
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelection and application of aptamers with high-affinity and high-specificity against dinophysistoxin-1†
Zhen Li‡
 a,
Bo Hu‡ab,
Rong Zhou‡a,
Xiaojuan Zhang‡ad,
Ruizhe Wangc,
Yun Gaoa,
Mingjuan Suna,
Binghua Jiao*ab and
Lianghua Wang*a
a,
Bo Hu‡ab,
Rong Zhou‡a,
Xiaojuan Zhang‡ad,
Ruizhe Wangc,
Yun Gaoa,
Mingjuan Suna,
Binghua Jiao*ab and
Lianghua Wang*a
aDepartment of Biochemistry and Molecular Biology, College of Basic Medical Sciences, Navy Medical University, Shanghai 200433, P. R. China. E-mail: bhjiao@smmu.edu.cn; lhwang@smmu.edu.cn
bDepartment of Marine Biomedicine and Polar Medicine, Naval Special Medical Center, Shanghai 200433, P. R. China
cSpine Center, Department of Orthopedics, Changzheng Hospital Affiliated to Second Military Medical University, 415th Feng Yang Road, Shanghai, 200003, P. R. China
dCollege of Medicine, Shaoxing University, 900th Chengnan Avenue, Shaoxing, Zhejiang Province 312000, P. R. China
First published on 26th February 2020
Abstract
Diarrhetic shellfish toxins (DSTs) are marine toxins distributed widely in the world, which pose a major threat to the health of mankind. Dinophysistoxin-1 (DTX-1) has the most potent toxicity in DSTs. However, the current detection methods have ethical problems and technical defects. Further research is needed, to develop a more suitable alternative to the supervision system. In this work, we successfully obtained an aptamer with high affinity and specificity bound to DTX-1 for the first time. After optimization, a core sequence of the aptamer with a higher KD of 64 nM was obtained, while the binding mode of the core sequence and DTX-1 was explored. Based on this aptamer, we developed a biolayer interferometry (BLI) biosensor platform for DTX-1 detection. The aptasensor exhibited a broad detection range from 40 to 600 nM DTX-1 (linear range from 80 to 200 nM), and the low detection limit was 614 pM. Morever, the aptasensor showed good reproducibility and stability, which indicated that this novel aptasensor had broad development prospects for the sensitive and rapid detection of DTX-1.
Introduction
With the first recognition of diarrhetic shellfish poisoning (DSP) in Japan, DSP has been considered a global public health problem. It is a gastrointestinal illness caused by the consumption of shellfish contaminated with diarrhetic shellfish toxins (DSTs).1 Traditionally, DSP has mainly occurred only in several coastal areas, such as Western Europe, Eastern Japan and Latin America.2,3 However, recent studies indicate that DSP is emerging in more regions in the world.4–6 The emergence of DSP also leads to the downturn of the shellfish industry, and the length of time is unexpected.7 Not only does it have a serious economic impact, but it also poses a great threat to human health. The major toxins causing severe diarrhea are okadaic acid (OA) and its analogues, dinophysistoxin-1 (DTX-1) and dinophysistoxin-2 (DTX-2), which are produced by marine dinoflagellates. These toxins have been testified to be potent inhibitors of several phosphatases, especially inhibitors of PP1 and PP2A.8,9 They are possibly carcinogenic,10 with symptoms such as diarrhea, nausea, and vomiting.11 They can also act on other protein phosphatases, including protein phosphatase 4 (PP4), protein phosphatase 5 (PP5) and protein phosphatase 2B (PP2B).12,13 In the early stage, OA has been thoroughly studied. In recent years, DTX-1 was found to be more toxic,14 especially with respect to oral toxicity.15 DTX-1 can destroy the integrity of epithelial cells, and play an important role in apoptosis,16 which induces the same cytotoxic effects with 1/5 concentrations of OA.17 While the effect of DTX-1 on pro-inflammatory and carcinogenicity of macrophages is ten times more than that of OA.16 Studies have shown that the deposition rate of DTX-1 in the shellfish digestive gland is higher than that of OA.18However, DTX-1 cannot be detected effectively. Although DSP was detected by the mouse bioassay (MBA) in early 1980s, it required 24–48 hours of observation on experimental animals.19 This method also lacks of specificity and sensitivity. Since 1997, LC-MS has already replaced MBA.20 However, this method requires the Temperature Equivalent Factor (TEF) of each compound to estimate the sample's total toxicity, in order to determine whether the regulatory limit has been exceeded,19,21 which is time-consuming and expensive. Another method is to use, enzyme linked immunosorbent assay (ELISA). But it has severe cross-reactivity, because the antibodies are mainly directed against inhibitors of PP.22
In summary, there is still a lack of a sensitive, reliable, real-time and inexpensive monitoring system for detecting DTX-1. The biosensors are ideal detection alternatives, which can avoid most of the shortcomings the other methods have. The development of a stable, high-sensitivity, inexpensive probe is an important basis for the preparation of biosensors.
Aptamers are functional single-stranded DNA (ssDNA) or RNA sequence that fold into complex tertiary structures and bind to specific molecular targets in a manner similar to that of antibodies.23,24 Thus, aptamers are called “chemical antibodies”.25 The vast majority of aptamers are obtained from the Systematic Evolution of Ligands by Exponential Enrichment (SELEX) method in vitro.26 Aptamers have a wide range of targets, including metal ions, compounds, proteins, cells, and even whole microorganisms, and can be easily modified by various functional tags to induce immobilization onto many surfaces.27 As a potential existence that can replace antibodies, aptamers have the advantages of high affinity, no batch to batch variations, chemical stability, low immunogenicity, and permissible modification.28,29 These have led to significant development in the field of environmental screening,30 therapeutics,31 drug delivery.32 However, one great potential of aptamers is that these nucleic acid sequences can be at the heart of some emerging devices, such as sensors and actuators.27 In recent years, a number of studies have shown that aptamers can be used as probes in combination with other techniques (e.g. surface enhanced Raman spectroscopy, quartz crystal microbalance, electrochemical methods, colorimetry, fluorescent modification and biolayer interferometry, etc.) to produce highly efficient aptamer-based sensors.23,33–38 Among them, biolayer interferometry (BLI) technology has a unique advantage as a new type of optical sensor, which is of label-free, highly specificity, real-time and inexpensive.38,39 These makes the method a suitable choice for on-site rapid detection and analysis of the marine biotoxin DTX-1.
Experimental
Materials and reagents
All nucleic acid sequences (Table S1†) were bespoke-synthesized by Sangon Biotech Co. Ltd (Shanghai, China). Dinophysistoxin-1 (DTX-1), okadaic acid (OA), nodularin-R (NOD-R), palytoxin (PTX), gonyautoxin (GTX), and saxitoxin (STX) were obtained from Taiwan Algal Science, Inc. (Taiwan, China). Dynabeads® M-270 Amine and the Qubit® ssDNA Assay Kit were purchased from Thermo Fisher Scientific (Massachusetts, USA). GoTaqHot® Start Colorless Master Mix was procured from the Promega Corporation (Wisconsin, USA). The QIAEX® IIGel Extraction Kit was purchased from Qiagen (Frankfurt, Germany). The Okadaic Acid Plate Kit purchased from Beacon Analytical Systems, Inc. (Maine, USA). 2-(N-Morpholino)-ethanesulfonic acid (MES), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), and the other chemical agents were purchased from Sigma-Aldrich Co. LLC (Shanghai, China). Oxalic acid (ultra-pure grade) was procured from BBI Life Sciences Corporation (Shanghai, China). Urea (ultra-pure grade) was procured from Amresco, Inc. (Ohio, USA). The 20 bp DNA Ladder (Dye Plus) was purchased from TaKaRa Bio, Inc. (Dalian, China). Bio Gel P-2 was procured from Bio-Rad (Hercules, USA). The BLI sensor chips were obtained from Forte Bio (Shanghai, China). DNA Urea-PAGE buffer, DNA PAGE buffer and binding buffer (pH 7.5, 50 mM Tris–HCl, 150 mM NaCl, 2 mM MgCl2) were procured from Tiandz (Beijing, China). All solutions were prepared using Milli-Q ultrapure water.Preparation of GTX1/4 magnetic beads
(1) Pre-treatment of magnetic beads: the magnetic beads were placed in an end-over-end rotation and mixed for 30 minutes. 600 μL of magnetic beads were taken therefrom and washed 4 times with 25 mM MES buffer (pH 5.0). Freshly prepared 500 μL EDC solution (50 mg mL−1) and 500 μL NHS solution (50 mg mL−1) were added to the magnetic beads and incubated for 30 min with rotation. (2) Incubation of magnetic beads with DTX-1: 150 μL DTX-1 solution (100 μg mL−1) was added to the above treated magnetic beads, and incubated at room temperature for 2 h. After the incubation, the beads were washed twice with 25 mM MES buffer and then washed 3 times with DTX-1 binding buffer (pH 7.5, 50 mM Tris–HCl, 150 mM NaCl, 2 mM MgCl2). (3) Preparation negative magnetic beads: 200 μL of magnetic beads were taken for pre-treatment in (1). They were then washed twice with 25 mM MES buffer and then washed 3 times with DTX-1 screening buffer (pH 7.5, 50 mM Tris–HCl, 150 mM NaCl, 2 mM MgCl2). (4) Magnetic beads blocking: the newly prepared EDC solution, NHS solution and 0.5 M oxalic acid solution were mixed at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. Two thousand and five hundred μL of a mixture of EDC, NHS, and oxalic acid was added to 600 μL of the positive selection magnetic beads; 750 μL of EDC, NHS, and oxalic acid mixed solution was added to 200 μL of the counter selection magnetic beads. The two magnetic beads were incubated for 30 min at room temperature, then washed 3 times with DTX-1 binding buffer, respectively. Six hundred μL and 200 μL of DTX-1 binding buffer were added and placed at 4 °C until use.
:1:1. Two thousand and five hundred μL of a mixture of EDC, NHS, and oxalic acid was added to 600 μL of the positive selection magnetic beads; 750 μL of EDC, NHS, and oxalic acid mixed solution was added to 200 μL of the counter selection magnetic beads. The two magnetic beads were incubated for 30 min at room temperature, then washed 3 times with DTX-1 binding buffer, respectively. Six hundred μL and 200 μL of DTX-1 binding buffer were added and placed at 4 °C until use.
In vitro selection of the DTX-1 aptamer
This selection uses Magnetic Bead SELEX, the specific selection strategy is shown in the Fig. 1. Appropriate amount of ssDNA (as shown in the selection scheme) was subjected to denaturation and renaturation treatment in a microcentrifuge tube (95 °C water bath for 10 min, ice bath for 5 min, room temperature for 5 min). The prepared magnetic beads were mixed with ssDNA, and the system was supplemented with a binding buffer to 600 μL and incubated at room temperature. After the incubation, rinse with binding buffer several times until the concentration of ssDNA in the supernatant could not be detected. 80 μL of ddH2O was added to the magnetic beads bound to the specific ssDNA, and the mixture was centrifuged at 95 °C for 20 min. The magnetic frame was magnetically adsorbed for 4 min to recover the ssDNA in supernatant, which was repeated 3 times. The recovered ssDNA was assayed by using Qubit® 2.0, and the recovery efficiency was calculated. The recovery library was amplified by PCR, and the amplification system was 50 μL, 40 tubes each time. The specific parameters were set as follows: 95 °C for 5 min, followed by 20 cycles of 95 °C for 30 s, 54 °C for 45 s, 72 °C for 30 s, and a final elongation step of 5 min at 72 °C. The dsDNA obtained by PCR was prepared by urea-denaturing polyacrylamide gel electrophoresis, and the length of the two strand primers was different, so that it could be easily separated during electrophoresis. Recover ssDNA from the gel excising from polyacrylamide (PAGE) gel electrophoresis selective.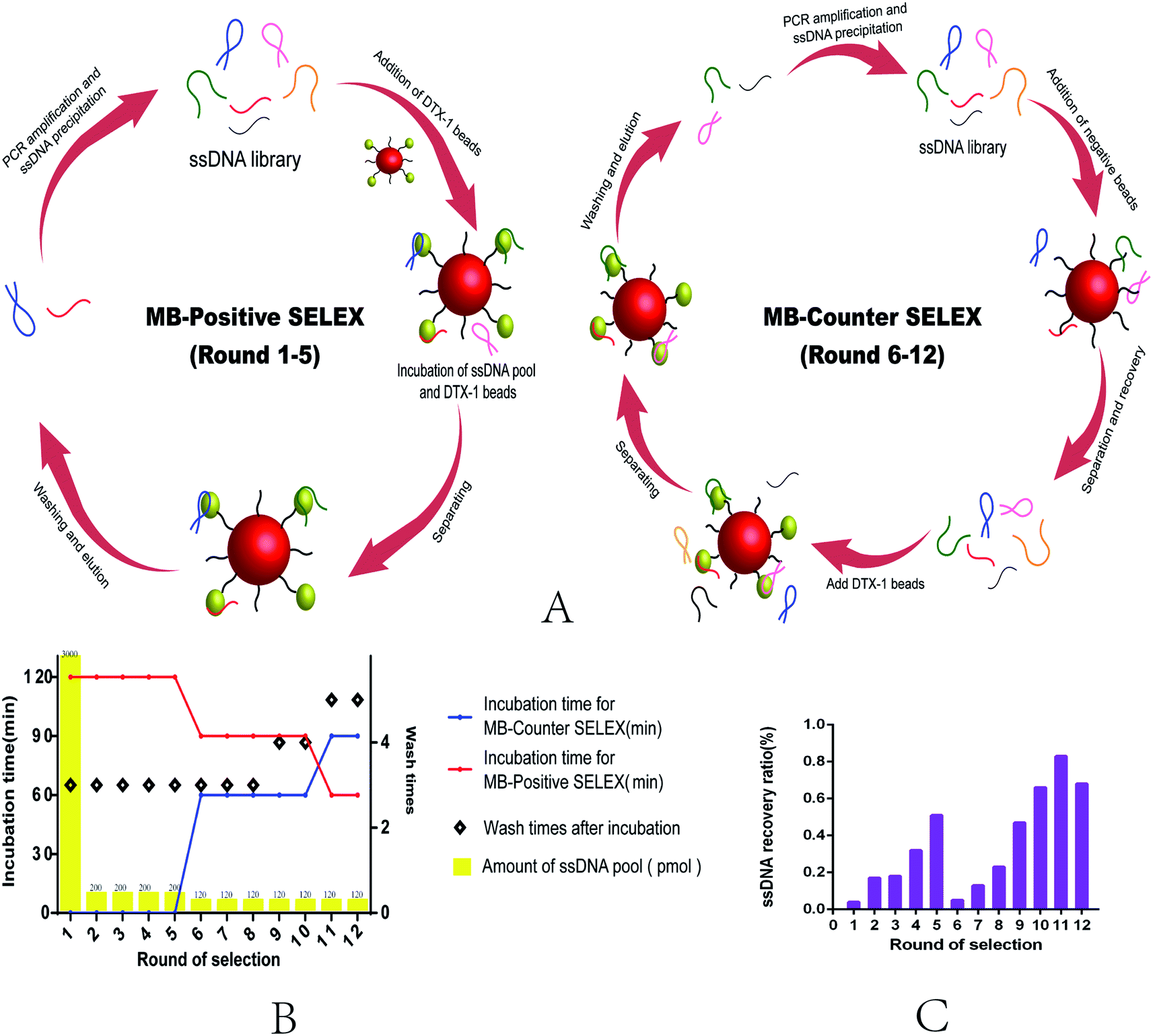 | ||
| Fig. 1 (A) Aptamer selection process: MB-positive SELEX (left) and MB-counter SELEX (right). (B) Summary of selection protocol for MB-SELEX. (C) Recovery ratio of ssDNA during MB-SELEX. The recovery efficiency is the ratio of the recovered amount of ssDNA to the added amount of ssDNA per round. | ||
The preparation of ssDNA
The ssDNA obtained from gel was purified and recovered. All procedures were strictly in accordance with the protocol of a gel recovery kit. (1) Added the same volume of diffusion buffer to the gel. Then incubated at 50 °C for 30 min. Centrifuged the sample for 1 min. (2) Pipetted the supernatant to a clean EP tube and added 6 volumes of Buffer QX1 to 1 volume of sample, then added 10 μL of QIAEX II and mix. (3) Incubated the mixture at room temperature for 10 min (kept QIAEX II in suspension). (4) Centrifuged and removed supernatant and washed the pellet twice with Buffer PE. (5) Air-dried the pellet until the pellet became white. (6) Added 20 μL of H2O and incubated for 5 min at room temperature. (7) Centrifuged and transfered the supernatant into a clean tube. (8) Repeated steps (6) and (7) and combined the eluates. Then the ssDNA was quantified and used in the next round of selection.Cloning and sequencing of selected DNA
When the recovery rate no longer rises, it is considered that the selection platform is reached and then the selection is stopped. The last round of ssDNA was taken for PCR amplification using normal primers. The amplified product was purified using a gel recovery kit and the dsDNA was sent to Sangon Biotech Co. Ltd (Shanghai, China) for cloning and sequencing.Determination of affinity and specificity of aptamer by BLI
The affinity and specificity of aptamer N59a were identified by BLI using an OctetRED 96 system (ForteBio, Shanghai). The operating principle and analysis procedure of the specific BLI has been clarified.40 The assay procedure covered five steps: baseline (2 min), loading (2 min), washing (3 min), association (5 min), and dissociation (5 min). The baseline solution (200 μL, pH 7.5, 50 mM Tris–HCl, 150 mM NaCl, 2 mM MgCl2), loading solution (i.e. baseline solution), washing solution (i.e. baseline solution), association solution (i.e. baseline solution) and dissociation solution (i.e. baseline solution) were added, respectively, into the corresponding wells of a 96-well microtiter plate. Considering the impact of non-specific binding and buffer-induced interferometry spectrum shifts, the response data obtained from the reaction surface were normalized by subtracting the signal simultaneously acquired from the control surface by the Octet Data Analysis Software CFR Part 11 Version 6.x. The affinity parameter KD was obtained in this way, a 1:1 binding mode with mass transfer fitting was used to obtain the kinetic data.
Results and discussion
In vitro selection of DTX-1 aptamer
The process of MB-SELEX includes forward selection and reverse selection (Fig. 1A). The detailed process strictly follows Fig. 1B (Table S2†). In order to improve the efficiency of SELEX, we adjusted to the incubation time and the number of elution times during the process, gradually increasing the selection pressure.41 From the sixth round, we introduced the negative magnetic beads to eliminate non-specific adsorption of ssDNA by magnetic beads. After 12 rounds of selection, the recovery rate of ssDNA reached a stable level, and this indicated that the selection terminal point was reached.24,26 The selection cycles were stopped (Fig. 1C).The sequences of final round were sent to sequence, 80 clones were opted randomly, and 80 sequences were obtained. We classified these sequences into 10 families (A–J) based on multiple sequence alignments (Fig. S3A†). Then, we choose the sequences with the highest homology or the minimum free energy by the mfold Web Server (http://unafold.rna.albany.edu/?q=mfold/DNA-Folding-Form) and the Guide trees (Fig. S3B†) for further binding affinity studies with DTX-1, as shown in Table 1. From the ten selected sequences, we found that seven of the sequences showed binding affinity for DTX-1 ranging from 0.17–1890 μM,42 while the three sequences showed no binding.
| Clone no. | Family | Aptamer sequence | KDa (μM) |
|---|---|---|---|
| a NB: no binding. | |||
| N11 | A | GAGGCAGCACTTCACACGATCGGACCCAAACTTTACCTTACCCCCATCTGCGTAATGACTGTAGTGATG | 0.498 |
| N26 | B | GAGGCAGCACTTCACACGATGCCTCCTACCCTGCTGCCTTAGCCGGTCTGCGTAATGACTGTAGTGATG | NB |
| N16 | C | GAGGCAGCACTTCACACGATATTTGGGGATCAGCCAGGTCAGTGCCACTGCGTAATGACTGTAGTGATG | 0.729 |
| N50 | D | GAGGCAGCACTTCACACGATCGTCCCTGCCCCTGCCTCCTTTCTATGCTGCGTAATGACTGTAGTGATG | 2.57 |
| N29 | E | GAGGCAGCACTTCACACGATCGCTGAAGTCAACCTCCCCTACCTGTGCTGCGTAATGACTGTAGTGATG | 2.2 |
| N59 | F | GAGGCAGCACTTCACACGATCCACCAGGCCAAACACGACCCCAAACACTGCGTAATGACTGTAGTGATG | 0.17 |
| N63 | G | GAGGCAGCACTTCACACGATTCCCTCCTCCTTTATATCCGGTCCGATCTGCGTAATGACTGTAGTGATG | 0.9 |
| N72 | H | GAGGCAGCACTTCACACGATCCCCGTTCCTTGTCCCCTTCTCTATATCTGCGTAATGACTGTAGTGATG | NB |
| N41 | I | GAGGCAGCACTTCACACGATCCCCCCCACACTCTTCCAACCCCCTCTCTGCGTAATGACTGTAGTGATG | 1890 |
| N8 | J | GAGGCAGCACTTCACACGATCACGGCAGGAGACCATCACCATTATCGCTGCGTAATGACTGTAGTGATG | NB |
Optimization of aptamer N59
Empirically speaking, the sequence with the highest affinity was chosen for further research.23 The results indicated N59 exhibited the highest affinity to DTX-1 of 0.17 μM, so N59 was chosen. First of all, we truncated the immobilized primer region of N59, and reserved the random sequence (N59a) to verify the influence of the primer region of the aptamer. Considering that the primer sequences were also likely to participate in the binding of the aptamer to the target, we predicted the secondary structures of N59 using the mfold Web Server, which was shown in Fig. S4a.† At the present stage, the special secondary structure such like stem-loop, bulge, pseudoknot and three-way helix structure, might mainly lead to the binding affinity.43 Based on this, we further cut off the stem loop A or B of N59 based on the secondary structure to obtain variants N59b, N59c.Upon examination, we found that when the immobilized primer region was truncated, the affinity of aptamer N59a had been significantly improved.44 This situation did not appear on the N59b or N59c, whose affinity had no significant difference with N59 (Table S3†). This result indicated that the primer region did not take part in the binding of the aptamer to the target, nor hindered the combination. However, the stem-loop A and B did not seem to have a crucial influence on the combination.39
The secondary structure analysis was also performed on N59a by the mfold Web Server, and the result was shown in Fig. S4b.† Based on this result, we truncated the stem loop C and got aptamer N59a1, which lost its binding ability to DTX-1. We believe that the structure of the stem-loop C in N59a might form a unique spatial structure, which was the key part of the aptamer that binds to DTX-1, while the remaining basic group improved the affinity of DTX-1 and aptamer.45
Identification of affinity and specificity of aptamer N59a
We used GTX, STX, NOD-R, PTX, OA (1 μM) as non-specific targets to analyse the association and dissociation process of DTX-1 (1 μM) (Fig. 2). GTX and STX are alkaloids.46,47 NOD-R is a peptide toxoid.48 PTX and DTX-1 are polyethers.49 OA is the homologue of DTX-1, which belongs to DSTs.50 In addition, random sequences acted as aptamer controls, blank samples containing running buffer were used as a reference.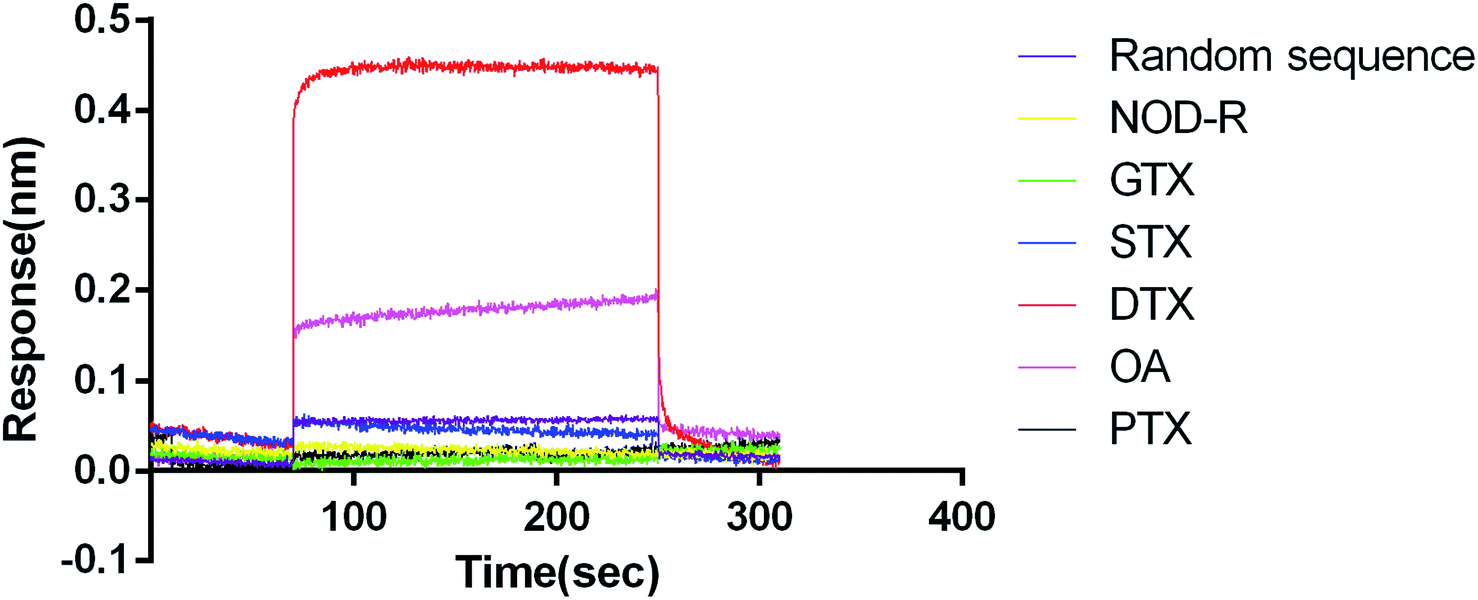 | ||
| Fig. 2 Characterization of affinity and specificity of aptamer N59a for DTX-1. The red line represents the interaction curve of aptamer N59a with DTX-1. The purple line represents the interaction curve of a random sequence with DTX-1. The yellow, green, blue, pink, and black lines represent the interaction curves of aptamer N59a with NOD-R, GTX, STX, OA and PTX, respectively. | ||
The results revealed that DTX-1 interacted with N59a with a KON (1/Ms) value of 5.39 × 107, a KDIS (1/s) value of 3.45 × 100, and a KD (M) value of 6.40 × 10−8. Furthermore, the random sequence showed no binding to DTX-1, might due to the fact that DTX-1 specifically bound only to N59a. At the same time, GTX, STX, NOD-R and PTX did not cause any reaction. However, OA interacted with N59a with a KD (M) value of 3.05 × 10−5, which has binding efficiency was much lower than that of DTX-1 and N59a. That is because the structures of OA and DTX-1 are same expect for one methyl.50 On the other hand, we did not introduce OA as counter-target during the selection. Therefore, there must be some cross-reaction between DTX-1 and OA, but such a huge difference in affinity can also indicate that the association of N59a aptamer to DTX-1 is specific.
Research on the core structure of aptamer N59a
Observing these above-described aptamers from selection, we noticed that all of them contained four pairs of cytosine bases. That indicated that N59a could form a unique spatial structure based on this unusual arrangement mode.51 G-quadruplexes scoring of the N59af (the reverse complement of N59) revealed that N59af had the potential to form several kinds of G-quadruplexes structures, and N59a itself had the potential to form an i-motif fold structure by itself theoretically.52,53 To verify these, we disrupted the arrangement mode of the bases. Therefore, N59a was truncated to obtain N59a2, N59a3, N59a4, N59a5, N59a6, N59a7 (Table S4†). Among them N59a5, N59a6 and N59a7 had already completely lost the potential to form into i-motif fold structure.It turned out that among these six aptamers, only N59a5, N59a6 and N59a7 did not have the ability to binding to DTX-1, which was in line with our inference. Therefore, we believe that the four pairs of cytosine bases in the sequence may be the basic skeleton for the formation of i-motif fold, which is also a key part of the binding of aptamers to DTX-1. At the same time, the affinity of N59a was much higher than that of N59c, indicating that the bases on both sides was able to accelerate the induction of binding of aptamers to DTX-1.
Analysis of the conformation of N59a
We investigated the conformational change of aptamer N59a in selection buffer (pH 7.5) by circular dichroism (APL, Chiascan). As shown in the Fig. 3A, the CD spectrum of N59a without DTX-1 is similar to the characteristic CD spectrum of the i-motif structure under neutral conditions,54 which might be due to that N59a forms an i-motif structure in this buffer. After the addition of DTX-1, the peak value of the CD spectrum was changed significantly from statistical point of view. The reason for this change is that the existence of DTX-1 induces a structural change of N59a, which also demonstrates that N59a can bind to DTX-1 specifically. We speculated the model diagram of N59a–DTX-1 complex according to the results of molecular calculation and simulation (Fig. 3B).55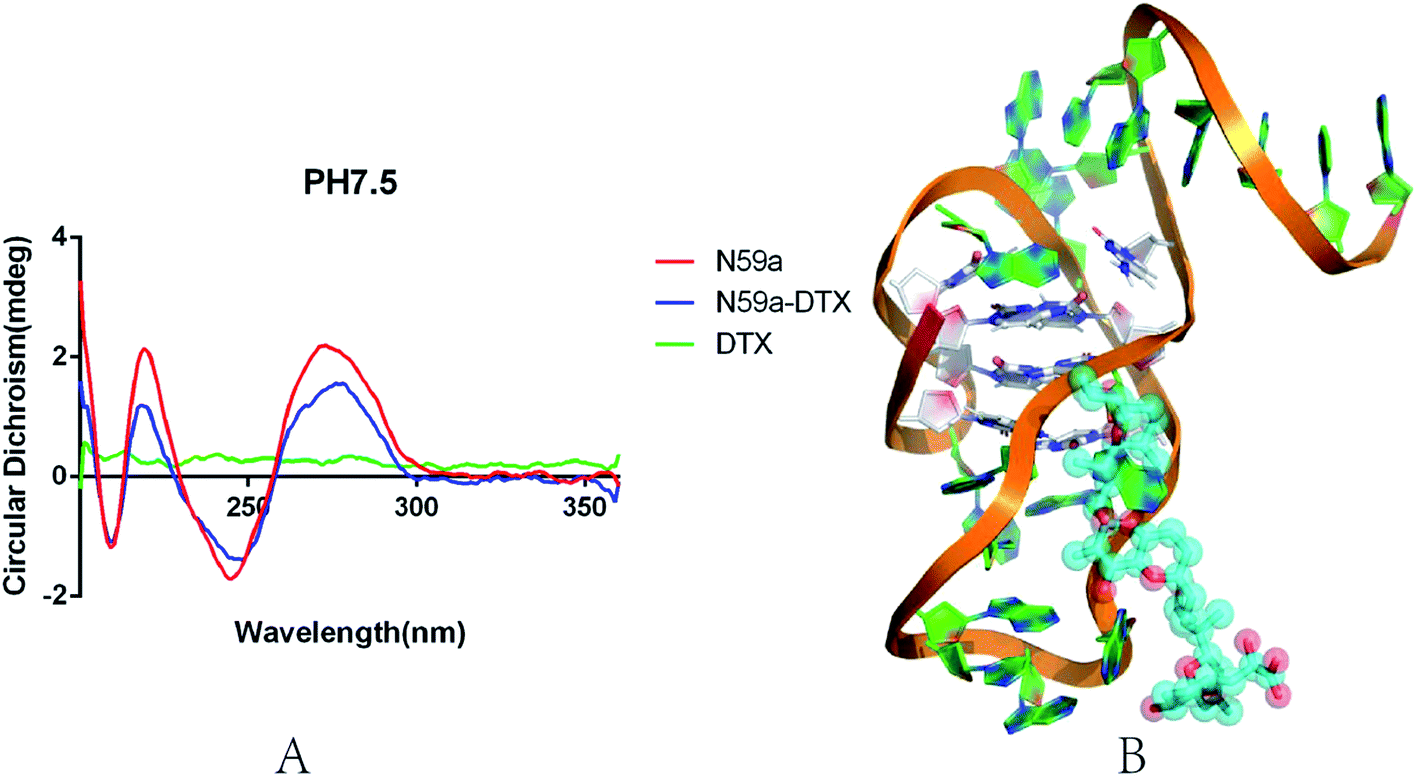 | ||
| Fig. 3 (A) The CD spectra of the DTX-1 and aptamer binding in binding buffer. The green line represents the CD spectra of the DTX-1. The red line represents the CD spectra of the N59a. The blue line represents the CD spectra of the DTX-1 and aptamer. The background of these samples is binding buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 2 mM MgCl2). (B) A preliminary estimate of the three-dimensional view of the interaction between N59a and DTX-1. | ||
BLI aptasensor for DTX-1 detection
In view of the current deficiency of DTX-1 detection, we established a an optical BLI sensor. Its key structure was based on aptamer N59a. BLI is an emerging sensor platform with great potential.56 It can monitor the surface property of the sensor, when the immobilized ligand is combined with the target molecule by biolayer interferometry technology in real time.57 At this stage, there have been reports of the BLI sensors used on small molecule detection.38,50We performed a series of measurements on the newly developed BLI sensor in the concentration range of 40–600 nM to evaluate the feasibility and stability of the sensor (Fig. 4A). As the concentration of DTX-1 increases, the optical thickness and mass density of the biofilm surface gradually increase. The wavelength shifts more obviously, eventually leading to an increase in response (Fig. 4A and B). The experiment was repeated several times for each sample. A calibration curve was obtained with DTX-1 concentration of BLI response 300 s. Fitting the curve to a sigmoidal logistic four-parameter equation: y = (Rmax − Rmin)/[(1 + (x/EC50)b)] + Rmin. Here, Rmax and Rmin are the maximal and minimal response. EC50 is the DTX-1 concentration leading to 50% of the maximum response. b is the correction factor. After repeated trials, we obtained the equation: y = (0.3842 − 0.00728)/[1 + (x/89.66)(−1.564)] + 0.00728. The correlation coefficient R2 was 0.9934 (Fig. 3B). In the concentration range of 60–200 nM (Fig. 3C), the biosensor displayed a good linear detection, which could be described by the liner regression equation: y = 0.0011x + 0.0928. The correlation coefficient R2 of this liner equation was 0.9988. The calculation of limit of detection (LOD, 614 pM) in accordance with the International Union of Pure and Applied Chemistry (IUPAC). Its noise level is the standard deviation of multiple measurements on blank samples (n = 20). Reports on the OA aptasensor at this stage indicated that it had high specificity for OA, while it is unable to detect DTX-1.57
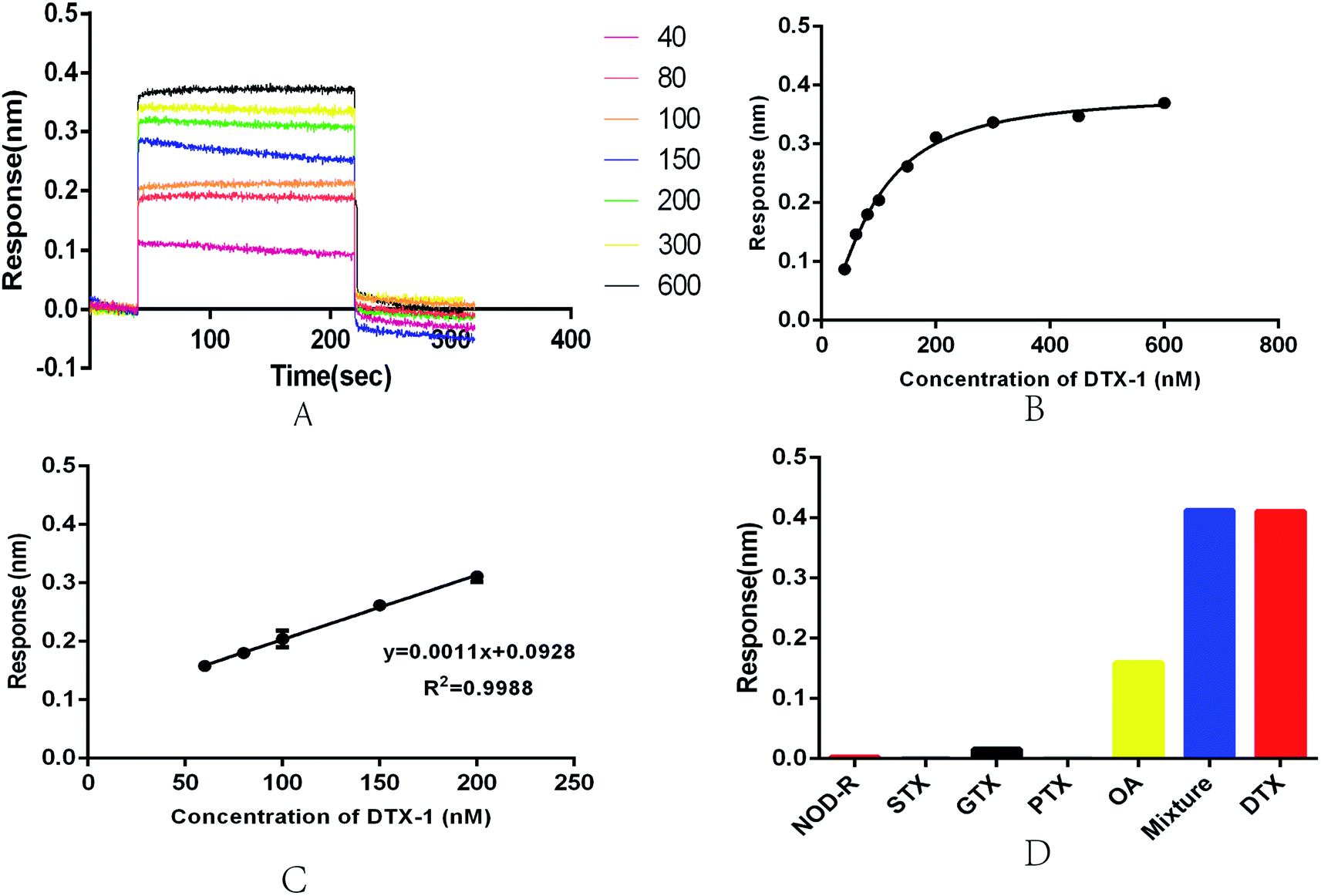 | ||
| Fig. 4 (A) Response plot for the aptamer-based BLI biosensor after addition of DTX-1 at different concentrations (40–600 nM). (B) The calibration curve for DTX-1 from 40 to 600 nM, a plot of the response as a function of the DTX-1 concentration. The error bars represent standard deviations. (C) The linear range of the calibration curve for DTX-1, a plot of the response as a function of the NOD-R concentration from 60 to 200 nM. (D) Specificity of the aptasensor with NOD-R, STX, GTX, PTX, and OA (each at 11 μM), and the mixture (including DTX-1, GTX, STX, NOD-R, PTX and OA, all toxin concentrations was 1 μM), respectively. | ||
Repeatability and specificity are essential for the aptasensor. In order to test the repeatability of the aptasensor, we executed repeated measurements on the 100 nM DTX-1 solution and collected the response. The coefficient of variation (CV) was 2.59%, indicating that the sensor had good repeatability. We designed cross-reactivity experiments though 1 μM GTX, STX, NOD-R, PTX, OA. The results were shown in Fig. 3D, DTX-1 caused a response of 0.411 nm, and OA caused a response of 0.16 nm. The other toxin samples caused responses of less than 0.01 nm. Because the aptamer has an affinity for OA, it will definitely cause a response. Meanwhile, the response of toxin mixtures (including DTX-1, GTX, STX, NOD-R, PTX and OA, all toxin concentrations was 1 μM, respectively) was 0.413 nm, indicating that the response was only caused by DTX-1. Even in the presence of OA, DTX-1 competitively inhibits the association of aptamers to OA, so the sensor can be specifically detected DTX-1 even in an environment with different kinds of toxins. The marine environment is complex and variable, so it is crucial to accurately detect DTX in a variety of toxin mixtures.38
Detection of DTX-1 in seawater
To assess the applied value of the newly developed BLI aptasensor in practical samples, we investigated the feasibility of detecting different concentrations of DTX-1 in seawater samples. Seawater samples (pH 7.8) including the different concentration of DTX-1 (80 nM, 100 nM, 200 nM) were detected, respectively. As shown in Table 2, a recovery percentage of 100.5 to 106.9% is obtained. In the meantime, the response value is also in line with expectations, indicating that the seawater does not significantly interfere with the detection of the aptasensor. In summary, we can conclude that the aptasensor has the potential for detection of DTX-1 in practical samples.| Seawater | DTX-1 (nM) | Recovery (%) | CV (%) |
|---|---|---|---|
| 1 | 80 | 100.5 | 4.7 |
| 2 | 100 | 106.9 | 5.2 |
| 3 | 200 | 101.0 | 1.2 |
Comparison with existing biosensors of DSP detection
We compared the existing reported biosensors for DTXs detection (Table 3) and found that three of the biosensors for detecting OA were cross-reactive with DTX-1. However, there have no reports of biosensors with high specificity for DTX-1 at this stage. Among the method capable of detecting DTX-1, this aptasensor has a lower detection line and a wider detection range, which can achieve the requirements of the national standard of DTX-1. Compared with the conventional detection method, the aptasensor has its unique merits. First of all, the nucleic acid and the probe used in the method are very stable and inexpensive, which can be conveniently used in large-scale transportation and preservation.27 Secondly, the method is efficient. The results can be obtained within half an hour. Next, its fixed-running program reduces labour costs, and aptasensor can be reused after regenerating with washing buffer for approximately 180 s; last but not least, the sensor is of high specificity and repeatability. However, none of the technologies are perfect. For this BLI aptasensor, the disadvantage is that the detection environment is slightly harsh. Therefore, some complex seawater samples may require pre-treatment which requires our further research and improvement. Therefore, we have no reason to query that this aptasensor has great potential in the detection of marine toxins.25,58| Analytical techniques | Linear range | LOD | Cross-reactivity of DTX-1a | Reference |
|---|---|---|---|---|
| a NM: not mention. | ||||
| Electrochemical | 100 pg mL−1 to 60 ng mL−1 | 70 pg mL−1 | No | 59 |
| Fluorometric | 0.5–250 ng mL−1 | 39 pg mL−1 | 70% | 60 |
| Immunoassay | 0.025–10 ng mL−1 | 10 pg mL−1 | No | 61 |
| Electrochemical | 10–250 nM | 8 pM | NM | 62 |
| Electrophysiology | — | 7.16 ng mL−1 | NM | 63 |
| Electrophysiology | 10–100 μg L−1 | 10 μg L−1 | NM | 64 |
| Immunoassay | 11.2–38.8 ng mL−1 | 1 ng mL−1 | 73% | 65 |
| Immunoassay | — | 20 ng g−1 | 40% | 66 |
Conclusions
In summary, this work is the first report of successful selection, optimization and identification of DNA aptamers that bind with high affinity and specificity to DTX-1. Meanwhile, we truncated DTX-1 aptamer and obtained the aptamer core sequence with a higher KD of 64 nM (R2 = 0.9832, X2 = 0.0082). We have conducted a preliminary investigation into the complicated structure formed by N59a and its binding mechanism of DTX-1. However, more evidences need to be further studied.We also constructed a BLI aptasensor for detection of DTX-1 with a detection limit as low as 614 pM, which demonstrated good linear response between 60 and 200 nM DTX-1. The high affinity and stability of the BLI aptasensor proves it may offer an alternative to traditional analytical methods for the rapid and sensitive detection of DTX-1.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported by the China National Major Scientific and Technological Special Project for Significant New Drugs Development (No. 2018ZX09J18112) and the National Natural Science Foundation of China (No. 41706156).Notes and references
- T. Yasumoto, Y. Oshima and M. Yamaguchi, Nippon Suisan Gakkaishi, 1978, 44, 1249–1255 CrossRef
.
- B. Reguera, P. Riobo, F. Rodriguez, P. A. Diaz, G. Pizarro, B. Paz, J. M. Franco and J. Blanco, Mar. Drugs, 2014, 12, 394–461 CrossRef PubMed
- P. Abal, M. C. Louzao, J. M. Cifuentes, N. Vilarino, I. Rodriguez, A. Alfonso, M. R. Vieytes and L. M. Botana, Food Chem. Toxicol., 2017, 102, 166–175 CrossRef CAS PubMed
- T. Chen, X. Xu, J. Wei, J. Chen, R. Miu, L. Huang, X. Zhou, Y. Fu, R. Yan, Z. Wang, B. Liu and F. He, PLoS One, 2013, 8, e65049 CrossRef PubMed
- B. Reguera and J. Blanco, Toxins, 2019, 11, 413 CrossRef PubMed
- X. Z. Wang, Y. Cheng, N. Li, H. M. Wen, R. Liu, C. X. Shan, C. Chai and H. Wu, Toxins, 2016, 8, 8 CrossRef PubMed
- S. L. Hinder, G. C. Hays, C. J. Brooks, A. P. Davies, M. Edwards, A. W. Walne and M. B. Gravenor, Environ. Health, 2011, 10, 54 CrossRef PubMed
- C. Bialojan and A. Takai, Biochem. J., 1988, 256, 283–290 CrossRef CAS PubMed
- A. Takai, M. Murata, K. Torigoe, M. Isobe, G. Mieskes and T. Yasumoto, Biochem. J., 1992, 284(Pt 2), 539–544 CrossRef CAS PubMed
- H. Fujiki, M. Suganuma, H. Suguri, S. Yoshizawa, K. Takagi, N. Uda, K. Wakamatsu, K. Yamada, M. Murata and T. Yasumoto, et al., Jpn. J. Cancer Res., 1988, 79, 1089–1093 CrossRef CAS PubMed
- K. Terao, E. Ito, T. Yanagi and T. Yasumoto, Toxicon, 1986, 24, 1141–1151 CrossRef CAS PubMed
- M. J. Twiner, G. J. Doucette, Y. Pang, C. Fang, C. J. Forsyth and C. O. Miles, Mar. Drugs, 2016, 14, 207 CrossRef PubMed
- M. C. Louzao, M. R. Vieytes and L. M. Botana, Mini-Rev. Med. Chem., 2005, 5, 207–215 CrossRef CAS PubMed
- H. Suzuki and Y. Okada, J. Vet. Med. Sci., 2018, 80, 616–619 CrossRef CAS PubMed
- D. A. Fernandez, M. C. Louzao, M. Fraga, N. Vilarino, M. R. Vieytes and L. M. Botana, Toxins, 2014, 6, 211–228 CrossRef CAS PubMed
- M. Del Campo, T. Y. Zhong, R. Tampe, L. Garcia and N. Lagos, Toxicon, 2017, 126, 23–31 CrossRef CAS PubMed
- P. J. Ferron, K. Hogeveen, V. Fessard and L. Le Hegarat, Mar. Drugs, 2014, 12, 4616–4634 CrossRef PubMed
- L. T. Nielsen, P. J. Hansen, B. Krock and B. Vismann, Toxicon, 2016, 117, 84–93 CrossRef CAS PubMed
- EFSA Panel on Contaminants in the Food Chain, EFSA J., 2008, 589, 1–62 Search PubMed
- L. L. Fu, X. Y. Zhao, L. D. Ji and J. Xu, Toxicon, 2019, 160, 1–7 CrossRef CAS PubMed
- L. M. Botana, A. Alfonso, I. Rodriguez, A. M. Botana, C. Louzao Mdel and M. R. Vieytes, Toxins, 2016, 8, 208 CrossRef PubMed
- R. Della Loggia, S. Sosa and A. Tubaro, Nat. Toxins, 1999, 7, 387–391 CrossRef CAS PubMed
- T. Wang, C. Chen, L. M. Larcher, R. A. Barrero and R. N. Veedu, Biotechnol. Adv., 2019, 37, 28–50 CrossRef CAS PubMed
- C. Tuerk and L. Gold, Science, 1990, 249, 505–510 CrossRef CAS PubMed
- Y. Gao, Z. Zhu, X. Xi, T. Cao, W. Wen, X. Zhang and S. Wang, Biosens. Bioelectron., 2019, 133, 177–182 CrossRef CAS PubMed
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346, 818–822 CrossRef CAS PubMed
- M. R. Dunn, R. M. Jimenez and J. C. Chaput, Nat. Rev. Chem., 2017, 1, 0076 CrossRef CAS
- K. Groff, J. Brown and A. J. Clippinger, Biotechnol. Adv., 2015, 33, 1787–1798 CrossRef CAS PubMed
- T. Wang, M. P. Gantier, D. Xiang, A. G. Bean, M. Bruce, S. F. Zhou, M. Khasraw, A. Ward, L. Wang, M. Q. Wei, H. AlShamaileh, L. Chen, X. She, J. Lin, L. Kong, S. Shigdar and W. Duan, Theranostics, 2015, 5, 1456–1472 CrossRef CAS PubMed
- S. Amaya-Gonzalez, N. de-los-Santos-Alvarez, A. J. Miranda-Ordieres and M. J. Lobo-Castanon, Sensors, 2013, 13, 16292–16311 CrossRef PubMed
- M. Jia, Y. Mao, C. Wu, S. Wang and H. Zhang, Anal. Chim. Acta, 2019, 1082, 136–145 CrossRef CAS PubMed
- K. D. Kovacevic, J. C. Gilbert and B. Jilma, Adv. Drug Delivery Rev., 2018, 134, 36–50 CrossRef CAS PubMed
- S. C. Gopinath, T. Lakshmipriya, Y. Chen, W. M. Phang and U. Hashim, Biotechnol. Adv., 2016, 34, 198–208 CrossRef CAS PubMed
- M. Roushani, Z. Rahmati, S. J. Hoseini and R. Hashemi Fath, Colloids Surf., B, 2019, 183, 110451 CrossRef PubMed
- F. Karimi and S. Dabbagh, Int. J. Biol. Macromol., 2019, 140, 842–850 CrossRef CAS PubMed
- S. Diaz-Amaya, L. K. Lin, A. J. Deering and L. A. Stanciu, Anal. Chim. Acta, 2019, 1081, 146–156 CrossRef CAS PubMed
- D. Sun, X. Lin, J. Lu, P. Wei, Z. Luo, X. Lu, Z. Chen and L. Zhang, Biosens. Bioelectron., 2019, 142, 111578 CrossRef PubMed
- S. Gao, B. Hu, X. Zheng, Y. Cao, D. Liu, M. Sun, B. Jiao and L. Wang, Biosens. Bioelectron., 2016, 79, 938–944 CrossRef CAS PubMed
- J. Concepcion, K. Witte, C. Wartchow, S. Choo, D. Yao, H. Persson, J. Wei, P. Li, B. Heidecker, W. Ma, R. Varma, L. S. Zhao, D. Perillat, G. Carricato, M. Recknor, K. Du, H. Ho, T. Ellis, J. Gamez, M. Howes, J. Phi-Wilson, S. Lockard, R. Zuk and H. Tan, Comb. Chem. High Throughput Screening, 2009, 12, 791–800 CrossRef CAS PubMed
- J. Macdonald, P. Houghton, D. Xiang, W. Duan and S. Shigdar, Nucleic Acid Ther., 2016, 26, 348–354 CrossRef CAS PubMed
- X. He, L. Guo, J. He, H. Xu and J. Xie, Anal. Chem., 2017, 89, 6559–6566 CrossRef CAS PubMed
- R. Matsushima, H. Uchida, S. Nagai, R. Watanabe, M. Kamio, H. Nagai, M. Kaneniwa and T. Suzuki, Toxins, 2015, 7, 5141–5154 CrossRef CAS PubMed
- S. Sedghi Masoud and K. Nagasawa, Chem. Pharm. Bull., 2018, 66, 1091–1103 CrossRef PubMed
- H. Abou Assi, M. Garavis, C. Gonzalez and M. J. Damha, Nucleic Acids Res., 2018, 46, 8038–8056 CrossRef PubMed
- T. A. Brooks, S. Kendrick and L. Hurley, FEBS J., 2010, 277, 3459–3469 CrossRef CAS PubMed
- J. Kypr, I. Kejnovska, D. Renciuk and M. Vorlickova, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed
- A. D. Protopopova, V. B. Tsvetkov, A. M. Varizhuk, N. A. Barinov, V. V. Podgorsky, D. V. Klinov and G. E. Pozmogova, Phys. Chem. Chem. Phys., 2018, 20, 3543–3553 RSC
- D. Verzijl, T. Riedl, P. Parren and A. F. Gerritsen, Biosens. Bioelectron., 2017, 87, 388–395 CrossRef CAS PubMed
- M. N. Kammer, I. R. Olmsted, A. K. Kussrow, M. J. Morris, G. W. Jackson and D. J. Bornhop, Analyst, 2014, 139, 5879–5884 RSC
- S. Ouyang, B. Hu, R. Zhou, D. Liu, D. Peng, Z. Li, Z. Li, B. Jiao and L. Wang, Analyst, 2018, 143, 4316–4322 RSC
- S. Eissa, A. Ng, M. Siaj, A. C. Tavares and M. Zourob, Anal. Chem., 2013, 85, 11794–11801 CrossRef CAS PubMed
- R. Chinnappan, R. AlZabn, T. A. Mir, M. Bader and M. Zourob, Mikrochim. Acta, 2019, 186, 406 CrossRef PubMed
- H. Gu, N. Duan, S. Wu, L. Hao, Y. Xia, X. Ma and Z. Wang, Sci. Rep., 2016, 6, 21665 CrossRef CAS PubMed
- S. Ramalingam, R. Chand, C. B. Singh and A. Singh, Biosens. Bioelectron., 2019, 135, 14–21 CrossRef CAS PubMed
- H. Li, X. Wei, C. Gu, K. Su, H. Wan, N. Hu and P. Wang, Anal. Sci., 2018, 34, 893–900 CrossRef CAS PubMed
- X. Zhang, J. Fang, L. Zou, Y. Zou, L. Lang, F. Gao, N. Hu and P. Wang, Biosens. Bioelectron., 2016, 77, 573–579 CrossRef CAS PubMed
- M. Le Berre, M. Kilcoyne and M. Kane, Toxicon, 2015, 103, 169–175 CrossRef CAS PubMed
- N. M. Llamas, L. Stewart, T. Fodey, H. C. Higgins, M. L. Velasco, L. M. Botana and C. T. Elliott, Anal. Bioanal. Chem., 2007, 389, 581–587 CrossRef CAS PubMed
- M. Darmostuk, S. Rimpelova, H. Gbelcova and T. Ruml, Biotechnol. Adv., 2015, 33, 1141–1161 CrossRef CAS PubMed
- A. Sultana and J. E. Lee, Curr. Protoc. Protein Sci., 2015, 79, 19.25.1–19.25.26 Search PubMed
- P. Nadal, M. Svobodova, T. Mairal and C. K. O'Sullivan, Anal. Bioanal. Chem., 2013, 405, 9343–9349 CrossRef CAS PubMed
- X. Luo, M. McKeague, S. Pitre, M. Dumontier, J. Green, A. Golshani, M. C. Derosa and F. Dehne, RNA, 2010, 16, 2252–2262 CrossRef CAS PubMed
- J. V. Mulcahy, J. R. Walker, J. E. Merit, A. Whitehead and J. Du Bois, J. Am. Chem. Soc., 2016, 138, 5994–6001 CrossRef CAS PubMed
- A. P. Thottumkara, W. H. Parsons and J. Du Bois, Angew. Chem., Int. Ed. Engl., 2014, 53, 5760–5784 CrossRef CAS PubMed
- S. Akter, M. Vehniainen, H. T. Kankaanpaa and U. Lamminmaki, Microorganisms, 2017, 5, 58 CrossRef PubMed
- A. Verma, G. S. Kohli, D. T. Harwood, P. J. Ralph and S. A. Murray, Environ. Microbiol., 2019, 21, 4196–4211 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra10600f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |