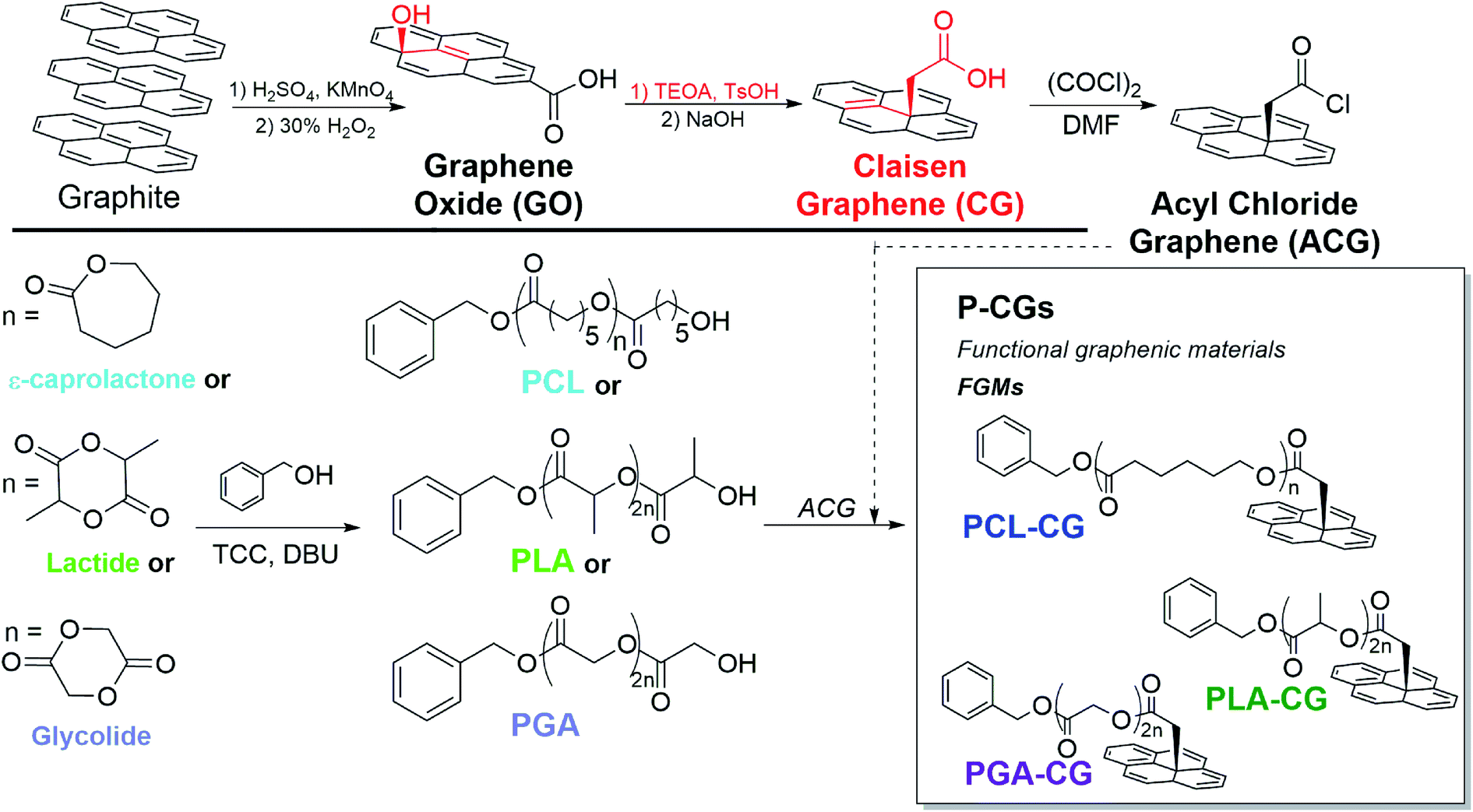 | ||
| Fig. 1 Progression of FGMs to achieve covalent polyester attachment. CG, with the basal methylene-bridged alcohols of GO oxidized to carboxylic acids, undergoes sequential conversion to ACG, replacing carboxylic acids with acyl chlorides (top). Polymerizations and sequential attachment of PCL, PLA, and PGA to ACG to yield P-CGs (bottom). Structural intricacies of the GO sheet have been simplified for clarity. | ||
With covalent functionalization, we retain the osteoinductivity of GO, fortify the mechanical properties compared to neat polyesters, and demonstrate tunable mechanical integrity of polyester-functionalized CG composites. This suggests that covalent attachment allows an effective transfer of mechanical stress between the graphenic backbone and the polymeric matrix. Furthermore, the FGM end-capped polymers may entangle more efficiently, providing another source of mechanical stability.28 This strategy could expand the utility of polyester-based hardware for application in load bearing biomedical implants.
Rationale behind material design
To create an ideal biodegradable implant, we considered two design criteria for our polyester-functionalized CG materials: the identities of the (1) polyester and (2) FGM.First, the polyester must be safely biodegradable with controlled mechanical properties, which we have achieved by synthesizing low molecular weight polyesters using organic catalysts.
Polyesters such as PCL, PLA, and PGA have many well-defined synthetic routes, and are also available for purchase from many chemical suppliers.1,29,30 We have elected to synthesize our polyesters for several reasons. When we construct the polyesters ourselves, we have precise control over the end groups, allowing modulation of ends groups as a future direction.31
We must also consider our target applications for P-CGs as a biotechnology when synthesizing our polyesters. Implants in the body that contain metals, such as inorganic catalysts, present challenges because metals can accumulate in body tissue.32 Abstaining completely from the use of metal catalysts supports the design for P-CGs as an implantable biotechnology. With purchased polymers, chemical suppliers may not disclose the catalyst(s) used, so the option of having an organic catalyst favors the synthesis of polyesters. Further, organic catalysts typically produce low molecular weight polymers, which were our target to increase end-capping efficiency with our FGM.33 In this report, we focus on one degree of polymerization for direct comparison between P-CGs, but by synthesizing the polymers, the possibility is available in the future for targeted molecular weights.
We end-cap the polymers onto the FGM as opposed to using grafting-from, grafting-to, or other grafting mechanisms. End-capping is functionalization of a living chain end before isolation and while the chain end is sterically accessible. This is contrast to grafting-to, which utilizes a dead polymer chain wherein reactive groups are potentially sterically inaccessible. Grafting-to strategy involves tethering a complete polymer to the graphenic surface, which adds unnecessary steps in the synthesis and decreases yields as the chain end is no longer accessible. Other similar grafting methods have emerged, but these require temperatures which would degrade polyesters.34 End-capping also offers advantages over grafting-from in control of molecular weight and dispersity of the polyesters, as polymers may freely grow in solution without being spatially fixed to a surface.
With the intention of end-capping the polymerization with an FGM, a suitable electrophile is necessary for the nucleophilic ring-opening polymerization, and acyl chloride functional groups are superior electrophiles. Acyl chloride groups could be attached to GO by treatment with a chlorinating agent such as oxalyl chloride.26 However, on GO, only edge-functionalization will be achieved.35,36
Edge-only functionalization limits the would-be resulting polyester-graphene oxide for several reasons. It is advantageous to convert GO to CG before treatment with a chlorinating agent since: (1) edge-only functionalization results in overall less polyester available, (2) basal-plane functionalization allows entanglement between adjacent polymers, which will likely reinforce the structure and strengthen P-CG mechanical properties,28 and (3) basal-plane functionalization includes a methylene bridge, which provides reduced steric effects to the FGM backbone, thus increasing functionality.
While edge versus basal plane functionalization is important to consider, it is also necessary to remark on oxidation level of the graphenic backbone in the FGM. When comparing the heavily oxidized backbone of GO and more reduced CG, reduced GO has been previously shown to have superior biocompatibility.37
Results and discussion
Analysis and characterization of polyesters
Polyesters (PCL, PLA, and PGA) were synthesized with low Ð and with targeted molecular weights via ring-opening polymerization.38 Molecular weights and dispersities were analyzed by GPC, NMR, and XPS and were found to be comparable (Table 1). PCL and PLA molecular weights were most reliably determined by THF GPC; however, the molecular weight of PGA was determined most accurately by 1H-NMR end-group analysis due to limited solubility in most solvents.| Đ | GPC | NMR | XPS | ||||
|---|---|---|---|---|---|---|---|
| Mn (kDa) | DP | Mn (kDa) | DP | Mn (kDa) | DP | ||
| PCL | 1.12 | 7.926 | 68 | 7.527 | 65 | 6.728 | 58 |
| PLA | 1.47 | 4.911 | 67 | 3.423 | 46 | 3.711 | 50 |
| PGA | — | — | — | 3.010 | 50 | 2.720 | 45 |
An organic coordination catalyst system was selected in order to avoid tin toxicity, while providing characteristics consistent with living polymerizations.38 To verify that the polymerization was controlled, PCL was observed by 1H-NMR to give a linear conversion to molecular weight (ESI Fig. S1†).
Analysis of covalent polyester attachment to Claisen graphene
Covalent functionalization of FGMs with controlled, synthetic polyesters was achieved by end-capping with an appropriate electrophilic FGM. We began by converting GO to Claisen graphene (CG) using the Johnson–Claisen rearrangement.26 CG is structurally similar to GO but is more reduced and also contains basal-plane, methylene-bridged carboxylic acids. These carboxylic acids were then transformed into more-reactive acyl chloride groups to create the electrophilic FGM, acyl chloride graphene (ACG). ACG was then added to quench the polymerization, resulting in FGMs with covalently attached polyesters (P-CGs, Fig. 1). P-CG conjugates were characterized by FTIR, TGA, and XPS.FTIR suggests covalent attachment by exhibiting peaks diagnostic of both polyesters and CG, even after exhaustive washing to remove unbound polyester. Methylene, carbonyl, and aromatic peaks may be monitored by FTIR to confirm the progression of functional groups from CG to PCL–CG (Fig. 2). CG contains mostly carboxylic acid functional groups confirmed by the broad peak (1) centered at 3282 cm−1 as well as the broad carbonyl stretch (2) at 1733 cm−1. PCL most notably contains a much sharper carbonyl stretch than CG (2) and no noticeable aromatic stretch (3). PCL–CG has the same major stretches as PCL and the appearance of stretches corresponding to the extra contribution from the FGM backbone (aromatic stretching (3) centered at 1575 cm−1, and greater CH stretches centered at approximately 3000 cm−1). Thus, FTIR shows PCL–CG contains functional groups from PCL and CG, suggesting successful synthesis of a covalent composite. PLA–CG and PGA–CG show similar peaks to those described for PCL–CG (ESI Fig. S2 and S3,† respectively).
 | ||
| Fig. 2 FTIR functional group comparison of polycaprolactone–Claisen graphene (PCL–CG) and components. | ||
Thermogravimetric analysis (TGA) thermograms of covalently bound P-CG materials clearly show a weight loss pattern that is distinct from non-covalent mixtures of CG with neat polyesters. These control experiments suggest covalent attachment and verify that the washing procedure is effective (ESI pp. S6–S8†).
TGA results are consistent with the observed XPS trend (discussed below) that functionalization efficiency of P-CG decreases with increasing persistence length and rigidity of the polyester backbone and may be used to make a prediction of functionalization efficiency. A higher persistence length, as defined by the average polymer chain length necessary to bend 90°, results in decreased rotational mobility.39 This appears to be related to reactivity. PCL, having the longest –CH2– chain in the repeat unit, allows the most vibrational degrees of freedom and thus gives the lowest persistence length. When comparing PGA and PLA, the difference may be found between the ester in the repeat unit. Since PGA simply has a methylene bridge, it has more vibrational degrees of freedom than the branched repeat unit of PLA. Thus, functionalization efficiency is most efficient in PCL–CG and least efficient for PLA–CG.
By examining the total weight percent degradation of the polymer vs. graphenic backbone, the approximate weight ratios are extracted (ESI Table S1†). This gives a good estimate of the weight percent of ACG necessary to quench each respective polymerization, which would otherwise be impossible to calculate without an exact known molecular weight of the ACG used.
PLA exhibits a lower attachment efficiency than PGA and PCL; this is likely due to a lower reactivity below the glass transition temperature for PLA (∼60 °C).40 Polymerizations occurred at 50 °C to reduce undesirable organic byproducts darkening the reaction mixture. The glass transitions of PCL (ESI Fig. S7†) and PGA are below 50 °C, allowing the polymers to be more flexible when ACG is added.
X-ray photoelectron spectroscopy (XPS) characterization
Materials were characterized by XPS using a new end-group analysis technique developed to complement 1H-NMR end-group analysis for polymers with limited solubility. This method relies on similar chemical principles to conventional end-group analysis by deconvoluting high resolution XPS spectra to isolate end-group carbons from backbone carbons. Using an appropriate system of equations based on the structure of the polymer, the degree of polymerization was determined (ESI eqn (S1)–(S5)†). The resulting molecular weights were comparable to 1H-NMR and GPC, verifying the efficacy of this method. This end-group analysis can be used even for polymers that do not contain unique atoms that are traditionally difficult to characterize by XPS. A full description of the method is available in the ESI.†End-group analysis by XPS revealed that functionalization efficiency of polyester functional graphenic materials decreases with increasing persistence length and rigidity of the polyester backbone. Using XPS, we were able to determine the percent of carboxylic acids on the functional graphenic material scaffolds that were utilized for end capping with the polyesters (Fig. 3 and ESI Fig. S8, and ESI Tables S2 and S3†). As the persistence length of the polymer increases, functionalization efficiency decreases.
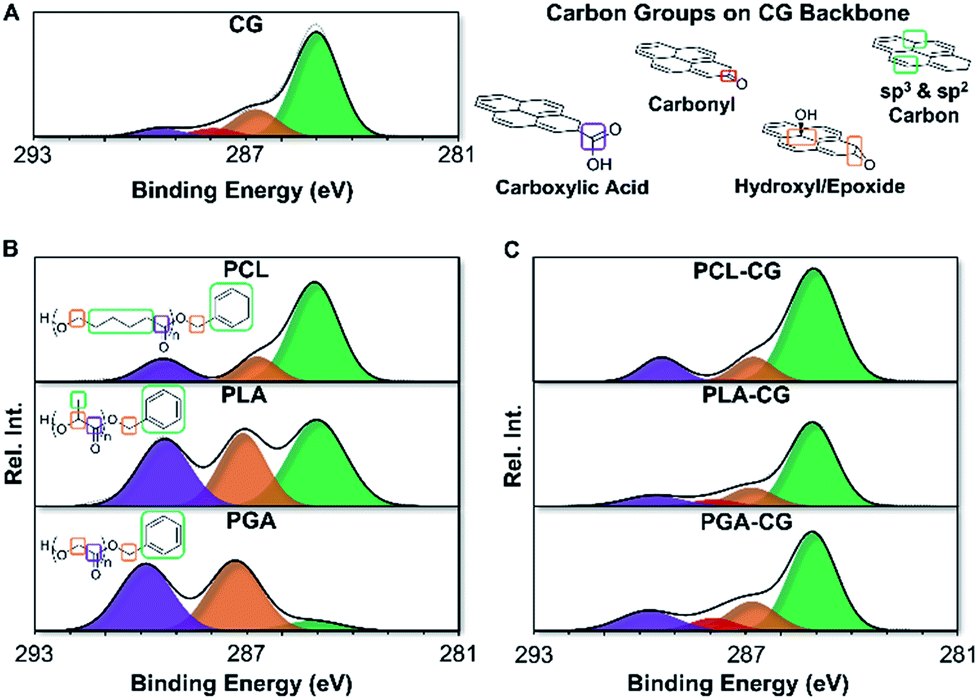 | ||
| Fig. 3 High resolution C 1s spectra obtained via X-ray photoelectron spectroscopy (XPS) of functional graphenic materials (FGMs), polyesters, and FGM-polyester composites. All the carbon groups in the representative structures are color coded to match the corresponding signal (peak) in the deconvoluted C 1s spectra. (A) deconvoluted C 1s spectra of Claisen graphene (CG) and structures demonstrating the chemically distinct carbon functional groups on the graphenic backbone. Note that the graphenic backbone is represented by a pyrene structure for simplicity. (B) deconvoluted C 1s spectra of polycaprolactone (PCL), polylactic acid (PLA), and polyglycolic acid (PGA). Chemically distinct carbon groups in the polymer backbone and chain ends are represented for each polymer. (C) deconvoluted C 1s spectra of polycaprolactone Claisen graphene (PCL–CG), polylactic acid Claisen graphene (PLA–CG), and polyglycolic acid Claisen graphene (PGA–CG). | ||
Cell vitality studies
NIH-3T3 fibroblasts and RAW 264.7 macrophages were exposed to P-CG materials for cell vitality studies. The motives for the selection of these cells are detailed in our previous reports on FGM cytocompatibility.41–45 In short, these cell lines mimic the environment experienced found following implantation of a synthetic implant. In general, when studied for cell vitality, higher concentrations give reduced vitality compared to the no treatment (NT) control, which is consistent with our prior FGM studies (Fig. 4A and B; fluorescence images 4C).42 This is due to the limited nutrient exchange of the cells by being covered by FGMs, rather than the effect of the materials themselves. Therefore, all P-CGs were found to be acceptably cytocompatible. This may also be attributed to the fact that no metal catalysts were used in production. | ||
| Fig. 4 Dose-dependent percent vitality ((A) top left, RAW 264.7 macrophages, and (B) bottom left, NIH-3T3 Fibroblasts) of P-CG materials compared to no treatment (NT) control after 48 h exposure. Error bars represent SEM of technical triplicates. (C) right, cell fluorescence overlay images of PLA-CG. Calcein AM (green) and Hoechst 33342 (blue) fluorophores used for assessing metabolic activity and as a DNA label, respectively. Propidium iodide (red) fluorophore labels the DNA of dying cells. Cell images for all polyester functional graphenic materials may be found in the ESI (Fig. S11†). | ||
The vitality of macrophages exposed to low (1 and 10 μg mL−1) PGA–CG is lower than expected. This may be explained by the decreased solubility of PGA. The minimal solubility makes separation of unreacted polymer extremely difficult. As a result, we found no effective and non-destructive means to separate unreacted PGA with PGA–CG; thus, nutrient exchange is limited further by the extra presence of material covering the cells.
Additional cell data quantifying cell counts and metabolism per cell may be found in the ESI (Fig. S9 and S10†).
Mechanical strength of P-CG scaffolds
We have developed a method to process powdered FGMs into three-dimensional scaffolds to test the mechanical properties of our materials (see Graphenic pellet processing in Methods). Our processing technique enables us to gauge the effect of covalent functionalization on the mechanical properties directly applicable to biomedical implants.Here, we tested our P-CG scaffolds using axial compressive and shear dynamic mechanical analysis (DMA), as well as compressive universal testing to failure to probe the strain at failure and stiffness. The mechanical testing was conducted in accordance with ASTM standards for bone regenerative engineering.46 ACG was excluded from measurement because the acyl chloride functional groups on ACG react with atmospheric water, thereby compromising the accuracy of the results.
In general, the mechanical properties of P-CGs exceeded that of precursor polyesters and met or exceeded that of precursor GO (Fig. 5). These enhanced properties illustrate the effectiveness of covalent functionalization in creating an avenue for strain transfer between the matrix and filler. The mechanical properties of PLA are reported in Fig. 5 as tensile. PLA is a soft, tacky solid, making measurements in compression difficult. While we saw enhanced properties compared with both our polyester and graphenic backbone precursors, it is important to comment on our material properties in comparison to the literature.
 | ||
| Fig. 5 (A) Compressive and shear storage moduli (E′ and G′, respectively), Young's modulus, and ultimate compressive strength (UCS) of all P-CG materials as well as precursors. *indicates tensile figures reported. (B) Comparative mechanical properties of polyester functional graphenic materials to Claisen graphene and graphene oxide. | ||
When comparing PCL and PLA to literature mechanical studies, our materials are weaker than the currently available PCL or PLA materials for biomedical applications, while the PGA homopolymer is not well documented.47,48 This is explained by molecular weights; our molecular weights are at least 10× less than polyesters synthesized with metal catalysts. While more work needs to be done to accomplish organically-produced polyesters with moduli in the GPa range to enable the full scope of biomedical applications, our materials may find applications in non-load bearing implants.
Mechanical integrity studies of PCL–CG, PLA–CG, and PGA–CG scaffolds in physiological conditions demonstrate that the mechanical tunability of our FGMs is dependent on the identity of the polyester. We monitored the mechanical properties of our scaffolds after exposure to water and phosphate buffered saline (PBS) at room temperature and 37 °C up to one month. The ultimate compressive strength of PCL–CG increased overtime in solutions, which is likely caused by the slow release of PCL, bringing the ultimate compressive strength closer to that of Claisen graphene (ESI Fig. S12†). The Young's modulus of PCL–CG and the ultimate compressive strength of PLA–CG each remain reasonably constant over a month in solution (ESI Fig. S13 and S14†). The Young's modulus of PLA–CG is somewhat depleted after a month in solution compared to initial moduli (ESI Fig. S15†). Fortunately, nearly all pressed pellets of PGA–CG dispersed after 24 h in any tested solution, which allows the opportunity for this material to be included in a short-term regeneration approach. These results suggest that our materials could be used to tailor treatment strategies for short- or long-term regeneration.
The mechanical integrity results support another facet of mechanical tunability based on the chosen polyester. PCL–CG shows a nominal increase in mechanical properties over time, PLA–CG remains acceptably constant, while PGA–CG loses integrity quickly. These mechanical results widen the scope of application of these materials further than originally anticipated. Longer regeneration strategies may call for materials containing PCL–CG, while short-term regeneration may call for materials such as PGA–CG.
Methods
Materials
Unless otherwise noted, the following reagents were prepared as described below. Acetone (reagent ACS/USP/NF grade, Pharmco, Brookfield, CT, USA) was dried under activated 3 Å sieves (Powder, Beantown Chemical, Hudson, NH, USA) before use. 3,4,4′-Trichlorocarbanilide (TCC, >98%, Tokyo Chemical Industry Co., Toshima, Kita-Ku, Tokyo, Japan) was recrystallized in dry acetone before use. Benzene (ACS 99.0%, Alfa Aesar, Ward Hill, MA, USA), benzyl alcohol (Alfa Aesar, Ward Hill, MA, USA), and ethyl acetate (GR ACS, EMD Milipore Corp., Billerica, MA, USA) were each dried under activated 4 Å sieves (Beads, Sigma-Aldrich, St. Louis, MO, USA) before use. Lactide (>98.0%, Tokyo Chemical Industry Co., Toshima, Kita-Ku, Tokyo, Japan) was recrystallized in dry ethyl acetate before use. All other materials were used as received.Example ring-opening polymerization using ε-caprolactone
PCL, PLA, and PGA were synthesized via a living ring-opening polymerization with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU, Alfa Aesar, Ward Hill, MA, USA) and 3,4,4′-trichlorocarbanilide (TCC) coordination co-catalysts.38 In a typical PCL preparation, recrystallized TCC (1.25 mol eq.) was dissolved in ε-caprolactone (100 mol eq.) (99%, Alfa Aesar, Ward Hill, MA, USA) and mixed with an equivalent volume of dry benzene. DBU (1.25 mol eq.) and dry benzyl alcohol (1 mol eq.) were combined separately and added together. All glassware was thoroughly dried by oven or flame prior to use and proper Schlenk technique with dry nitrogen was used in all steps. After stirring for 4–8 hours at 50 °C, the polymerization was quenched with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 w/w ACG:ε-caprolactone in situ in dry acetone. For example, if 10 g of ε-caprolactone was added for the polymerization, 1 g of ACG was used to quench. It is worth noting that addition of a non-nucleophilic base such as triethylamine does not significantly improve functionalization of CG. It is advantageous to have a lower relative amount of ACG compared to polymer, as purification after the reaction washes out extra polymer, but not unreacted ACG. The P-CG product was washed separately with each of hexane (Reagent Grade ACS, Pharmco-Aaper, Brookfield, CT, USA), methanol (Reagent ACS/USP/NF Grade, Pharmco, Brookfield, CT, USA), and THF (HPLC Grade, Meets ACS Spec, Fisher Chemical, Fair Lawn, NJ, USA) and separated by 3600 × g centrifugation for 5 min each. The supernatants were discarded, apart from THF, which then 10× v/v hexane was added to precipitate PCL for analysis. Example procedures for PLA and PGA may be found in the ESI (pp. S2 and S3†). Polymers were characterized by gel permeation chromatography (GPC), proton-nuclear magnetic resonance (1H-NMR) spectroscopy, and X-ray photoelectron spectroscopy (XPS). The polymerizations were monitored via 1H-NMR end group analysis. 1H-NMR end-group analysis is discussed in the ESI (Fig. S15–S17 and eqn (S6)–(S8)†).
:10 w/w ACG:ε-caprolactone in situ in dry acetone. For example, if 10 g of ε-caprolactone was added for the polymerization, 1 g of ACG was used to quench. It is worth noting that addition of a non-nucleophilic base such as triethylamine does not significantly improve functionalization of CG. It is advantageous to have a lower relative amount of ACG compared to polymer, as purification after the reaction washes out extra polymer, but not unreacted ACG. The P-CG product was washed separately with each of hexane (Reagent Grade ACS, Pharmco-Aaper, Brookfield, CT, USA), methanol (Reagent ACS/USP/NF Grade, Pharmco, Brookfield, CT, USA), and THF (HPLC Grade, Meets ACS Spec, Fisher Chemical, Fair Lawn, NJ, USA) and separated by 3600 × g centrifugation for 5 min each. The supernatants were discarded, apart from THF, which then 10× v/v hexane was added to precipitate PCL for analysis. Example procedures for PLA and PGA may be found in the ESI (pp. S2 and S3†). Polymers were characterized by gel permeation chromatography (GPC), proton-nuclear magnetic resonance (1H-NMR) spectroscopy, and X-ray photoelectron spectroscopy (XPS). The polymerizations were monitored via 1H-NMR end group analysis. 1H-NMR end-group analysis is discussed in the ESI (Fig. S15–S17 and eqn (S6)–(S8)†).
Graphenic pellet processing
All graphenic and polyester powders were dried for 48 h under vacuum prior to material processing. Powder (20 mg) was added to a custom, stainless-steel mold with an inner diameter of 3.75 mm at room temperature. The powder was pressed for 30 s on a Columbian D63 1/2 bench vise to approximately 1000 PSI and then removed from the mold. All pellets had an average diameter-to-thickness ratio of 3.Compressive universal testing
Data was acquired on an Instron 4469 with a load cell of 50 kN. Testing was carried out using strain rates of 0.001, 0.01, and 0.1 s−1 at room temperature until construct failure. Raw data was analyzed and corrected for instrument artifacts according to ASTM D695 (ref. 49) using Trios software (TA Instruments). Stress–strain curves were truncated at the ultimate stress point to eliminate artifacts from the universal testing geometries. The Young's moduli were determined at the onset of the linear region of the stress–strain curve.Gel permeation chromatography
GPC measurements were performed on a Waters Instrument equipped with a 717 plus autosampler, a Waters 2414 refractive index (RI) detector and two SDV columns (porosity 1000 and 100000 Å; polymer standard services) with THF as the eluent (flow rate 1 mL min−1, 40 °C). A 10-point calibration based on polystyrene standards (Polystyrene, ReadyCal Kit, Polymer Standard Services) was applied for determination of molecular weights.
1H-nuclear magnetic resonance spectroscopy (1H-NMR)
A 500 MHz NMR (Bruker AvanceTM 500) was used to acquire 1H-NMR spectra. Analysis was done on Mestrenova software.13C-nuclear magnetic resonance spectroscopy (13C-NMR)
A Bruker NEO NMR instrument equipped with a Prodigy Broadband Observe (BBO) cryoprobe operating at 125.73 MHz was used to acquire 13C-NMR data. Analysis was done on Mestrenova software.Dynamic mechanical analysis (DMA)
DMA was measured on a Discovery HR-2 Hybrid Rheometer (TA Instruments, New Castle, DE). Compressive DMA testing and torsional shear were performed at room temperature by applying a 0.1% strain at 1 Hz with a 1 N pre-force.Fourier transform infrared spectroscopy (FTIR) spectroscopy
A PerkinElmer Frontier FT-IR Spectrometer with an attenuated total reflectance attachment containing a germanium crystal was used to perform FTIR spectroscopy. Raw spectra were obtained over a range of 4000–700 cm−1 with 4 cm−1 resolution and analyzed on Spectrum software (PerkinElmer).Thermogravimetric analysis (TGA)
A PerkinElmer TGA 4000 was used to perform TGA under N2 from 50–800 °C with a ramp rate of 10 °C min−1. The raw data was analyzed using TRIOS software.Dynamic light scattering (DLS) and zeta potential
A Zetasizer Nano ZS (Malvern Instruments Ltd., Worcestershire, UK) was used to perform DLS and zeta potential measurements using Zetasizer Software v7.12 (Malvern, Inc.). Five DLS measurements of 100 μg mL−1 dispersions consisting of 10 scans of 10 s each were acquired in backscatter (173°) mode. The instrument automatically determined the best attenuation factor and measurement position. The mean count rate was ∼200 kcps.Dispersions of graphenic materials (100 μg mL−1) were loaded in Malvern disposable folded capillary cells (DTS1070) for zeta potential measurements. Five measurements were acquired using the optimal scanning parameters of the instrument (ranging from 10–100 scans per measurement).
Differential scanning calorimetry (DSC)
A PerkinElmer DSC 4000 equipped with a PerkinElmer Intracooler was used to perform DSC of PCL under N2 from −80 to 100 °C with a ramp rate of 10 °C min−1. The raw data was analyzed using TRIOS software.Lyophilization
A Labconco FreeZone 2.5+ Benchtop Freeze Dry System was used for lyophilization. Pressure and temperature parameters for sublimation were below 0.07 mBar and −80 °C, respectively.X-ray photoelectron spectroscopy (XPS)
All XPS data was acquired on a Thermo Fisher ESCALAB 250 Xi instrument with an Al K-alpha source gun and a flood gun in charge compensation standard mode. Spectra were obtained using a Constant Analyzer Energy (CAE) scan mode, a standard lens mode (angle and field of view of 32000 steps), and a 200 μm spot size. Peak fitting details are described in the ESI.†Cell culture
NIH-3T3 murine fibroblasts were cultured in complete cell culture media. The basal media was Dulbecco's Modified Eagle Medium having concentrations of 4500 mg L−1 of D-glucose, 584 mg L−1 of L-glutamine, and 100 mg L−1 of sodium pyruvate (Thermo Fisher Scientific, #11995065). The basal media was completed by adding calf serum (Thermo Fisher Scientific, #16010159) at a final concentration of 10% v/v and penicillin–streptomycin (Thermo Fisher Scientific, #15140122) at a final concentration of 100 U mL−1.RAW 264.7 murine macrophages were cultured in complete cell culture media. The basal media was Dulbecco's Modified Eagle Medium having concentrations of 4500 mg L−1 of D-glucose, 584 mg L−1 of L-glutamine, and 100 mg L−1 of sodium pyruvate (Thermo Fisher Scientific, #11995065). The basal media was completed by adding fetal bovine serum (Thermo Fisher Scientific, #26140079) at a final concentration of 10% v/v and penicillin–streptomycin (Thermo Fisher Scientific, #15140122) at a final concentration of 100 U mL−1.
Cells were cultured in tissue-culture, filter-cap, 25 cm2 flasks (Greiner Bio-One CELLSTAR®, #690175). The temperature was 37 °C, and the atmosphere was humidified and 5% CO2.
Cytocompatibility – experimental procedures
For cytocompatibility analysis, cells were seeded into tissue-culture, 96-well plates (Greiner Bio-One CELLSTAR®, #655180). To ensure accuracy, stock cell suspensions of NIH-3T3 fibroblasts and RAW 264.7 macrophages were added to the interior wells of 96-well plates at a density of at 3 × 104 and 2 × 104 cells per cm2. These cell lines were selected as they approximate the initial immune response encountered by a foreign body. This is expected for a surgical implant, and these cell lines can serve as an initial cytocompatibility study for use in a biodegradable scaffold.Before exposure to cells, powders of functionalized graphenic materials were weighed out on an analytical balance into microcentrifuge tubes. The powders were sterilized by exposure to the 254 nm ultraviolet light of the biosafety cabinet for 10 min. Then, the powders were diluted to 1 mg mL−1 in complete cell culture media. To disrupt/break up large flocculants, the mixtures were bath sonicated (240 W, 42 kHz, ultrasonic cleaner, Kendal) for ∼10 s to destabilize the flocs. Then, the remaining large agglomerates were aseptically vigorously pipetted. Finally, the samples were again subjected to ∼10 s of bath sonication. Ultimately, this resulted in dispersed functional graphenic materials.
To accurately expose cells to smaller concentrations, 10-fold serial dilutions were performed. Briefly, an aliquot of the stock suspension of 1 mg mL−1 was diluted in fresh, complete cell culture media at 1:9 v/v, resulting in a stock solution of 0.1 mg mL−1. This process was repeated to generate 0.01 mg mL−1. By design, each stock solution was of large enough volume to be used for all the technical replicate per cell line.
2 h after seeding, the cells were well adhered, and then they were exposed to functional graphenic materials. The cell media was aspirated from the adhered cells and replaced with 200 μL of functional graphenic material-containing complete cell culture media, as indicated. Also “no treatment” control samples were prepared by adding fresh media without any functional graphenic materials.
After 48 h of exposure to the functional graphenic materials, the cellular enumeration, metabolism, and late apoptosis/necrosis assays were performed. To do so, a staining solution was prepared that contained Hoechst 33342 at 20 μM (ThermoFisher Scientific, #62249), Calcein AM at 2.5 μM (PromoKine, #PK-CA707-80011-2), and propidium iodide at 2 μg mL−1 (Alfa Aesar, #J66584) in phosphate buffered saline (PBS, Thermo Fisher Scientific, #10010049). Hoechst 33342 labels the DNA of all cells, enabling quantification of cell number, which reflects proliferation, through the fluorescence labeling of the nuclei. Upon intracellular esterase conversion, Calcein AM becomes brightly fluorescent, enabling a measure of cellular metabolism. Propidium iodide is excluded from the nucleus of live, healthy cells; however, late apoptotic and necrotic cells whose membranes are compromised enable propidium iodide to infiltrate their nuclei and label the DNA within. These fluorescently labeled nuclei report the number of dying cells.
For the cytocompatibility assay, the cell media was aspirated, and the cells were exposed to the staining solution for 10 min in the incubator. Then, the staining solution was aspirated, and the cells were maintained in fresh PBS for the duration of the experiment. To quantify the fluorescence intensity of these fluorescent reporters, we used a fluorescence microplate reader with excitations of 350/20 nm, 485/15 nm, and 530/20 nm and emissions of 450/20 nm, 525/15 nm, and 620/20 nm for Hoechst 33342, Calcein AM, and propidium iodide, respectively. Further, absorbance spectra were acquired from 350 to 700 nm, with a step size of 10 nm.
Since graphenic materials may alter fluorescence assays and quench intensity, we also performed direct fluorescence imaging using an EVOS® FL Auto Cell Imaging System (Thermo Fisher Scientific, #AMAFD1000) with a 10×, 0.30 numerical aperture objective (Thermo Fisher Scientific, #AMEP4681) and DAPI (Ex: 357/44 nm, Em: 447/60 nm; Thermo Fisher Scientific, #AMEP4650), GFP (Ex: 470/22 nm, Em: 510/42 nm; Thermo Fisher Scientific, #AMEP4651), and RFP (Ex: 531/40 nm, Em: 593/40 nm; Thermo Fisher Scientific, #AMEP4652) light cubes. Fluorescence and phase contrast images were acquired at the same imaging parameters for all samples for each cell line.
Conclusions and future directions
A synthesis was devised to covalently functionalize graphene oxide with biocompatible polyesters to develop a new type of scaffold for biomedical applications. Our synthetic procedures optimize the materials used to match the attachment efficiencies. Polymer characterizations are consistent across FTIR, TGA, and XPS. Novel analysis with high-resolution XPS was developed to determine degree of polymerization even for insoluble materials. These results were consistent with GPC and NMR end-group analysis, verifying the efficacy of this new XPS end-group analysis technique.Polyester functional graphenic materials were shown to be biocompatible, suggesting our materials hold promise as clinical scaffolds. The biocompatibility is supported further as no metal catalysts were used. The ultimate compressive strengths and Young's moduli were improved for every polyester functional graphenic material compared to the corresponding polyester. Therefore, with enhanced mechanical properties compared to neat polyesters, this technology expands the scope of injuries that could be treated with polyester hardware.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the following Carnegie Mellon University laboratories: the Noonan lab for use of the gel permeation chromatograph, the Washburn lab for use of the lyophilizer, the Matyjaszewski lab for use of the Zetasizer, the Whitehead lab for the NIH-3T3 and RAW 264.7 cells, and the Material Science and Engineering lab for compressive testing. NMR instrumentation at Carnegie Mellon University was partially supported by the NSF (CHE-0130903, CHE-1039870 and CHE-1726525). We thank J. Gillespie for providing training and use of the XPS (Materials Characterization Laboratory at the University of Pittsburgh). The authors would like to thank Wojdyr et al. for the use of Fityk, an open-source software for XPS analysis.50References
- B. Azimi, P. Nourpanah, M. Rabiee and S. Arbab, J. Eng. Fibers Fabr., 2014, 9, 17 Search PubMed.
- B. Tyler, D. Gullotti, A. Mangraviti, T. Utsuki and H. Brem, Adv. Drug Delivery Rev., 2016, 107, 163–175 CrossRef CAS PubMed.
- Poly(Lactic Acid): Synthesis, Structures, Properties, Processing, and Applications, ed. R. Auras, L.-T. Lim, S. E. M. Selke and H. Tsuji, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010 Search PubMed.
- C. M. Agrawal, G. G. Niederauer and K. A. Athanasiou, Tissue Eng., 1995, 1, 241–252 CrossRef CAS PubMed.
- E. Malikmammadov, T. E. Tanir, A. Kiziltay, V. Hasirci and N. Hasirci, J. Biomater. Sci., Polym. Ed., 2018, 29, 863–893 CrossRef CAS PubMed.
- M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- J. V. Andhariya, R. Jog, J. Shen, S. Choi, Y. Wang, Y. Zou and D. J. Burgess, J. Controlled Release, 2019, 308, 1–13 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- K. Garvin and C. Feschuk, Clin. Orthop. Relat. Res., 2005, NA, 105–110 CrossRef PubMed.
- V. Pavot, M. Berthet, J. Rességuier, S. Legaz, N. Handké, S. C. Gilbert, S. Paul and B. Verrier, Nanomedicine, 2014, 9, 2703–2718 CrossRef CAS PubMed.
- N. Ashammakhi and P. Rokkanen, Biomaterials, 1997, 18, 3–9 CrossRef CAS.
- S. Park, J. H. Kim, I. H. Kim, M. Lee, S. Heo, H. Kim, E. H. Kim, Y. B. Choy and C. Y. Heo, J. Craniofac. Surg., 2013, 24, 1021–1025 CrossRef PubMed.
- T. C. Schumacher, K. Tushtev, U. Wagner, C. Becker, M. große Holthaus, S. B. Hein, J. Haack, C. Heiss, M. Engelhardt, T. El Khassawna and K. Rezwan, The Knee, 2017, 24, 933–939 CrossRef.
- K. A. Hing, Int. J. Appl. Ceram. Technol., 2005, 2, 184–199 CrossRef CAS.
- S. Li, J. R. de Wijn, J. Li, P. Layrolle and K. de Groot, Tissue Eng., 2003, 9, 535–548 CrossRef CAS PubMed.
- S. Duan, X. Yang, F. Mei, Y. Tang, X. Li, Y. Shi, J. Mao, H. Zhang and Q. Cai, J. Biomed. Mater. Res., Part A, 2015, 103, 1424–1435 CrossRef PubMed.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. Y. Lee, Z. Khatun, J.-H. Lee, Y. Lee and I. In, Biomacromolecules, 2011, 12, 336–341 CrossRef CAS PubMed.
- W. C. Lee, C. H. Y. X. Lim, H. Shi, L. A. L. Tang, Y. Wang, C. T. Lim and K. P. Loh, ACS Nano, 2011, 5, 7334–7341 CrossRef CAS PubMed.
- Y. Luo, H. Shen, Y. Fang, Y. Cao, J. Huang, M. Zhang, J. Dai, X. Shi and Z. Zhang, ACS Appl. Mater. Interfaces, 2015, 7, 6331–6339 CrossRef CAS PubMed.
- S. Kumar and K. Chatterjee, ACS Appl. Mater. Interfaces, 2016, 8, 26431–26457 CrossRef CAS PubMed.
- A. E. Jakus, E. B. Secor, A. L. Rutz, S. W. Jordan, M. C. Hersam and R. N. Shah, ACS Nano, 2015, 9, 4636–4648 CrossRef CAS PubMed.
- S. A. Sydlik, J.-H. Lee, J. J. Walish, E. L. Thomas and T. M. Swager, Carbon, 2013, 59, 109–120 CrossRef CAS.
- O. J. Yoon, C. Y. Jung, I. Y. Sohn, H. J. Kim, B. Hong, M. S. Jhon and N.-E. Lee, Composites, Part A, 2011, 42, 1978–1984 CrossRef.
- Graphene oxide: fundamentals and applications, ed. S. Eigler, Wiley, Chichester, West Sussex, United Kingdom, 2017 Search PubMed.
- S. A. Sydlik and T. M. Swager, Adv. Funct. Mater., 2013, 23, 1873–1882 CrossRef CAS.
- L. Kürti and B. Czakó, Strategic applications of named reactions in organic synthesis: background and detailed mechanisms, Elsevier Academic Press, Amsterdam; Boston, 2005 Search PubMed.
- J. T. Seitz, J. Appl. Polym. Sci., 1993, 49, 1331–1351 CrossRef CAS.
- C. L. Sungyeap Hong, Mod. Chem. Appl., 2014, 2 DOI:10.4172/2329-6798.1000144.
- M. Baghaban-Eslaminejad, A. Oryan, A. Kamali and A. Moshiri, in Nanostructures for Oral Medicine, Elsevier, 2017, pp. 777–832 Search PubMed.
- D. Pholharn, Y. Srithep and J. Morris, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 213, 012022 Search PubMed.
- I. Gotman, J. Endourol., 1997, 11, 383–389 CrossRef CAS PubMed.
- N. Rubio, H. Au, H. S. Leese, S. Hu, A. J. Clancy and M. S. P. Shaffer, Macromolecules, 2017, 50, 7070–7079 CrossRef CAS.
- Z. Xu and C. Gao, Macromolecules, 2010, 43, 6716–6723 CrossRef CAS.
- Z. M. Wright, A. M. Arnold, B. D. Holt, K. E. Eckhart and S. A. Sydlik, Regener. Eng. Transl. Med., 2019, 5, 190–209 CrossRef CAS.
- G. Shao, Y. Lu, F. Wu, C. Yang, F. Zeng and Q. Wu, J. Mater. Sci., 2012, 47, 4400–4409 CrossRef CAS.
- S. A. Sydlik, S. Jhunjhunwala, M. J. Webber, D. G. Anderson and R. Langer, ACS Nano, 2015, 9, 3866–3874 CrossRef CAS PubMed.
- N. U. Dharmaratne, J. U. Pothupitiya, T. J. Bannin, O. I. Kazakov and M. K. Kiesewetter, ACS Macro Lett., 2017, 6, 421–425 CrossRef CAS.
- P. C. Hiemenz and T. Lodge, Polymer chemistry, CRC Press, Boca Raton, 2nd edn, 2007 Search PubMed.
- A. Södergård and M. Stolt, Prog. Polym. Sci., 2002, 27, 1123–1163 CrossRef.
- B. D. Holt, A. M. Arnold and S. A. Sydlik, Adv. Healthcare Mater., 2016, 5, 3056–3066 CrossRef CAS PubMed.
- A. M. Arnold, B. D. Holt, L. Daneshmandi, C. T. Laurencin and S. A. Sydlik, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4855–4860 CrossRef CAS PubMed.
- K. E. Eckhart, B. D. Holt, M. G. Laurencin and S. A. Sydlik, Biomater. Sci., 2019, 7, 3876–3885 RSC.
- Z. M. Wright, B. D. Holt and S. A. Sydlik, J. Mater. Chem. B, 2017, 5, 7743–7755 RSC.
- B. D. Holt, A. M. Arnold and S. A. Sydlik, Polym. Int., 2017, 66, 1190–1198 CrossRef CAS.
- Bone graft substitutes and bone regenerative engineering, ed. C. T. Laurencin and T. Jiang, ASTM International, 2014 Search PubMed.
- S. Eshraghi and S. Das, Acta Biomater., 2010, 6, 2467–2476 CrossRef CAS PubMed.
- T. Kasuga, Y. Ota, M. Nogami and Y. Abe, Biomaterials, 2000, 22, 19–23 CrossRef.
- Committee D20, Test Method for Compressive Properties of Rigid Plastics, ASTM International, 2015 Search PubMed.
- M. Wojdyr, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10646d |
| This journal is © The Royal Society of Chemistry 2020 |